

Abstract
Large-scale genomic data have been utilized to generate unprecedented biological findings and new hypotheses. To delineate functional elements in the human genome, the Encyclopedia of DNA Elements (ENCODE) project has generated an enormous amount of genomic data, yielding around 7,000 data profiles in different cell and tissue types. In this article, we reviewed the systematic analyses that have integrated ENCODE data with other data sources to reveal new biological insights, ranging from human genome annotation to the identification of new candidate drugs. These analyses demonstrate the critical impact of ENCODE data on basic biology and translational research.
Graphical Abstract
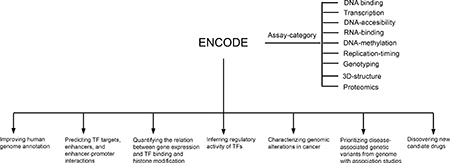
Introduction
The development of high-throughput technologies has generated enormous amounts of data which allow biologists to examine numerous biological hypotheses. In this review, we discuss how the Encyclopedia of DNA Elements (ENCODE) project has contributed to our understanding of many aspects of biology. The ENCODE project is an international collaborative project funded by the National Human Genome Institute (NHGRI). It aims to identify and characterize functional regions in the human genome by utilizing a variety of high-throughput approaches [1].
The pilot phase (2003–2007) of the ENCODE project was launched to explore 1% of the human genome to establish protocols for scaling up analyses to the entire genome [2–4]. During the pilot phase, a variety of experimental and computational methods were compared and refined for large-scale analyses. Then, the ENCODE project was expanded to the entire human genome during the production phase (2007–2012). In alignment with ENCODE, the modENCODE project [5] and the mouseENCODE project [6] were launched to systematically identify the DNA elements in the genomes of three model organisms including fly, worm and mouse. To date, ENCODE has generated abundant genomic data of different types as well as various tools and software. In total, 7,694 profiles of different assay categories have been released to the public so far as summarized in Table 1 [7].
Table 1.
Summary of ENCODE data [8].
| Assay category | DNA binding | Transcription | DNA-accessibility RNA-binding | DNA-methylation Replication timing | Genotyping | 3D-structure | Proteomics | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Assay name | ChIP-seq | shRNA RNA-seq, toal RNA-seq, RNA microarray, small RNA- seq, RAMPAGE, polyA RNA-seq, CAGE, CRISPRi RNA-seq, si RNA RNA-seq, CRISPR RNA-seq, single cell RNA-seq, microRNA counts, microRNA-seq, polyA depleted RNA-seq, RNA-PET | DNase-seq, ATAC-seq, genetic modification DNase-seq, FAIRE-seq, MNase-seq | eCLIP, RIP- seq, RIP- chip, ¡CLIP, Switchgear | DNAme array, RRBS, WGBS, MRE-seq, MeDIP-seq | Repli-seq, Repli- chip | genotyping array, DNA- PET, genotyping HTS | ChlA-PET, Hi-C, 5C | MS-MS | |
| Tier1 | K562 | 697 | 506 | 52 | 237 | 8 | 6 | 5 | 11 | 6 |
| GM1287B | 249 | 60 | 5 | 25 | 10 | 6 | 5 | 4 | 5 | |
| H1-hESC | 106 | 21 | 3 | 3 | 6 | 1 | 1 | 1 | 2 | |
| Tier2 | 978 | 481 | 38 | 172 | 34 | 40 | 13 | 16 | 1 | |
| Tier3 | 1065 | 400 | 255 | 6 | 252 | 61 | 86 | 40 | 0 | |
| Other | 926 | 382 | 190 | 0 | 116 | 35 | 32 | 34 | 0 | |
The number of datasets for each assay category across different Tiers, cell types and tissues are shown until May, 2018 (ENCODE data portal: https://www.encodeproject.org). The Tiers refer to the ENCODE classification system of cell lines according to priority of performing the assays. Tier1 cell lines were considered of the highest priority in ENCODE project and therefore the included cell lines (K562, GM12878 and H1-hESC) are presented individually. The assay category refers to the type of genomic features described by the assay.
