

Abstract
Rickets was a common metabolic disease of bone a century ago in Europe, North America, and East Asia (mainly due to vitamin D deficiency) but was largely eradicated in growing children by use of cod liver oil and the introduction of vitamin D fortification of milk in the 1930s in the United States. Vitamin D deficiency (VDD) remains the most common form of metabolic bone disease that is entirely preventable and treatable. Historically, rickets has appeared in sporadic epidemics and, despite the introduction of numerous preventive strategies, VDD has remained a global health problem amongst children. Moreover, developed countries such as Canada, Australia, the United Kingdom, and the United States have not been exempt from this.
The radiological and histological features of rickets are both distinctive and characteristic and they reflect the underlying pathophysiological issue of decreased mineralization of bone as a result of VDD. The radiological features include 1) metaphyseal cupping and fraying, 2) poor mineralization of epiphyseal centers, 3) irregular and widened epiphyseal plates, 4) increased distance between the end of shaft and epiphyseal center, 5) cortical spurs at right angles to the metaphysis, 6) coarse trabeculation, and 7) periosteal reactions. Fractures may also be evident.
The histological features of rickets reflect the failure of cartilage to mineralize and undergo resorption. This results in 1) disordered proliferation of chondrocytes in the hypertrophic zone secondary to a lack of apoptosis, 2) loss of the columnar arrangement of chondrocytes that results in thickening and disorganization of the hypertrophic zone, 3) tongue-like projections of cartilage that extend into the spongiosa, 4) irregularity of the limit between the proliferative and hypertrophic zones, and 5) penetration of blood vessels into the hypertrophic zone.
The case of a premature 3-month-old female infant, born in the winter months in the arctic region of Canada who died from a lobar pneumonia with an incidental finding of radiological and pathological evidence of rickets, is presented. The case is used to review the entity of rickets from historical, pathophysiological, radiological, and histological perspectives.
Keywords: Forensic pathology, Vitamin D deficiency, VDD, Rickets, Radiological findings, Immunomodulation
Case Report
The decedent was a 3-month-old female infant of indigenous origin who was born prematurely (corrected gestational age of 2.5 months) in the height of winter (February) in the arctic region of Canada. The details of her antenatal care, pregnancy, and delivery were not known. She had exhibited flu-like symptoms in the two-day period preceding her death but no medical attention was sought. She had coslept on a couch with her 6-year-old sister and had rolled off and fallen onto the floor without the knowledge of the parents. Her sister had picked her up and placed her back on the couch. She was subsequently found unresponsive the following morning and taken to a local health center where resuscitation was unsuccessful and death was certified. There were no criminal suspicions.
A postmortem skeletal survey was performed three days after as part of the sudden unexpected death in infancy (SUDI) protocol of the Ontario Forensic Pathology Service, which includes routine histology, bacteriology, virology, vitreous biochemistry, routine toxicology, screen for inherited metabolic diseases, and extraction and storage of DNA for targeted genetic testing, if indicated. Additionally, bone pathology consultation and postmortem biochemistry of the blood for vitamin D and parathyroid hormone were undertaken.
The postmortem skeletal survey was reported by a pediatric radiologist (with quality assurance by a second reader) who identified radiological features of rickets through the identification of 1) diffuse decrease in the mineralization of the bones, 2) mild cupping and fraying of the distal radial, ulnar, tibial, and fibular metaphyses, 3) periosteal reaction of the posterior aspect of the left 8th rib (suggestive of a previous fracture), and 4) periosteal reaction of the mid-diaphysis of the right femur and right fibula (Images 1–5).
Image 1.
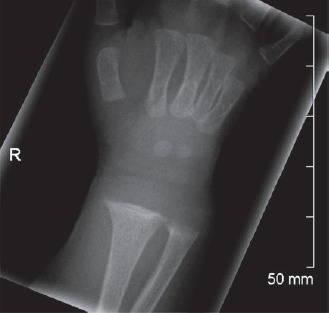
Radiograph of right wrist.
Image 2.
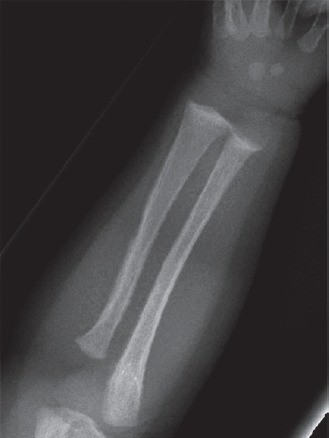
Radiograph of left forearm and wrist.
Image 3.
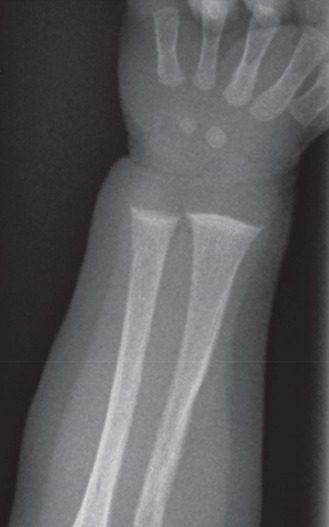
Radiograph of right forearm and wrist.
Image 4A).
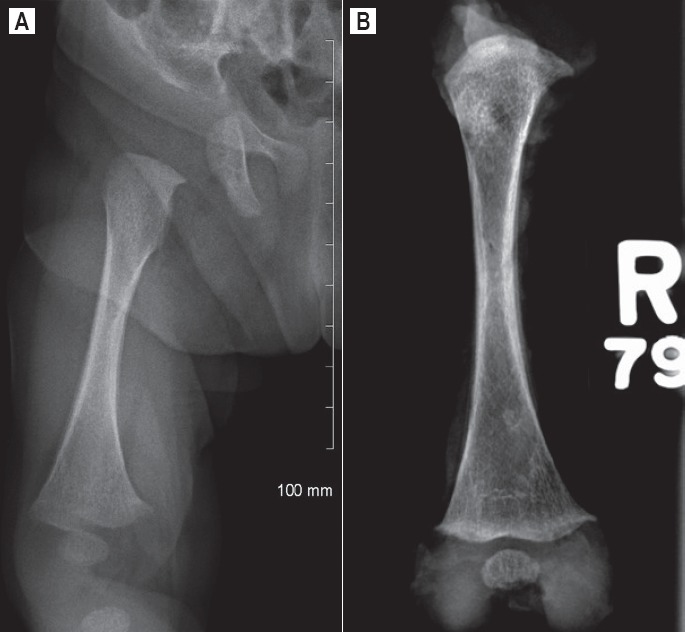
Radiograph of right femur – periosteal reaction. B) Radiograph of excised right femur – periosteal reaction.
Image 5A).

Radiograph of right tibia and fibula. B) Radiograph of excised right tibia and fibula.
External examination at postmortem revealed a normally developed female infant with no external injuries. The growth parameters (for corrected gestational age) were weight less than the 3rd percentile, height between the 50th and 85th percentiles, and head circumference between the 15th and 50th percentiles.
Internal examination excluded a head injury as the cause of death. There was a firm consistency of the posterior aspect of the lower lobe of the right lung with features of a lobar pneumonia on histological examination to account for death (Image 6). Streptococcus pneumoniae and Hemophilus influenzae were both isolated from the lung. The middle ears contained a mucoid exudate and Streptococcus pneumoniae was isolated from both middle ears, in addition to Hemophilus influenzae in the right middle ear.
Image 6A).

Lower lobe of the right lung (H&E, x50). B) Lower lobe of the right lung (H&E, x400).
There were bulbous swellings of the costochondral junctions of the left 6th through 10th ribs and right 5th through 9th ribs together with a fracture callus of the posterior aspect of the left 8th rib that were not detected on initial radiological survey but were evident on subsequent radiography of the excised rib segments (Images 7–9).
