

Abstract
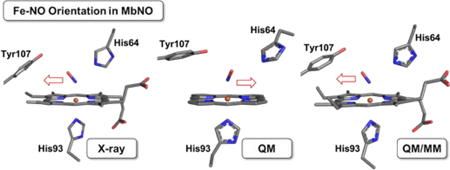
The globular dioxygen-binding heme protein myoglobin (Mb) is present in several species. Its interactions with the simple nitrogen oxides, namely nitric oxide (NO) and nitrite, have been known for decades, but the physiological relevance has only recently become more fully appreciated. We previously reported the O-nitrito binding mode of nitrite to ferric horse heart wild-type (wt) MbIII and human hemoglobin. We have expanded on this work and report the interactions of nitrite with wt sperm whale (sw) MbIII and its H64A, H64Q and V68A/I107Y mutants whose dissociation constants increase in the order H64Q < wt < V68A/I107Y < H64A. We also report their X-ray crystal structures that reveal the O-nitrito binding mode of nitrite to these derivatives. The MbII-mediated reductions of nitrite to NO and structural data for the wt and mutant MbII–NOs are described. We show that their FeNO orientations vary with distal pocket identity, with the FeNO moieties pointing towards the hydrophobic interiors when the His64 residue is present, but pointing towards the hydrophilic exterior in the absence of this His64 residue. This correlates with the nature of H-bonding to the bound NO ligand (nitrosyl O vs. N atom). Quantum mechanics and hybrid quantum mechanics/molecular mechanics calculations help elucidate the origin of the experimentally preferred NO orientations. In a few cases, the calculations reproduce the experimentally observed orientations only when the whole protein is taken into consideration.
Table of Contents Graphic
INTRODUCTION
Since its first mention as the red protein “myochrome” by Mörner in 1897,1 the globular heme protein myoglobin (Mb) has continued to intrigue researchers across a broad range of fields.2 The interaction of Mb with the simple nitrogen oxides (NOx; x = 1, 2), including the use of nitrite (NO2-) in the meat curing industry, has a long history.3–5 The now widely-recognized vasodilatory property of nitrite continues to attract intense interest, especially in its interactions with heme proteins.6–8
Nitrite is an intermediate in denitrification,9–13 and its enzymatic reduction to NO (eq. 1) is of direct relevance to the global N-cycle.14, 15 Much of nitrite biology involves its interactions with metals,15, 16 necessitating consideration of its coordination chemistry.
| (1) |
Three main binding modes of nitrite to metals have been structurally characterized (Figure 1),17 although only the first two (N-nitro and O-nitrito) have been determined for heme proteins and heme models to date.
Figure 1.
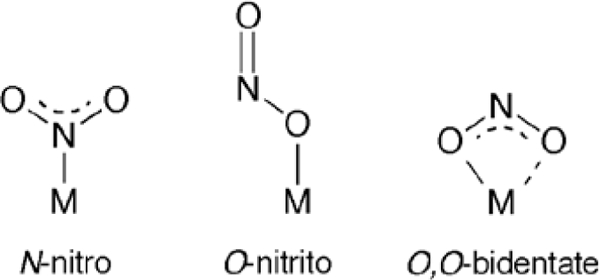
The three common binding modes for nitrite to metals.
The N-nitro binding mode remains the most common binding mode of nitrite to heme Fe centers,18–31 despite the differences in proximal ligation to heme (e.g., His for cyt cd1 NiR, Cys for SiR, and Lys for cyt c NiR), macrocycle saturation (e.g., d vs c heme), and distal pocket amino acid identities. The first suggestions of a possible O-nitrito binding mode to a heme protein was that for ferric HbIII–ONO by Hartridge in 192032 and Barnard in 1937,33 although this binding mode was not structurally confirmed until almost a century later with our reports on the structures of the nitrite adducts of horse heart (hh) MbIII and derivatives,34–36 and ferric human hemoglobin (HbIII).37, 38
Support for low energy differences between the N-nitro and O-nitrito binding modes in heme proteins and their truncated active site models comes from density functional theory (DFT) calculations that predict the N-nitro mode to be lower in energy by a few kcals/mol.19, 31, 39–46 A recent detailed quantum chemical analysis of EPR spectral data for MbIII–nitrite,47 however, supports the O-nitrito binding mode to the metal center as observed experimentally. These results suggest that the Mb (and Hb) distal pocket environments with the single His64 H-bonding residue in these proteins are somewhat unique in their interactions with nitrite. Indeed, we reported that although the hh wt MbIII–nitrite complex displays the O-nitrito binding mode, its hh H64V mutant displayed a long Fe–N distance of ~2.6 Å suggestive of an electrostatic interaction with nitrite (~65% nitrite occupancy) in an apparent N-nitro form (Figure 2, left).48 The recently obtained structure of the sperm whale (sw) MbIII–nitrite analog (Figure 2, right) displays a more defined N-nitro mode (Fe–N = 2.1 Å) illustrating the importance of the His64 ligand (or lack thereof) in this nitrite orientation.
Figure 2.
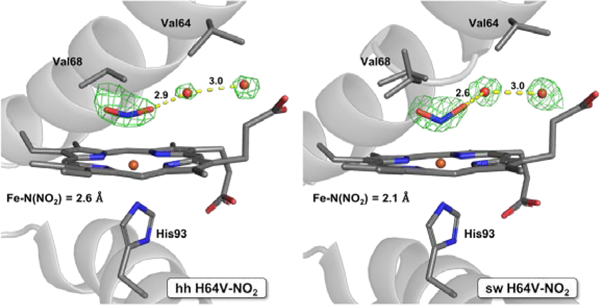
Models and Fo–Fc omit maps showing nitrite in the distal pockets of the H64V mutants of ferric hh MbIII–nitrite (left; PDB ID: 3HEP) and ferric sw MbIII–nitrite (right; PDB ID: 6CF0).
The reduction of nitrite to NO in denitrification as part of the global N-cycle proceeds via the reaction shown in eq. 2 that is catalyzed by the NiR enzymes.9, 10, 12, 16 Haldane first
| (2) |
suggested in 1901, based on his studies on salted meat using visible spectroscopy, that ferrous HbII reduced nitrite to the nitrosyl derivative HbII–NO,5 and this was later elaborated by Brooks in 1937.49 Interestingly, ferrous MbIIs from several species have now been shown to participate in nitrite reduction to generate NO (eqs. 3–4).3, 50 Other metalloproteins such as Hb,51 endothelial NO synthase,52, 53 xanthine oxidase,54 neuroglobin,55, 56 cytoglobin,57–59 and some plant Hbs60 similarly reduce nitrite to NO.
| (3) |
| (4) |
It is somewhat surprising that although MbNO and HbNO have biological relevance, not much is currently known about the FeNO conformational preferences in these proteins as a function of varied distal pocket H-bonding environments. Indeed, only a small handful of MbII–NO and HbII–NO crystal structures have been reported to date.61,34, 62,63,64, 65 It is important to note that the NO vectors point towards the hydrophobic interiors in all these MbII–NO and HbII–NO structures reported to date.66 Given that Mbs from several species reduce nitrite to generate their MbII–NO derivatives, we were interested in determining the effects of distal pocket mutations on both the binding modes of the parent Mb-nitrite adducts and the orientations of the Fe–NO moieties in their MbII–NO derivatives. In particular, we note that although distal pocket H-bonding to the nitrosyl N-atom has been demonstrated in MbII–NO, the corresponding (sole) H-bonding to the nitrosyl O-atom has not yet been reported.
In this paper, we report the preparation and crystal structures of the nitrite and NO complexes of wt and mutant sw Mbs, with the goal of examining the nature of nitrite binding (N-nitro vs. O-nitrito) and determining the FeNO orientation preferences in these wt and mutant Mbs. We chose the mutants shown in Figure 3 that differ in H-bonding capacity to the bound NOx ligands (wt with 1 H-bonding residue; H64A with 0 H-bonding residues; H64Q with 1 H- bonding residue; V68A/I107Y with 2 H-bonding residues), which to the best our knowledge, is the first systematic structural investigation of different H-bonding capacities on NO orientations in the heme distal pocket, including some new patterns not reported previously. The H64Q mutant is an analog of the wt Mbs from certain species of worms,67, 68 elephants,69, 70 and sharks,71–73 and of the α chains of opossum Hbs.74 Alternatively, the H64A mutant is related to the Mbs that have hydrophobic residues (Val and Leu) at this E7 position.75–78 We hypothesized that the introduction of the potential second H-bond interaction in the V68A/I107Y double mutant would help stabilize the V-shaped N-nitro binding mode as observed in cyt cd1 NiRs. In addition to the experimental characterization of the MbIII–nitrite and MbII–NO products, we sought to probe the factors that influence the FeNO H-bonding and orientational preferences in the wt and mutant MbII–NOs by quantum mechanics (QM) and hybrid quantum mechanics and molecular mechanics (QM/MM) calculations. Importantly, such a combined experimental and computational study shows for the first time that the FeNO orientations vary as a function of distal pocket identity.
Figure 3.
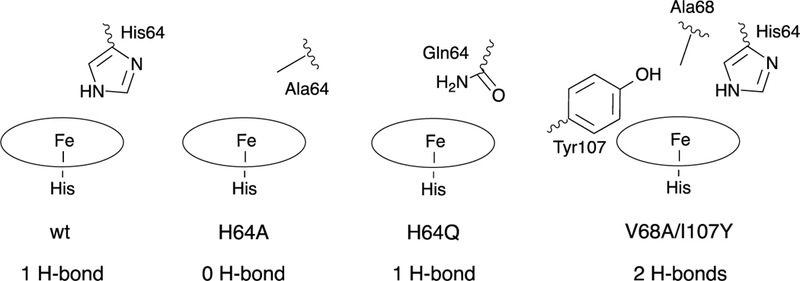
The wt and mutant Mbs used in this study.
MATERIALS AND METHODS
Expression, purification and crystallization of wild-type and mutant sperm whale myoglobins
The recombinant wt sw Mb plasmid was a kind gift from Dr. Mario Rivera (Univ. of Kansas); the recombinant wt protein contains a D122N mutation (due to an error in the former’s sequence determination)79 and a Met residue at the N-terminus. Mutagenesis was performed using the Quick-Change method (Strategene). For the double mutant V68A/I107Y, the V68A mutation was performed first followed by the I017 mutation. The wt, H64A, H64Q and V68A/I107Y mutants were expressed in E. coli BL21(DE3) and purified as described by Springer and Sligar.80 The mutant H64A and H64Q proteins had higher and more stable expression levels (~100 mg/mL) compared with those of the wt and double-mutant V68A/I107Y proteins (~10 mg/mL). Crystals of the recombinant ferric MbIII proteins were grown as described previously.81, 82
Reactions of the sw Mb proteins with NOx
Sodium nitrite, potassium ferricyanide, and sodium dithionite were purchased from Sigma-Aldrich and used as received.
UV-vis monitoring of the reactions:
A Hewlett Packard 8453 spectrophotometer was used to obtain the UV-vis spectra of the complexes to determine the reaction times and the stabilities of the complexes. The protein samples in 20 mM Tris, 1 mM EDTA, pH 7.4 were purified as stated above.80 UV-vis spectra were recorded for all the protein derivatives; 10–15 μL of protein (10–20 mg/mL) were added into 2.5 mL 0.1 M phosphate buffer at pH 7.4. Next, 5 μL of 1 M sodium nitrite in phosphate buffer were added to each of the protein samples to obtain the spectra of sw MbIII–nitrite adducts. To obtain the spectra of the sw MbII–NO derivatives, the MbIII samples were reduced by the addition of 5 μL of 1.0 M dithionite, followed by the addition of 5 μL of 1.0 M sodium nitrite into the respective cuvettes. The spectra were recorded at different time points.
