

Abstract
Herein is reported a case of granulomatosis with polyangiitis (GPA), formerly known as Wegener's granulomatosis, diagnosed at forensic autopsy in an 83-year-old woman with a history of Alzheimer disease. Significant findings at autopsy were cardiac hypertrophy, nephroarteriolosclerosis, hemorrhagic lungs, and necrotizing granulomatous inflammation with vasculitis involving the lungs and kidneys. Semiquantitative immunofluorescence testing was positive for cytoplasmic antineutrophil cytoplasm antibodies at a titer of 1:40. An enzyme-linked immunosorbent assay for proteinase 3 was strongly positive at 134 units (reference range: positive greater than 30 units). The cause of death was therefore determined to be granulomatosis with polyangiitis. Presentation of GPA at such an advanced age is rare, especially with the presentation at death.
Keywords: Forensic pathology, Wegener granulomatosis, Granulomatosis with polyangiitis, ANCA-associated vasculitidies
Introduction
Granulomatosis with polyangiitis (GPA) was formerly known by the name Wegener's granulomatosis, named after Friedrich Wegener, who described the disease in 1936. In recent years, Woywodt and Matteson have promoted the abandonment of eponyms in the classification of diseases not only for Wegener's granulomatosis but also for other diseases (1, 2), resulting in the modern name, granulomatosis with polyangiitis, being adopted (3).
Granulomatosis with polyangiitis is one of the antineutrophil cytoplasm antibody (ANCA)-associated vasculitidies as defined by the 2012 Chapel Hill consensus conference, together with microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA; also known as Churg-Strauss syndrome) (4).
Most cases of ANCA-associated vasculitidies present between the ages of 40 and 50 years (5). Herein is described a case of GPA in an elderly woman who died of GPA, in which the diagnosis was made at autopsy.
Case Report
The case involves an 83-year-old woman with a history of Alzheimer disease, hypertension, and seasonal allergies. Her prescription medications included aripiprazole and donepezil for her dementia, hydrochlorothiazide for hypertension, and loratadine for her allergies. She had been complaining of sinus pain and had seen her primary care physician, who had prescribed amoxicillin for a presumed sinus infection, which she had taken the night before she died. She was last known to be alive the prior evening when she and her husband went to bed. Her husband awoke the next morning and found her unresponsive; emergency medical services were called and she was pronounced dead at the scene. The case was referred to the medical examiner's office for autopsy.
The body measured 163 cm (64 in) long and weighed 55.3 kg (122 pounds). The external examination was unremarkable; her body appeared well developed and well nourished. Internal examination revealed an enlarged heart (550 g). The coronary arteries all exhibited less than 25% luminal stenosis by atherosclerotic plaque. The cortical surfaces of the kidneys were markedly granular, with pitted scars. The right and left lungs weighed 1425 and 800 g, respectively. Cut surfaces of the pulmonary parenchyma exuded bloodtinged fluid and although markedly heavy, did not appear to be otherwise abnormal.
Microscopically, the lungs exhibited prominent and widespread alveolar hemorrhage (Image 1) with focal hyaline membranes (Image 2). The pulmonary parenchyma was largely overtaken by epithelioid granulomas with multinucleated giant cells; several were necrotizing (Images 3 through 5). Small arterioles exhibited acute vasculitis with fibrinoid necrosis (Image 3). Acid fast (Ziehl-Nielsen) and Gomori methenamine silver stains did not reveal the presence of acid fast bacilli or fungal organisms, respectively. Microscopic sections of kidney demonstrated loosely formed necrotizing granulomas with multinucleated giant cells (Image 6) as well as multifocal vasculitis with infiltration of vessel walls, predominantly by neutrophils, and fibrinoid necrosis (Images 7 and 8). No other organs exhibited similar microscopic findings.
Image 1.

Diffuse alveolar hemorrhage (H&E, x40).
Image 2.