Image 7.

Bulbous swelling of the costochondral junctions of the left 6th through10th ribs (white arrows) and callus of the posterior aspect of the left 8th rib (yellow arrow).
Image 8.

Radiograph of excised left 6 through 10 rib segment with bulbous swellings of the costochondral junctions and fracture callus of the posterior aspect of the left 8th rib.
Image 9 A and B.

Excised right 5th through 9th rib segment with bulbous swellings of the costochondral junctions.
The left radius and ulna, right radius and ulna, right femur, left tibia and fibula, right tibia and fibula, left 6th through10th ribs, and right 5th through 9th ribs were excised for fixation, radiography, decalcification, and targeted histological examination (hematoxylin and eosin). A separate segment of fixed rib was used to prepare undecalcified sections that were stained with Toluidine Blue and Goldner's Trichrome.
The bone histology slides were reviewed by a pediatric pathologist with specialist training in metabolic bone disease. The morphological findings of the long bones and the undecalcified sections of the ribs were diagnostic of rickets (Images 10–12), with a healing fracture callus of the posterior segment of the left 8th rib (Image 13) and periosteal reactions of the diaphyses of the right femur (Image 14) and fibula (Image 15), as reported radiologically.
Image 10.

Hypertrophic cartilage in the distal left tibia (H&E, x40).
Image 11.
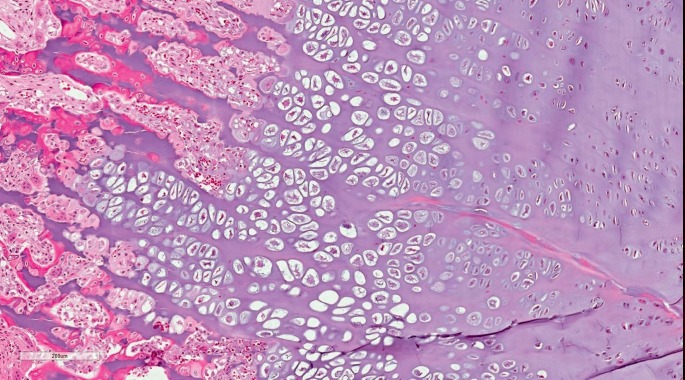
Irregular tongues of hypertrophic cartilage with capillarization of the growth plate in the distal ulnar metaphysis (H&E, x400).
Image 12.

Marked irregularity of the growth plate with tongues of hypertrophic cartilage (H&E, x40).
Image 13.

Healing fracture of the posterior aspect of the left 8th rib (H&E, x400).
Image 14A and B.

Cross section of right femur with periosteal reaction (H&E, x100 and x400).
Image 15A and B.

Cross section of right fibula with periosteal reaction (H&E, x100 and x400).
The metaphyses of the long bones showed marked thickening of the epiphyseal plate with both enlargement in height and width, which was mostly attributed to the markedly distorted hypertrophic cell zone with enlargement of chondrocytes and increase in axial zonal height (Image 10). There was marked loss of the columnar arrangement with disordered organization of cells with only a small amount of intervening matrix. There was poor definition of the zone of provisional calcification, and in several areas there were tongues of viable hypertrophic cartilage without active endochondral replacement that extended beyond the zone of primary spongiosa into the newly formed medullary bone with capillarization (Images 11 and 12). The resting and proliferative zones were unremarkable and normal. The medullary bone consisted of irregular, thin trabeculae.
The section from the posterior aspect of the left 8th rib exhibited a healing fracture with cortical thickening and an admixture of cellular fibrous tissue, cartilage, and spicules of woven bone (Image 13).
The undecalcified sections of the rib revealed a layer of universalized bone (osteoid seam) surrounding the mineralized segment of trabecular bone, which were evident beyond and far away from the epiphyseal plate, in keeping with a failure of mineralization (Image 16).
Image 16 A and B.
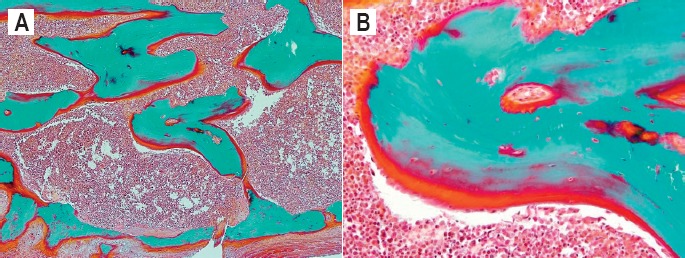
Undecalcified sections of rib (Goldner's Trichrome, x50 and x400).
Vitamin D determination in the postmortem blood revealed a total concentration of 14 ng/mL (35 nmol/L), which was reported as constituting mild to moderate deficiency. It must be recalled that Scheimberg and Perry classified postmortem vitamin D concentrations as normal (>80 nmol/L), suboptimal (50-79 nmol/L), insufficiency (25-49 nmol/L) and deficiency (<25nmol/L) (1). Unfortunately, it was not possible to obtain a parathyroid hormone (PTH) concentration as the blood sample was hemolyzed.
Interpretation of the postmortem vitamin D concentration and the radiological and histological features of rickets led to the conclusion that the decedent had vitamin D deficiency (VDD) rickets, the most common form of rickets.
This case was discussed with two other forensic pathologists and the postmortem examination report was formally peer-reviewed before final issuance. The case was also presented at a multidisciplinary meeting that consisted of practitioners with expertise in pediatric radiology, pediatric bone pathology, pediatric metabolic bone disease, forensic pathology, and child protection. The pervading view was that in the context of the diagnosis of rickets of the radiological and pathological severity seen, the single, healing posterior rib fracture and periosteal reactions were primarily attributed to rickets and not to deliberately inflicted, abusive injury, as congenital rickets was the likely underlying predisposition.
The infant had died of a Streptococcus pneumoniae lobar pneumonia. Streptococcal pneumonia is the most common cause of community-acquired pneumonia. Vitamin D deficiency was considered an ancillary contributor to its development since, apart from its role in the maintenance of the musculoskeletal system, vitamin D has important immune-modulating functions (2).
Although bacteriological culture of the blood isolated Streptococcus pneumoniae, this did not correlate with histological features of sepsis and its detection is interpreted as being reflective of the three-day postmortem interval rather than clinical septicemia. The results of the other ancillary investigations were noncontributory to death.
Discussion
Rickets is a metabolic bone disease that is most commonly associated with deficiency of vitamin D and its attendant reduced availability of circulating calcium, principally due to the inability to absorb calcium. Rickets was essentially unknown until the 1650s when cases appeared in northern Europe. It was first described in association with skeletal deformities during the mid 17th century in London, England. O'Riordan and Bijvoet had reported that the first clear descriptions of rickets were published between 1645 and 1668 by Whistler, Boot, Glisson, and Mayow in that chronological sequence (3) and that the longest and most detailed account of rickets by far was in a book published by Glisson in 1650 (4).
O'Riordan and Bijvoet also stated that although the term rickets was prevalent, its origin is obscure but words similar to the term meaning “crooked” existed in middle English, middle French, the dialect of Devonshire, and ancient Greek (3). Harrison reported that the prevalence of rickets had increased during the 18th and 19th centuries and had reached as high as 40-60% in children of the inner city neighborhoods of England, Germany, Poland, Scandinavia, Canada, and the United States (5). It was reported that Whistler, Boot, and Glisson had all used the English word “rickets” in their writings and both Whistler and Boot had discussed the origin of the word rickets without saying how it had been derived (3). It has been reported that their suggestions included that the term was coined after an apothecary named Rickets (which was not an uncommon family name that still exists) who had successfully treated the disease but there was no evidence to support that theory (3). Another reported possibility that they had entertained was that the word had been derived from “rachitis,” which indicated inflammation of the vertebral column but in the end, it appears that they had concluded that its origin was unknown (3). It was reported that both Glisson and Boot had performed postmortem examinations on patients with rickets who had died and they had believed that it was a new disease that had only appeared some 20 years before they had written about it (3). All three authors had described the disease in London, but Boot had also seen it in Ireland and Paris (3).