Determination of nitrite reductase activity of ferrous MbII and nitrite binding constants of MbIII:
The Mb protein samples were fully oxidized by the addition of excess of potassium ferricyanide,83 and the mixtures passed through Sephadex G25 columns to remove the excess reagent. The protein fractions were concentrated to ~20 mg/mL. The measurement of nitrite reductase activity of ferrous MbIIs and the dissociation constant KD for Mbs followed published protocols.55, 84
Preparation of the crystalline derivatives:
Crystals of the Mb–NOx complexes were obtained by the soaking method using crystals of the respective MbIIIs obtained by the method of Phillips et al.79 Prior to reactions with the NOx, a suitably sized crystal of the ferric MbIII–H2O precursor was harvested for X-ray diffraction data collection and its crystal structure solved. The structures of the deoxy MbIIs without any ligands were solved using crystals derived from the dithionite reduction of the MbIII–H2O precursors. Specifically, appropriately sized MbIII–H2O crystals were transferred into a 4 μL droplet (well solution containing 100 mM Tris, 1 mM EDTA, pH 7.4, 2.6 M (NH4)2SO4, with 10% glycerol) on a cover slide. The cover slide with the drop was placed in a layer of light mineral oil to minimize the introduction of air. A few crystals of solid dithionite were added into the droplet, and the color changed from red to pink during the ensuing ~10 min period, signifying the formation of the deoxyMbII. The treated crystals were harvested using cryo-loops and frozen in liquid nitrogen prior to X-ray diffraction data collection.
The MbIII–nitrite complexes were obtained using a similar method as described above. A few solid crystalline particles of sodium nitrite were added into the droplet to react with the respective MbIII–H2O crystals for 20–30 min. The treated crystals were harvested using cryo-loops at different time periods and frozen in liquid nitrogen prior to X-ray diffraction data collection.
Crystals of the ferrous MbII–NO derivatives were obtained by soaking the respective sw MbIII–H2O crystals with nitrite for 20–30 min, followed by the addition of excess solid dithionite to reduce the MbIII–nitrite intermediate. In these reactions, a clear color change from brown to pink was observed. The treated crystals were harvested as above.
X-ray diffraction data collection, processing, structure solution, and refinement
The diffraction data were collected at home source using a Rigaku RU-H3R and/or MicroMax 007HF microfocus X-ray generator, coupled to a R-AXIS IV++ detector or a PILATUS 200K detector. The data were collected at 100 K with CuKα radiation (λ = 1.54178 Å) from the generator operated at 50 kV/100 mA (Rigaku RU-H3R generator) or 40 kV/30 mA (Rigaku MicroMax 007HF generator).
The data were processed using iMOSFLM85, 86 or HKL3000.87 The output mtz files from iMOSFLM were scaled by Aimless or Scala.88 When HKL3000 was used, the sca output files were transformed into mtz files by Scalepack2mtz as implemented in the CCP4 program suite.89
Initial phases were obtained by molecular replacement using PHASER (CCP4).90 The starting model used was wt sw MbIII–H2O at 1.5 Å resolution (PDB ID: 2MBW) with the heme, sulfates, and water molecules removed from the structure. Refinements were performed using either Refmac5 (CCP4) or Phenix.refine (PHENIX).91, 92 Models were rebuilt using COOT.93 The MolProbity server was used for structure validation.94 The details are described below for each of the structures.
The figures were generated using PYMOl.95 2Fo–Fc electron density maps were initially calculated by Fast Fourier Transform (FFT) in the CCP4 software package.96 The resulting map files were converted to ccp4 files and displayed in PYMOL. In general, each Fo–Fc electron density map was generated as follows: (i) a new PDB file in which ligands were removed from the pocket was initially refined for 5 cycles in Refmac5; (ii) the new mtz file that was generated from the first step was input into FFT to generate the map that was then displayed in PYMOL.
A note on previously published crystal structures:
Sixteen sw Mb crystal structures are reported in this paper. Five reference (MbIII, deoxyMbII) structures used in this work have been reported previously: wt MbIII–H2O,97 wt deoxyMbII,97 H64A MbIII–H2O (PDB ID: 102M),82 H64Q MbIII–H2O (PDB ID: 2MGH),79 and H64Q deoxyMbII (PDB ID: 2MGG).79 We reproduced these five structures for a more realistic comparison (see Supporting Information) with our new structures detailed below. Only one sw Mb–NOx structure has been reported to date, namely sw wt MbII–NO (PDB ID: 1HJT) prepared from gaseous NO addition (after sodium fluoride addition) to ferric MbIII.61 The new structures reported in this work are described below.
wt MbIII–nitrite:
Ten initial cycles of restrained refinement were run with Refmac, and the R factor decreased from 0.470 to 0.318. Ligands and water were added to the model depending on the Fo–Fc electron density map in the subsequent refinement cycles. Three sulfate anions, seven nitrite anions and one glycerol were added iteratively using COOT. Two conformations for each of the sidechains of Lys133 and Lys145 were modeled with 50% occupancy each. The final model was refined to 1.85 Å resolution with an R factor of 0.148 and Rfree of 0.187.
wt MbII–NO:
Ten initial cycles of restrained refinement were run with Refmac, and the R factor decreased from 0.444 to 0.307. Ligands and water were added to the model depending on the Fo–Fc electron density map in the subsequent refinement cycles. One sulfate anion, one nitrite anion, one nitric oxide and one glycerol molecule were added iteratively using COOT. The C-terminal residues Gln152 and Gly153 were omitted due to the lack of electron density. The final model was refined to 1.70 Å resolution with an R factor of 0.138 and Rfree of 0.191.
H64A deoxyMbII:
Ten initial cycles of restrained refinement were run with Refmac, and the R factor decreased from 0.372 to 0.255. Ligands and water were added to the model depending on the Fo–Fc electron density map in the subsequent refinement cycles. Six sulfate anions and one glycerol molecule were added iteratively using COOT. Two conformations for each of the sidechains of Leu115, Asn132 and Tyr151 were modeled with 50% occupancy each. The N-terminal Met residue was omitted due to the lack of electron density. The final model was refined to 1.78 Å resolution with an R factor of 0.148 and Rfree of 0.171.
H64A MbIII–nitrite:
Ten initial cycles of restrained refinement were run with Refmac, and the R factor decreased from 0.347 to 0.255. Ligands and water were added to the model depending on the Fo–Fc electron density map in the subsequent refinement cycles. Three sulfate anions, eight nitrite anions and one glycerol molecule were added iteratively using COOT. Two conformations for each of the sidechains of Asn122 were modeled with 50% occupancy each. The N-terminal Met residue was omitted due to the lack of electron density. The final model was refined to 1.85 Å resolution with an R factor of 0.148 and Rfree of 0.187.
H64A MbII–NO:
Ten initial cycles of restrained refinement were run with Refmac, and the R factor decreased from 0.334 to 0.245. Ligands and water were added to the model depending on the Fo–Fc electron density map in the subsequent refinement cycles. Three sulfate anions, seven nitrite anions, one nitric oxide and two glycerol molecules were added iteratively using COOT. Two conformations for each of the sidechains of His12, Asn122, and Lys133 were modeled with 50% occupancy each. The N-terminal Met residue was omitted due to the lack of electron density. The final model was refined to 1.78 Å resolution with an R factor of 0.149 and Rfree of 0.186.
H64Q MbIII–nitrite:
Ten initial cycles of restrained refinement were run with Refmac, and the R factor decreased from 0.294 to 0.222. Ligands and water were added to the model depending on the Fo–Fc electron density map in the subsequent refinement cycles. Two sulfate anions, ten nitrite anions and two glycerol molecules were added iteratively using COOT. Two conformations for each of the sidechains of Lys16, Val68 and Asn122 were modeled with 50% occupancy each. The N-terminal Met residue was omitted due to the lack of electron density. The final model was refined to 1.81 Å resolution with an R factor of 0.160 and Rfree of 0.201.
H64Q MbII–NO:
Ten initial cycles of restrained refinement were run with Refmac, and the R factor decreased from 0.317 to 0.240. Ligands and water were added to the model depending on the Fo–Fc electron density map in the subsequent refinement cycles. Three sulfate anions, seven nitrite anions, one nitric oxide and three glycerol molecules were added iteratively using COOT. Two conformations for each of the sidechains of Asn122 were modeled with 50% occupancy each. The N-terminal Met residue was omitted due to the lack of electron density. The final model was refined to 1.78 Å resolution with an R factor of 0.153 and Rfree of 0.193.
V68A/I107Y MbIII–H2O:
Ten initial cycles of restrained refinement were run with Refmac, and the R factor decreased from 0.371 to 0.240. Ligands and water were added to the model depending on the Fo–Fc electron density map in the subsequent refinement cycles. Three sulfate anions and one glycerol molecule were added iteratively using COOT. The C-terminal residues Gln152 and Gly153 were omitted due to the lack of electron density. The final model was refined to 1.78 Å resolution with an R factor of 0.160 and Rfree of 0.199.
V68A/I107Y deoxyMbII:
Ten initial cycles of restrained refinement were run with Refmac, and the R factor decreased from 0.384 to 0.244. Ligands and water were added to the model depending on the Fo–Fc electron density map in the subsequent refinement cycles. Two sulfate anions and two glycerol molecules were added iteratively using COOT. The N-terminal Met residue and the C-terminal residues Gln152 and Gly153 were omitted due to the lack of electron density. The final model was refined to 1.80 Å resolution with an R factor of 0.186 and Rfree of 0.250.
V68A/I107Y MbIII–nitrite:
Ten initial cycles of restrained refinement were run with Refmac, and the R factor decreased from 0.455 to 0.276. Ligands and water were added to the model depending on the Fo–Fc electron density map in the subsequent refinement cycles. One sulfate anion and two nitrite anions were added iteratively using COOT. The N-terminal Met residue and the C-terminal residues Gln152 and Gly153 were omitted due to the lack of electron density. The final model was refined to 1.57 Å resolution with an R factor of 0.175 and Rfree of 0.247.
V68A/I107Y MbII–NO:
Ten initial cycles of restrained refinement were run with Refmac, and the R factor decreased from 0.433 to 0.282. Ligands and water were added to the model depending on the Fo–Fc electron density map in the subsequent refinement cycles. One sulfate anion, one nitrite anion and one nitric oxide were added iteratively using COOT. The N-terminal Met residue was omitted due to the lack of electron density. The final model was refined to 1.79 Å resolution with an R factor of 0.157 and Rfree of 0.205.
The X-ray data collection and refinement statistics are collected in Table S1 in the Supporting Information.
A note on the reproducibility of the O-nitrito conformation:
We previously reported an apparent N-nitro to O-nitrito linkage isomerization upon exposure of chlorin-substituted MbChl(NO2) complex to multiple cycles of home-source X-ray data collection.98 To probe if such a linkage isomerization was occurring in the MbIII–nitrite complexes reported here, we collected multiple X-ray diffraction data sets for each MbIII–nitrite complex using variable total X-ray radiation exposure times. Only the O-nitrito binding mode to the heme centers was observed in each case.
Computational Details
Two sets of calculations were used to investigate the FeII–NO orientations in the nitrosyl derivatives of the wt and mutant Mbs: (i) quantum mechanics (QM) calculations on active site models, and (ii) hybrid quantum mechanics and molecular mechanics (QM/MM) calculations on the whole protein derivatives. All calculations were performed using the program Gaussian 09.99
In the first set of QM calculations, in order to account for any H-bonding and van der Waals interactions involving the NO ligand, all residues with any atoms within a 7 Å distance to Fe were included in the active site models, except for the case of the H64Q MbII–NO compound, in which the Thr67 residue was not included to avoid the artificial H-bond formation with Gln64 in this truncated model (this H-bonding interaction was not present in the experimentally determined X-ray structure). The specific residues accounted for in the active site models are listed in Table 1. A non-substituted porphyrin macrocycle was used. The partial geometry optimizations, with Fe and Cα atoms of residues fixed during the geometry optimization to mimic the protein environment, were conducted for all the active site models based on similar studies.100, 101 Frequency calculations were subsequently performed to verify the nature of the corresponding stationary states on their potential energy surfaces and provide zero-point energy corrected electronic energies (EZPE’s), enthalpies (H’s), and Gibbs free energies (G’s). Based on a recent methodology study for similar model systems,102, 103 geometries were optimized using the mPW1PW91 method,104 together with the self-consistent reaction field method using the PCM approach105–108 with a dielectric constant of 4.0 to simulate the protein environment. For the geometry optimization, we utilized (i) the Wachters’ basis109 for Fe, (ii) 6–311++G(2d,2p) for first shell atoms (the four coordinated porphine N atoms and the axial histidine N atom, the NO ligand, and the heavy atom forming the H-bond with NO), and (iii) 6–31G(d) for the other atoms.