Based on these data, many important biological analyses have been performed, including genome annotation, chromatin state classification, and the identification of regulatory regions in the genome [2,3]. Moreover, ENCODE data has been integrated with other data sources such as cancer data to gain new insights into cancer development and drug discovery. Up to May, 2018, 2563 articles have been published by the ENCODE project and its community, and 6,683 articles that cited the ENCODE landmark paper have been published (Figure 1) [2]. In this review, we focus on systematic analyses that have integrated ENCODE with other data sources to better understand complex biological processes and human diseases.
Figure 1.

Publications and citations based on the ENCODE project [2]. (A) The number of publications from ENCODE and its community until May, 2018 (ENCODE data portal: https://www.encodeproject.org). ~2007: all publications before 2007; 2015 ~: all publications after 2015. (B) The number of publications that have cited the ENCODE landmark paper until May, 2018 [2]. Citations are based on Google Scholar (https://scholar.google.com). ~2013: all publications before 2013.
Improving human genome annotation
The Human Genome Project has completed the sequence of the human genome in 2003, however, at that time the annotation of the genome was far from accurate [8]. Based on ENCODE data, protein-coding and noncoding transcripts, long non-coding RNAs and pseudogenes have been carefully annotated through a combination of computational analyses, manual annotation, and experiment validation [9,10]. According to the refined annotation, it is now estimated that a total of 74.7% of the human genome is covered by primary transcripts [3,9]. High-quality annotation would facilitate functional investigation of any genomic sequence in the human genome. For example, highthroughput annotations of long non-coding RNAs make it possible to explore their properties and functions with high efficiency [*11,12].
Using ChIP-seq data for eight histone modifications and one sequence-specific insulator protein, CTCF, Ernst et al. developed an algorithm named ChromHMM that segmentized the genomes of nine human cell lines into 15 different chromosome states [13,14]. Additionally, Hoffman et al. used 31 ChIP-seq, DNase-seq and FAIRE-seq profiles to develop a method called Segway that defined 25 chromosome states [15]. Using the integration of these two segmentation methods, the ENCODE project established a consensus set of seven principle genomic states: three related to gene transcription, three related to distal regulatory elements, and one related to actively repressed or inactive genomic regions [2]. Subsequent experimental studies confirmed the defined histone modification states of distal regulatory elements [16]. Additional segmentation methods have utilized this initial segmentation effort and expanded it in various ways, including methods that integrate cell type specificity [17], evaluate species-specific confounding factors [18], or take into account higher-order chromatin structure [19,**20]. Undoubtedly, the ENCODE project has significantly improved human genome annotation and segmentation.
Predicting transcription factor targets, enhancers, and enhancer-promoter interactions
Transcription factors (TFs) regulate gene transcription by binding to specific DNA elements in open chromatin regions. Human genome annotation based on ENCODE data has identified chromatin regions highly enriched for TF binding sites (TFBSs) [2]. Many promoters contain TF binding motifs, but only a small fraction of these motifs is actually bound by TFs and thus contain TFBSs. Additionally, enhancers that play critical roles in gene transcriptional regulation are also enriched for TFBSs. ENCODE data, including chromatin accessibility, histone modification, and TF binding data, provide an excellent opportunity to systematically identify TF targets, enhancers, and enhancerpromoter interactions.
ChIP-seq assays provide a powerful method to identify TFBSs throughout the entire genome. However, it may not be feasible to apply this assay to all human TFs across all cell types under various conditions (e.g., medium, drug treatment, etc.), due to its timeconsuming properties. To overcome this challenge, a number of computational methods have been developed to predict context-specific TF target genes by integrating diverse genomic data, such as histone modifications, Position Weight Matrix (PMW) motif information and DNase I hypersensitive sites (DHSs) [21–24]. The CENTIPEDE method developed by Pique-Regi et al. constructed prediction models that integrate DHS data and PMW motif information [22]. In this initial study, CENTIPEDE identified a total of 827,000 TF binding motifs in human lymphoblastoid cell lines [22]. Follow-up studies further expanded on this knowledge or adapted CENTIPEDE to increase prediction accuracy [25–27]. Another method called protein interaction quantitation (PIQ), developed by Sherwood et al., is also based on the integration of DHS data and PMW motif information, and reported improved prediction accuracy [28].