The discovery of vitamin D and its role in causing rickets when deficient revolves around the observations of a number of biochemists and clinical scientists, but the major breakthrough is ascribed to German physicians who noted that rickets could be reversed by the administration of oral doses of cod liver oil. E.V. McCollum, who had previously identified vitamin A in cod liver oil, also showed that cod liver oil contained an antirachitic factor that he called vitamin D (5).
Vitamin D or calcitriol, is a steroid hormone and one of the fat-soluble vitamins (vitamins A, D, E, K). Windas, in 1922, demonstrated that vitamin D was a secosteroid, a steroid with a broken ring (6). Vitamin D is an imprecise term that refers to one or more members of a group of steroid molecules. It has important roles in regulating body levels of calcium and phosphorus and thus the mineralization of bone, but receptors for vitamin D are present in a wide variety of cells. It also possesses immune modulating functions (7, 8) and regulates 3% of the human genome (9).
It is now believed that VDD is a risk factor for the development of cardiovascular disease, diabetes mellitus, infections, multiple sclerosis, and some forms of cancer (8–11). Recent evidence suggests that VDD is associated with increased mortality in adults and possibly sudden infant death syndrome (SIDS) (10, 12–13). Although some researchers have investigated if it has a role in sudden infant death, it is unknown if it is linked to SIDS (14).
The active form of vitamin D is 1,25 dihydroxyvitamin D, also known as calcitriol. The form of vitamin D found in cod liver oil is a prohormone that must be converted to its active form through a series of sequential hydroxylation steps in the liver and kidney.
The plant form of vitamin D is an isomer called vitamin D2 or ergosterol. Vitamin D3 (also known as cholecalciferol) is generated in the skin of animals when light energy is absorbed by a precursor molecule, 7-dehydrocholesterol. A previtamin D3 compound, 7-dehydrocholesterol, is found in human skin and its conversion to vitamin D is dependent on photoisomerization by ultraviolet (UV) light at a wavelength of 288 nm (5).
Vitamin D can be acquired from multiple sources, including ingestion of vitamin D2 or D3 and the endogenous production of vitamin D3, which requires exposure to sunlight or ultraviolet irradiation (5). Vitamin D is, therefore, not a true vitamin because individuals with adequate exposure to sunlight do not require dietary supplementation. The dietary sources of vitamin D include egg yolks, fish oil, and a number of plants. However, natural diets do not contain adequate quantities of vitamin D and exposure to sunlight or consumption of vitamin D supplemented food is necessary to prevent deficiencies.
Biochemistry and Physiology of Vitamin D
Vitamin D is primarily involved in mineral metabolism and bone growth. It facilitates the intestinal absorption of calcium and stimulates absorption of phosphate and magnesium ions. In the absence of vitamin D, dietary calcium is not absorbed efficiently. It also stimulates the expression of a number of proteins involved in transporting calcium from the lumen of the intestine, across the epithelial cells, and into blood. Calbindin is an intracellular protein that ferries calcium across the intestinal epithelial cell. Vitamin D is a transcriptional regulator of bone matrix proteins that induces the expression of osteocalcin and suppresses synthesis of type I collagen. In cell culture, it stimulates the differentiation of osteoclasts.
Vitamin D, as either D3 or D2, does not have significant biological activity and must be metabolized within the body to the hormonally active form known as 1,25-dihydroxycholecalciferol. The major endogenous synthetic source of vitamin D in humans is the epidermis. Vitamin D3 is produced in the skin by the UV-B mediated reaction that converts 7-dehydroxycholesterol to previtamin D3 and then previtamin D3 to vitamin D3. Vitamin D3 then finds its way into the circulation and is converted to 25-OH vitamin D3 in the liver where cholecalciferol is hydroxylated to 25-hydroxycholecalciferol by the enzyme, 25-hydroxylase. In the kidney, 25-hydroxycholecalciferol serves as a substrate for 1-alpha-hydroxylase, yielding 1,25-dihydroxycholecalciferol, the biologically active form of vitamin D (15). The conversions occur in the steps as illustrated in Figure 1.
In nutritional rickets, the serum calcium concentration is subnormal secondary to the combined effects of 1) VDD, 2) the inadequate degree of intestinal absorption of calcium, and 3) inadequate resorption of bone to release its calcium stores (6, 16). The decreased serum calcium causes secondary hyperparathyroidism with parathyroid hormone-induced phosphaturia. The rachitic child, therefore, has biochemical features of hypocalcemia, hypophosphatemia, increased alkaline phosphatase, hypocalciuria, and hyperphosphaturia (17). These biochemical abnormalities are reversed by the administration of calcitriol, either directly or via vitamin D administration, and become associated with bone healing as the extracellular calcium and phosphate levels return to normal and hydroxyapatite is deposited on osteoid tissue (18).
Vitamin D deficiency may cause apnea, hypocalcemic seizures, tetany, delayed motor milestones, skeletal deformities, and fractures. In older children, it has been associated with an increased risk of frequent lower respiratory tract infections, tuberculosis, bronchial asthma, and type 1 diabetes mellitus (1).
Vitamin D deficiency remains the most common form of metabolic bone disease that is entirely preventable and treatable. Its incidence is on the rise in the UK and other western countries, especially among ethnic minority groups. In 2006, Callaghan et al. reported an incidence of 7.5 per 100 000 children in the UK but more recent studies suggest 29%-40% (19).
Historical Perspective
Historically, epidemics of rickets occurred in waves. The first wave occurred during the industrial revolution in the western world as a result of a lack of sunlight from smog, and utilization of cod liver oil was an effective remedy (20). The work of Steenbock and Hess revealed that irradiated ergosterol could be added to milk or children could receive vitamin D supplementation, as was done in western Europe, and the rather gigantic first wave of the northern hemispheric epidemic of nutritional rickets disappeared and a major public health problem from 1900-1925 became a rarity (3, 5, 16).
There has been a significant increase in the frequency and duration of breast feeding over the decades, both of which are risk factors for the development of nutritional infantile VDD (21). The second wave of nutritional rickets occurred during the 1960s to 1980s as a result of breastfeeding women who did not get enough exposure to sunlight from full body coverage secondary to religious beliefs (20, 22, 23). At least three at-risk groups of women were identified and consisted of 1) Asian migrants from India and Uganda to the UK, 2) Muslim women in the Middle East and Central Asia, and 3) female members of certain religious groups in the inner city areas of northeastern cities of the US. All three groups wore heavy robes that covered the majority of their bodies, with often only their hands and faces exposed, in compliance with religious beliefs. These women were vitamin D deficient themselves and their fetuses were exposed to reduced circulation of 25-OH vitamin D in utero, giving rise to the development of congenital rickets. Postnatally, additional factors conspired to facilitate the development of rickets as 1) human breast milk contains inadequate amounts of vitamin D (unless maternal exposure to sunshine occurs), 2) the infants were not taken outside to receive sunlight or supplemented with vitamin D, and 3) photocutaneous synthesis of vitamin D is less efficient in dark skinned individuals.
Human milk typically contains a vitamin D concentration of 25 IU per liter or less. The amount of daily vitamin D required to prevent nutritional rickets is not fully known but may be in the range of 75-100 IU. Although breastfeeding is the recommended method of infant feeding and provides infants with necessary nutrients and immune factors, breast milk alone does not provide infants with an adequate intake of vitamin D. The average lactating woman must feed her infant 8 L (about 89 three-ounce bottles) of breast milk per day to provide the recommended daily intake of 200 IU of vitamin D (24). Even infants born to mothers replete with vitamin D develop decreasing levels within eight weeks if they are not supplemented (25).