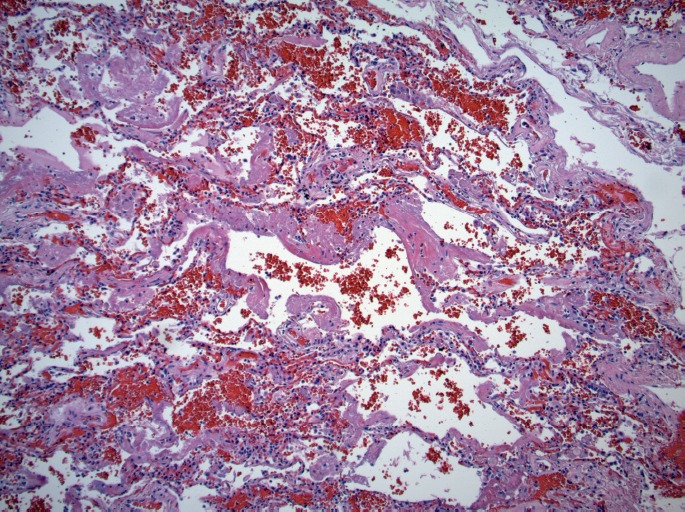
Alveoli with hyaline membranes (H&E, x100).
Image 3.

Pulmonary parenchyma with multiple granulomas containing multinucleated giant cells, some of which exhibit necrosis. Vasculitis involving small vessel (arrow) (H&E, x200).
Image 5.
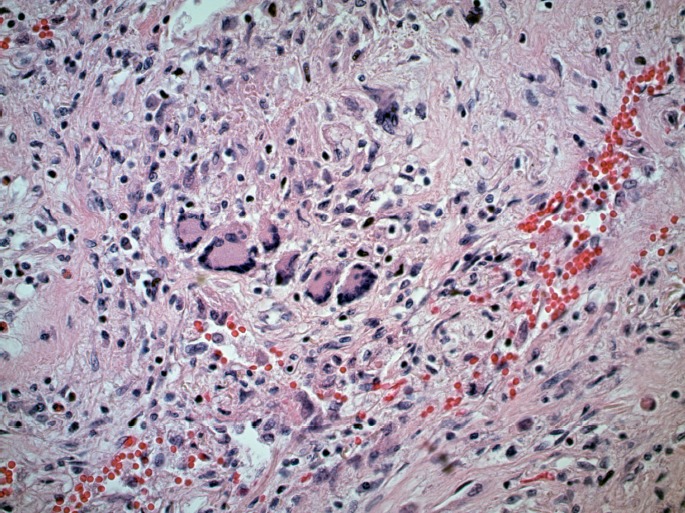
Pulmonary granuloma with several multinucleated giant cells (H&E, x400).
Image 6.
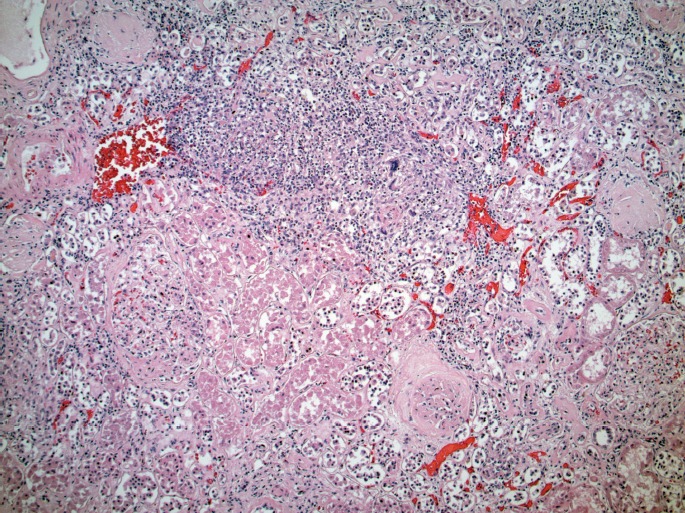
Kidney with necrotic granuloma containing multinucleated giant cells (H&E, x200).