Enhancers constitute another class of regulatory elements in the genome, which physically bind to specific TFs and interact with promoters of target genes via distant chromatin interactions. A number of computational methods have been developed to predict enhancers in the genomes [29–31]. In one study, Yip et al. identified 13,539 potential enhancers based on gene-distal regulatory modules using TF ChIP-seq data and then validated some of them experimentally [30]. In another study, Rajagopal et al. constructed a random forest-based algorithm that identifies enhancers in multiple cell lines based on histone modification patterns from ENCODE data [29]. Many other enhancer prediction methods have been developed into programs or packages that are publicly available from bioinformatics websites such as OMICtools (https://omictools.com/enhancer-prediction-category).
The integration of ENCODE data and other data sources allows computational prediction of enhancer-promoter (EP) interactions in a systematic manner. In a largescale study, 1,046 regulatory elements, including enhancers, were connected to their target promoters based on data from DHS, ChIP-seq and Chromosome Conformation Capture Carbon Copy (5C) experiments [23]. Cao et al. developed a computational method called joint effect of multiple enhancers (JEME) and applied it to predict EP interactions in 935 human primary cell types, cell lines and tissues by the integration of histone modification, DNase-seq, RNA-seq and other data types [*32]. These analyses have revealed critical insights into EP interactions, including that i) they are not simply determined by genomic proximity [33–35]; ii) multiple enhancers might control the same promoter [36]; and iii) EPs are cell-type specific [37,*38].
Quantifying the relation between gene expression, transcription factor binding and histone modifications
Inappropriate gene regulation underlies a variety of human diseases and therefore there has been a long-standing interest in the prediction of gene expression. TFs are essential in gene expression regulation and different approaches have been undertaken to utilize TFs as an indicator of gene expression.
The work of Ouyang et al. was one of the first studies to systematically investigate the contribution of TF binding signals captured by ChIP-seq assay to gene expression [39]. Using the intensities of ChIP-seq peaks near genes of interest as a measure of TF association strength, they suggested that the activity of 12 TFs explains a large portion of the gene expression variation observed in mouse embryonic stem cells [39]. A subsequent study used a similar log-linear regression-based approach to infer TF regulation and also incorporated histone modification data, which further improved gene expression prediction [40]. Following this, other studies based on different machine learning approaches have confirmed the quantitative relationship between gene expression levels, TF binding and histone modification signals [41,42]. As reported in the ENCODE landmark paper [2], more than 50% variation of gene expression can be explained by TF binding and/or histone modification signals in the promoter proximal DNA regions in the human genome. The same conclusion has also been made in several other species including yeast, fly, worm and mouse [5]. Thus, by utilizing genomic data from ENCODE and modENCODE, it has become possible to model transcriptional regulation of genomes in a quantitative, and likely, a dynamic manner.
Inferring regulatory activity of transcription factors
Inferring the regulatory activity of TFs has been another application of ENCODE data. TF genes tend to have relatively low expression levels and TF activity is intensively regulated at the post-transcriptional and post-translational level [45,46]. As such, the mRNA levels of TFs often do not accurately reflect their regulatory activity and therefore may not manifest their functions [43,44]. To overcome this issue, computational methods have been developed to infer TF activity based on the expression level of their target genes. The ENCODE project has provided 1,864 ChIP-seq profiles for over 100 human TFs in different cell lines, enabling the identification of TF target genes in an unbiased and systematic fashion.
