

Abstract
PURPOSE
Broad-panel sequencing of tumors facilitates routine care of people with cancer as well as clinical trial matching for novel genome-directed therapies. We sought to extend the use of broad-panel sequencing results to survival stratification and clinical outcome prediction.
METHODS
By using sequencing results from a cohort of 1,054 patients with advanced lung adenocarcinomas, we developed OncoCast, a machine learning tool for survival risk stratification and biomarker identification.
RESULTS
With OncoCast, we stratified this patient cohort into four risk groups on the basis of tumor genomic profile. Patients whose tumors harbored a high-risk profile had a median survival of 7.3 months (95% CI, 5.5 to 10.9 months) compared with a low-risk group with a median survival of 32.8 months (95% CI, 26.3 to 38.5 months) with a hazard ratio of 4.6 (P < .001), far superior to any individual gene predictor or standard clinical characteristics. We found that comutations of both STK11 and KEAP1 are strong determinants of unfavorable prognosis with currently available therapies. In patients with targetable oncogenes (eg, EGFR, ALK, ROS1) who received targeted therapies, the tumor genetic background additionally differentiated survival with mutations in TP53 and ARID1A, which contributed to a higher risk score for shorter survival.
CONCLUSION
A mutational profile derived from broad-panel sequencing presents an effective genomic stratification for patient survival in advanced lung adenocarcinoma. OncoCast is available as a public resource that facilitates the incorporation of mutational data to predict individual patient prognosis and compare risk characteristics of patient populations.
INTRODUCTION
With the growth of precision medicine programs driven by genomic testing as well as recent US Food and Drug Administration clearance of next-generation sequencing (NGS) platforms for clinical use, there has been rapid growth in the availability of broad-panel sequencing data for patients with cancer. Broad-panel sequencing facilitates clinical trial matching of novel genome-directed therapies. Zehir et al1 delineated the molecular landscape of 10,000 metastatic cancers in a pretreated real-world cohort sequenced by the Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) platform, a hybridization capture-based NGS panel that can detect mutations and copy number alterations in 341 or more cancer-associated genes.2 This study showed that 37% of patients harbored at least one therapeutically actionable alteration, and 11% were matched to genome-directed clinical trials.
In patients with lung adenocarcinoma, tumor genotyping is now an essential step in routine clinical decision making. To determine treatment, patients with lung adenocarcinomas are currently categorized on the basis of the presence of mutated driver oncogenes (eg, EGFR, ALK, ROS1, and BRAF). A multi-institutional study characterized genetic aberrations across 10 genes in 733 tumor samples and identified an oncogenic driver in 64% of the patients.3 By using broad-panel sequencing, Jordan et al4 reported on a single-institution experience of 860 patients with metastatic lung adenocarcinoma. More than 37% of patients received a matched therapy, and the use of matched therapy was strongly influenced by the level of pre-existent clinical evidence that the mutation identified predicts the drug response.
Although the focus of tumor genotyping has been on ascertaining mutations that identify therapeutic targets, there is considerable unexplained variability in clinical outcomes, even within specific molecular subsets of patients with metastatic cancer. Similarly, association between high mutational load and clinical benefit has been observed in patients treated with programmed cell death protein 1/programmed death-ligand 1 (PD-1/PD-L1) inhibitors.5 However, additional markers are needed to predict durable benefit and long-term survival among these patients. Previous attempts to evaluate the effects of comutations have had a relatively limited scope. Some studies have explored the effects of single co-occurring alterations on outcome in patients with EGFR-mutant and KRAS-mutant lung adenocarcinoma, and they observed that the presence or absence of pairs of co-occurring events could be used to identify those patients with a poor prognosis most in need of novel therapeutic approaches.3,6-8 However, a systematic approach is needed to additionally improve our understanding of survival and treatment outcome of patients.
Thus, we developed OncoCast, a computational tool for survival stratification, and applied it to a large clinical series of patients with metastatic lung adenocarcinomas to improve the understanding of heterogeneity in clinical outcome for these patients and the mutation and comutational patterns that underlie such heterogeneity. Analysis of this large clinical cohort provides real-world evidence for understanding survival outcome for patients undergoing current standard care for advanced lung adenocarcinomas. Such real-world evidence can supplement the information from randomized clinical trials in that the results are more generalizable to patients treated outside randomized clinical trials, and the larger sample sizes of real-world data sets allow subset analysis that clinical trials are not powered for. The open-source computational pipeline that we have developed can facilitate the application of statistical and machine learning approaches for clinico-genomics analysis of precision medicine data sets.
METHODS
The OncoCast method is described in the Data Supplement. R package software is available at https://github.com/shenmskcc/OncoCast. An interactive Web interface was developed by using R Shiny with two main functions: GeneView and PatientView. In GeneView, users can interactively explore gene importance and comutation patterns by risk groups. In PatientView, users can type in a patient’s mutational profile and specify the clinical characteristics. The genomic risk score along with the predicted probability of survival at different time marks will be calculated and will be viewable in a dynamic plot. The Shiny app is available at https://github.com/shenmskcc/LungIMPACT.