Table 1.
Residues involved in optimization in the QM calculations.
| Protein | Residues |
|---|---|
| wt MbII–NO | Leu29, Phe43, His64, Val68, His93, Ile107 |
| H64A MbII–NO | Leu29, Phe43, Ala64, Thr67, Val68, His93, Ile107 |
| H64Q MbII–NO | Leu29, Phe43, Gln64, Val68, His93, Ile107 |
| V68A/I107Y MbII–NO | Leu29, Phe43, His64, Ala68, His93, Tyr107 |
The QM/MM calculations were performed using the ONIOM approach.110 All the protein X-ray structures were first inspected for any potential problems and edited with the solvent, NO2-, SO42-, and glycerol molecules removed. The QM region consists of Fe, the truncated porphine macrocycle, the axial His93 and the NO ligand. In the ONIOM treatment of the QM/MM boundary atoms, His93 was cut at the Cα–Cβ bond. The QM region was treated with the same method and basis set as described above for the QM calculations for the active site models. The MM region was described using the Amber force field111 as used in typical ONIOM calculations.112–114 The Merz-Singh-Kollman115 charge of NO and Amber111, 116 charges for other atoms were used as the MM charges. The details of treatment of a few missing Amber parameters for the current MbII–NO systems have been described recently in similar work for MbII–HNO.117
RESULTS AND DISCUSSION
Nitrite binding to the ferric wt and mutant sw MbIIIs
Heme nitrite complexes such as MbIII–nitrite are generally prepared from adduct formation between nitrite and the ferric heme protein (eq. 5). An alternate but far less common route to their formation occurs from attack of NO on ferryl heme species,118, 119, 120 where the O-nitrito formulations for Mb118 and Hb119 (eq. 6) were based on UV-vis spectroscopy.
| (5) |
| (6) |
Solution studies:
In this current work, we added excess nitrite to the ferric MbIII–H2O precursors to generate the MbIII–nitrite derivatives. UV-vis spectral monitoring of the reaction (eq. 5) reveals changes in the spectra similar to those reported previously121 for the reaction of ferric wt sw MbIII–H2O with nitrite. Specifically, addition of sodium nitrite to a solution of ferric sw MbIII–H2O in phosphate buffer at pH 7.4 results in a red-shift of the Soret band from 408 nm to 409 nm, as well as a change in the band profile in the 450–650 nm region (Figures 4A and S1). The absorbance changes were plotted versus the concentration of nitrite to fit to the hyperbolic equation for a 1:1 binding model, with a KD of 12.2 ± 2.2 mM.
Figure 4.
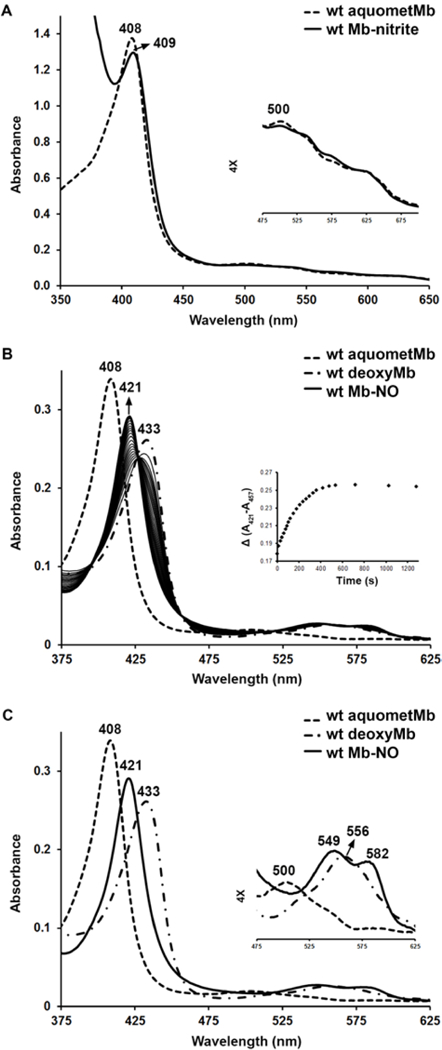
UV-vis spectral monitoring of the reactions of ferric wt sw MbIII–H2O. (A) changes after addition of nitrite. (B) changes after addition of dithionite followed by the addition of nitrite as well as the plot of ΔAbs(421–457) against time. (C) UV-vis spectra of the three forms of wt sw Mb (ferric, deoxy ferrous, and NO) with the enlarged Q region. Conditions: 0.1 M phosphate buffer, pH 7.4, [protein] = 1.8–7.4 μM, [nitrite] in (A) = 40 mM, [dithionite] = 5 mM, [nitrite] in (B) and (C) = 4 mM.
In contrast, no significant changes in the UV-vis spectra were observed when nitrite was added to the ferric H64A MbIII–H2O mutant even with concentrations up to 80 mM in [nitrite] (Figures S2A and S3), indicating very weak binding of nitrite to the heme. Spectral changes for the H64Q mutant are shown in Figures S4A and S5 with an observed red-shift of the Soret band from 408 nm to 412 nm; a KD of 1.45 ± 0.30 mM was obtained. The related spectral changes for the V68A/I107Y mutant and resulting KDs are shown in Figures S6A and S7, and Table 2, respectively. The dissociation constants for nitrite binding to the heme centers of the wt and mutant MbIIIs thus increase in the order H64Q < wt < V68A/I107Y < H64A.
Table 2.
Dissociation constants for the binding of nitrite to the ferric sw wt and mutant MbIII–H2O proteins.
| Protein | KD (mM)a |
|---|---|
| wt | 12.2 ± 2.2 |
| H64Q | 1.45 ± 0.30 |
| V68A/I107Y | 19.4 ± 0.6 |
| H64A | >> 20 |
In 0.1 M phosphate, pH 7.4, at 25 °C.
Crystallographic studies:
Much controversy still surrounds the binding mode of the nitrite anion to Mb. As mentioned in the Introduction, heme protein–nitrite complexes generally display the N-binding mode of nitrite in their X-ray crystal structures.18–31 Our published crystal structures for hh wt MbIII–nitrite (and for human HbIII) displaying the nitrite O-binding mode were thus unexpected.34, 36–38, 48 Most computational studies have also supported an N-binding ground state conformation in heme-nitrite complexes,19, 31, 39–46 although a recent thorough computational analysis supports the crystallographically determined O-binding mode in ferric MbIII–ONO.47 We thus sought to determine the X-ray crystal structures of the wt, H64A, H64Q, and V68A/I107Y MbIII–nitrite complexes to examine if the distal pocket changes in these sw Mbs would affect their nitrite binding modes.
The four wt, H64A, H64Q, and V68A/I107 MbIII–nitrite adducts were prepared by soaking nitrite into droplets containing crystals of the respective MbIII–H2O precursors for 20–30 mins. The crystal structures revealed the O-binding modes of the nitrite ligands in all four MbIII–nitrite adducts as shown in Figure 5, with the nitrite ligands refined at 100% occupancy in each structure.
Figure 5.
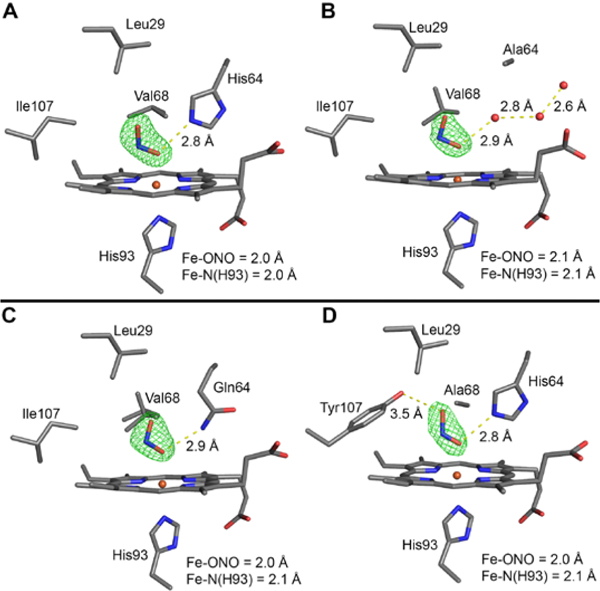
The Fo–Fc omit electron density maps (green mesh, contoured at 3σ) and final models of the heme active sites of the ferric sw MbIII–nitrite adducts. (A) wt MbIII–nitrite at 1.85 Å resolution (PDB ID: 5UT7), (B) H64A MbIII–nitrite at 1.85 Å resolution (PDB ID: 5UT9), (C) H64Q MbIII–nitrite at 1.81 Å resolution (PDB ID: 5UTA), (D) V68A/I107Y MbIII–nitrite at 1.57 Å resolution (PDB ID: 5UTD). The 2Fo–Fc maps are shown in Figure S9.
The O-binding mode in the sw wt MbIII–nitrite complex (Figure 5A), with a H-bond between the nitrite O1 atom (i.e., Fe–ONO) and the distal His64 residue (O1…Nε(His64) = 2.8 Å), is analogous to that determined for the hh MbIII–nitrite and human HbIII–nitrite derivatives.34, 36–38, 48, 98, 122 The distance between the nitrite terminal O2 atom and Nε(His64) is 3.2 Å, with the next closest distances between the nitrite and distal pocket residues being that with the Val68 residue; at ~3.3 Å between the nitrite N and O1 atoms to Cγ2 of Val68, and at ~3.4 Å between the nitrite O2 atom and Cγ2 of Val68. Selected geometrical data for the heme–nitrite moieties for this and the other structurally characterized Mb–NOx derivatives from this work are collected in Table S2. Only minor structural changes in the heme pocket occur when nitrite replaces the bound H2O ligand in wt MbIII–H2O to give the MbIII–nitrite adduct (Figure 6A).
Figure 6.
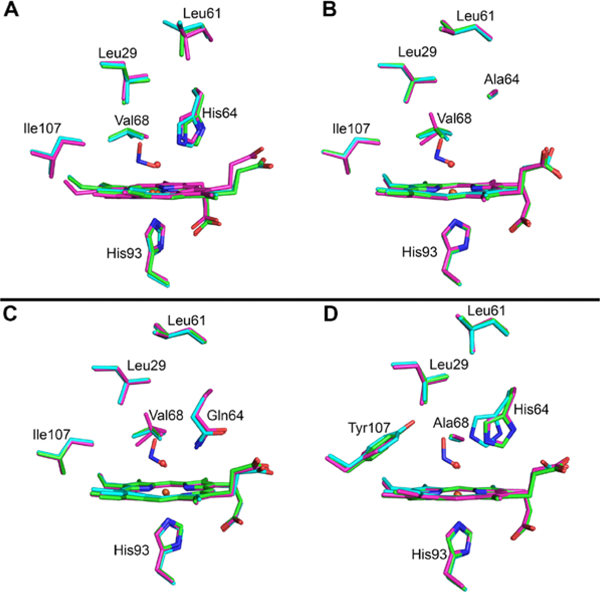
Comparisons of the heme pockets of the sw ferric MbIII–H2O (in green), ferrous deoxyMbII (in magenta) and ferric MbIII–nitrite (in cyan) adducts. A) the wt adducts (rmsd 0.51 Å), (B) the H64A adducts (rmsd 0.13 Å), (C) the H64Q adducts (rmsd 0.14 Å), and (D) the V68A/I107Y adducts (rmsd 0.70 Å).