In their investigation of iron levels in breastfed infants, Zieglar et al. incidentally discovered that the majority of unsupplemented infants had VDD, which was severe in the winter (26). Hollis and Wagner suggested that supplementation of lactating women with 2000-4000 IU of vitamin D per day, rather than the recommended 400 IU per day, was required to provide the mother and the breastfed infant with adequate vitamin D to prevent deficiency (27). Most breastfed infants are able to synthesize additional vitamin D through routine sunlight exposure.
The American Academy of Pediatrics (AAP) initially published guidelines for vitamin D intake in 2003 with the recommendation that all infants receive a minimum intake of 200 IU of vitamin D per day, beginning during the first two months of life but that guideline was updated in 2008 with a new recommendation of an increased daily intake of vitamin D of 400 IU for all infants and children beginning in the first few days of life (28).
Although there was evidence that limited sun exposure may prevent rickets in some breastfed infants (29, 30), in Canada, concern over the health risks of sun exposure have led to the recommendation that all breastfed infants receive supplemental vitamin D (400 IU/day) (31, 32). It was recommended that breast-fed infants who reside above the 55th latitude in Canada, or in areas at lower latitudes that have a high incidence of VDD, receive 800 IU/day during the winter months (33).
The second wave of rickets receded as a consequence of vitamin D supplementation. However, rickets in children in the Middle East is still recognized there and in Europe. Exclusive breast feeding, moderate vitamin D deficiency, and lack of sunshine exposure are among some of the important contributing factors that perpetuate the problem (23, 34–36).
Within recent time, VDD has been on the rise and a third outbreak of rickets with several unusual features was noticed in the early 1990s (6). This latest wave was identified in the US, particularly in the southern region (where the incident angle of the sun favors photocutaneous conversion of vitamin D) and was amongst dark skinned infants, particularly African-American children, whose mothers breastfed. Exclusive breastfeeding, moderate VDD, and lack of sunshine exposure are among some of the important contributing factors (23, 34, 35).
There were some unusual features of the US component of the third wave. In the affected southern region of the USA, despite the incident angle of the sun favoring photocutaneous conversion of vitamin D, the outbreak still developed. The mothers exclusively breastfed and seldom took their children outside. The reasons purported for the latter are 1) concerns over sunshine exposure, 2) fear of violence, 3) degeneration of the neighborhood, and 4) mothers who work at home, either on the telephone and/or personal computer.
Although it was initially believed that VDD primarily affected the elderly and dark skinned populations in the US, in 2008 Keller and Barnes reported that it was then being demonstrated in otherwise healthy young adults, children, and infants of all races (21, 36). That same year, Gordon et al. reported an insufficiency rate of 40% and a deficiency rate of 12.1% (37).
A number of articles on congenital rickets were published in 2007, which included an international perspective on maternal and newborn VDD (38). Concentrations of 25-hydroxyvitamin D (25-OHD) less than 25 nmol/L (10 ng/mL) were found in 18%, 25%, 80%, 42% and 61% of pregnant women in the UK, United Arab Emirates, Iran, northern India, and New Zealand, respectively, and in 60-80% of nonwestern women in the Netherlands. Most US experts defined VDD as less than 50 nmol/L (20 ng/mL) and insufficiency as between 20-30 ng/mL (36). As neonatal concentrations approximate two-thirds of the maternal level, congenital deficiency is inevitable in the setting of high prevalence of VDD in healthy young women (39).
Bodnar et al. in 2007 investigated maternal and newborn vitamin D levels in what can be considered the largest US study of its kind, which demonstrated deficiency or insufficiency in 83% of black women and 92% of their newborns (40). In white women and their newborns, the corresponding figures were 47% and 66%, respectively. Deficiency was worse in winter than in summer. It was striking that over 90% of the sample population was on prenatal vitamins.
The reemergence of VDD as a common occurrence has developed from the interaction of multiple factors (21). Many foods were fortified with vitamin D in the past, but in 2008 Keller and Barnes reported that milk was the only product required to be fortified in the US; however, studies have shown that up to 70% of sampled milk does not contain the required amount of vitamin D. Other natural food sources of vitamin D are few.
Exposure to UV-B rays in sunlight is the best natural source of obtaining vitamin D through cutaneous synthesis, but UV-B rays are lost during the winter months above the 35° latitude. Concomitant increased use of sunscreen and protective clothing to prevent skin cancer and to retard skin aging have also contributed to a marked reduction in cutaneous synthesis of vitamin D. A sun protection factor of 8 blocks out about 95% of the UV-B rays and produces the same effect on vitamin D synthesis as a burka worn by certain Islamic women, who continue to exhibit high rates of VDD even in the sunny climate of the middle east. As vitamin D is fat soluble, maternal obesity predisposes to poorer vitamin D status in neonates as fat storage reduces its availability (41).
The factors that affect vitamin D synthesis from sunlight are summarized in Table 1.
Table 1.
Factors That Affect Vitamin D Synthesis From Sunlight
| Living at high latitudes (closer to the polar regions), particularly during winter months |
| Air quality conditions: high levels of air pollution |
| Weather conditions: dense cloud covering |
| The degree to which clothing covers the skin |
| Use of sunscreen |
| Skin pigmentation: darker skin types |
Congenital Rickets
Fetal development can be adversely affected by VDD during pregnancy. Apart from its role in skeletal development, calcium is essential for many biological pathways inclusive of muscular contraction, second messenger formation, secretion of neurotransmitters and hormones, and blood coagulation (1). Skeletal growth is a dynamic process that requires 25-30 g of calcium to be transferred from the mother to the fetus during pregnancy (42), and is achieved mainly by increased intestinal absorption of calcium facilitated by vitamin D. Nutrients will be leached from the bones and tissues of the pregnant mother to maintain fetal development, but the net amount of vitamin D transferred to the fetus will be reduced and will affect bone mineralization and growth. Low maternal vitamin D status during pregnancy is associated with splaying of the distal metaphysis of the fetal femur (43). Intrauterine fractures due to congenital rickets have been described in the literature (44). The last trimester bears the burden of fetal skeletal mineralization as approximately 80% is deposited then. A fetus that was VDD during pregnancy will likely be born with a near normal serum calcium concentration and normal skeletal morphology but risks developing rickets within the first weeks and months of life, especially if exclusively breastfed (45).
The radiological appearance of VDD in early infancy will vary with many factors that include age at presentation, prenatal VDD, nutritional history, geographic location, sun exposure, skin color, and season (21). Case reports of congenital rickets have ranged from normal appearing bones to diffuse rarefaction, fractures, fractures at birth, metaphyseal fraying, and cupping (46–51).
The cranium has been identified as the best location to seek out radiographic evidence of congenital rickets and nutritional rickets in infants under three months of age (52), as the skull is the most affected bone at this age, reflective of rapid brain growth. Craniotabes, the softening of the skull in rickets that especially affects the occipital region and is associated with palpable enlargement of the sutures and fontanelles, was a common clinical finding in congenital rickets in the past but is once again being described in association with VDD in the newborn (53). On plain radiographs, the appearance is of pseudodiastasis with a diastatic appearance and poorly defined margins as a result of demineralization of the sutures. The skull is better evaluated on computed tomography scan of the head to exclude sutural diastasis from increased intracranial pressure (54).
The metaphyseal changes of rickets are first evident in the most recently formed metaphyseal margins in the fastest growing long bones (i.e., femora, tibiae, fibulae) and are usually symmetrical. The distal femur is the fastest growing long bone and changes occur at that site with rickets (55–57).