Cheng et al. developed a rank-based statistical framework that summarizes the relative expression levels of genes regulated by a TF to infer its activity in each sample. In this framework, target genes of a TF are modeled as probabilistic events rather than being defined as a definite gene set [45]. Jiang et al. developed a multivariate linear model to integrate all ENCODE ChIP-seq TF binding profiles with 7,484 tumor gene expression profiles to systemically estimate the activities of 150 human TFs in different cancer types. Some of the inferred TF activities were then validated using knock out experiments in the K562 and HL60 cell lines [46]. Additionally, several other methods that rely on gene expression to infer TF activities have been developed based on different statistical models [47,48]. In summary, the integration of ENCODE ChIP-seq data and gene expression profiles has enabled statistical inference of TF regulatory activities under different biological settings, and therefore led to more accurate investigation of TF functions and transcriptional regulatory mechanisms.
Characterizing genomic alterations in cancer
The studies described in the previous sections have been instrumental to our understanding of genomic alterations in human diseases, including cancer. ENCODE data have been used in various pivotal studies that have characterized cancer-specific alterations in regulatory elements through the development of methods and accumulation of sequence-based studies. For example, global enhancer activation has been shown in almost all cancer types and might play a role in genomic rearrangements that are characteristic of cancers [**49]. The elucidation of EP interactions can identify enhancer-target networks for specific cancer types and point to cancer-specific alterations [*32].
Additionally, it is now recognized that alterations affecting regulatory regions are potentially as important in cancer progression as alterations in protein coding regions or those that directly alter functional RNA molecules [*50]. Epigenetic changes have been proposed to play a major role in the mutational landscape of cancers and might explain up to 86% of mutational rates in cancer genomes together with replication timing [51]. This association between chromatin features and mutation density is highly cell-type specific and mutations tend to be enriched in heterochromatin [52]. Accordingly, coding regions and regulatory elements seem to contain fewer mutations compared to intergenic, non-regulatory regions [53,54]. Due to the complexity of mutations in noncoding regions, several algorithms have been developed to assess tumor-specific somatic variants from tumor genomes and obtain a short list of candidate driver mutations or cancer-specific TFs [46,55–57].
The activities of specific TFs have been linked to cancer development and prognosis based on ENCODE data. For example, the activity but not the expression of E2F4, a TF involved in cell cycle regulation, has been reported to be associated with prognosis of breast cancer patients [58]. In this study, E2F4 activity in tumor samples was inferred based on the expression of its target genes, which were determined based on ENCODE ChIP-seq data. A single-cell sequencing study in glioblastoma has revealed stemnessrelated expression states that might be driven by a set of core TFs, including POU3F2, SOX2, SALL2 and OLIG2 [59]. Lastly, the integration of ENCODE ChIP-seq data and data from The Cancer Genome Atlas (TCGA) has proposed new TF oncogenes in several cancer types [46].
These integrative analyses of ENCODE data in the cancer field showed that large data compendia like ENCODE could identify genomic regions which are potentially more strongly linked to the biology of cancer. ENCODE data served as an essential component, and through the integration of data from other sources, such as TCGA, have moved cancer research forward.
Prioritizing disease-associated genetic variants from genome wide association studies
Disease- and trait-associated genetic variants are rapidly being identified with genomewide association studies (GWAS) and related strategies. Almost 85 million single nucleotide polymorphisms (SNPs) have been identified in the human genome so far [60]. Interestingly, most disease-associated SNPs are located in non-coding regions of the genome, and are equally distributed between intronic and intergenic regions [61–63].
The ENCODE Consortium has been one of the pioneers in integrating GWAS associations with genomic data to annotate GWAS associations [2]. Up to 71% of GWAS SNPs may have a potential causative SNP overlapping a DHS, and 31% of loci have a candidate SNP that overlaps a binding site occupied by a TF [2], which is consistent with the suggestion of positive selection in TFBSs [64]. However, it has been suggested that SNPs proposed by GWAS are often not the associated functional SNP in regions of interest [65]. Additionally, disease-associated SNPs have been shown to be significantly enriched in cell-type-specific enhancers and regulatory regions [14,66]. For example, SNPs associated with hematological disorders were most enriched in erythrocyte leukemia cell enhancers [14], whereas SNPs in TFs associated with similar functions, such as glucose homeostasis or immune regulation, might predispose individuals to diabetes or autoimmune diseases [63,67]. Thus, the integration of GWAS and ENCODE data has provided intriguing results and the expansion of both platforms will hopefully lead to a better understanding of the role of SNPs in human diseases.