Image 7.
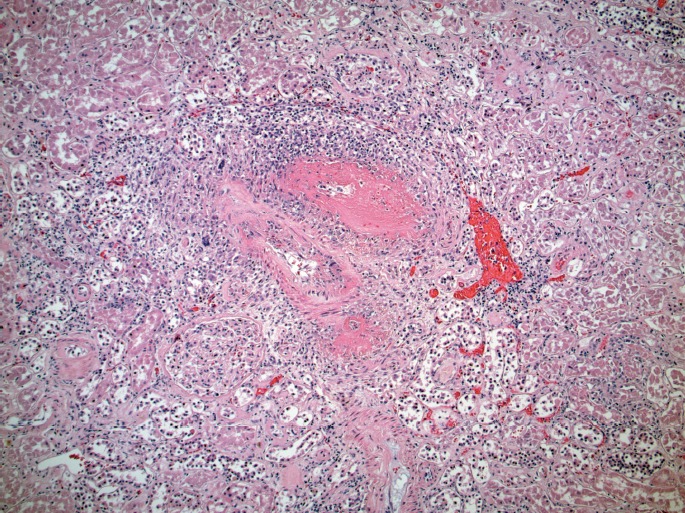
Renal arteriole with acute vasculitis (H&E, x200).
Image 8.
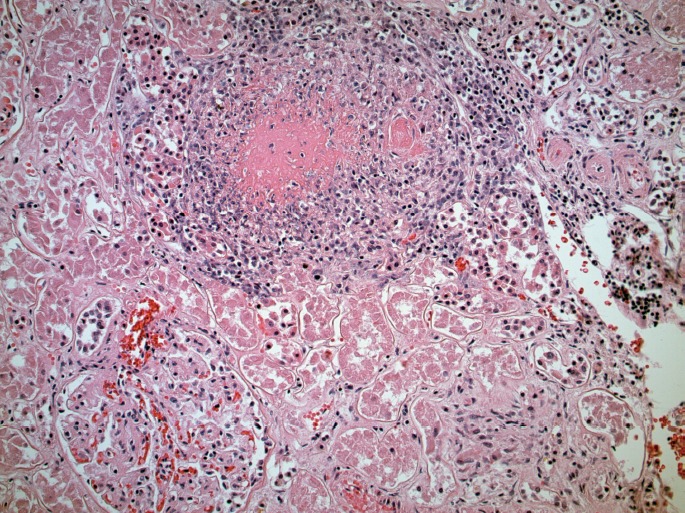
Renal arteriole with acute vasculitis (H&E, x400).
Image 4.

Pulmonary granulomas with multinucleated giant cells and prominent necrosis (H&E, x200).
Indirect semiquantitative immunofluorescence staining of neutrophils mixed with the decedent's serum demonstrated positivity in a cytoplasmic distribution (c-ANCA) at a titer of 1:40. Reflex enzyme-linked immunosorbent assay (ELISA) testing for proteinase 3 (PR3)-ANCA was positive, with a result of 134 Units (reference range: negative < 20 Units; weak positive 21-30 Units; and positive > 30 Units). Myeloperoxidase (MPO)-ANCA testing by ELISA was negative.
The microscopic findings and ANCA testing results are diagnostic of GPA, which was determined to be the cause of death.
Discussion
Epidemiology
The differential diagnosis of the ANCA-positive vasculitidies includes GPA, MPA, and EGPA. The ANCA-associated vasculitidies affect males and females approximately equally, with most patients in between 40 and 50 years of age (5, 6). The vast majority of patients are Caucasian or of Hispanic descent (5). The incidence of GPA has been reported to be between two and 12 per million per year (7).
Clinical Features
Patients with GPA generally exhibit pneumonitis, chronic sinusitis, nasopharyngeal mucosal ulceration, and renal disease (6).