The metaphyseal changes in the distal femurs and proximal tibiae are first seen in the medial aspects (58), and the early metaphyseal changes in the distal ulnar are detectable even when the distal radial metaphyses appear normal (59). The physeal and metaphyseal changes can be subtle in early infancy but may become increasingly sclerotic and deformed with treatment (59). These early metaphyseal changes may mimic Salter-Harris II fractures but are asymptomatic and usually resolve without the interval changes of fracture healing (i.e., subperiosteal new bone formation, callus, and remodelling) within a few weeks. Early bowing deformity, particularly of the tibiae, may be evident when subperiosteal changes are present. These can be demonstrated on lateral views. Bowing deformities of the long bones (such as the “saber shin deformity”), when present, are representative of positional stress or musculotendinous forces where the pull of the Achilles tendon on the calcaneus causes anterior bowing of the tibiae (52).
In the rare disorder of congenital rickets, fractures are exceedingly uncommon and occur on the background of the typical rachitic changes (60).
Cohen et al. reported on a cohort study of 41 cases of sudden unexpected death in infancy and childhood (SUDIC) in which postmortem vitamin D levels were measured and compared with reviews of the radiology, rib histology, and cause of death (13). There were 25 cases of unexplained SUDIC after full autopsy. Seventy-six percent of the unexplained SUDIC cases had inadequate vitamin D levels with abnormal rib histology in 69%, but abnormal radiology in only 19%. The high incidence of bone abnormalities on histology compared with radiology (69% vs. 19%) was explained by the fact that bone calcium content needs to be reduced by at least 30% before reduced bone density can be appreciated on radiography (7), and the fact that some histological features of rickets (thickening of the cartilaginous growth plate and blood vessel penetration) cannot be detected on imaging. The findings indicated that histological examination of bones was more sensitive in detecting changes of rickets than radiology.
Histology of Normal Growth Plate and in Rickets
The normal growth plate exhibits an orderly continuation of three successive zones. These are: 1) the zone of resting cartilage; 2) the hypertrophic zone; and 3) the zone of mineralization.
The zone of resting cartilage consists of small individually scattered chondrocytes followed by a zone of proliferative cartilage in which chondrocytes become aligned in the direction of growth as they enlarge and divide. The hypertrophic zone consists of enlarged chondrocytes with more prominent nuclei in maintained alignment in the direction of growth. The zone of mineralization results from apoptosis of the hypertrophic chondrocytes with associated calcification of the cartilage and invasion by blood vessels to form bony trabeculae.
This is manifested as a zone of normal hypertrophic chondrocytes in columnar alignment with a smooth, sharp metaphyseal to diaphyseal line with no extension of any of the hypertrophic cartilage, once there are capillaries of the primary spongiosa and empty lacunae at the interface (which denotes that Ca2+ rich capillary blood has diffused and killed the chondrocytes) (Image 17A).
Figure 1.

Steps of metabolization of Vitamin D.
Image 17 A and B.

Comparison of growth plates. A = Normal growth plate. B = Growth plate in rickets (H&E, x100).
In rickets, the histological features reflect the failure of cartilage to mineralize and undergo resorption and is manifested by 1) disordered proliferation of chondrocytes in the hypertrophic zone secondary to a lack of apoptosis with 2) loss of the columnar arrangement of chondrocytes and resultant 3) thickening and disorganization of the hypertrophic zone with 4) tongue-like projections of cartilage extending into the spongiosa, 5) irregularity of the limit between the proliferative and hypertrophic zones, and 6) penetration of blood vessels into the hypertrophic zone (61). The histological comparisons are depicted below (Image 17A and B).
Postmortem Biochemistry in Rickets
Vitamin D is stable in postmortem blood for several days after death (1) and valid results can therefore be obtained. The serum 25-OHD concentration is the best marker for vitamin D status. It is stable in the serum (12) but a degree of postmortem degradation cannot be excluded. There is no consensus on the optimal level of circulating 25-OHD and different studies quote different definitions for VDD and insufficiency (8–10, 36, 37, 62, 64). The WHO definition defines VDD as <25 nmol/L and insufficiency as <50 nmol/L. Measurement of postmortem vitamin D is easy and inexpensive.
In their retrospective analysis of 52 postmortem cases (age range: 2 days to 10 years) with determination of vitamin D concentrations, Scheimberg and Perry defined normal postmortem vitamin D as >80 nmol/L, suboptimal as 50-79 nmol/L, insufficiency as 25-49 nmol/L and deficiency as <25 nmol/L (1). Seventeen children were found to be VDD, 24 children were vitamin D insufficient and ten children had suboptimal vitamin D. Only one child had a normal vitamin D concentration. Fractures were present in three cases. In the VDD cases, abnormal growth plate histology was identified in ten cases (59%) with abnormal radiology in only three cases. For the vitamin D insufficiency cases, eight cases (33%) had abnormal histology with normal radiology in all cases. Calcium, phosphate, and PTH are not stable after death and no valid results can be obtained from their analysis. However, attempts should be made to determine the postmortem concentrations of vitamin D and alkaline phosphatase.
Rickets and Child Abuse
Rickets-associated fractures can invoke suspicions of child abuse, and the presence of a vitamin disorder with fractures does not exclude child abuse as the cause of the fracture, as it is difficult to differentiate between fractures due to deliberately inflicted injury from those sustained by minimal force on a weakened bone structure. Vitamin D deficiency can also be the result of neglect, which is a form of abuse.
Although injury to the non-weight bearing parts of the skeleton (i.e., clavicles, ribs, lower arms, and hands) are suspicious in children with rickets (65), the possibility of congenital rickets must be considered as an alternative explanation. Congenital rickets can be prevented through the administration of prenatal vitamins to pregnant women, exposure of infants to sunlight for 90-300 minutes/week, and daily supplementation of 400 IU of vitamin D (6). In the present case, underlying congenital rickets was highly likely and the most probable explanation of the bone pathology findings. Neither the pregnancy history nor the details of the postpartum nutrition were available, given the geographically remote place of origin of the infant.
Duncan and Chandy reported a case of a 3-month-old female infant who was born at 28 weeks gestation via C-section for maternal preeclampsia (66). She had presented with multiple fractures and review of the radiographs gave a diagnosis of rickets. It was felt that the appearances were consistent with trauma in weakened bones from severe rickets associated with prematurity. Two case conferences could not reach a decision on the likelihood of child abuse as it was felt that a diagnosis of non-accidental injury would be unsafe. She died at five months of age and postmortem examination confirmed the histological features of rickets and no further injuries were found on either further skeletal survey or at autopsy. Three years later, her sister presented with a spiral fracture of the left femur. No evidence of metabolic bone disease was found and an unconvincing explanation for the injury was provided. However, this does not provide proof that the first child had suffered abusive injury.
Two other case reports described rickets simulating child abuse but in neither case were both diagnoses thought to coexist (65, 67).
The bone changes of VDD rickets have been invoked as a possible alternate diagnosis for non-accidental fractures in children identified on medical imaging studies (especially classic metaphyseal fractures). There have been excellent studies published characterizing the radiologic, macroscopic, and histologic features of child abuse-related fractures, including classic metaphyseal fractures, other fractures of long bones, and rib fractures. As the bone pathology of VDD rickets is distinct, this should not pose a diagnostic dilemma to the forensic pathologist who is confronted with an autopsy on a suspected child abuse-related death. The sampling of a costochondral junction should be routine in all autopsies of infants and young children, as the histologic abnormalities of rickets may precede the radiologic abnormalities (13, 68). In any pediatric case where fractures are identified on pre-autopsy medical imaging studies, it is imperative that the abnormal bones be excised and examined histologically. In addition, measurement of the vitamin D level in postmortem serum should be considered in all pediatric cases, especially when fractures are present.