Interestingly, whereas an N-nitro binding mode was observed in the sw H64V MbIII–nitrite adduct (lacking the distal His64 residue; Figure 2, right), the newly determined H64A sw MbIII–nitrite structure (also lacking the distal His64 residue) unexpectedly reveals an O-binding mode of the nitrite anion to the heme Fe center (Figure 5B). Fixed water molecules in the vicinity of the distal pocket near the protein exterior provide a “water bridge” H-bonding interaction with the nitrite ligand with a (nitrite)O1…OH2 distance of 2.9 Å. This “water bridge” is also present in the crystal structure of the H64A MbIII–H2O precursor. In this H64A MbIII–nitrite complex, the related closest interaction between the nitrite ligand and distal pocket residues is with the Cζ of Phe43 ((nitrite)O2…Cζ = 3.5 Å); the Val68 sidechain is flipped from its original horizontal orientation in its H64A MbIII–H2O precursor to a vertical orientation in the nitrite adduct. Importantly, the apparent discrepancy between the observed N-nitro binding in the sw H64V MbIII–nitrite adduct (Figure 2, right) and the sw H64A MbIII–nitrite adduct reported here can be rationalized by the observation that the crystal structure of the precursor H64A sw MbIII–H2O contains an Fe-bound H2O ligand;97 thus, the O-bound nitrite appears to essentially simply replace another O-bound ligand (H2O) as illustrated in Figure 6B. In contrast, however, the sw H64V MbIII structure does not contain a fixed H2O ligand to Fe,79 implying there may be a lower affinity for an added O-bound ligand such as nitrite in the O-nitrito form.
Introducing a more flexible Gln distal residue in the H64Q mutant also generates an O-bound nitrite conformation as shown in Figure 5C. The distances between the O1 and O2 atoms of the bound nitrite and the N atom of Gln64 are 2.9 Å and 3.3 Å, respectively. Two conformations (horizontal and vertical) of the Val68 sidechain were modeled to fit the electron density in this nitrite adduct (Figures 5C and 6C). Such Val68 conformation flips have been observed previously in organometallic Mbs with direct Fe–aryl bonds.123
Contrary to our initial expectations, the mutation of I107 to Tyr to provide a second H-bonding residue in the distal pocket (Figure 3, right) did not alter the O-binding mode of the nitrite ligand to an anticipated N-bound form (Figure 5D) in this V68A/I107Y MbIII–nitrite adduct. As seen in Figure 6D, the His64 residue in this double-mutant swings inward (Nε movement of ~1.1 Å) with retention of the H-bond with the nitrite O1 atom as was the case in the wt MbIII–ONO derivative; the O1 and O2 distances from Nε(His64) being 2.8 Å and 3.1 Å, respectively. The other closest contacts between nitrite and distal pocket residues are with the nearby Phe43 and Ala68 (at ~3.4 Å with the closest C atoms of these residues). The closest interaction between the nitrite ligand and the Tyr107 sidechain is between the nitrite O2 atom and the Oη of Tyr107 at a distance of 3.5 Å, a distance too long to form a substantial H-bond interaction.
Nitrite reduction to generate the nitrosyl MbII–NO derivatives
Nitrite reduction to NO mediated by deoxyMbII has been reported for sw wt Mb (eqs. 3 and 4),55 H64A mutant sw Mb,55 and other sw and hh Mb proteins.48, 55, 66 Mechanisms of
| (3) |
| (4) |
such heme-based nitrite reduction have normally invoked the N-nitro bound form of nitrite as the starting point in the reaction pathway (Figures 1 and 7 (left)). Silaghi-Dumitrescu first proposed the possibility of a viable O-nitrito intermediate for heme-based nitrite reduction which would involve NO release and Fe–OH formation (Figure 7, right).40 This concept has been extended to the possible involvement of a viable O-nitrito intermediate in Hb-based nitrite reduction.43 In either pathway, the final end-product is the ferrous nitrosyl MbII–NO derivative (eqs. 3–4).
Figure 7.
Proposed pathways for heme-based nitrite reduction involving the N-nitro (left) and O-nitrito (right) isomers.
Solution studies:
Following established procedures, we performed the nitrite reduction experiments with the sw MbII H64Q and V68A/I107Y mutants. The ferric MbIII–H2O precursors were first reduced to their deoxyMbIIs using dithionite followed by reaction with nitrite.
The reactions of nitrite with the H64Q and V68A/I107 deoxyMbIIs in phosphate buffer at pH 7.4 and at 37 °C are illustrated in Figure S8, and yielded bimolecular reaction rates of 0.57 ± 0.06 M−1s−1 (H64Q) and 6.3 ± 0.4 M−1s−1 (V68A/I107). These may be compared with the reaction rates for wt sw Mb (5.6 ± 0.6 M−1s−1) and H64A sw Mb (1.8 ± 0.3 M−1s−1) reported previously.55
The UV-vis spectra of the wt deoxyMbII and MbII–NO derivatives are shown in Figures 4B-C; the mutant deoxyMbIIs and MbII–NO spectra are shown in Figure S2 (H64A), Figure S4 (H64Q), and Figure S6 (V68A/I107Y). With these wt and mutant sw MbII–NOs in hand, we then sought to characterize them by X-ray crystallography to determine distal pocket effects on the heme–NO geometry.
Crystallographic studies:
Despite the biological relevance of MbII–NO complexes, there are only four reported X-ray crystal structures of MbII–NO derivatives.61,34, 62, 63 Brucker et al. first reported the crystal structure of wt sw MbII–NO at 1.7 Å resolution (∠FeNO = 112°) prepared from gaseous NO addition (after sodium fluoride addition) to crystalline MbIII–H2O;61 the preliminary structure of the L29F mutant MbII–NO derivative (∠FeNO = 130°) was only briefly mentioned. We reported the crystal structures of wt hh MbII–NO with varied but reproducible ∠FeNO angles of ~120° or ~144° dependent on the preparative conditions.34, 62 The crystal structure of blackfin tuna wt MbII–NO was also reported, with an ∠FeNO angle of 134°.63 A few related ferrous HbII–NO structures are also known.64, 65 In any event, the availability of the four sw MbII–NO derivatives prepared in this work led us to determine their crystal structures to systematically examine distal pocket effects on the FeNO conformations in this series.
All four wt, H64A, H64Q, and V68A/I107Y MbII–NO derivatives were prepared from dithionite reduction of the in situ generated crystalline ferric MbIII–nitrite precursors described above. Their crystal structures were determined, and the heme sites are shown in Figure 8. Modeling the FeNO group as nitrosyl (Fe–N–O) rather than isonitrosyl (Fe–O–N) stems from spectroscopic and theoretical (QM, QM/MM) results on MbII–NO124, 125 and model heme–NO systems126–128 that show the isonitrosyl linkage isomers to be highly unstable even at low temperature and ~18–37 kcal/mol higher in energy.
Figure 8.
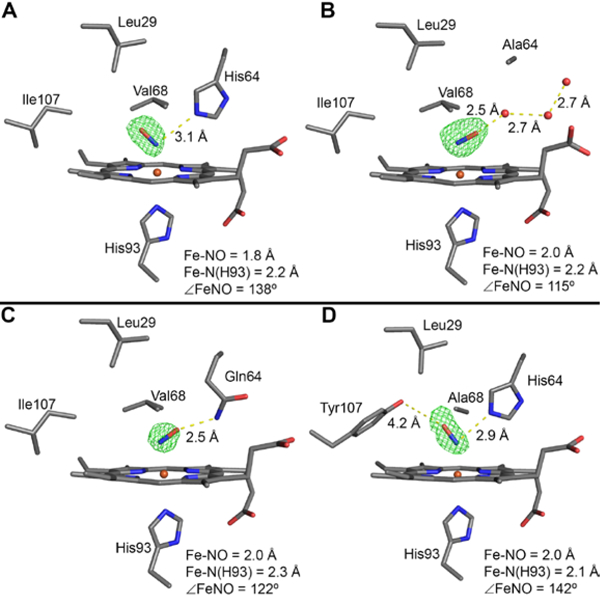
The Fo–Fc omit electron density map (green mesh, contoured at 3σ) and final model of the heme active site of the ferrous sw MbII–NO derivatives. (A) wt MbII–NO at 1.70 Å resolution (PDB ID: 5JNG), (B) H64A MbII–NO at 1.78 Å resolution (PDB ID: 5JNI), (C) H64Q MbII–NO at 1.78 Å resolution (PDB ID: 5JNJ), (D) V68A/I107Y MbII–NO at 1.79 Å resolution (PDB ID: 5JNK). The 2Fo–Fc maps are shown in Figure S10.
The sw wt MbII–NO heme site structure (Figure 8A) exhibits an ∠FeNO bond angle of 138°, which is consistent with the canonical ∠FeNO bond angle (138.5°) expected for synthetic six-coordinate ferrous nitrosyl porphyrins with N-base trans ligands,129 and is also consistent with the previous quantum chemically predicted value (138.1°) for MbII–NO.130
The ∠FeNO bond angle of 138° in sw wt MbII–NO prepared from the nitrite/dithionite method is larger than that observed for the complex prepared from the NO gas method (112°)61 but is closer to the 144° seen in ferrous hh MbII–NO prepared from the nitrite/dithionite method.34 A H-bonding interaction between the nitrosyl N-atom and the distal His64 residue is present in this structure, with a (nitrosyl)N… Nε(His64) distance of 3.1 Å. The next closest distances between the NO ligand and distal pocket residues are with the Cγ2(Val68) atom; 3.2 Å distance from nitrosyl O atom, and 3.3 Å from the nitrosyl N-atom. Selected geometrical parameters for the wt and mutant sw MbII–NO derivatives described below are listed in Table S2.
The NO ligand in the H64A MbII–NO derivative is oriented towards the hydrophilic distal pocket exterior, in the general direction of the heme propionates (Figure 8B). This NO orientation towards the protein exterior is unprecedented in MbII–NO and HbII–NO protein structures, with no example reported in the literature or in the Protein Data Bank to date. In the absence of the H-bonding distal H64 residue, three fixed water molecules provide a H-bonding network from the nitrosyl O-atom to the protein exterior, with a (nitrosyl)O…OH2 distance of 2.5 Å. The nitrosyl N and O atoms are 3.0 Å and 3.2 Å, respectively, from the Cγ2(Val68) atom.
The crystal structure of the H64Q MbII–NO derivative (Figure 8C) also reveals that the NO ligand is oriented towards the hydrophilic exterior, similar to that seen for the H64A derivative described above. Although the Gln64 residue provides a H-bonding interaction with the bound NO ligand, this H-bonding interaction is with the nitrosyl O-atom rather than the N-atom, which is quite unusual for Mb-like heme protein–NO derivatives. Interestingly, such heme nitrosyl ambidentate O-atom vs. N-atom H-bonding in heme models has been calculated to have an effect on Fe–N(O) and FeN–O vibrational spectral (normal vs. inverse) correlations.131
The structure of the V68A/I107Y MbII–NO derivative is similar to that of the wt MbII–NO in terms of the orientation of the NO ligand that points towards the hydrophobic protein interior (Figure 8D). Both the nitrosyl N and O-atoms are at a 2.9 Å distance from the Nε atom of His64. However, similarly to what was observed in the nitrite adduct, the Tyr107 sidechain is not in H-bonding contact with the NO ligand. The closest distance is between the nitrosyl O-atom and the Tyr sidechain O-atom (at 4.2 Å). Apart from the His64 residue, the closest distance between the NO ligand and distal pocket residues is 3.6 Å, between the Cβ atom of Ala68 and the nitrosyl O-atom.