RESULTS
Cohort Characteristics
Consecutive patients with metastatic or recurrent lung adenocarcinomas for which MSK-IMPACT data were available were included. Electronic medical records were used to identify patient clinical factors as well as survival outcomes as previously described.4 Overall survival (OS) was defined as the time from date of diagnosis of advanced disease (stage IV or recurrent cancer) until date of death or last follow-up. In this cohort, the majority (68%) of the tumors in our cohort were biopsied and sequenced within 30 days of diagnosis of metastatic disease. However, a fraction (21%) of the tumors were sampled and sequenced more than 6 months from the date of metastatic disease, with 16% sequenced at more than 1 year and 8% sequenced at more than 2 years, which represents older samples used for sequencing analysis. Those patients with older samples which had been taken at initial diagnosis of advanced lung cancer were immortal from their initial sampling time to the time of referral for MSK-IMPACT sequencing. This interval can be long for a small fraction of patients (8% with a delayed interval of more than 2 years, as mentioned earlier), which introduces survival bias. Left truncation was used to adjust for this bias. Details of left-truncation analysis are described in the Data Supplement. The most frequently mutated genes were TP53 (55.1%), KRAS (30%), EGFR (29.4%), STK11 (17.7%), and KEAP1 (17.7%; Data Supplement). Among the frequently comutated gene pairs, STK11 and KEAP1 were comutated in 10% of the tumors, and KRAS and STK11 were comutated in 9% of the tumors (Data Supplement).
Prognostic Relevance and Clonality of Cancer Genes
To define the prognostic significance of MSK-IMPACT panel genes that were sequenced, we developed OncoCast, a machine learning tool for survival risk stratification and biomarker identification by implementing a lasso-penalized proportional hazards regression for deriving prediction rules for OS and feature selection (Data Supplement). OncoCast uses an ensemble learning strategy by repeatedly splitting the cohort into training and test sets that generate an ensemble of classifiers with varying selection of genes and gene combinations (Data Supplement).
The median number of prognostic genes selected was 19 (range, 4-67 genes), with a total of 169 cancer genes selected at least once by the ensemble learner. The relative prognostic importance of each gene was measured by how often it was selected (fraction of the models containing the gene) and its average regression coefficients, which determined the corresponding weights for individual genes in the scoring rule. Figure 1A shows frequency of selection and average coefficient value for individual genes. STK11 and KEAP1 were observed to be highly prognostically relevant. They were followed in importance by TP53, KRAS, SMARCA4, and EGFR. An additional 21 genes were selected in at least 20% of the models (Fig 2A), including ALK fusion (58%), ROS1 fusion (22%), and ERBB2 mutation (31%), additionally highlighting the power of our approach to aggregate effects from rare events, which otherwise would not be included in standard association analyses.
FIG 1.
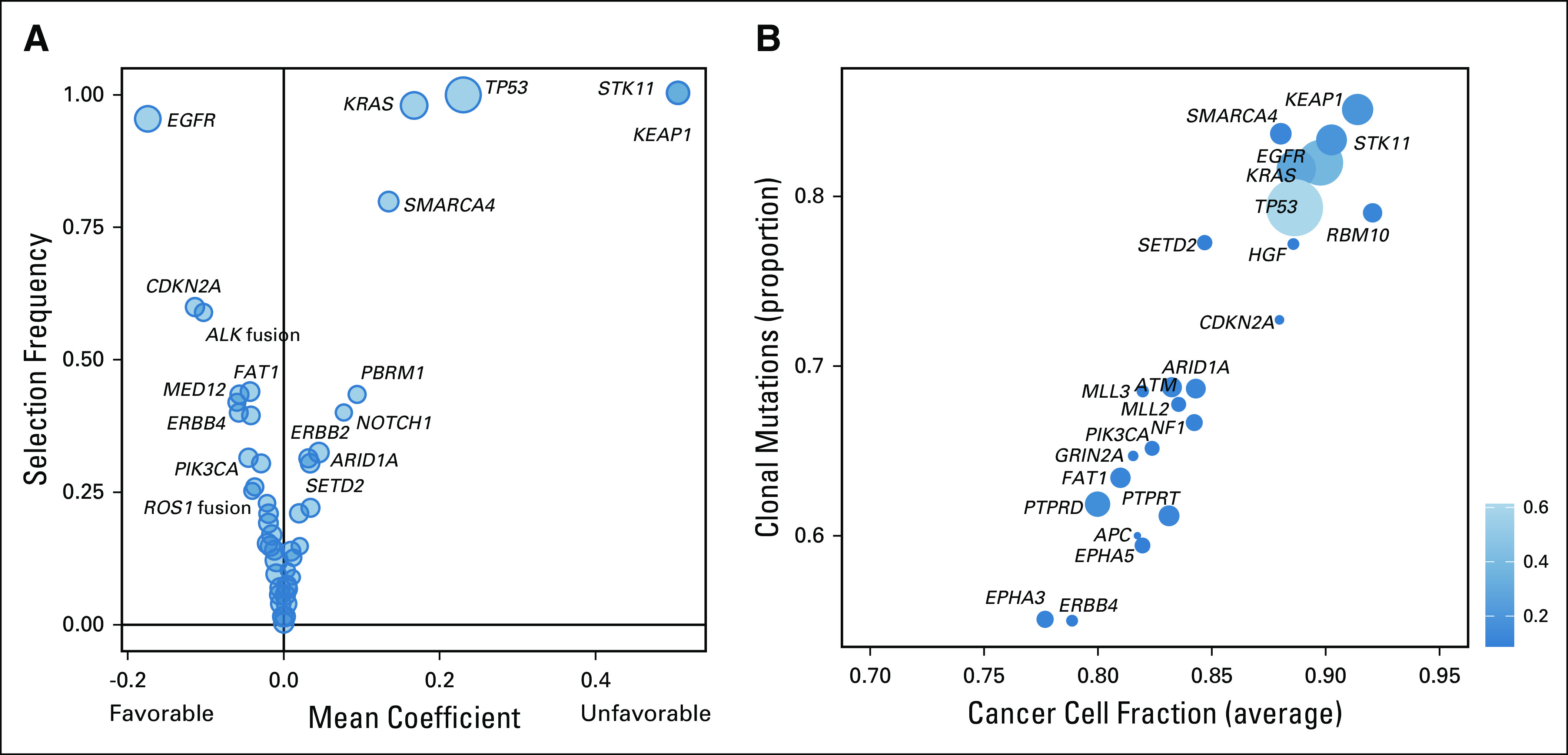
Prognostic relevance and clonality of cancer genes. (A) Plot of selection frequency in each model and regression coefficients for individual genes; the black vertical bar shows favorable versus unfavorable association with overall survival. (B) Clonality analysis of the cancer gene alterations. Circle size is proportional to mutation frequency.