Discovering new candidate drugs
Finally, ENCODE data have also been used to predict new candidate drugs, providing an example of translational research that connects genomic data with biomedical studies. In one study, ENCODE ChIP-seq data were integrated with RNA-seq data to identify drugs that might alter TF activity in specific diseases [**68]. In another study, Chen et al. showed that they could use ENCODE data to identify genome-wide signatures of TF activity and proposed that these signatures could be utilized to identify new candidate drugs [69]. They validated their framework by showing that commonly used drugs against estrogen receptor positive (ER+) breast cancer, such as tamoxifen and fulvestrant, displayed the highest inhibitory potential in ER+ breast cancer and proposed novel drugs with benefit in this disease, such as the anti-inflammatory drug oxaprozin [69]. Additionally, Gayvert et al. developed a method, called Computational drug-Repositioning Approach For Targeting Transcription factors (CRAFTT), which identifies molecules that can indirectly modulate TFs of interest. For example, they validated the prediction of dexamethasone to inhibit the TF ERG, which is associated with several oncogenic translocation events [70]. Although examples of pharmacogenomic analysis using ENCODE data remain limited, these studies demonstrated the great potential of using ENCODE data to promote translational researches.
Conclusions
In summary, high-throughput data from ENCODE have provided us with an unprecedented wealth of genomic information. These data have not only significantly enhanced our understanding of the human genome, but also provided new insights into other research areas from transcriptional regulation to disease mechanisms and drug development. Although our discussion only touched upon a few examples, we envision even more applications of ENCODE data through integrations with other data sources.
Highlights.
The ENCODE project has produced abundant genomic data of different categories.
Systematic analyses based on ENCODE data have revealed critical biological insights.
The integration of ENCODE with other data sources has provided novel knowledge on human diseases.
Acknowledgements
We thank Dr. Mark. Gerstein for helpful comments on the manuscript.
Funding
This work was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number KL2TR001088, the Center of Biomedical Research Excellence (COBRE) grant under award number GM103534, and the Dartmouth Geisel School of Medicine Start-up Fund.
Footnotes
Declarations of interest: none.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.ENCODE Project Consortium, Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, et al. : Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447:799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.ENCODE Project Consortium: An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qu H, Fang X: A brief review on the Human Encyclopedia of DNA Elements (ENCODE) project. Genomics Proteomics Bioinformatics 2013, 11:135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skipper M: Genomics: users’ guide to the human genome. Nat Rev Genet 2012, 13:678. [DOI] [PubMed] [Google Scholar]
- 5.Celniker SE, Dillon LAL, Gerstein MB, Gunsalus KC, Henikoff S, Karpen GH, Kellis M, Lai EC, Lieb JD, MacAlpine DM, et al. : Unlocking the secrets of the genome. Nature 2009, 459:927–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stamatoyannopoulos JA, Snyder M, Hardison R, Ren B, Gingeras T, Gilbert DM, Groudine M, Bender M, Kaul R, Canfield T, et al. : An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol 2012, 13:418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sloan CA, Chan ET, Davidson JM, Malladi VS, Strattan JS, Hitz BC, Gabdank I, Narayanan AK, Ho M, Lee BT, et al. : ENCODE data at the ENCODE portal. Nucleic Acids Res 2016, 44:D726–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.International Human Genome Sequencing Consortium: Finishing the euchromatic sequence of the human genome. Nature 2004, 431:931–945. [DOI] [PubMed] [Google Scholar]