It is thus interesting to note that for the four MbII–NO structures reported in this work, the two that contain the distal His64 residue display NO ligand orientations in the direction of the hydrophobic protein interior (i.e., away from the heme propionates). In contrast, the two mutants that do not contain the distal H64 residue (for any Mb and Hb structures) display unprecedented NO ligand orientations towards the hydrophilic exterior. These observations are summarized in the heme site structural overlays shown in Figure S11.
We found that this orientational preference for the NO ligand in the wt and mutant MbII–NOs in this work was reproducible over the several (>10) independent compound preparations, using two different orders of addition of dithionite and nitrite, and X-ray diffraction data collection experiments. Silvernail et al.132 have determined, using computational methods for a series of six-coordinate model (por)Fe(NO)(imidazole) compounds, that there is no significant difference in potential energy between the two observed (opposite) NO orientations in the simple non-protein models. With an expanded set of protein MbII–NO derivatives with varying NO ligand orientations available to us, we proceeded to probe these orientational preferences with both QM and QM/MM methods.
Computational studies: Probing the experimentally observed differential FeNO orientations:
In order to help understand why the NO adopts different orientations in the different wt and mutant MbII–NO derivatives, we probed, using QM and QM/MM calculations, both the original NO orientations as observed in the experimentally derived X-ray crystal structures and the alternative NO orientation models. The active site models of the X-ray crystal structures and optimized geometries are shown in Figure 9. The relative energies of the systems containing the hypothetical alternative NO orientations (i.e., compared with the experimentally observed X-ray orientations) are listed in Table 3. For the wt MbII–NO, the QM optimized structure with the X-ray NO orientation (wt_b in Figure 9) is similar to that in the original X-ray structure (wt_a), with the H-bonding between NO and distal His64 maintained in the optimized structure. The QM optimized structure with the alternative NO orientation (wt_c), however, reveals significant differences. In particular, the H-bond to NO is lost (in wt_c) and there is also ample displacement regarding the relative positions of the distal pocket residues His64 and Val68.
Figure 9.
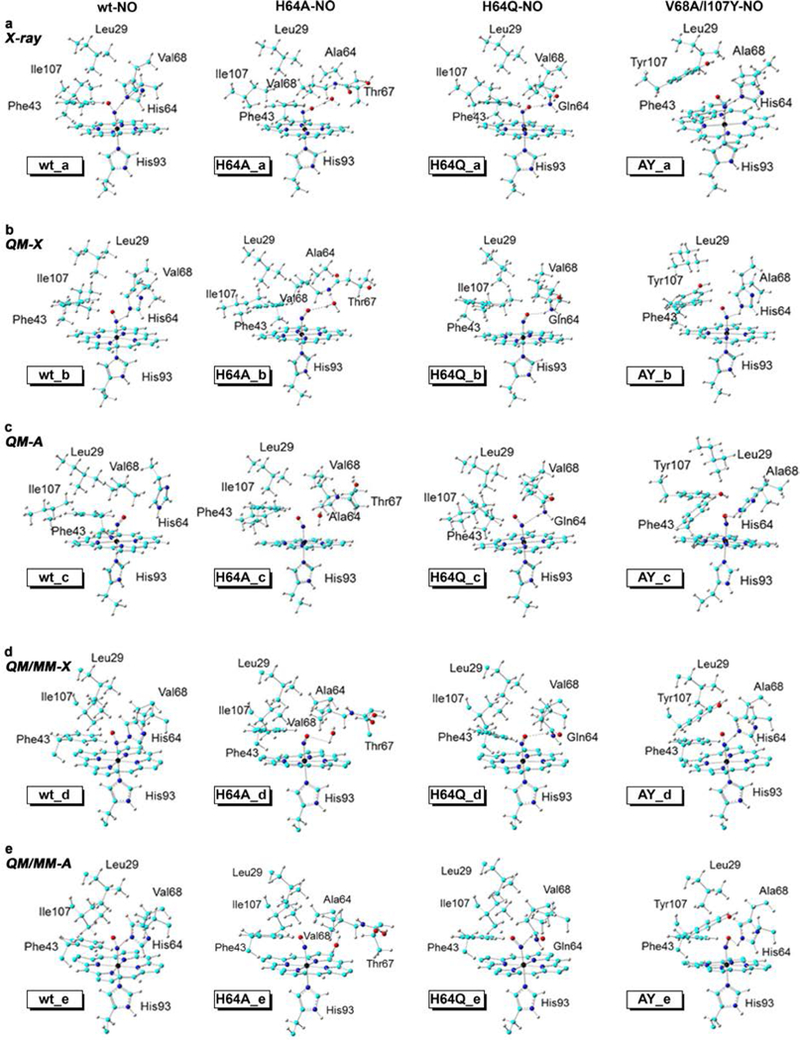
(a) Active site models from X-ray structures. (b) QM-X: QM optimized structures using X-ray orientations of NO. (c) QM-A: QM optimized structures of alternate NO orientations. (d) QM/MM-X: QM/MM optimized structures using X-ray orientations of NO. (e) QM/MM-A: QM/MM optimized structures of alternate NO orientations.
Table 3.
Relative energies (in kcal/mol) and Fe–NO bond lengths (in Å) of proteins with the alternative NO orientations compared to the X-ray orientations in the wild-type (wt) and mutant MbII–NO derivatives.
| Method | Protein |
Figure 9
labela |
ΔE | ΔEZPE | ΔH | ΔG | ΔRFe-NO |
|---|---|---|---|---|---|---|---|
| QM | MbII–NO | wt_c | 0.47 | 0.26 | –0.75 | 1.82 | 0.026 |
| H64A MbII–NO | H64A_c | 5.17 | 3.57 | 3.39 | 3.26 | 0.009 | |
| H64Q MbII–NO | H64Q_c | –0.19 | –0.41 | –0.93 | 0.82 | 0.009 | |
| V68A/I107Y MbII–NO | AY_c | 0.60 | 0.48 | 1.09 | –2.01 | –0.001 | |
| QM/MM | MbII–NO | wt_e | / | / | / | / | / |
| H64A MbII–NO | H64A_e | 22.63 | 19.06 | 21.34 | 11.60 | 0.006 | |
| H64Q MbII–NO | H64Q_e | 4.09 | 3.35 | 3.85 | 3.86 | 0.001 | |
| V68A/I107Y MbII–NO | AY_e | 0.09 | 6.56 | 6.41 | 5.19 | 0.004 | |
Structure labels as indicated in Figure 9.
These features result in a Gibbs free energy penalty of 1.82 kcal/mol for this alternative NO orientation (Table 3). As this alternative orientation triggers big changes in the positions of the His64 and Val68 residues in these truncated active site models, which may not be possible to be accommodated in the actual protein due to the protein environment, attempts at the QM/MM geometry optimization of the whole protein with this alternative orientation, indeed, led to the same observed X-ray orientation (i.e., wt_e = wt_d).
For the H64A mutant (H64A_a in Figure 9), the QM optimized X-ray NO orientation (H64A_b) structure with the NO ligand pointing towards the protein exterior is close to the original X-ray structure (H64A_a), while the alternative NO orientation optimized structure (H64A_c; with NO pointing towards the hydrophobic interior) reveals large differences. In particular, the original H-bond between NO and the active site water molecule is again lost (H64A_c); similar to the loss of H-bonding with the distal His64 residue in wt MbII–NO (wt_c). For this system, the alternative orientation (H64A_c) not only costs 3.26 kcal/mol in Gibbs free energy, but also has a significant cost around 3–5 kcal/mol in other energy terms. Consistent with this relatively larger energy difference compared with the wt MbII–NO case, the positions of three more active site residues are significantly affected, including Phe43, Thr67, and Ile107, besides Ala64 and Val68 corresponding to His64 and Val68 in wt MbII–NO. This extensive effect on the active site residues eventually affects the whole protein structure more significantly, with the QM/MM optimized alternative NO orientation (H64A_e) being less stable than the X-ray orientation (H64A_d) by 11.60 kcal/mol in Gibbs free energy. The large difference in the QM versus the QM/MM results emphasize the importance of considering the whole protein even when focusing on only heme site ligand conformations.
In contrast with above obvious favorable energies in the QM calculations for the X-ray NO orientations for wt and H64A MbII–NO compounds, the H64Q MbII–NO derivative has a Gln64 residue that is relatively more flexible than His64 in forming H-bonds. Indeed, NO can form a H-bond with Gln64 in both the X-ray NO orientation (H64Q_b; H-bond with the nitrosyl O atom) and the alternative orientation (H64Q_c; H-bond with the nitrosyl N atom). However, the H-bond in the X-ray orientation (H64Q_b; involving the nitrosyl O atom) is still slightly more favorable, as indicated by a shorter H-bond distance of 2.250 Å vs. 2.476 Å in the alternative orientation. Accordingly, the X-ray orientation active site structure is more stable by 0.82 kcal/mol in Gibbs free energy. The corresponding QM/MM optimized structure with the X-ray orientation (H64Q_d; H-bond with the nitrosyl O atom) is also more favorable than the alternative orientation (H64Q_e; H-bond with the nitrosyl N atom) by a more significant energy difference of ΔG of 3.86 kcal/mol, but is still the smallest penalty for the alternative NO orientation among all QM/MM calculations here (Table 3).
For the double mutant V68A/I107Y MbII–NO, the QM optimized alternative orientation (AY_c; H-bond between His64 and the nitrosyl O atom) is surprisingly more stable than the X-ray orientation (AY_b; H-bond with the nitrosyl N atom) by ΔG of 2.01 kcal/mol. The H-bond distances for NO with His64 in both structures are similar; 2.457 Å (AY_b; X-ray orientation) vs. 2.499 Å (AY_c; calculated alternative orientation). However, a closer look at the optimized structures (AY_b vs. AY_c) in the truncated models used in the QM calculations reveals unexpected large differences in the positions of Phe43 and Tyr107 in the optimized X-ray orientation structure (AY_b) when compared with the experimentally determined X-ray structure (AY_a). Nevertheless, once the whole protein was used, the QM/MM calculations show a clear energy preference for the X-ray orientation (AY_d; H-bond with the nitrosyl N atom) by ΔG of 5.19 kcal/mol (Table 3). In addition, these active site residues’ positions are now closer to the X-ray structure (AY_a) than those from the QM truncated model studies (AY_b). The H-bond distance of NO with His64 of 2.146 Å in the X-ray orientation (AY_d) is also more favorable than that (at 2.335 Å) in the alternative orientation (AY_e). Results for this protein system suggests that one should be careful about results in truncated model studies and sometimes the large protein models are necessary to obtain more reliable data.
Overall, above investigations show that NO prefers the orientations as observed in the X-ray structures with more stable energies. Interestingly, as shown in Table 3, in the X-ray orientation, the Fe–NO bond length is relatively shorter (ΔRFe-NO) than that in the alternative orientation from both QM and QM/MM calculations with correct energy orders. This again suggests a more stable NO binding. In addition, these results indicate that when there is a non-His64 distal residue such as Ala64 or Gln64, NO adopts an orientation toward this residue, while if there is a His64 residue present, NO prefers an orientation point away from this residue in the direction of the protein interior, to achieve more stable binding overall in the protein environment.
CONCLUSIONS
In this work, we have clarified the preferred O-nitrito binding of nitrite to the heme center in wt and mutant MbIIIs differing in the number of potential H-bonds (i.e., 0, 1, or 2) to the bound ligand. The dissociation constants increase in the order H64Q < wt < V68A/I107Y < H64A. This well-defined O-nitrito binding to MbIIIs (and HbIII) remains unique in heme-nitrite structural biology and presents opportunities for further nitrite structural biology using other heme proteins with different active site architectures. Replacement of the bound H2O ligand in the MbIII–H2O precursors with the O-bound nitrite does not result in substantial changes in the overall Mb protein fold. Minor changes in side-chain orientations in the active site were, however, observed, such as the orientations of the Val68 residue in the H64A and H64Q mutants (i.e., without the distal His64 residue) and the inward movement of the His64 residue in the V68A/I107 double mutant derivative to maintain its H-bond with the bound nitrite.