FIG 2.

An integrated prognostic scoring system for metastatic lung adenocarcinomas. (A) Histogram of the prognostic risk score computed using the OncoCast model in 1,054 metastatic lung adenocarcinomas. The risk score is scaled from 0 to 10, with a higher score indicating higher likelihood of shorter survival. Dashed lines indicate the percentile cutoffs used to stratify patients into four risk subgroups (low, intermediate-low, intermediate-high, and high). (B) Boxplots of concordance index for predicting overall survival using clinical demographic factors including age, sex, smoking, and the OncoCast risk score. CN, copy number; mut, mutation.
Several recent studies examining tumor heterogeneity have shown that clonal or truncal drug targets and/or tumor neoantigens are more predictive of response to systemic targeted and immunotherapies than subclonal events.9-11 We thus used the high depth of coverage afforded by our sequencing data (mean coverage, 758 ×) to determine the clonality of the gene alterations found to be associated with prognosis in the analysis we outlined earlier. Clonality analysis revealed that all of the most prognostic alterations were predominantly clonal, with average cancer cell fraction above 85% (Fig 1B).
OncoCast: An Integrated Prognostic Scoring System Based on NGS Tumor Profiling
OncoCast aggregates prognostic effects across the sequenced cancer genes to derive a genomic risk score for each patient (scaled from 0 to 10), with a higher score indicating a greater likelihood of shorter survival. The distribution of the risk scores revealed a wide spread within the cohort (Fig 2A). To evaluate prognostic performance, we calculated the C-index which measures the concordance between the risk score and survival (Fig 2B). A three-fold cross-validation was used for unbiased assessment. Clinical demographic factors (including age, sex, smoking) were weakly concordant with survival, with median C-indices ranging from 0.53 to 0.57. By contrast, the OncoCast risk score as determined by tumor genomic profiling demonstrated a significantly better concordance with a median C-index above 0.65. In advanced or metastatic lung cancers, studies have shown only weak to moderate strength of survival association for clinical factors.12,13 The genomic classifier we developed here presents a significant improvement. In addition, the OncoCast classifier on the basis of clinical-grade sequencing as part of routine care highlights the immediate application and strong practical utility of this system. The prediction strength is comparable to established gene expression–based genomic classifiers in early-stage lung cancer from a large multisite study.14 Inclusion of copy number alteration (CNA) data in the analysis revealed no discernible improvement in C-index when the CNA data were incorporated (Fig 2B).