- 9.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. : Landscape of transcription in human cells. Nature 2012, 489:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. : GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 2012, 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *11.Hon C-C, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJL, Gough J, Denisenko E, Schmeier S, Poulsen TM, Severin J, et al. : An atlas of human long non-coding RNAs with accurate 5’ ends. Nature 2017, 543:199–204. This work generated 27,919 human IncRNA genes and further identified 19,175 potentially functional lncRNAs in the human genome by integrating GENCODE and ENCODE data with other databases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lagarde J, Uszczynska-Ratajczak B, Carbonell S, Pérez-Lluch S, Abad A, Davis C, Gingeras TR, Frankish A, Harrow J, Guigo R, et al. : High-throughput annotation of full-length long noncoding RNAs with capture long-read sequencing. Nat Genet 2017, 49:1731–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ernst J, Kellis M: ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 2012, 9:215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. : Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffman MM, Buske OJ, Wang J, Weng Z, Bilmes JA, Noble WS: Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat Methods 2012, 9:473–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwasnieski JC, Fiore C, Chaudhari HG, Cohen BA: High-throughput functional testing of ENCODE segmentation predictions. Genome Res 2014, 24:1595–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biesinger J, Wang Y, Xie X: Discovering and mapping chromatin states using a tree hidden Markov model. BMC Bioinformatics 2013, 14 Suppl 5:S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sohn K-A, Ho JWK, Djordjevic D, Jeong H-H, Park PJ, Kim JH: hiHMM: Bayesian non-parametric joint inference of chromatin state maps. Bioinforma Oxf Engl 2015, 31:2066–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larson JL, Huttenhower C, Quackenbush J, Yuan G-C: A tiered hidden Markov model characterizes multi-scale chromatin states. Genomics 2013, 102:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **20.Marco E, Meuleman W, Huang J, Glass K, Pinello L, Wang J, Kellis M, Yuan G-C: Multi-scale chromatin state annotation using a hierarchical hidden Markov model. Nat Commun 2017, 8:15011 This work proposed an algorithm, diHMM, which can annotate chromatin states at multiple length scales and might provide more insights into long-range chromatin interactions and the effect of spatial organization within the nucleus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK, et al. : An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 2012, 489:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pique-Regi R, Degner JF, Pai AA, Gaffney DJ, Gilad Y, Pritchard JK: Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res 2011, 21:447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. : The accessible chromatin landscape of the human genome. Nature 2012, 489:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Natarajan A, Yardimci GG, Sheffield NC, Crawford GE, Ohler U: Predicting celltype-specific gene expression from regions of open chromatin. Genome Res 2012, 22:1711–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hosoya T, D’Oliveira Albanus R, Hensley J, Myers G, Kyono Y, Kitzman J, Parker SCJ, Engel JD: Global dynamics of stage-specific transcription factor binding during thymocyte development. Sci Rep 2018, 8:5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moyerbrailean GA, Kalita CA, Harvey CT, Wen X, Luca F, Pique-Regi R: Which Genetics Variants in DNase-Seq Footprints Are More Likely to Alter Binding? PLoS Genet 2016, 12:e1005875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raj A, Shim H, Gilad Y, Pritchard JK, Stephens M: msCentipede: Modeling Heterogeneity across Genomic Sites and Replicates Improves Accuracy in the Inference of Transcription Factor Binding. PloS One 2015, 10:e0138030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sherwood RI, Hashimoto T, O’Donnell CW, Lewis S, Barkal AA, van Hoff JP, Karun V, Jaakkola T, Gifford DK: Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat Biotechnol 2014, 32:171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rajagopal N, Xie W, Li Y, Wagner U, Wang W, Stamatoyannopoulos J, Ernst J, Kellis M, Ren B: RFECS: a random-forest based algorithm for enhancer identification from chromatin state. PLoS Comput Biol 2013, 9:e1002968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yip KY, Cheng C, Bhardwaj N, Brown JB, Leng J, Kundaje A, Rozowsky J, Birney E, Bickel P, Snyder M, et al. : Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcriptionrelated factors. Genome Biol 2012, 13:R48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitaker JW, Nguyen TT, Zhu Y, Wildberg A, Wang W: Computational schemes for the prediction and annotation of enhancers from epigenomic assays. Methods San Diego Calif 2015, 72:86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *32.Cao Q, Anyansi C, Hu X, Xu L, Xiong L, Tang W, Mok MTS, Cheng C, Fan X, Gerstein M, et al. : Reconstruction of enhancer-target networks in 935 samples of human primary cells, tissues and cell lines. Nat Genet 2017, 49:1428–1436. This work developed the JEME method and systematically applied this method to predict enhancer-gene interactions in human primary cells, tissues and cell lines. The JEME method is more accurate than existing method in predicting enhancer targets in unseen samples. [DOI] [PubMed] [Google Scholar]
