

Abstract
The diagnosis of adrenal insufficiency is often delayed, as the presenting symptoms of fatigue, abdominal pain, and anorexia are vague and nonspecific. However, timely diagnosis and treatment with replacement steroids are needed to prevent fatal adrenal crisis. While the most common cause of primary adrenal insufficiency in childhood is congenital adrenal hyperplasia, a significant minority (13-23%) is caused by autoimmune destruction of the gland. We present a case of a 4-year-old, previously healthy child who had a one-day history of nausea and vomiting, and was found unresponsive by her caretaker. Despite emergency rescue and transport to the hospital, she was pronounced dead. At autopsy, the adrenal glands were atrophied. Histologic examination revealed lymphocytic infiltration of the adrenal glands consistent with autoimmune adrenal insufficiency. Fecal viral antigen testing was positive for rotavirus. The cause of death was determined to be adrenal crisis in the setting of rotavirus gastroenteritis due to adrenal insufficiency (Addison disease).
Keywords: Forensic pathology, Addison disease, Autoimmune, Pediatric, Rotavirus, Adrenal
Introduction
Adrenal insufficiency (AI) is an uncommon disease in adults and even more rare in children (1). However, prompt recognition and diagnosis of AI is needed to begin treatment and prevent a potentially fatal adrenal crisis (2). Adrenal insufficiency can be subdivided into either primary or secondary/tertiary. In primary AI, the production of mineralocorticoids and glucocorticoids is impaired due to destruction of the adrenal cortex. In contrast, secondary or tertiary AI results from diseases affecting the pituitary or hypothalamus. In these, the production of mineralocorticoids is usually intact because the renin-angiotensin-aldosterone pathway still functions; however, production of corticosteroids is impaired due to impaired production of adrenocorticotropic hormone (ACTH) (3). Secondary AI is most common form of AI and is typically due to the abrupt withdrawal of pharmacologic treatment with corticosteroids (4).
The term “Addison disease” is nonspecific, referring to any chronic form of primary AI (2). While autoimmune adrenalitis (also known as idiopathic Addison disease) is the most common cause of primary AI in adults in developed countries, the most common cause of primary AI in children is congenital adrenal hyperplasia (CAH). However, a significant minority (13 to 23%) of primary AI in children is due to autoimmune adrenalitis. Unlike autoimmune AI, CAH most commonly presents in the neonatal period (1, 5) and it has been proposed that autoimmune AI is the most common cause of primary AI presenting after the neonatal period (6). We present a case report of a 4-year-old child with subclinical autoimmune adrenalitis who experienced a fatal adrenal crisis precipitated by rotavirus infection.
Case Report
A 4-year-old previously healthy child was found unresponsive by her caregiver after a one- to two-day history of an illness characterized by nausea and vomiting. Emergency medical services were called and she was transported to the hospital where she was pronounced dead after attempted resuscitation.
Autopsy revealed an underweight, otherwise well-developed 4-year-old. Weight-for-stature was 3rd percentile, while weight-for-age was 25th percentile. No areas of hyperpigmentation on the skin were observed. The right and left adrenal glands were markedly atrophic, weighing less than 1 g each (expected combined weight of 6 g). The pituitary, pancreas, and thyroid glands were grossly unremarkable.
Microscopic evaluation of the adrenals showed lymphoplasmacytic inflammation (Image 1) consisting predominantly of T cells with a slight predominance of CD8 to CD4 positive cells. Focal follicle formation by CD20 positive B cells was also observed (Image 2).
Image 1.