We categorized the patient cohort into low (0 to ≤ 25th percentile), low-intermediate (> 25th to 75th percentile), high-intermediate (> 75th to 90th percentile), and high (> 90th percentile and above) risk groups informed by the multiple modes of the risk score distribution (Fig 3A). For the low-risk group, median OS was 32.8 months (95% CI, 26.3 to 38.5 months). By contrast, for the high-risk group, the median survival was 7.3 months (95% CI, 5.5 to 10.9 months). There was a difference of more than 6 units in average risk score between the high- and low-risk groups. The OncoCast classification substantially outperformed all of the individual genes as a predictor of OS (Fig 3B). The hazard ratio (HR) was 4.6 (95% CI, 3.2 to 6.5) for the OncoCast risk score for the high-risk versus the low-risk group, far superior to clinical factors or any individual gene. OncoCast risk score remained a highly significant predictor after adjusting for clinical variables and treatment types as potential confounding factors in a multivariable Cox regression model (Data Supplement).
FIG 3.
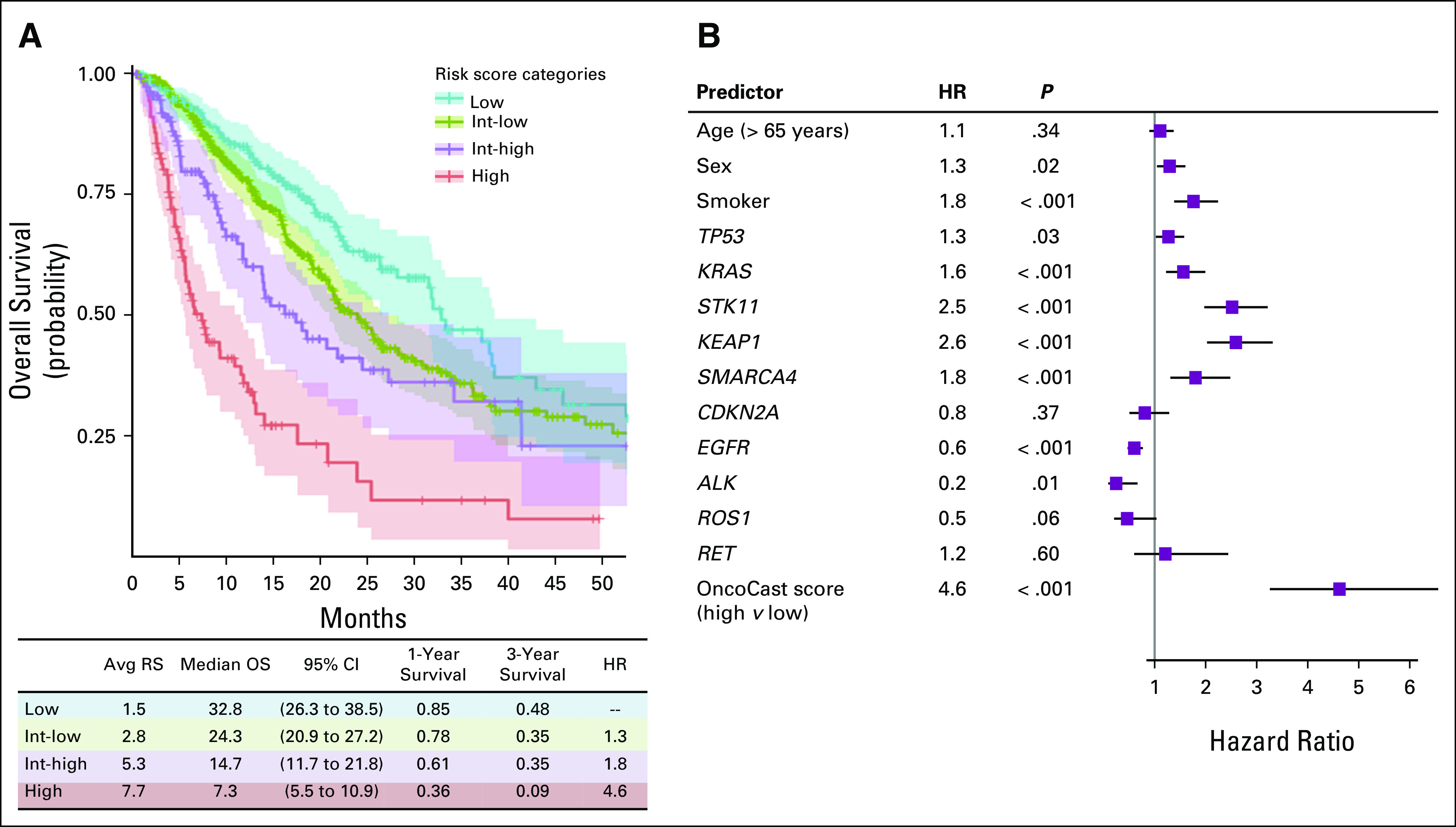
Outcome prediction on the basis of risk score. (A) Kaplan-Meier plot of survival curves for the four risk subgroups (Low, intermediate-low [Int-low], intermediate-high [Int-high], and high). Colored areas represent 95% CIs. The average risk score (Avg RS), median overall survival (OS), and 1-year and 3-year survival probabilities are reported for each group. (B) Forest plot of hazard ratios (HRs) for age, sex, smoking status, and individual driver gene alterations compared with the OncoCast integrated scoring approach.
To confirm the validity of the OncoCast survival stratification, we applied it to a separate data set of patients with mostly early-stage non–small-cell lung cancer obtained from The Cancer Genome Atlas lung adenocarcinoma analysis. We saw a similar stratification of the risk groups (Data Supplement). The high-risk group showed significantly worse OS with an HR of 1.9 (P = .03), and it was highly enriched for concurrent STK11 and KEAP1 mutations.
Prognostic Mutation and Comutation Patterns
Overlaying the OncoCast risk score with the mutational landscape in an OncoPrint plot highlights that the tumors with the highest-risk profile were enriched for comutation of STK11 and KEAP1 (Fig 4). STK11 is a tumor suppressor gene that encodes for the serine/threonine kinase LKB1 that functions as a negative regulator of mammalian target of rapamycin (mTOR) signaling. Consistent with its functional role as a tumor suppressor, the majority of STK11 mutations were truncating and frameshift indels. To examine the status of the other allele in such cases, we performed allele-specific CNA using the Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing (FACETS) algorithm.15 Strikingly, more than 90% of STK11 mutant tumors had evidence of biallelic inactivation through loss of heterogeneity (LOH) of chromosome 19p (Data Supplement). KEAP1 also resides in the 19p region with 89% of KEAP1-mutant tumors demonstrating LOH. The majority of KEAP1 mutations were missense, and the mutant copy was frequently duplicated (present as copy-neutral LOH and uniparental gains) as reflected in the average total copy number.
FIG 4.