Radiologically, features that can assist in differentiating abuse from rickets include normal bone density with sharp metaphyseal breaks in metaphyseal fractures from abuse whilst reduced bone density and metaphyseal splaying and fraying will be seen in rickets. Localized periosteal reaction related to fractures will be seen in non-accidental injury. In contrast, generalized periosteal reaction may occur in metabolic bone disease but can also be seen in normal or premature infants and is, therefore, not a diagnostic feature (66).
Conclusion
In a 2007 publication, Ward et al. published on the incidence of VDD rickets amongst children in Canada whom they had studied over a two-year period through the Canadian Pediatric Surveillance Programme (69). One hundred and four confirmed cases of VDD rickets were identified, with an incidence rate of 2.9 cases per 100 000. The highest incidence rates were amongst children who resided in the North (Yukon Territory, Northwest Territory, and Nunavut). The mean age of diagnosis was 1.4 years (range of 2 weeks – 6.3 years). Eighty-nine percent of cases had intermediate or darker skin, 94% had been breastfed, and 2.9% had received standard infant formula. None of the breastfed infants had received vitamin D supplementation according to the guideline of 400 IU/day. The maternal risk factors included limited sun exposure and lack of vitamin D from diet or supplements during pregnancy and lactation.
Rickets has been identified as a persistent global health concern through many publications in the literature (70–75). The question has been raised as to whether rickets has reemerged in recent years or has simply remained a persistent problem. Cases have been reported not only from regions with more limited sunshine (New Zealand, United Kingdom, and United States) (75–77) but also from sunnier regions such as Africa, Saudi Arabia, and Australia (71, 78, 79). As has been observed worldwide, rickets has continued to be an important child health issue in Canada despite simple measures for its prevention (69).
Footnotes
ETHICAL APPROVAL
As per Journal Policies, ethical approval was not required for this manuscript
STATEMENT OF HUMAN AND ANIMAL RIGHTS
This article does not contain any studies conducted with animals or on living human subjects
STATEMENT OF INFORMED CONSENT
No identifiable personal data were presented in this manuscsript
DISCLOSURES & DECLARATION OF CONFLICTS OF INTEREST
The authors, reviewers, editors, and publication staff do not report any relevant conflicts of interest
FINANCIAL DISCLOSURE The authors have indicated that they do not have financial relationships to disclose that are relevant to this manuscript
References
- 1).Scheimberg I., Perry L. Does low vitamin D have a role in pediatric morbidity and mortality? An observational study of vitamin D in a cohort of 52 postmortem examinations. Pediatr Dev Pathol. 2014. Nov-Dec; 17(6): 455–64. PMID: 25019937. 10.2350/14-05-1491-OA.1. [DOI] [PubMed] [Google Scholar]
- 2).Di Rosa M., Malaguarnera M., Nicoletti F., Malaguarnera L. Vitamin D3: a helpful immuno-modulator. Immunology. 2011. Oct; 134(2): 123–39. PMID: 21896008. PMCID: PMC3194221. 10.1111/j.1365-2567.2011.03482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).O'Riordan J.L., Bijvoet O.L. Rickets before the discovery of vitamin D. Bonekey Rep. 2014. Jan 8; 3: 478 PMID: 24466409. PMCID: PMC3899557. 10.1038/bonekey.2013.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Glisson F. De rachitide sive Morbo puerili, qui vulgò The Rickets dicitur, tractatus/operâ primò ac potissimùm Francisci Glissonii conscriptus; adscitis in operis societatem Georgio Bate, & Ahasuero Regemortero London: Guil. Du-Gardi; 1650. 427 p. [Google Scholar]
- 5).Harrison H.E. A tribute to the first lady of public health (Martha M. Eliot). V. The disappearance of rickets. Am J Public Health Nations Health. 1966. May; 56(5): 734–7. PMID: 5327833. PMCID: PMC 1257027. 10.2105/ajph.56.5.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Chesney R.W. Rickets: the third wave. Clin Pediatr (Phila). 2002. Apr; 41(3): 137–9. PMID: 11999675. 10.1177/000992280204100301. [DOI] [PubMed] [Google Scholar]
- 7).Bagnoli F., Casucci M., Rossetti A. et al. Vitamin D as a drug. J Matern Fetal Neonatal Med. 2011. Oct; 24 Suppl 1: 7–11. PMID: 21942582. 10.3109/14767058.2011.607559. [DOI] [PubMed] [Google Scholar]
- 8).Absoud M., Cummins C., Lim M.J. et al. Prevalence and predictors of vitamin D insufficiency in children: a Great Britain population based study. PLoS One. 2011; 6(7): e22179 PMID: 21799790. PMCID: PMC3142132. 10.1371/journal.pone.0022179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Davies J.H., Shaw N.J. Preventable but no strategy: vitamin D deficiency in the UK. Arch Dis Child. 2011. Jul; 96(7): 614–5. PMID: 20656737. 10.1136/adc.2010.191627. [DOI] [PubMed] [Google Scholar]
- 10).Melamed M.L., Michos E.D., Post W., Astor B. 25-hydroxyvitamin D levels and the risk of mortality in the general population. Arch Intern Med. 2008. Aug 11; 168(15): 1629–37. PMID: 18695076. PMCID: PMC2677029. 10.1001/archinte.168.15.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Autier P., Gandini S. Vitamin D supplementation and total mortality: a meta-analysis of randomized controlled trials. Arch Intern Med. 2007. Sep; 167(16): 1730–7. PMID: 17846391. 10.1001/archinte.167.16.1730. [DOI] [PubMed] [Google Scholar]
- 12).Hillman L.S., Ericson M., Haddad J.G. Serum 25-hydroxyvitamin D concentrations in sudden infant death syndrome. Pediatrics. 1980. Jun; 65(6): 1137–9. PMID: 6966389. [PubMed] [Google Scholar]
- 13).Cohen M.C., Offiah A., Sprigg A., Al-Adnani M. Vitamin D deficiency and sudden unexpected death in infancy and childhood: a cohort study. Pediatr Dev Pathol. 2013. Jul-Aug; 16(4): 292–300. PMID: 23600989. 10.2350/13-01-1293-OA.1. [DOI] [PubMed] [Google Scholar]
- 14).Adams J.S., Hewison M. Update in vitamin D. J Clin Endocrinol Metab. 2010. Feb; 95(2): 471–8. PMID: 20133466. PMCID: PMC2840860. 10.1210/jc.2009-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Vieth R. Vitamin D supplementation, 25-hydroxyvitamin D concentrations, and safety. Am J Clin Nutr. 1999. May; 69(5): 842–56. PMID: 10232622. [DOI] [PubMed] [Google Scholar]
- 16).Kreiter S.R., Schwartz R.P., Kirkman H.N. Jr. et al. Nutritional rickets in African American breast-fed infants. J Pediatr. 2000. Aug; 137(2): 153–7. PMID: 10931404. 10.1067/mpd.2000.109009. [DOI] [PubMed] [Google Scholar]