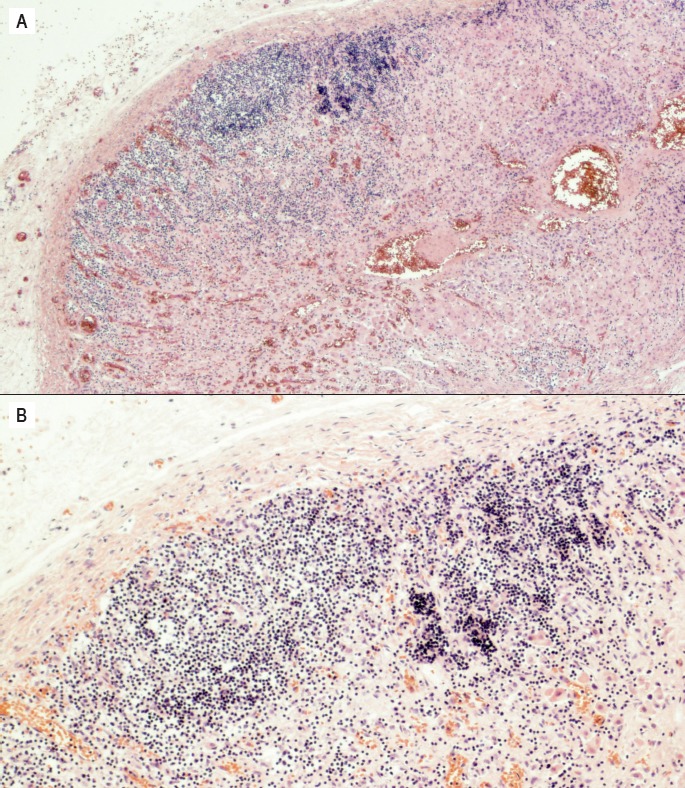
Photomicrograph of the left adrenal gland demonstrating diffuse infiltration and destruction of the adrenal cortex by lymphocytes and plasma cells. A) H&E, x40 B) H&E, x100.
Image 2.

Immunohistochemistry of left adrenal gland demonstrating proportion of CD8+ T cells, CD4+ T cells, and B cells. This series of photomicrographs depicts the results of immunohistochemical staining of the left adrenal glands for A) CD8 (x100), B), CD4 (x100), and C) CD20 (x100). This demonstrates a slight predominance of CD8+ to CD4+ T cells, and highlights scattered follicle formation by B cells.
The contents of the small intestine were mucoid and bilious. Microscopic examination showed superficial necrosis of the mucosa, with increased lymphocytes in the lamina propria. The pancreas was microscopically unremarkable.
Postmortem serum cortisol was 1 μg/dL (reference range, 3-22 μg/dL). Fecal viral antigen testing detected rotavirus. Vitreous sodium was 136 mmol/L and vitreous glucose was <35 mg/dL. Hemoglobin A1C was 5.2%.
The gross and histologic features of the adrenal glands, combined with the low serum cortisol and clinical history, are essentially diagnostic of an adrenal crisis due to autoimmune adrenalitis. The cause of death was determined to be adrenal crisis in the setting of rotavirus gastroenteritis due to adrenal insufficiency (Addison disease).
Discussion
Although timely diagnosis of adrenal insufficiency is needed to initiate treatment and prevent adrenal crisis, it is often delayed due to the nonspecific nature of the presenting symptoms. Initial symptoms and signs may include nausea and vomiting, behavioral changes, poor weight gain, hypoglycemia, weakness, and hypotension (1, 6). Further confounding the issue is the inconsistent presence of specific findings to suggest primary AI. Skin hyperpigmentation, often thought of as a classic sign of primary AI, may be absent in up to one third of patients (and was absent in the presented case). Hyperkalemia is also absent in up to 50% of children and usually only mild when present (1, 7). One study that examined children with non-CAH primary AI found a delay in diagnosis of up to 0.5-5 years, and 25% of these children presented in frank adrenal crisis (8). Adrenal crises can also be precipitated by infection, as in the case presented here, surgery, or trauma, as these physiologic stresses increase demand for cortisol and the adrenal glands are unable to respond appropriately (2).