Nitrite reduction to NO is enabled by the ferrous MbIIs to generate the MbII–NO derivatives. Importantly, we report the first systematic structural investigation of the FeNO geometries in wt and mutant Mbs that led to unexpected but reproducible FeNO conformational preferences in these proteins. We found that when the natural His64 residue was present (wt and V68A/I107Y), the NO vector was pointed towards the hydrophobic protein interior as previously noted for other wt MbII–NO and HbII–NO compounds. However, in the absence of this His64 residue (H64A and H64Q), the NO vector was oriented towards the hydrophilic protein exterior. Detailed QM and hybrid QM/MM theoretical investigations were employed to rationalize these varied FeNO orientations observed experimentally by X-ray crystallography. In addition, these calculations underpin the caution that in probing such FeNO orientations in heme proteins, sometimes the whole protein needs to be taken into account when examining such heme-ligand conformational preferences in protein derivatives.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the National Science Foundation (CHE-1566509 to GBR-A) and the National Institutes of Health (GM085774 to YZ, and HL096973 to MTG and SS) for funding for this work. This manuscript reports data obtained in the University of Oklahoma Macromolecular Crystallography Laboratory, which is supported, in part, by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103640.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.8b00542.
Crystallography experimental details and tables, additional spectral and crystal structural figures.
The authors declare no competing financial interest.
REFERENCES
- [1].Mörner KAH (1897) Beobachtungen über den Muskelfarbstoff. Nord. Med. Ark. 30, 1–8. [Google Scholar]
- [2].Suzuki T, and Imai K (1998) Evolution of Myoglobin. Cell Mol. Life Sci. 54, 979–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Møller JKS, and Skibsted LH (2002) Nitric Oxide and Myoglobins. Chem. Rev. 102, 1167–1178. [DOI] [PubMed] [Google Scholar]
- [4].Binkerd EF, and Kolari OE (1975) The History and Use of Nitrate and Nitrite in the Curing of Meat. Food Cosmet. Toxicol. 13, 655–661. [DOI] [PubMed] [Google Scholar]
- [5].Haldane J (1901) The Red Colour of Salted Meat. J. Hyg. (London) 1, 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dejam A, Hunter CJ, Schechter AN, and Gladwin MT (2004) Emerging Role of Nitrite in Human Biology. Blood Cells Mol. Disease 32, 423–429. [DOI] [PubMed] [Google Scholar]
- [7].Gladwin MT, Grubina R, and Doyle MP (2008) The New Chemical Biology of Nitrite Reactions with Hemoglobin: R-State Catalysis, Oxidative Denitrosylation, and Nitrite Reductase/Anhydrase. Acc. Chem. Res. 42, 157–167. [DOI] [PubMed] [Google Scholar]
- [8].Helms CC, Liu XH, and Kim-Shapiro DB (2017) Recent Insights into Nitrite Signaling Processes in Blood. Biol. Chem. 398, 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Eady RR, and Hasnain SS (2004) Denitrification, In Comprehensive Coordination Chemistry II (Que L Jr., and Tolman WB, Eds.), Elsevier, San Diego, CA: Vol. 8, pp 759–786. [Google Scholar]
- [10].Averill BA (1996) Dissimilatory Nitrite and Nitric Oxide Reductases. Chem. Rev. 96, 2951–2964. [DOI] [PubMed] [Google Scholar]
- [11].Hollocher TC, and Hibbs JB Jr. (1996) Enzymes of Bacteria, Plants and Fungi that Process Free Nitrogen Oxides, In Methods in Nitric Oxide Research (Feelish M, and Stamler JS, Eds.), John Wiley and Sons, Chichester, pp 119–146. [Google Scholar]
- [12].Tavares P, Pereira AS, and Moura JJG (2006) Metalloenzymes of the Denitrification Pathway. J. Inorg. Biochem. 100, 2087–2100. [DOI] [PubMed] [Google Scholar]
- [13].Brittain T, Blackmore R, Greenwood C, and Thomson AJ (1992) Bacterial Nitrite-reducing Enzymes. Eur. J. Biochem. 209, 793–802. [DOI] [PubMed] [Google Scholar]
- [14].Zumft WG, and Körner H (1997) Enzyme Diversity and Mosaic Gene Organization in Denitrification. Anton van Leewenhock 71, 43–58. [DOI] [PubMed] [Google Scholar]
- [15].Maia LB, and Moura JJG (2014) How Biology Handles Nitrite. Chem. Rev. 114, 5273–5357. [DOI] [PubMed] [Google Scholar]
- [16].Wasser IM, de Vries S, Moenne-Loccoz P, Schroder I, and Karlin KD (2002) Nitric Oxide in Biological Denitrification: Fe/Cu Metalloenzyme and Metal Complex NOx Redox Chemistry. Chem. Rev. 102, 1201–1234. [DOI] [PubMed] [Google Scholar]
- [17].Hitchman MA, and Rowbottom GL (1982) Transition Metal Nitrite Complexes. Coord. Chem. Rev. 42, 55–132. [Google Scholar]
- [18].Williams PA, Fulop V, Garman EF, Saunders NFW, Ferguson SJ, and Hajdu J (1997) Haem-ligand Switching During Catalysis in Crystals of a Nitrogen-Cycle Enzyme. Nature 389, 406–412. [DOI] [PubMed] [Google Scholar]
- [19].Einsle O, Messerschmidt A, Huber R, Kroneck PMH, and Neese F (2002) Mechanism of the Six-Electron Reduction of Nitrite to Ammonia by Cytochrome c Nitrite Reductase. J. Am. Chem. Soc. 124, 11737–11745. [DOI] [PubMed] [Google Scholar]
- [20].Lukat P, Rudolf M, Stach P, Messerschmidt A, Kroneck PMH, Simon J, and Einsle O (2008) Binding and Reduction of Sulfite by Cytochrome c Nitrite Reductase. Biochemistry 47, 2080–2086. [DOI] [PubMed] [Google Scholar]
- [21].Polyakov KM, Boyko KM, Tikhonova TV, Slutsky A, Antipov AN, Zvyagilskaya RA, Popov AN, Bourenkov GP, Lamzin VS, and Popov VO (2009) High-Resolution Structural Analysis of a Novel Octaheme Cytochrome c Nitrite Reductase from the Haloalkaliphilic Bacterium Thioalkalivibrio nitratireducens. J. Mol. Biol. 389, 846–862. [DOI] [PubMed] [Google Scholar]
- [22].Trofimov AA, Polyakov KM, Tikhonova TV, Tikhonov AV, Safonova TN, Boyko KM, Dorovatovskii PV, and Popov VO (2012) Covalent Modifications of the Catalytic Tyrosine in Octahaem Cytochrome c Nitrite Reductase and their Effect on the Enzyme Activity. Acta Crystallogr. Sect. D 68, 144–153. [DOI] [PubMed] [Google Scholar]
- [23].Trofimov AA, Polyakov KM, Lazarenko VA, Popov AN, Tikhonova TV, Tikhonov AV, and Popov VO (2015) Structural Study of the X-ray-Induced Enzymatic Reaction of Octahaem Cytochrome c Nitrite Reductase. Acta Crystallogr. Sect. D 71, 1087–1094. [DOI] [PubMed] [Google Scholar]
- [24].Crane BR, Siegel LM, and Getzoff ED (1997) Probing the Catalytic Mechanism of Sulfite Reductase by X-ray Crystallography: Structures of the Escherichia coli Hemoprotein in Complex with Substrates, Inhibitors, Intermediates, and Products. Biochemistry 36, 12120–12137. [DOI] [PubMed] [Google Scholar]
- [25].Parey K, Warkentin E, Kroneck PMH, and Ermler U (2010) Reaction Cycle of the Dissimilatory Sulfite Reductase from Archaeoglobus fulgidus. Biochemistry 49, 8912–8921. [DOI] [PubMed] [Google Scholar]
- [26].Nakano S, Takahashi M, Sakamoto A, Morikawa H, and Katayanagi K (2012) The Reductive Reaction Mechanism of Tobacco Nitrite Reductase Derived from a Combination of Cystal Structures and Ultraviolet-Visible Microspectroscopy. Proteins 80, 2035–2045. [DOI] [PubMed] [Google Scholar]
- [27].Nakano S, Takahashi M, Sakamoto A, Morikawa H, and Katayanagi K (2012) X-Ray Crystal Structure of a Mutant Assimilatory Nitrite Reductase That Shows Sulfite Reductase-Like Activity. Chem. Biodivers. 9, 1989–1999. [DOI] [PubMed] [Google Scholar]
- [28].He C, Ogata H, and Knipp M (2010) Complex Formation of Nitrite with the Ferriheme b β-Barrel Proteins Nitrophorin 4 and Nitrophorin 7. Biochemistry 49, 5841–5851. [DOI] [PubMed] [Google Scholar]
- [29].He CM, Ogata H, and Knipp M (2012) Insertion of an H-Bonding Residue into the Distal Pocket of the Ferriheme Protein Nitrophorin 4: Effect on Nitrite-Iron Coordination and Nitrite Disproportionation. Chem. Biodivers. 9, 1761–1775. [DOI] [PubMed] [Google Scholar]
- [30].He CM, Ogata H, and Lubitz W (2016) Elucidation of the Heme Active Site Electronic Structure Affecting the Unprecedented Nitrite Dismutase Activity of the Ferriheme b Proteins, the Nitrophorins. Chem. Sci. 7, 5332–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nilsson ZN, Mandella BL, Sen K, Kekilli D, Hough MA, Moenne-Loccoz P, Strange RW, and Andrew CR (2017) Distinguishing Nitro vs Nitrito Coordination in Cytochrome c’ Using Vibrational Spectroscopy and Density Functional Theory. Inorg. Chem. 56, 13205–13212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hartridge H (1920) Nitrite Methaemoglobin and Related Pigments. J. Physiol. 54, 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Barnard RD (1937) The Reactions of Nitrite with Hemoglobin Derivatives. J. Biol. Chem. 120, 177–191. [Google Scholar]
- [34].Copeland DM, Soares A, West AH, and Richter-Addo GB (2006) Crystal Structures of the Nitrite and Nitric Oxide Complexes of Horse Heart Myoglobin. J. Inorg. Biochem. 100, 1413–1425. [DOI] [PubMed] [Google Scholar]
- [35].Heinecke J, Yi J, Richter-Addo GB, and Ford PC (2012) Nitrite Reduction by Co(II)- and Mn(II)-Substituted Myoglobins. Towards Understanding the Necessary Components of Mb Nitrite Reductase Activity. J. Inorg. Biochem. 107, 47–53. [DOI] [PubMed] [Google Scholar]
- [36].Yi J, Orville AM, Skinner JM, Skinner MJ, and Richter-Addo GB (2010) Synchrotron X-ray-Induced Photoreduction of Ferric Myoglobin Nitrite Crystals Gives the Ferrous Derivative with Retention of the O-Bonded Nitrite Ligand. Biochemistry 49, 5969–5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yi J, Safo MK, and Richter-Addo GB (2008) The Nitrite Anion Binds to Human Hemoglobin via the Uncommon O-Nitrito Mode. Biochemistry 47, 8247–8249. [DOI] [PubMed] [Google Scholar]
- [38].Yi J, Thomas LM, Musayev FN, Safo MK, and Richter-Addo GB (2011) Crystallographic Trapping of Heme Loss Intermediates during the Nitrite-Induced Degradation of Human Hemoglobin. Biochemistry 50, 8323–8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bykov D, and Neese F (2015) Six-Electron Reduction of Nitrite to Ammonia by Cytochrome c Nitrite Reductase: Insights from Density Functional Theory Studies. Inorg. Chem. 54, 9303–9316. [DOI] [PubMed] [Google Scholar]