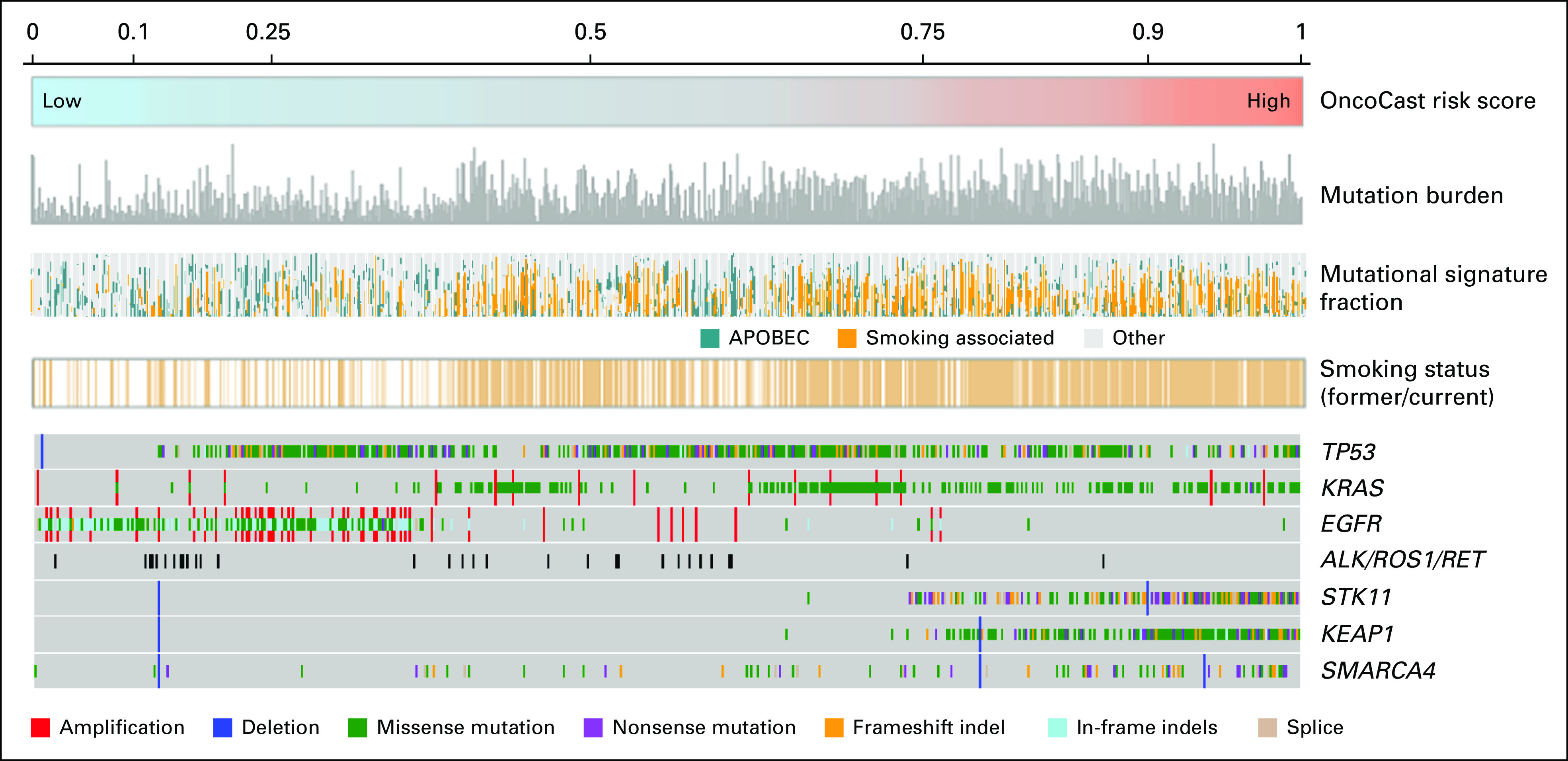
Overlaying OncoCast risk score with tumor mutation burden, mutational signature, smoking status, and mutation status for commonly mutated genes. APOBEC, apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like.
We also explored whether other tumor characteristics such as tumor mutational signature or intratumor heterogeneity were prognostic and associated with OncoCast risk score. Previously defined smoking-associated and apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) signatures were the most prevalent mutational signatures in this cohort. The identification of a smoking signature was highly concordant with patient reported smoking status (Fig 4). An APOBEC signature was enriched in low-risk groups and in patients who self-reported as never smokers. The median mutation burden in the overall data set was 9.95 mutations per Mb (interquartile range, 4.97-19.07). Tumor mutation burden was associated with risk groups that had medians of 16.58, 16.05, 9.95, and 5.76 mutations per Mb in the high-, high-intermediate-, low-intermediate-, and low-risk groups, respectively (Kruskal-Wallis test P < .001; Data Supplement). However, mutation burden did not provide additional prognostic value beyond OncoCast risk score in a multivariable Cox model (Data Supplement).
Clonal diversity was calculated for each tumor by summarizing the cancer cell fraction for all somatic mutations using the Shannon index. A diversity index of zero represents homogeneity, in which case all of the mutations detected in the tumor are clonal (cancer cell fraction, 100%). The median diversity index in the cohort was 1.19 (range, 0.00-56.32). A trend of increased clonal heterogeneity was observed in high-risk groups (Data Supplement). Again, clonal heterogeneity did not provide additional prognostic value on top of the OncoCast risk score.
Genotypes Associated With Risk Groups
To better understand the association between targetable cancer driver mutations and OncoCast risk score, we explored the distribution of genotypes within four major risk categories (Fig 5). The low-risk group was highly enriched for EGFR mutants and tumors harboring oncogenic fusions in ALK, RET, and ROS1. However, 30% of the low-risk group tumors lacked targetable alterations in these four genes. The defining characteristic for the patients in the low-risk group without EGFR, ALK, RET, and ROS1 alterations was an absence of any of the poor prognostic gene alterations. In the high-risk groups, common comutation patterns were observed. Strikingly, more than 95% of the patients in the high-risk group had tumors that harbored comutations of KEAP1 and STK11. The top three major genotype categories in the high-risk group were KRAS-STK11-KEAP1, TP53-STK11-KEAP1, and KRAS-STK11-KEAP1-SMARCA4. Furthermore, when compared with The Cancer Genome Atlas resected lung adenocarcinoma cohort, STK11 and KEAP1 comutation was two-fold more common in the metastatic lung cohort (10.2% v 5.2%; P < .001). This suggests that STK11 and KEAP1 comutation defines a cohort of patients with resected lung adenocarcinoma at higher risk of disease progression and cancer-specific mortality.
FIG 5.

Genomic risk stratification and distribution of common mutation and comutation patterns by each risk group.