At autopsy, the adrenal glands in autoimmune AI can be normal-sized, atrophic, or nearly unidentifiable. Other common causes of primary AI in children include traumatic adrenal hemorrhage, adrenal hemorrhage due to infection (i.e., Waterhouse-Friderichsen Syndrome), thrombosis from antiphospholipid antibody syndrome, direct infection of the gland (e.g., tuberculosis), X-linked adrenoleukodystrophy, and other congenital syndromes (7, 13). However, these etiologies do not cause atrophy of the adrenals. Histologic exam of the adrenal cortex in autoimmune AI typically shows a lymphocyte-predominant infiltrate with associated follicle formation and fibrosis. In the late stages of disease, the glands may be predominantly fibrotic with only minimal inflammation (6, 9).
Postmortem laboratory studies supportive of primary AI include a depressed serum cortisol concentration, often <3 μg/dL. The cut-off for “low” cortisol may even be raised to 20 μg/dL in patients who are critically ill, and should be demonstrating elevated cortisol as a stress response. Clearly, there is room for uncertainty; some authors suggest a cutoff of <5 μg/dL to increase sensitivity, and additionally, cortisol concentrations are known to fluctuate during the day (2, 10). The concentrations measured, therefore, needs to be interpreted in the clinical context in order to understand its significance. Serum cortisol has been described to be stable at least up to 18-20 hours postmortem, and possibly up until the onset of recognizable decomposition (2, 10, 11). In the case presented, the postmortem interval between death and autopsy was only 18 hours, so this should not significantly confound the interpretation of the decreased cortisol concentration.
While hyponatremia can be detected in the vitreous fluid seen due to loss of aldosterone, vitreous sodium does decrease with increasing decomposition and this must be considered. Hypoglycemia may be detected in the vitreous fluid as well, due to the loss of cortisol, but vitreous glucose is also known to drop after death and so a low value must be interpreted with some scrutiny. Evaluating hyperkalemia in vitreous fluid is entirely unreliable, as potassium begins to increase immediately and unpredictably after death. If antemortem samples are available for testing, then these electrolyte derangements could be confirmed (2, 12). Adrenocorticotropic hormone concentrations and plasma renin activity can also be measured on antemortem samples, and should both be elevated in primary AI (7). In the case presented, no antemortem samples were available for testing.
The detection of auto-antibodies against 21-hydroxylase is specific for autoimmune AI, and can be done on postmortem samples (2).
Other associated endocrinopathies need to be considered when investigating a potential case of autoimmune AI. Autoimmune AI can occur in children in isolation, or may be part of the Autoimmune polyglandular syndromes (types 1, 2, or 4). Autoimmune polyglandular syndromes Type 1 is defined by a germline mutation in the AIRE-1 gene, and presents with adrenal insufficiency, hypoparathyroidism, and mucocutaneous candidiasis. Autoimmune polyglandular syndrome Type 2 has no single specific mutation, but is defined as adrenal insufficiency with immune-mediated diabetes mellitus and autoimmune thyroiditis. Type 4 is simply any constellation of autoimmune diseases that doesn't fit into the other categories (6). In this patient, there was no indication of other endocrinopathies or chronic Candida infections, although it is possible other autoimmune diseases may have developed, had she survived.
In the case presented, the low serum cortisol is very supportive of an adrenal crisis. Although 21-hydroxylase antibodies were not tested in this case, the finding of atrophic adrenal glands with dense lymphoplasmacytic infiltration is specific for autoimmune Addison Disease. It is likely that this child initially had subclinical AI; no skin hyperpigmentation was seen on exam, and she had been previously healthy (although her underweight status may have been partially due to AI). The clinical history and viral antigen testing suggest that a gastrointestinal rotavirus infection unmasked her disease, causing an adrenal crisis.
Conclusion
This case re-emphasizes that although adrenal insufficiency is a rare diagnosis in childhood, it must be suspected in cases of sudden death, particularly those with a preceding illnesses or surgery.