- 33.Sanyal A, Lajoie BR, Jain G, Dekker J: The long-range interaction landscape of gene promoters. Nature 2012, 489:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whalen S, Truty RM, Pollard KS: Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat Genet 2016, 48:488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu Y, Chen Z, Zhang K, Wang M, Medovoy D, Whitaker JW, Ding B, Li N, Zheng L, Wang W: Constructing 3D interaction maps from 1D epigenomes. Nat Commun 2016, 7:10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He B, Chen C, Teng L, Tan K: Global view of enhancer-promoter interactome in human cells. Proc Natl Acad Sci U S A 2014, 111:E2191–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roy S, Siahpirani AF, Chasman D, Knaack S, Ay F, Stewart R, Wilson M, Sridharan R: A predictive modeling approach for cell line-specific long-range regulatory interactions. Nucleic Acids Res 2015, 43:8694–8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *38.Thormann V, Rothkegel MC, Schöpflin R, Glaser LV, Djuric P, Li N, Chung H-R, Schwahn K, Vingron M, Meijsing SH: Genomic dissection of enhancers uncovers principles of combinatorial regulation and cell type-specific wiring of enhancer-promoter contacts. Nucleic Acids Res 2018, 46:2868–2882. This work investigated how transcription factors interact with enhancers to affect enhancer-promoter interactions in a cell type-specific manner using the glucocorticoid receptor (GR) as an example. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ouyang Z, Zhou Q, Wong WH: ChIP-Seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells. Proc Natl Acad Sci U S A 2009, 106:21521–21526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McLeay RC, Lesluyes T, Cuellar Partida G, Bailey TL: Genome-wide in silico prediction of gene expression. Bioinforma Oxf Engl 2012, 28:2789–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng C, Gerstein M: Modeling the relative relationship of transcription factor binding and histone modifications to gene expression levels in mouse embryonic stem cells. Nucleic Acids Res 2012, 40:553–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng C, Alexander R, Min R, Leng J, Yip KY, Rozowsky J, Yan K-K, Dong X, Djebali S, Ruan Y, et al. : Understanding transcriptional regulation by integrative analysis of transcription factor binding data. Genome Res 2012, 22:1658–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Filtz TM, Vogel WK, Leid M: Regulation of transcription factor activity by interconnected post-translational modifications. Trends Pharmacol Sci 2014, 35:76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM: A census of human transcription factors: function, expression and evolution. Nat Rev Genet 2009, 10:252–263. [DOI] [PubMed] [Google Scholar]
- 45.Cheng C, Yan X, Sun F, Li LM: Inferring activity changes of transcription factors by binding association with sorted expression profiles. BMC Bioinformatics 2007, 8:452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang P, Freedman ML, Liu JS, Liu XS: Inference of transcriptional regulation in cancers. Proc Natl Acad Sci U S A 2015, 112:7731–7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma’ayan A: ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinforma Oxf Engl 2010, 26:2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang K, Saito M, Bisikirska BC, Alvarez MJ, Lim WK, Rajbhandari P, Shen Q, Nemenman I, Basso K, Margolin AA, et al. : Genome-wide identification of posttranslational modulators of transcription factor activity in human B cells. Nat Biotechnol 2009, 27:829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **49.Chen H, Li C, Peng X, Zhou Z, Weinstein JN, Cancer Genome Atlas Research Network, Liang H: A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples. Cell 2018, 173:386–399.e12. This work systematically examined the function of different enhancers in various cancer contexts and provided clinical insights in the role of enhancers in cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *50.Shar NA, Vijayabaskar MS, Westhead DR: Cancer somatic mutations cluster in a subset of regulatory sites predicted from the ENCODE data. Mol Cancer 2016, 15:76 This work integrated transcription factor binding sites, DNase I hypersensitive sites and RNA-seq to show that somatic mutations in cancer occur preferentially in potential regulatory regions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schuster-Böckler B, Lehner B: Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 2012, 488:504–507. [DOI] [PubMed] [Google Scholar]