Survival Stratification in Specific Treatment Subsets
We then sought to demonstrate the ability of the OncoCast technique to explore tumor genomic predictor outcomes in the subset of patients with mutated driver oncogenes treated with kinase inhibitors. The model, OncoCast-TR (targeted therapies model), was derived using the genomic profiles and survival outcomes from the start of therapy for the 387 patients who received targeted therapies (EGFR, ALK, ROS). Survival time was calculated from the start of treatment to death or last follow-up. Late entry was accounted for by using left-truncation analysis and incorporating time-to-tumor sampling and sequencing. The risk score from the OncoCast-TR model clearly indicated two groups with distinct survival differences (Fig 6A and 6B). TP53 is strongly associated with worse survival outcome (Fig 6C), with 96% of patients in the TR2 group harboring concurrent TP53 mutations and 0% concurrent TP53 mutations in the TR1 group. ARID1A also showed association with poor survival with 11 (78%) of 14 ARID1A mutations in TR2.
FIG 6.
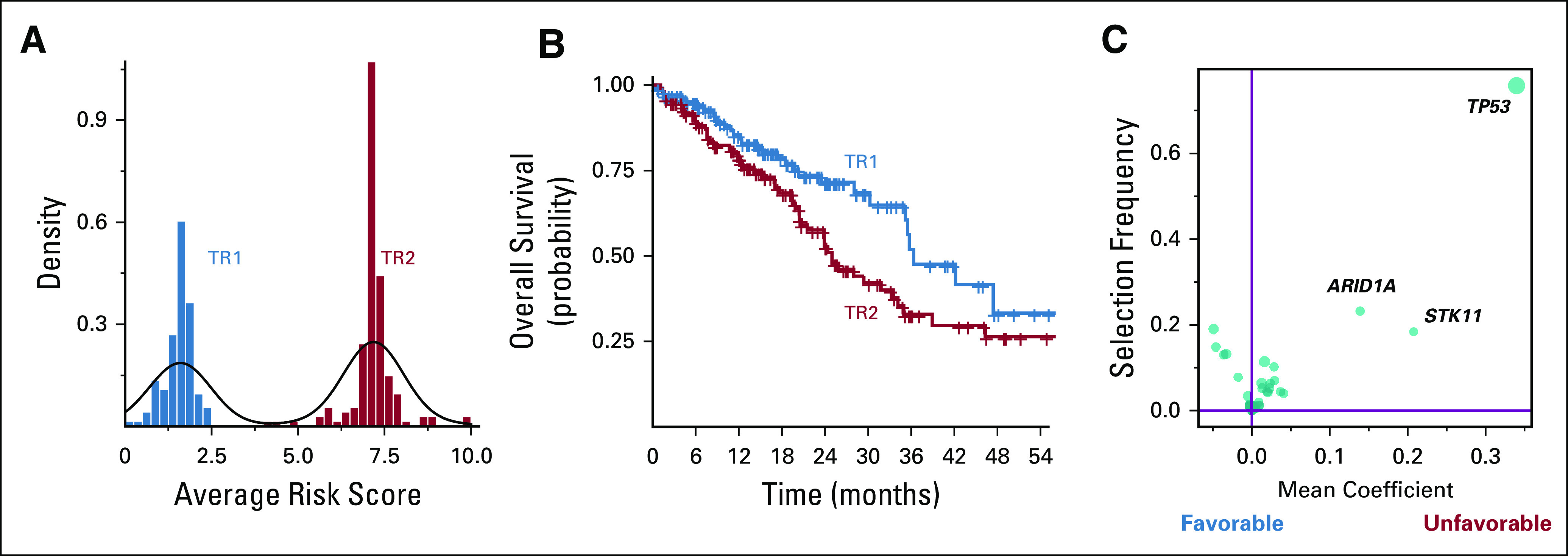
(A) Risk score distribution from the OncoCast-TR (targeted therapies) model stratifies patients into two subsets. (B) Overall survival probability for the two TR patient subsets (TR1 and TR2). (C) Gene importance plot for the OncoCast-TR model.
Interactive Tool for Visualizing and Exploring Mutation Pattern and Survival
To facilitate clinical translation and research use of the data, we created an interactive Web application (Data Supplement) that allows for visualization of mutation patterns and individualized prediction of OS to be generated on the basis of a user-defined mutational profile and clinical characteristics. There are two main functional modules: GeneView and PatientView. In GeneView, the user can input the genes of interest and interactively explore the prognostic effects in a dynamic volcano plot and with genotype pie charts. In PatientView, an OncoCast risk score will be calculated from a user supplied mutational profile. Along with a patient’s clinical profile, the application outputs the predicted survival probabilities and 95% CIs. Although the tool was built using the largest metastatic lung adenocarcinoma cohort to date, it was designed to be dynamically updated as new data are incorporated into the model.
DISCUSSION
Tumors from patients with lung adenocarcinoma have a high frequency of actionable oncogenic drivers.16,17 The introduction of targeted therapy has transformed the clinical care of patients with lung adenocarcinoma by incorporating tumor genotyping into therapeutic decision making. However, the prognostic value of the additional information from broad genomic profiling has not been explored. We developed OncoCast, a statistical learning tool, for integrating broad genomic profiling data and clinical outcomes for survival stratification and the identification of associated biomarkers. By using an ensemble learning approach, we demonstrated the prognostic utility of data from a large panel NGS assay interrogating 341 cancer-associated genes in 1,054 patients with metastatic lung adenocarcinoma treated with currently available therapies. We show that it is a practical approach to molecular stratification in metastatic lung adenocarcinoma and provides the ability to identify biomarkers that predict survival outcome.