Footnotes
ETHICAL APPROVAL
As per Journal Policies, ethical approval was not required for this manuscript
STATEMENT OF HUMAN AND ANIMAL RIGHTS
This article does not contain any studies conducted with animals or on living human subjects
STATEMENT OF INFORMED CONSENT
No identifiable personal data were presented in this manuscsript
DISCLOSURES & DECLARATION OF CONFLICTS OF INTEREST
This work was presented at the 2016 NAME Annual Meeting.The authors, reviewers, editors, and publication staff do not report any relevant conflicts of interest
FINANCIAL DISCLOSURE The authors have indicated that they do not have financial relationships to disclose that are relevant to this manuscript
References
- 1.Hsieh S., White P.C. Presentation of primary adrenal insufficiency in childhood. J Clin Endocrinol Metab. 2011. Jun; 96(6): E925–8. PMID: 21470994. 10.1210/jc.2011-0015. [DOI] [PubMed] [Google Scholar]
- 2.Kemp W.L., Koponen M.A., Meyers S.E. Addison Disease: the first presentation of the condition may be at autopsy. Acad Forensic Pathol. 2016. Jun; 6(2): 249–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shulman DI, Palmert MR, Kemp SF; Lawson Wilkins Drug and Therapeutics Committee. Adrenal insufficiency: still a cause of morbidity and death in childhood. Pediatrics. 2007. Feb; 119(2): e484–94. PMID: 17242136. 10.1542/peds.2006-1612. [DOI] [PubMed] [Google Scholar]
- 4.Levy-Shraga Y., Pinhas-Hamiel O. Novel insights into adrenal insufficiency in childhood. Minerva Pediatr. 2014. Dec; 66(6): 517–32. PMID: 25058175. [PubMed] [Google Scholar]
- 5.Perry R., Kecha O., Paquette J. et al. Primary adrenal insufficiency in children: twenty years experience at the Sainte-Justine Hospital, Montreal. J Clin Endocrinol Metab. 2005. Jun; 90(6): 3243–50. PMID: 15811934. 10.1210/jc.2004-0016. [DOI] [PubMed] [Google Scholar]
- 6.Ten S., New M., Maclaren N. Clinical review 130: Addison's disease 2001. J Clin Endocrinol Metab. 2001. Jul; 86(7): 2909–22. PMID: 11443143. 10.1210/jcem.86.7.7636. [DOI] [PubMed] [Google Scholar]
- 7.Caplan M.J. A practical forensic approach to fatal pediatric endocrinopathies. Acad Forensic Pathol. 2016. Jun; 6(2): 258–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simm P.J., McDonnell C.M., Zacharin M.R. Primary adrenal insufficiency in childhood and adolescence: advances in diagnosis and management. J Paediatr Child Health. 2004. Nov; 40(11): 596–9. PMID: 15469526. 10.1111/j.1440-1754.2004.00482.x. [DOI] [PubMed] [Google Scholar]
- 9.Betterle C., Morlin L. Autoimmune Addison's disease. Endocr Dev. 2011; 20: 161–72. PMID: 21164269. 10.1159/000321239. [DOI] [PubMed] [Google Scholar]
- 10.Clapper A., Nashelsky M., Dailey M. Evaluation of serum cortisol in the postmortem diagnosis of acute adrenal insufficiency. Am J Forensic Med Pathol. 2008. Jun; 29(2): 181–4. PMID: 18520491. 10.1097/PAF.0b013e318174e7c8. [DOI] [PubMed] [Google Scholar]
- 11.Finlayson N.B. Blood cortisol in infants and adults: a postmortem study. J Pediatr. 1965. Feb; 67(2): 248–52. 10.1016/s0022-3476(65)80247-5. [DOI] [Google Scholar]
- 12.Palmiere C., Mangin P. Postmortem chemistry update part I. Int J Legal Med. 2012. Mar; 126(2): 187–98. PMID: 21947676. 10.1007/s00414-011-0625-y. [DOI] [PubMed] [Google Scholar]
- 13.Uçar A., Baş F., Saka N. Diagnosis and management of pediatric adrenal insufficiency. World J Pediatr. 2016. Aug; 12(3): 261–74. PMID: 27059746. 10.1007/s12519-016-0018-x. [DOI] [PubMed] [Google Scholar]