Diagnosis
Staining of neutrophils with ANCA antibodies in a cytoplasmic distribution in GPA patients was reported over 30 years ago (8) and was later demonstrated to exhibit high sensitivity and specificity (93% and 97%, respectively) in clinical studies (8, 9). In contrast, the other ANCA-positive vasculitidies, MPA and EGPA, generally exhibit immunofluorescence in a perinuclear pattern. Combining the results of indirect immunofluorescence for ANCA with ELISAs detecting specific antibodies to PR3 and myeloperoxidase (MPO) increased the diagnostic specificities to 99%; the combination of c-ANCA staining and positivity for PR3 was demonstrated to be highly specific for GPA (10).
Pathology
Granulomatosis with polyangiitis is characterized by a pathologic triad: 1) a necrotizing granulomatous inflammation that can involve any portion of the respiratory tract; 2) a small- to medium-sized vessel granulomatous vasculitis that may or may not be necrotizing, which is commonly seen in the respiratory system but can also affect other sites, including the kidney; and 3) necrotizing glomerulonephritis (in some but not all cases), that may progress to crescentic glomerulonephritis (6), which was not seen in the presented case.
The histopathologic features and associations with ANCA are summarized in Table 1.
Table 1.
Pathologic Diagnosis of the Antineutrophil Cytoplasm Antibody (ANCA)-Associated Vasculitidies
| Granulomatosis with Polyangiitis (GPA) | Microscopic Polyangiitis (MPA) | Eosinophilic Granulomatosis with Polyangiitis (EGPA) | |
|---|---|---|---|
| Typical histopathology | Granulomatous, necrotizing vasculitis | Nongranulomatous, necrotizing vasulitis | Granulomatous, necrotizing vasculitis with eosinophils |
| Typical ANCA immunofluroesence pattern | Cytoplasmic | Perinuclear | Perinuclear |
| Typical ANCA specificity | PR3 | MPO | MPO |
PR3 - Proteinase 3
MPO - Myeloperoxidase
Pathogenesis
The disease is thought to represent an autoimmune disease involving an abnormal T-cell response to inhaled antigens. Indeed, the disease responds to immunosuppressive therapy (see below). The autoantibodies to MPO and PR3, in addition to being disease markers, have also been hypothesized to play a pathogenic role in the ANCA-associated vasculitidies; however, the precise mechanisms are not entirely clear, especially in the case of PR3-ANCA and GPA, and are reviewed elsewhere (11, 12).
Treatment and Prognosis
Granulomatosis with polyangiitis is rapidly fatal without treatment (6); however, advances in medical treatment include steroids, chemotherapy, and anticytokine therapies that have greatly prolonged survival (5, 11).
Conclusion
The presented case is an atypical presentation of a relatively rare disease. The decedent in this case presented at 83 years of age, which is several decades later than is typical for GPA, although small series of cases have been reported in the elderly, indicating higher mortality rates in this population as well as higher rates of renal and central nervous system involvement (13, 14) The presented case was especially unique given the fact the individual did not carry a diagnosis of GPA; rather, her first clinical presentation was at death.