- 17).Baroncelli G.I., Bertelloni S., Ceccarelli C. et al. Bone turnover in children with vitamin D deficiency rickets before and during treatment. Acta Paediatr. 2000. May; 89(5): 513–8. PMID: 10852183. 10.1080/080352500750027763. [DOI] [PubMed] [Google Scholar]
- 18).Chesney R. Bone structure, growth and hormonal regulation. In: Behrman R., Kleugman R., Jenson H. eds. Nelson Textbook of Pediatrics. Philadelphia: WB Saunders; 2002 [Google Scholar]
- 19).Callaghan A.L., Moy R.J., Booth I.W. et al. Incidence of symptomatic vitamin D deficiency. Arch Dis Child. 2006. Jul; 91(7): 606–7. PMID: 16595644. PMCID: PMC2082851. 10.1136/adc.2006.095075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Bachrach S., Fisher J., Parks J.S. An outbreak of vitamin D deficiency rickets in a susceptible population. Pediatrics. 1979. Dec; 64(6): 871–7. PMID: 574626. [PubMed] [Google Scholar]
- 21).Keller K.A., Barnes P.D. Rickets vs. abuse: a national and international epidemic. Pediatr Radiol. 2008. Nov; 38(11): 1210–6. PMID: 18810424. 10.1007/s00247-008-1001-z. [DOI] [PubMed] [Google Scholar]
- 22).Bishop N. Rickets today—children still need milk and sunshine. N Engl J Med. 1999. Aug 19; 341(8): 602–4. PMID: 10451468. 10.1056/NEJM199908193410810. [DOI] [PubMed] [Google Scholar]
- 23).Welch T.R., Bergstrom W.H., Tsang R.C. Vitamin D-deficient rickets: the reemergence of a once-conquered disease. J Pediatr. 2000. Aug; 137(2): 143–5. PMID: 10931400. 10.1067/mpd.2000.109008. [DOI] [PubMed] [Google Scholar]
- 24).Gartner LM, Greer FR; Section on Breastfeeding and Committee on Nutrition. American Academy of Pediatrics. Prevention of rickets and vitamin D deficiency: new guidelines for vitamin D intake. Pediatrics. 2003. Apr; 111(4 Pt 1): 908–10. PMID: 12671133. 10.1542/peds.111.4.908. [DOI] [PubMed] [Google Scholar]
- 25).Hoogenboezem T., Degenhart H.J., de Muinck Keizer-Schrama S.M. et al. Vitamin D metabolism in breast-fed infants and their mothers. Pediatr Res. 1989. Jun; 25(6): 623–8. PMID: 2740153. 10.1203/00006450-198906000-00014. [DOI] [PubMed] [Google Scholar]
- 26).Ziegler E.E., Hollis B.W., Nelson S.E., Jeter J.M. Vitamin D deficiency in breastfed infants in Iowa. Pediatrics. 2006. Aug; 118(2): 603–10. PMID: 16882813. 10.1542/peds.2006-0108. [DOI] [PubMed] [Google Scholar]
- 27).Hollis B.W., Wagner C.L. Vitamin D deficiency during pregnancy: an ongoing epidemic. Am J Clin Nutr. 2006. Aug; 84(2): 273. PMID: 16895872. [DOI] [PubMed] [Google Scholar]
- 28).Wagner CL, Greer FR; American Academy of Pediatrics Section on Breastfeeding; American Academy of Pediatrics Committee on Nutrition. Prevention of rickets and vitamin D deficiency in infants, children, and adolescents. Pediatrics. 2008. Nov; 122(5): 1142–52. PMID: 18977996. 10.1542/peds.2008-1862. [DOI] [PubMed] [Google Scholar]
- 29).Specker B.L., Valanis B., Hertzberg V. et al. Sunshine exposure and serum 25-hydroxyvitamin D concentrations in exclusively breast-fed infants. J Pediatr. 1985. Sep; 107(3): 372–6. PMID: 3839846. 10.1016/s0022-3476(85)80509-6. [DOI] [PubMed] [Google Scholar]
- 30).Greer F.R., Marshall S. Bone mineral content, serum vitamin D metabolite concentrations, and ultraviolet light exposure in infants fed human milk and without vitamin D2 supplements. J Pediatr. 1989. Feb; 114(2): 204–12. PMID: 2783734 10.1016/s0022-3476(89)80784-x. [DOI] [PubMed] [Google Scholar]
- 31).Health Canada [Internet]. Ottawa: Health Canada; 2010. Vitamin D supplementation of breastfed infants in Canada: key statistics and graphics (2007-2008); 2010. Oct 17 [cited 2017 Feb 27]. Available from: http://www.hc-sc.gc.ca/fn-an/surveill/nutrition/commun/prenatal/vitamin-eng.php. [Google Scholar]
- 32).Vitamin D supplementation: recommendations for Canadian mothers and infants. Paediatr Child Health [Internet]. 2007. Sep [cited 2017 Feb 27]; 12(7): 583–98. PMID: 19030432. PMCID: PMC2528771. 10.1093/pch/12.7.583. Available from: http://www.cps.ca/en/documents/position/vitamin-d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Vitamin D supplementation in northern Native communities. Paediatr Child Health. 2002. Sep; 7(7): 459–63. PMID: 20046323. PMCID: PMC2795675. 10.1093/pch/7.7.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Mughal M.Z., Salama H., Greenaway T. et al. Lesson of the week: florid rickets associated with prolonged breast feeding without vitamin D supplementation. BMJ. 1999. Jan 2; 318(7175): 39–40. PMID: 9872885. PMCID: PMC1114534. 10.1136/bmj.318.7175.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Pal B.R., Shaw N.J. Rickets resurgence in the United Kingdom: improving antenatal management in Asians. J Pediatr. 2001. Aug; 139(2): 337–8. PMID: 11487770. 10.1067/mpd.2001.114877. [DOI] [PubMed] [Google Scholar]
- 36).Holick M.F. Vitamin D deficiency. N Engl J Med. 2007. Jul 19; 357(3): 266–81. PMID: 17634462. 10.1056/NEJMra070553. [DOI] [PubMed] [Google Scholar]
- 37).Gordon C.M., Feldman H.A., Sinclair L. et al. Prevalence of vitamin D deficiency among healthy infants and toddlers. Arch Pediatr Adolesc Med. 2008. Jun; 162(6): 505–12. PMID: 18524739. PMCID: PMC3206624. 10.1001/archpedi.162.6.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Dawodu A, Wagner CL. Mother-child vitamin D deficiency: an international perspective. Arch Dis Child. 2007. Sep; 92(9): 737–40. PMID: 17715433. PMCID: PMC2084036. 10.1136/adc.2007.122689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39).Hillman L.S., Haddad J.G. Human perinatal vitamin D metabolism I: 25-Hydroxyvitamin D in maternal and cord blood. J Pediatr. 1974. May; 84(5): 742–9. PMID: 4820711. 10.1016/s0022-3476(74)80024-7. [DOI] [PubMed] [Google Scholar]
- 40).Bodnar L.M., Simhan H.N., Powers R.W. et al. High prevalence of vitamin D insufficiency in black and white pregnant women residing in the northern United States and their neonates. J Nutr. 2007. Feb; 137(2): 447–52. PMID: 17237325. PMCID: PMC4288960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41).Bodnar L.M., Catov J.M., Roberts J.M., Simhan H.N. Prepregnancy obesity predicts poor vitamin D status in mothers and their neonates. J Nutr. 2007. Nov; 137(11): 2437–42. PMID: 17951482. PMCID: PMC2556251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42).Lewis S., Lucas R.M., Halliday J., Ponsonby A-L. Vitamin D deficiency and pregnancy: from preconception to birth. Mol Nutr Food Res. 2010. May 3; 54(8): 1092–102. PMID: 20440696. 10.1002/mnfr.201000044. [DOI] [PubMed] [Google Scholar]