- [40].Silaghi-Dumitrescu R (2004) Linkage Isomerism in Nitrite Reduction by Cytochrome cd1 Nitrite Reductase. Inorg. Chem. 43, 3715–3718. [DOI] [PubMed] [Google Scholar]
- [41].Silaghi-Dumitrescu R, Svistunenko DA, Cioloboc D, Bischin C, Scurtu F, and Cooper CE (2014) Nitrite Binding to Globins: Linkage Isomerism, EPR Silence and Reductive Chemistry. Nitric Oxide 42, 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Basu S, Grubina R, Huang J, Conradie J, Huang Z, Jeffers A, Jiang A, He X, Azarov I, Seibert R, Mehta A, Patel R, King SB, Hogg N, Ghosh A, Gladwin MT, and Kim-Shapiro DB (2007) Catalytic Generation of N2O3 by the Concerted Nitrite Reductase and Anhydrase Activity of Hemoglobin. Nat. Chem. Biol. 3, 785–794. [DOI] [PubMed] [Google Scholar]
- [43].Perissinotti LL, Marti MA, Doctorovich F, Luque FJ, and Estrin DA (2008) A Microscopic Study of the Deoxyhemoglobin-Catalyzed Generation of Nitric Oxide from Nitrite Anion. Biochemistry 47, 9793–9802. [DOI] [PubMed] [Google Scholar]
- [44].Hopmann KH, Cardey B, Gladwin MT, Kim-Shapiro DB, and Ghosh A (2011) Hemoglobin as a Nitrite Anhydrase: Modeling Methemoglobin-Mediated N2O3 Formation. Chem. Eur. J. 17, 6348–6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Berto TC, and Lehnert N (2011) Density Functional Theory Modeling of the Proposed Nitrite Anhydrase Function of Hemoglobin in Hypoxia Sensing. Inorg. Chem. 50, 7361–7363. [DOI] [PubMed] [Google Scholar]
- [46].Zhang TT, Liu YD, and Zhong RG (2015) Iron(II) Porphyrins Induced Conversion of Nitrite into Nitric Oxide: A Computational Study. J. Inorg. Biochem. 150, 126–132. [DOI] [PubMed] [Google Scholar]
- [47].Sundararajan M, and Neese F (2015) Distal Histidine Modulates the Unusual O-Binding of Nitrite to Myoglobin: Evidence from the Quantum Chemical Analysis of EPR Parameters. Inorg. Chem. 54, 7209–7217. [DOI] [PubMed] [Google Scholar]
- [48].Yi J, Heinecke J, Tan H, Ford PC, and Richter-Addo GB (2009) The Distal Pocket Histidine Residue in Horse Heart Myoglobin Directs the O-Binding Mode of Nitrite to the Heme Iron. J. Am. Chem. Soc. 131, 18119–18128‥ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Brooks J (1937) The Action of Nitrite on Haemoglobin in the Absence of Oxygen. Proc. Roy. Soc. Lond. B 123, 368–382. [Google Scholar]
- [50].Helbo S, Weber RE, and Fago A (2013) Expression Patterns and Adaptive Functional Diversity of Vertebrate Myoglobins. Biochim. Biophys. Acta- Prot. Proteom. 1834, 1832–1839. [DOI] [PubMed] [Google Scholar]
- [51].Kim-Shapiro DB, and Gladwin MT (2014) Mechanisms of Nitrite Bioactivation. Nitric Oxide 38, 58–68 and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gautier C, van Faassen E, Mikula I, Martasek P, and Slama-Schwok A (2006) Endothelial Nitric Oxide Synthase Reduces Nitrite Anions to NO under Anoxia. Biochem. Biophys. Res. Commun. 341, 816–821. [DOI] [PubMed] [Google Scholar]
- [53].Vanin AF, Bevers LM, Slama-Schwok A, and van Faassen EE (2007) Nitric Oxide Synthase Reduces Nitrite to NO under Anoxia. Cell. Mol. Life Sci. 64, 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Li H, Samouilov A, Liu X, and Zweier JL (2003) Characterization of the Magnitude and Kinetics of Xanthine Oxidase-Catalyzed Nitrate Reduction: Evaluation of Its Role in Nitrite and Nitric Oxide Generation in Anoxic Tissues. Biochemistry 42, 1150–1159. [DOI] [PubMed] [Google Scholar]
- [55].Tiso M, Tejero J, Basu S, Azarov I, Wang XD, Simplaceanu V, Frizzell S, Jayaraman T, Geary L, Shapiro C, Ho C, Shiva S, Kim-Shapiro DB, and Gladwin MT (2011) Human Neuroglobin Functions as a Redox-regulated Nitrite Reductase. J. Biol. Chem. 286, 18277–18289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tejero J, Sparacino-Watkins CE, Ragireddy V, Frizzell S, and Gladwin MT (2015) Exploring the Mechanisms of the Reductase Activity of Neuroglobin by Site-Directed Mutagenesis of the Heme Distal Pocket. Biochemistry 54, 3716–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li HT, Hemann C, Abdelghany TM, El-Mahdy MA, and Zweier JL (2012) Characterization of the Mechanism and Magnitude of Cytoglobin-mediated Nitrite Reduction and Nitric Oxide Generation under Anaerobic Conditions. J. Biol. Chem. 287, 36623–36633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Corti P, Ieraci M, and Tejero J (2016) Characterization of Zebrafish Neuroglobin and Cytoglobins 1 and 2: Zebrafish Cytoglobins Provide Insights into the Transition from Six-coordinate to Five-coordinate globins. Nitric Oxide 53, 22–34. [DOI] [PubMed] [Google Scholar]
- [59].Reeder BJ, and Ukeri J (2018) Strong Modulation of Nitrite Reductase Activity of Cytoglobin by Disulfide Bond Oxidation: Implications for Nitric Oxide Homeostasis. Nitric Oxide 72, 16–23. [DOI] [PubMed] [Google Scholar]
- [60].Tiso M, Tejero J, Kenney C, Frizzell S, and Gladwin MT (2012) Nitrite Reductase Activity of Nonsymbiotic Hemoglobins from Arabidopsis thaliana. Biochemistry 51, 5285–5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Brucker EA, Olson JS, Ikeda-Saito M, and Philips GN Jr. (1998) Nitric Oxide Myoglobin: Crystal Structure and Analysis of Ligand Geometry. Proteins: Struct. Func. Genet. 30, 352–356. [PubMed] [Google Scholar]
- [62].Copeland DM, West AH, and Richter-Addo GB (2003) Crystal Structures of Ferrous Horse Heart Myoglobin Complexed with Nitric Oxide and Nitrosoethane. Proteins: Struct. Func. Genet. 53, 182–192. [DOI] [PubMed] [Google Scholar]
- [63].Schreiter ER, Rodriguez MM, Weichsel A, Montfort WR, and Bonaventura J (2007) S-Nitrosylation-induced Conformational Change in Blackfin Tuna Myoglobin. J. Biol. Chem. 282, 19773–19780. [DOI] [PubMed] [Google Scholar]
- [64].Chan NL, Kavanaugh JS, Rogers PH, and Arnone A (2004) Crystallographic Analysis of the Interaction of Nitric Oxide with Quaternary-T Human Hemoglobin. Biochemistry 43, 118–132. [DOI] [PubMed] [Google Scholar]
- [65].Yi J, Soares AS, and Richter-Addo GB (2014) Crystallographic Characterization of the Nitric Oxide Derivative of R-State Human Hemoglobin. Nitric Oxide 39, 46–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Crystal structures of a double-mutant F43H/H64A Mb-nitrite and its MbNO derivative have been published, but these do not display direct NOx binding to the heme Fe center: Wu LB, Yuan H, Gao SQ, You Y, Nie CM, Wen GB, Lin YW, and Tan XS (2016) Regulating the Nitrite Reductase Activity of Myoglobin by Redesigning the Heme Active Center. Nitric Oxide 57, 21–29. [DOI] [PubMed] [Google Scholar]
- [67].Blaxter ML, Vanfleteren JR, Xia J, and Moens L (1994) Structural Characterization of an Ascaris Myoglobin. J. Biol. Chem. 269, 30181–30186. [PubMed] [Google Scholar]
- [68].Kloek AP, Sherman DR, and Goldberg DE (1993) Novel Gene Structure and Evolutionary Context of Caenorhabditis elegans Globin. Gene 129, 215–221. [DOI] [PubMed] [Google Scholar]
- [69].Romeroherrera AE, Goodman M, Dene H, Bartnicki DE, and Mizukami H (1981) An Exceptional Amino-Acid Replacement on the Distal Side of the Iron Atom in Proboscidean Myoglobin. J. Mol. Evol. 17, 140–147. [DOI] [PubMed] [Google Scholar]
- [70].Tada T, Watanabe Y, Matsuoka A, Ikeda-Saito M, Imai K, Yukio N, and Shikama K (1998) African Elephant Myoglobin with an Unusual Autoxidation Behavior: Comparison with the H64Q Mutant of Sperm Whale Myoglobin. Biochim. Biophys. Acta - Prot. Struct. Mol. Enzymol. 1387, 165–176. [DOI] [PubMed] [Google Scholar]
- [71].Fisher WK, Koureas DD, and Thompson EOP (1980) Myoglobins of Cartilaginous Fishes. 2. Isolation and Amino-Acid-Sequence of Myoglobin of the Shark Mustelus-Antarcticus. Aus. J. Biol. Sci. 33, 153–167. [DOI] [PubMed] [Google Scholar]
- [72].Fisher WK, Koureas DD, and Thompson EOP (1981) Myoglobins of Cartilaginous Fishes. 3. Amino-Acid-Sequence of Myoglobin of the Shark Galeorhinus-Australis. Aust. J. Biol. Sci. 34, 5–10. [PubMed] [Google Scholar]
- [73].Suzuki T, Suzuki T, and Yata T (1985) Shark Myoglobins. 2. Isolation, Characterization and Amino-Acid Sequence of Myoglobin from Galeorhinus-Japonicus. Aust. J. Biol. Sci. 38, 347–354. [DOI] [PubMed] [Google Scholar]
- [74].Stenzel P, Brimhall B, Jones RT, Black JA, Mclachlan A, and Gibson D (1979) Opossum Hemoglobin - Amino-Acid Sequences of the Alpha and Beta Chains. J. Biol. Chem. 254, 2071–2076. [PubMed] [Google Scholar]
- [75].Conti E, Moser C, Rizzi M, Mattevi A, Lionetti C, Coda A, Ascenzi P, Brunori M, and Bolognesi M (1993) X-ray Crystal Structure of Ferric Aplysia limacina Myoglobin in Different Liganded States. J. Mol. Biol. 233, 498–508. [DOI] [PubMed] [Google Scholar]
- [76].Suzuki T (1986) Amino Acid Sequence of Myoglobin from the Mollusc Dolabella auricularia. J. Biol. Chem. 261, 3692–3699. [PubMed] [Google Scholar]
- [77].Suzuki T, and Furukohri T (1990) Amino Acid Sequence of Myoglobin from the Mollusc Bursatella leachii. J. Protein Chem. 9, 69–73. [DOI] [PubMed] [Google Scholar]
- [78].Blaxter ML, Ingram L, and Tweedie S (1994) Sequence, Expression and Evolution of the Globins of the Parasitic Nematode Nippostrongylus-Brasiliensis. Mol. Biochem. Parasit. 68, 1–14. [DOI] [PubMed] [Google Scholar]
- [79].Quillin ML, Arduini RM, Olson JS, and Philips GN Jr. (1993) High-Resolution Crystal Structures of Distal Histidine Mutants of Sperm Whale Myoglobin. J. Mol. Biol. 234, 140–155. [DOI] [PubMed] [Google Scholar]
- [80].Springer BA, and Sligar SG (1987) High-Level Expression of Sperm Whale Myoglobin in Escherichia coli. Proc. Natl. Acad. Sci. USA 84, 8961–8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Phillips GN, Arduini RM, Springer BA, and Sligar SG (1990) Crystal-Structure of Myoglobin from a Synthetic Gene. Proteins: Struc. Func. Genet. 7, 358–365. [DOI] [PubMed] [Google Scholar]
- 82.[] Smith RD (1999) Correlations Between Bound N-alkyl Isocyanide Orientations and Pathways for Ligand Binding in Recombinant Myoglobins, Ph.D. Thesis, Rice University, Houson, TX. [Google Scholar]
- [83].Antonini E, and Brunori M (1971) Hemoglobin and Myoglobin In Their Reactions With Ligands in Frontiers of Biology, (Neuberger A and Tatum EL Eds), North-Holland, Amsterdam, Vol. 21, pp 19–39. [Google Scholar]