Footnotes
ETHICAL APPROVAL
As per Journal Policies, ethical approval was not required for this manuscript
STATEMENT OF HUMAN AND ANIMAL RIGHTS
This article does not contain any studies conducted with animals or on living human subjects
STATEMENT OF INFORMED CONSENT
No identifiable personal data were presented in this manuscsript
DISCLOSURES & DECLARATION OF CONFLICTS OF INTEREST
J. Keith Pinckard is the Editor-In-Chief of Academic Forensic Pathology: The Official Publication of the National Association of Medical Examiners. The author, reviewers, editors, and publication staff do not report any other relevant conflicts of interest
FINANCIAL DISCLOSURE The authors have indicated that they do not have financial relationships to disclose that are relevant to this manuscript
References
- 1).Woywodt A., Matteson E.L. Wegener's granulomatosis–probing the untold past of the man behind the eponym. Rheumatology (Oxford). 2006. Oct; 45(10): 1303–6. PMID: 16887845. 10.1093/rheumatology/ke1258. [DOI] [PubMed] [Google Scholar]
- 2).Woywodt A., Matteson E. Should eponyms be abandoned? Yes. BMJ. 2007. Sep 1; 335(7617): 424 PMID: 17762033. PMCID: PMC1962844. 10.1136/bmj.39308.342639.AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Falk R.J., Gross W.L., Guillevin L. et al. Granulomatosis with polyangiitis (Wegener's): an alternative name for Wegener's granulomatosis. Ann Rheum Dis. 2011. Apr; 70(4): 704 PMID: 21372195. 10.1136/ard.2011.150714. [DOI] [PubMed] [Google Scholar]
- 4).Jennette J.C., Falk R.J., Bacon P.A. et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013. Jan; 65(1): 1–11. PMID: 23045170. 10.1002/art.37715. [DOI] [PubMed] [Google Scholar]
- 5).Pagnoux C. Updates in ANCA-associated vasculitis. Eur J Rheumatol. 2016. Sep; 3(3): 122–33. PMID: 27733943. PMCID: PMC5058451. 10.5152/eurjrheum.2015.0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Robbins S.L., Kumar V., Cotran R.S. Robbins and Cotran pathologic basis of disease. 8th ed. Philadelphia: Saunders/Elsevier; 2010. 1450 p. [Google Scholar]
- 7).Mohammad A.J., Jacobsson L.T., Westman K.W. et al. Incidence and survival rates in Wegener's granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome and polyarteritis nodosa. Rheumatology (Oxford). 2009. Dec; 48(12): 1560–5 PMID: 19797309. 10.1093/rheumatology/kep304. [DOI] [PubMed] [Google Scholar]
- 8).van der Woude F.J., Rasmussen N., Lobatto S. et al. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener's granulomatosis. Lancet. 1985. Feb 23; 325(8426): 425–9. PMID: 2857806. 10.1016/s0140-6736(85)91147-x. [DOI] [PubMed] [Google Scholar]
- 9).Tervaert J.W., van der Woude F.J., Fauci A.S. et al. Association between active Wegener's granulomatosis and anticytoplasmic antibodies. Arch Intern Med. 1989. Nov; 149(11): 2461–5. PMID: 2684074. 10.1001/archinte.1989.00390110055012. [DOI] [PubMed] [Google Scholar]
- 10).Hagen E.C., Daha M.R., Hermans J. et al. Diagnostic value of standardized assays for anti-neutrophil cytoplasmic antibodies in idiopathic systemic vasculitis. EC/BCR Project for ANCA Assay Standardization. Kidney Int. 1998. Mar; 53(3): 743–53. PMID: 9507222. 10.1046/j.1523-1755.1998.00807.x. [DOI] [PubMed] [Google Scholar]
- 11).Seo P., Stone J.H. The antineutrophil cytoplasmic antibody-associated vasculitides. Am J Med. 2004. Jul 1; 117(1): 39–50. PMID: 15210387. 10.1016/j.amjmed.2004.02.030. [DOI] [PubMed] [Google Scholar]
- 12).Kallenberg C.G. Pathogenesis of PR3-ANCA associated vasculitis. J Autoimmun. 2008. Feb-Mar; 30(1-2): 29–36. PMID: 18162369. 10.1016/j.jaut.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 13).Weiner S.R., Paulus H.E., Weisbart R.H. Wegener's granulomatosis in the elderly. Arthritis Rheum. 1986. Sep; 29(9): 1157–9. PMID: 3753542. 10.1002/art.1780290915. [DOI] [PubMed] [Google Scholar]
- 14).Krafcik S.S., Covin R.B., Lynch JP 3rd, Sitrin R.G. Wegener's granulomatosis in the elderly. Chest. 1996. Feb; 109(2): 430–7. PMID: 8620718. 10.1378/chest.109.2.430. [DOI] [PubMed] [Google Scholar]