Overlaying the OncoCast risk score with the tumor genomic landscape revealed novel biologic and clinical insights. The major prognostic genes associated with poor survival included STK11, KEAP1, TP53, KRAS, and SMARCA4. Remarkably, comutations of both STK11 and KEAP1 defined an exceptionally high-risk profile with a short median OS of 7.3 months and an increase of more than 6 units in risk score compared with a low-risk group, corresponding to an HR of 4.6. The low-risk group had no mutations in these major poor prognostic genes. Some of the favorable prognostic genes may reflect the availability of effective targeted therapies in the case of EGFR, ALK, and ROS1. However, in patients with these targetable oncogenes, there is heterogeneity in the additional mutations their tumors harbor. Our model is novel in that it outputs a continuous risk score on the basis of the specific genetic background of the patient’s tumor, including additional genetic alterations beyond the driver oncogene. This provides finer granularity for understanding the heterogeneity in clinical outcome. For example, our model revealed that mutations in TP53 and ARID1A define a high-risk subgroup with shorter survival in the tyrosine kinase inhibitor–treated patient cohort.
Conversely, some of the unfavorable prognostic genes may reflect negative associations with treatment responses (eg, the recent observation that STK11 inactivation is associated with poor responses to immunotherapy18). Our results additionally highlight the significant risk imparted by concurrent mutations in genes such as KEAP1, STK11, and SMARCA4. KEAP1 is a negative regulator of NRF2. Mutations in KEAP1 are associated with chemotherapy resistance and poor survival. Some studies have suggested that targeting NRF2 may enhance chemotherapy sensitization.19,20 mTOR is a kinase downstream of LKB1 (STK11), and mTOR inhibitors have been proposed as a potential therapeutic approach in STK11-mutant tumors.21 SMARCA4 was also identified as a major driver of a poor prognosis factor in metastatic lung adenocarcinoma in our study. It is a core factor in switch/sucrose nonfermentable (SWI/SNF) chromatin remodeling complexes that regulate genomic instability and DNA repair. SMARCA4 was mutated in 9.4% of lung adenocarcinomas in the MSK-IMPACT cohort, with 42% of the mutations being truncating and residing in regions of LOH. It has recently been shown that SMARCA4-inactivating mutations increase sensitivity to Aurora kinase A inhibitor in NSCLCs.22
In a multivariable analysis (Data Supplement) that included treatment covariables, the OncoCast readout remained highly significant, which suggests that the genomic profile provides more than simple readout of genotype-matched therapies. In addition, this model can be dynamically updated over time. With increasing sample sizes, we will be poised to identify outcome associations with rare alterations, and we will be better able to explain the heterogeneity in clinical outcomes for patients with advanced lung adenocarcinoma.
The OncoCast risk score described here provides a valuable tool for more accurately determining the prognosis of patients enrolled in clinical trials or included in real-world data sets. A key component of any analysis of clinical research data is a clear description of the patient population being explored. Although conventional clinical factors such as age, sex, and performance status have long been used, our data indicate that we can significantly improve the description of patient populations by incorporating the genomic risk scores in our understanding to allow better comparisons of groups of patients enrolled in clinical trials or included in real-world data sets.
With the growth of precision medicine programs driven by genomic testing as well as recent US Food and Drug Administration clearance of NGS platforms for clinical use, there will be a rapid growth in the availability of broad sequencing data for patients with a variety of cancers and growing integration with clinical data through institutional databases and electronic health records. OncoCast, the computational tool discussed here, will facilitate the incorporation of mutational data as a stratification factor in both prospective clinical trials and retrospective/real-world data collections to more precisely describe patient populations, which will allow better generalization of the results from such research efforts.
Footnotes
Supported in part by Grant No. P01 CA129243 from the National Cancer Institute (NCI), by Grant No. P30CA008748 from NCI (Memorial Sloan Kettering Cancer Center), and the Dallepezze Family Foundation.
AUTHOR CONTRIBUTIONS
Conception and design: Ronglai Shen, Ai Ni, Matthew Hellmann, Mark G. Kris, Maria Arcila, Gregory J. Riely
Financial support: David B. Solit, Gregory J. Riely
Administrative support: Mark G. Kris, David B. Solit, Gregory J. Riely
Provision of study materials or patients: Mark G. Kris, David B. Solit, Gregory J. Riely
Collection and assembly of data: Ronglai Shen, Matthew Hellmann, Emmet Jordan, Ryan Ptashkin, Ahmet Zehir, Mark G. Kris, Charles M. Rudin, David B. Solit, Maria Arcila, Marc Ladanyi, Gregory J. Riely
Data analysis and interpretation: Ronglai Shen, Axel Martin, Ai Ni, Matthew Hellmann, Kathryn C. Arbour, Arshi Arora, Ryan Ptashkin, Ahmet Zehir, Mark G. Kris, Michael F. Berger, David B. Solit, Venkatraman E. Seshan, Marc Ladanyi, Gregory J. Riely
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST AND DATA AVAILABILITY STATEMENT
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or po.ascopubs.org/site/ifc.