- [84].Nicolis S, Monzani E, Ciaccio C, Ascenzi P, Moens L, and Casella L (2007) Reactivity and Endogenous Modification by Nitrite and Hydrogen Peroxide: Does Human Neuroglobin Act Only as a Scavenger? Biochem. J. 407, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Leslie AG, and Powell HR (2007) Processing Data with MOSFLM, In Evolving Methods for Macromolecular Crystallography, Springer, pp 41–51. [Google Scholar]
- [86].Leslie AGW (1992) Recent Changes to the MOSFLM Package for Processing Film and Image Plate Data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography No. 26, pp 27–33. [Google Scholar]
- [87].Otwinowski Z, and Minor W (1997) Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- [88].Evans PR, and Murshudov GN (2013) How Good are My Data and What is the Resolution? Acta Crystallogr. D 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, and Wilson KS (2011) Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. D 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) PHASER Crystallographic Software. J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Murshudov GN, Vagin AA, and Dodson EJ (1997) Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Cryst. D 53, 240–255. [DOI] [PubMed] [Google Scholar]
- [92].Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012) Towards Automated Crystallographic Structure Refinement with Phenix.refine. Acta Cryst. D 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Emsley P, and Cowtan K (2004) COOT: Model-Building Tools for Molecular Graphics. Acta Cryst. D 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- [94].Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: All-atom Structure Validation for Macromolecular Crystallography. Acta Cryst. D 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Schrödinger LLC. (2010) The PYMOL Molecular Graphics System 2.0 ed., New York, NY, 2018. [Google Scholar]
- [96].Read RJ, and Schierbeek AJ (1988) A Phased Translation Function. J. Appl. Crystallogr. 21, 490–495. [Google Scholar]
- [97].Vojtechovsky J, Chu K, Berendzen J, Sweet RM, and Schlichting I (1999) Crystal Structures of Myoglobin-Ligand Complexes at Near-Atomic Resolution. Biophys. J. 77, 2153–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Yi J, Thomas LM, and Richter-Addo GB (2012) Distal Pocket Control of Nitrite Binding in Myoglobin. Angew. Chem. Int. Ed. Engl. 51, 3625–3627. [DOI] [PubMed] [Google Scholar]
- [99].Frisch MJ, et al. (2010) Gaussian 09; Revision B.01, Gaussian, Inc., Wallingford CT. [Google Scholar]
- [100].Yang L, Ling Y, and Zhang Y (2011) HNO Binding in a Heme Protein: Structures, Spectroscopic Properties, and Stabilities. J. Am. Chem. Soc. 133, 13814–13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Span I, Wang K, Eisenreich W, Bacher A, Zhang Y, Oldfield E, and Groll M (2014) Insights into the Binding of Pyridines to the Iron-Sulfur Enzyme IspH. J. Am. Chem. Soc. 136, 7926–7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Abucayon EG, Khade RL, Powell DR, Zhang Y, and Richter-Addo GB (2016) Hydride Attack on a Coordinated Ferric Nitrosyl: Experimental and DFT Evidence for the Formation of a Heme Model-HNO Derivative. J. Am. Chem. Soc. 138, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Abucayon EG, Khade RL, Powell DR, Shaw MJ, Zhang Y, and Richter-Addo GB (2016) Over or Under: Hydride Attack at the Metal versus the Coordinated Nitrosyl Ligand in Ferric Nitrosyl Porphyrins. Dalton Trans. 45, 18259–18266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Adamo C, and Barone V (1998) Exchange Functionals with Improved Long-Range Behavior and Adiabatic Connection Methods Without Adjustable Parameters: The mPW and mPW1PW Models. J. Chem. Phys. 108, 664–675. [Google Scholar]
- [105].Cossi M, Barone V, Cammi R, and Tomasi J (1996) Ab initio Study of Solvated Molecules: A New Implementation of the Polarizable Continuum Model. Chem. Phys. Lett. 255, 327–335. [Google Scholar]
- [106].Mennucci B, and Tomasi J (1997) Continuum Solvation Models: A New Approach to the Problem of Solute’s Charge Distribution and Cavity Boundaries. J. Chem. Phys. 106, 5151–5158. [Google Scholar]
- [107].Cossi M, Barone V, Mennucci B, and Tomasi J (1998) Ab initio Study of Ionic Solutions by a Polarizable Continuum Dielectric Model. Chem. Phys. Lett. 286, 253–260. [Google Scholar]
- [108].Cossi M, Scalmani G, Rega N, and Barone V (2002) New Developments in the Polarizable Continuum Model for Quantum Mechanical and Classical Calculations on Molecules in Solution. J. Chem. Phys. 117, 43–54. [Google Scholar]
- [109].Wachters AJ (1970) Gaussian Basis Set for Molecular Wavefunctions Containing Third-Row Atoms. J. Chem. Phys. 52, 1033–1036. [Google Scholar]
- [110].Vreven T, Byun KS, Komaromi I, Dapprich S, Montgomery JA, Morokuma K, and Frisch MJ (2006) Combining Quantum Mechanics Methods with Molecular Mechanics Methods in ONIOM. J. Chem. Theor. Comput. 2, 815–826. [DOI] [PubMed] [Google Scholar]
- [111].Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, and Kollman PA (1995) A 2nd Generation Force-Field for the Simulation of Proteins, Nucleic-Acids, and Organic-Molecules. J. Am. Chem. Soc. 117, 5179–5197. [Google Scholar]
- [112].Torrent M, Vreven T, Musaev DG, Morokuma K, Farkas O, and Schlegel HB (2002) Effects of the Protein Environment on the Structure and Energetics of Active Sites of Metalloenzymes. ONIOM Study of Methane Monooxygenase and Ribonucleotide Reductase. J. Am. Chem. Soc. 124, 192–193. [DOI] [PubMed] [Google Scholar]
- [113].Chung LW, Li X, Sugimoto H, Shiro Y, and Morokuma K (2010) ONIOM Study on a Missing Piece in Our Understanding of Heme Chemistry: Bacterial Tryptophan 2,3-Dioxygenase with Dual Oxidants. J. Am. Chem. Soc. 132, 11993–12005. [DOI] [PubMed] [Google Scholar]
- [114].Kawatsu T, Lundberg M, and Morokuma K (2011) Protein Free Energy Corrections in ONIOM QM:MM Modeling: A Case Study for Isopenicillin N Synthase (IPNS). J. Chem. Theor. Comput. 7, 390–401. [DOI] [PubMed] [Google Scholar]
- [115].Besler BH, Merz KM, and Kollman PA (1990) Atomic Charges Derived from Semiempirical Methods. J. Comput. Chem. 11, 431–439. [Google Scholar]
- [116].AMBER Parameter Database. Contributed by D. A. Giammona, Ph.D. Thesis, University of California at Davis, U.S.A.,1984. http://www.pharmacy.manchester.ac.uk/bryce/amber/. [Google Scholar]
- [117].Khade RL, Yang YW, Shi YL, and Zhang Y (2016) HNO-Binding in Heme Proteins: Effects of Iron Oxidation State, Axial Ligand, and Protein Environment. Angew. Chem. Int. Ed. 55, 15058–15061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Herold S, and Rehmann F-JK (2001) Kinetic and Mechanistic Studies of the Reactions of Nitrogen Monoxide and Nitrite with Ferry Myoglobin. J. Biol. Inorg. Chem. 6, 543–555. [DOI] [PubMed] [Google Scholar]
- [119].Herold S, and Rehmann F-JK (2003) Kinetics of the Reactions of Nitrogen Monoxide and Nitrite with Ferryl Hemoglobin. Free Radical Biol. Med. 34, 531–545. [DOI] [PubMed] [Google Scholar]
- [120].Borisov VB, Forte E, Sarti P, Brunori M, Konstantinov AA, and Giuffre A (2006) Nitric Oxide Reacts with the Ferryl-Oxo Catalytic Intermediate of the CuB-lacking Cytochrome bd Terminal Oxidase. FEBS Lett. 580, 4823–4826. [DOI] [PubMed] [Google Scholar]
- [121].Wanat A, Gdula-Argasinska J, Rutkowska-Zbik D, Witko M, Stochel G, and van Eldik R (2002) Nitrite Binding to Metmyoglobin and Methemoglobin in Comparison to Nitric Oxide Binding. J. Biol. Inorg. Chem. 7, 165–176. [DOI] [PubMed] [Google Scholar]
- [122].Yi J, and Richter-Addo GB (2012) Unveiling the Three-Dimensional Structure of the Green Pigment of Nitrite-Cured Meat. Chem. Commun. 48, 4172–4174. [DOI] [PubMed] [Google Scholar]
- [123].Wang B, Thomas LM, and Richter-Addo GB (2016) Organometallic Myoglobins: Formation of Fe-Carbon Bonds and Distal Pocket Effects on Aryl Ligand Conformations. J. Inorg. Biochem. 164, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Nutt DR, Karplus M, and Meuwly M (2005) Potential Energy Surface and Molecular Dynamics of MbNO: Existence of an Unexpected FeON Minimum. J. Phys. Chem. B 109, 21118–21125. [DOI] [PubMed] [Google Scholar]
- [125].Nutt DR, and Meuwly M (2007) Ferric and Ferrous Iron in Nitroso-Myoglobin: Computer Simulations of Stable and Metastable States and their Infrared Spectra. ChemPhysChem 8, 527–536. [DOI] [PubMed] [Google Scholar]
- [126].Cheng L, Novozhilova I, Kim C, Kovalevsky A, Bagley KA, Coppens P, and Richter-Addo GB (2000) First Observation of Photoinduced Nitrosyl Linkage Isomers of Iron Nitrosyl Porphyrins. J. Am. Chem. Soc. 122, 7142–7143. [Google Scholar]
- [127].Lee J, Kovalevsky AY, Novozhilova IV, Bagley KA, Coppens P, and Richter-Addo GB (2004) Single- and Double-Linkage Isomerism in a Six-Coordinate Iron Porphyrin Containing Nitrosyl and Nitro Ligands. J. Am. Chem. Soc. 126, 7180–7181. [DOI] [PubMed] [Google Scholar]
- [128].Novozhilova IV, Coppens P, Lee J, Richter-Addo GB, and Bagley KA (2006) Experimental and Density Functional Theoretical Investigations of Linkage Isomerism in Six-Coordinate {FeNO}6 Iron Porphyrins with Axial Nitrosyl and Nitro Ligands. J. Am. Chem. Soc. 128, 2093–2104. [DOI] [PubMed] [Google Scholar]
- [129].Wyllie GRA, Schultz CE, and Scheidt WR (2003) Five- to Six-Coordination in (Nitrosyl)iron(II) Porphyrinates: Effects of Binding the Sixth Ligand. Inorg. Chem. 42, 5722–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Zhang Y, Gossman W, and Oldfield E (2003) A Density Functional Theory Investigation of Fe-N-O Bonding in Heme Proteins and Model Systems. J. Am. Chem. Soc. 125, 16387–16396. [DOI] [PubMed] [Google Scholar]
- [131].Xu C, and Spiro TG (2008) Ambidentate H-Bonding by Heme-Bound NO: Structural and Spectral Effects of -O versus -N H-Bonding. J. Biol. Inorg. Chem. 13, 613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Silvernail NJ, Barabanschikov A, Sage JT, Noll BC, and Scheidt WR (2009) Mapping NO Movements in Crystalline [Fe(Porph)(NO)(1-MeIm)]. J. Am. Chem. Soc. 131, 2131–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.