- 52.Polak P, Karlić R, Koren A, Thurman R, Sandstrom R, Lawrence M, Reynolds A, Rynes E, Vlahoviček K, Stamatoyannopoulos JA, et al. : Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 2015, 518:360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee W, Jiang Z, Liu J, Haverty PM, Guan Y, Stinson J, Yue P, Zhang Y, Pant KP, Bhatt D, et al. : The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature 2010, 465:473–477. [DOI] [PubMed] [Google Scholar]
- 54.Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W: Genome-wide analysis of noncoding regulatory mutations in cancer. Nat Genet 2014, 46:1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fu Y, Liu Z, Lou S, Bedford J, Mu XJ, Yip KY, Khurana E, Gerstein M: FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol 2014, 15:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khurana E, Fu Y, Colonna V, Mu XJ, Kang HM, Lappalainen T, Sboner A, Lochovsky L, Chen J, Harmanci A, et al. : Integrative annotation of variants from 1092 humans: application to cancer genomics. Science 2013, 342:1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Plaisier CL, O’Brien S, Bernard B, Reynolds S, Simon Z, Toledo CM, Ding Y, Reiss DJ, Paddison PJ, Baliga NS: Causal Mechanistic Regulatory Network for Glioblastoma Deciphered Using Systems Genetics Network Analysis. Cell Syst 2016, 3:172–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khaleel SS, Andrews EH, Ung M, DiRenzo J, Cheng C: E2F4 regulatory program predicts patient survival prognosis in breast cancer. Breast Cancer Res BCR 2014, 16:486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. : Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344:1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. : A global reference for human genetic variation. Nature 2015, 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJH, Shishkin AA, et al. : Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518:337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA: Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 2009, 106:9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. : Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Melton C, Reuter JA, Spacek DV, Snyder M: Recurrent somatic mutations in regulatory regions of human cancer genomes. Nat Genet 2015, 47:710–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M: Linking disease associations with regulatory information in the human genome. Genome Res 2012, 22:1748–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H, Chen H, Liu F, Ren C, Wang S, Bo X, Shu W: Functional annotation of HOT regions in the human genome: implications for human disease and cancer. Sci Rep 2015, 5:11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Onengut-Gumuscu S, Chen W-M, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, Farber E, Bonnie JK, Szpak M, Schofield E, et al. : Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet 2015, 47:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **68.Garcia-Alonso L, Iorio F, Matchan A, Fonseca N, Jaaks P, Peat G, Pignatelli M, Falcone F, Benes CH, Dunham I, et al. : Transcription Factor Activities Enhance Markers of Drug Sensitivity in Cancer. Cancer Res 2018, 78:769–780. Large-scale analysis in which 265 compounds were tested in 1,056 cancer cell lines and 9,250 primary tumors to uncover transcription factors whose activity is affected by anticancer drugs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen J, Hu Z, Phatak M, Reichard J, Freudenberg JM, Sivaganesan S, Medvedovic M: Genome-wide signatures of transcription factor activity: connecting transcription factors, disease, and small molecules. PLoS Comput Biol 2013, 9:e1003198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gayvert KM, Dardenne E, Cheung C, Boland MR, Lorberbaum T, Wanjala J, Chen Y, Rubin MA, Tatonetti NP, Rickman DS, et al. : A Computational Drug Repositioning Approach for Targeting Oncogenic Transcription Factors. Cell Rep 2016, 15:2348–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]