Ronglai Shen
Research Funding: GRAIL
Matthew Hellmann
Stock and Other Ownership Interests: Shattuck Labs
Honoraria: AstraZeneca, Bristol-Myers Squibb
Consulting or Advisory Role: Bristol-Myers Squibb, Merck, Genentech, AstraZeneca/MedImmune, Novartis, Janssen, Nektar Therapeutics, Syndax Pharmaceuticals, Mirati Therapeutics, Shattuck Labs
Research Funding: Bristol-Myers Squibb (Inst)
Patents, Royalties, Other Intellectual Property: A patent has been filed by Memorial Sloan Kettering (PCT/US2015/062208) for the use of tumor mutation burden for prediction of immunotherapy efficacy, which is licensed to Personal Genome Diagnostics (Inst)
Travel, Accommodations, Expenses: AstraZeneca, Bristol-Myers Squibb
Kathryn C. Arbour
Consulting or Advisory Role: AstraZeneca
Ryan Ptashkin
Stock and Other Ownership Interests: Loxo Oncology, Array BioPharma
Mark G. Kris
Consulting or Advisory Role: AstraZeneca, Regeneron Pharmaceuticals, Pfizer
Travel, Accommodations, Expenses: AstraZeneca
Other Relationship: Memorial Sloan Kettering Cancer Center
Charles M. Rudin
Consulting or Advisory Role: Bristol-Myers Squibb, AbbVie, Seattle Genetics, Harpoon Therapeutics, Genentech, AstraZeneca, Ascentage Pharma, Bicycle Therapeutics, Celgene, Daiichi Sankyo, Ipsen, Loxo Oncology, PharmaMar, Elucida Oncology
Research Funding: AbbVie/Stemcentrx (Inst), Viralytics (Inst), Merck (Inst), Daiichi Sankyo (Inst)
Michael F. Berger
Consulting or Advisory Role: Roche
Research Funding: Illumina
David B. Solit
Stock and Other Ownership Interests: Loxo Oncology
Consulting or Advisory Role: Pfizer, Loxo Oncology, Illumina, Intezyne Technologies, Vivideon Therapeutics
Travel, Accommodations, Expenses: Merck
Maria Arcila
Consulting or Advisory Role: AstraZeneca
Travel, Accommodations, Expenses: AstraZeneca, Invivoscribe Technologies, Raindance Technologies
Marc Ladanyi
Honoraria: Merck (I)
Consulting or Advisory Role: National Comprehensive Cancer Network/AstraZeneca Tagrisso Request for Proposal Advisory Committee, Takeda Pharmaceuticals, Bristol-Myers Squibb, Bayer AG, Merck (I)
Research Funding: Loxo Oncology (Inst), Helsinn Therapeutics
Gregory J. Riely
Research Funding: Novartis (Inst), Genentech (Inst), Millennium Pharmaceuticals (Inst), GlaxoSmithKline (Inst), Pfizer (Inst), Infinity Pharmaceuticals (Inst), ARIAD Pharmaceuticals (Inst)
Patents, Royalties, Other Intellectual Property: Patent application submitted covering pulsatile use of erlotinib to treat or prevent brain metastases (Inst)
Travel, Accommodations, Expenses: Merck Sharp & Dohme
No other potential conflicts of interest were reported.
REFERENCES
- 1.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311:1998–2006. doi: 10.1001/jama.2014.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017;7:596–609. doi: 10.1158/2159-8290.CD-16-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hellmann MD, Nathanson T, Rizvi H, et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell. 2018;33:843–852.e4. doi: 10.1016/j.ccell.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arbour KC, Jordan E, Kim HR, et al. Effects of co-occurring genomic alterations on outcomes in patients with KRAS-mutant non-small cell lung cancer. Clin Cancer Res. 2018;24:334–340. doi: 10.1158/1078-0432.CCR-17-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu HA, Suzawa K, Jordan E, et al. Concurrent alterations in EGFR-mutant lung cancers associated with resistance to EGFR kinase inhibitors and characterization of MTOR as a mediator of resistance. Clin Cancer Res. 2018;24:3108–3118. doi: 10.1158/1078-0432.CCR-17-2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu HA, Sima CS, Shen R, et al. Prognostic impact of KRAS mutation subtypes in 677 patients with metastatic lung adenocarcinomas. J Thorac Oncol. 2015;10:431–437. doi: 10.1097/JTO.0000000000000432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Bruin EC, McGranahan N, Mitter R, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251–256. doi: 10.1126/science.1253462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizvi H, Sanchez-Vega F, La K, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36:633–641. doi: 10.1200/JCO.2017.75.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology: Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Azzoli CG, Temin S, Giaccone G. 2011 Focused update of 2009 American Society of Clinical Oncology Clinical Practice Guideline update on chemotherapy for stage IV non-small-cell lung cancer. J Oncol Pract. 2012;8:63–66. doi: 10.1200/JOP.2011.000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerber DE, Dahlberg SE, Sandler AB, et al. Baseline tumour measurements predict survival in advanced non-small cell lung cancer. Br J Cancer. 2013;109:1476–1481. doi: 10.1038/bjc.2013.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Director’s Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma. Shedden K, Taylor JM, et al. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat Med. 2008;14:822–827. doi: 10.1038/nm.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen R, Seshan VE. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clinical Lung Cancer Genome Project. Network Genomic Medicine A genomics-based classification of human lung tumors. Sci Transl Med. 2013;5:209ra153. doi: 10.1126/scitranslmed.3006802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12:175–180. doi: 10.1016/S1470-2045(10)70087-5. [DOI] [PubMed] [Google Scholar]
- 18.Skoulidis F, Goldberg ME, Greenawalt DM, et al. STK11/LKB1 mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018;8:822–835. doi: 10.1158/2159-8290.CD-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chian S, Thapa R, Chi Z, et al. Luteolin inhibits the Nrf2 signaling pathway and tumor growth in vivo. Biochem Biophys Res Commun. 2014;447:602–608. doi: 10.1016/j.bbrc.2014.04.039. [DOI] [PubMed] [Google Scholar]
- 20.Ren D, Villeneuve NF, Jiang T, et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci U S A. 2011;108:1433–1438. doi: 10.1073/pnas.1014275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klümpen H-J, Queiroz KCS, Spek CA, et al. mTOR inhibitor treatment of pancreatic cancer in a patient with Peutz-Jeghers syndrome. J Clin Oncol. 2011;29:e150–e153. doi: 10.1200/JCO.2010.32.7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tagal V, Wei S, Zhang W, et al. SMARCA4-inactivating mutations increase sensitivity to Aurora kinase A inhibitor VX-680 in non-small cell lung cancers. Nat Commun. 2017;8:14098. doi: 10.1038/ncomms14098. [DOI] [PMC free article] [PubMed] [Google Scholar]