- 43).Mahon P., Harvey N., Crozier S. et al. Low maternal vitamin D status and fetal bone development: Cohort study. J Bone Miner Res. 2010. Jan; 25(1): 14–9. PMID: 19580464. PMCID: PMC4768344. 10.1359/jbmr.090701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44).Soler-Bel J., Veganzones I., Navarro A. et al. [Fatal rickets in the fetus and undiagnosed maternal celiac disease]. Gastroenterol Hepatol. 2011. Dec; 34(10): 678–82. Spanish. PMID: 21757262. 10.1016/j.gastrohep.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 45).Balasubramanian S., Ganesh R. Vitamin D deficiency in exclusively breast-fed infants. Indian J Med Res. 2008. Mar; 127(3): 250–5. PMID: 18497439. [PubMed] [Google Scholar]
- 46).al-Senan K., al-Alaiyan S., al-Abbad A., LeQuesne G. Congenital rickets secondary to untreated maternal renal failure. J Perinatol. 2001. Oct-Nov; 21(7): 473–5. PMID: 11894521. 10.1038/sj.jp.7210597. [DOI] [PubMed] [Google Scholar]
- 47).Moncrieff M., Fadahunsi T.O. Congenital rickets due to maternal vitamin D deficiency. Arch Dis Child. 1974. Oct; 49(10): 810–1. PMID: 4429363. PMCID: PMC1649156. 10.1136/adc.49.10.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48).Zeidan S., Bamford M. Congenital rickets with maternal pre-eclampsia. J R Soc Med. 1984. May; 77(5): 426–7. PMID: 6726758. PMCID: PMC1439916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49).Mohapatra A., Sankaranarayanan K., Kadam S.S. et al. Congenital rickets. J Trop Pediatr. 2003. Apr; 49(2): 126–7. PMID: 12729298. 10.1093/tropej/49.2.126. [DOI] [PubMed] [Google Scholar]
- 50).Kirk J. Congenital rickets–a case report. Aust Paediatr J. 1982. Dec; 18(4): 291–3. PMID: 7165596. [PubMed] [Google Scholar]
- 51).Ford J.A., Davidson D.C., McIntosh W.B. et al. Neonatal rickets in Asian immigrant population. Br Med J. 1973. Jul 28; 3(5873): 211–2. PMID: 4541627. PMCID: PMC1586268. 10.1136/bmj.3.5873.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52).Resnick D. Bone and joint imaging. 2nd ed. Philadelphia: Saunders, 1996. Chapter 42, Rickets and osteomalacia; p 511–24. [Google Scholar]
- 53).Yorifuji J., Yorifuji T., Tachibana K. et al. Craniotabes in normal newborns: the earliest sign of subclinical vitamin D deficiency. J Clin Endocrinol Metab. 2008. May; 93(5): 1784–8. PMID: 18270256. 10.1210/jc.2007-2254. [DOI] [PubMed] [Google Scholar]
- 54).Swischuk L.E., Hayden C.K. Jr.. Seizures and demineralization of the skull. A diagnostic presentation of rickets. Pediatr Radiol. 1977. Sep 1; 6(2): 65–7. PMID: 896352. 10.1007/bf00973524. [DOI] [PubMed] [Google Scholar]
- 55).Jaffe H.L. Rickets and osteomalacia. In: Metabolic, degenerative and i nflammatory diseases of bones and joints. Lea and Febiger, Philadelphia; 1972. [Google Scholar]
- 56).Shore R., Chesney R.W. Rickets: Part II. Pediatr Radiol. 2013. Jan; 43(2): 152–72. PMID: 23179485. 10.1007/s00247-012-2536-6. [DOI] [PubMed] [Google Scholar]
- 57).Muller H. Ueber die entwicklung der knochensubstanz nebst bemerkungen uber den bau rachitischer knochen. Zeitschr f wissensch zool. 1858; 9: 147–233. German. [Google Scholar]
- 58).Thacher T.D., Fischer P.R., Pettifor J.M. et al. Radiographic scoring method for the assessment of the severity of nutritional rickets. J Trop Pediatr. 2000. Jun; 46(3): 132–9. PMID: 10893912. 10.1093/tropej/46.3.132. [DOI] [PubMed] [Google Scholar]
- 59).Silverman F.N. Metabolic abnormalities of the skeleton. In: Caffey's pediatric x-ray diagnosis: an integrated imaging approach. Mosby: St. Louis; 1993. p. 1746–54. [Google Scholar]
- 60).Teotia S.P.S., Teotia M. Author's response: Dietary calcium deficiency & rickets. Indian J Med Res. 2008. Nov; 128(5): 674–6. [PubMed] [Google Scholar]
- 61).Teitelbaum S.L. Pathological manifestations of osteomalacia and rickets. Clin Endocrinol Metab. 1980. Mar; 9(1): 43–62. PMID: 6998610. 10.1016/s0300-595x(80)80020-x. [DOI] [PubMed] [Google Scholar]
- 62).Prentice A. Vitamin D deficiency: a global perspective. Nutr Rev. 2008. Oct; 66(10 Suppl 2): S153–64. PMID: 18844843. 10.1111/j.1753-4887.2008.00100.x. [DOI] [PubMed] [Google Scholar]
- 63).Stoian C.A., Lyon M., Cox R.G. et al. Vitamin D concentrations among healthy children in Calgary, Alberta. Paediatr Child Health. 2011. Feb; 16(2): 82–6. PMID: 22294867. PMCID: PMC3043039. 10.1093/pch/16.2.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64).Tolppanen A.M., Fraser A., Fraser W.D., Lawlor D.A. Risk factors for variation in 25-hydroxyvitamin D3 and D2 concentrations and vitamin D deficiency in children. J Clin Endocrinol Metab. 2012. Apr; 97(4): 1202–10. PMID: 22278429. 10.1210/jc.2011-2516. [DOI] [PubMed] [Google Scholar]
- 65).Zeiss J., Wycliffe N.D., Cullen B.J. et al. Radiological case of the month. Simulated child abuse in drug-induced rickets. Am J Dis Child. 1988. Dec; 142(12): 1367–8. PMID: 3195536. [PubMed] [Google Scholar]
- 66).Duncan A.A., Chandy J. Case report: multiple neonatal fractures—dietary or deliberate? Clin Radiol. 1993. Aug; 48(2): 137–9. PMID: 8004894. 10.1016/s0009-9260(05)81090-6. [DOI] [PubMed] [Google Scholar]
- 67).Paterson C.R. Vitamin D deficiency rickets simulating child abuse. J Pediatr Orthop. 1981; 1(4): 423–5. PMID: 7334122. 10.1097/01241398-198112000-00012. [DOI] [PubMed] [Google Scholar]
- 68).Kepron C., Pollanen M.S. Rickets or abuse? A histologic comparison of rickets and child abuse-related fractures. Forensic Sci Med Pathol. 2015. Mar; 11(1): 78–87. PMID: 25557084. 10.1007/s12024-014-9639-3. [DOI] [PubMed] [Google Scholar]
- 69).Ward L.M., Gaboury I., Ladhani M., Zlotkin S. Vitamin D–deficiency rickets among children in Canada. CMAJ. 2007. Jul 17; 177(2): 161–6. PMID: 17600035. PMCID: PMC1913133. 10.1503/cmaj.061377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70).Binet A., Kooh S.W. Persistence of vitamin D-deficiency rickets in Toronto in the 1990s. Can J Public Health. 1996. Jul-Aug; 87(4): 227–30. PMID: 8870299. [PubMed] [Google Scholar]
- 71).Robinson P.D., Högler W., Craig M.E. et al. The re-emerging burden of rickets: a decade of experience from Sydney. Arch Dis Child. 2006. Jul; 91(7): 564–8. PMID: 15956045. PMCID: PMC2082843. 10.1136/adc.2004.069575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72).Strand M.A., Perry J., Jin M. et al. Diagnosis of rickets and reassessment of prevalence among rural children in northern China. Pediatr Int. 2007. Apr; 49(2): 202–9. PMID: 17445039. 10.1111/j.1442-200X.2007.02343.x. [DOI] [PubMed] [Google Scholar]
- 73).Haworth J.C., Dilling L.A. Vitamin D-deficient rickets in Manitoba, 1972–84. CMAJ. 1986. Feb 1; 134(3): 237–41. PMID: 3942930. PMCID: PMC1490697. [PMC free article] [PubMed] [Google Scholar]
- 74).Mylott B.M., Kump T., Bolton M.L., Greenbaum L.A. Rickets in the Dairy State. WMJ. 2004; 103(5): 84–7. PMID: 15553572. [PubMed] [Google Scholar]
- 75).Weisberg P., Scanlon K.S., Li R., Cogswell M.E. Nutritional rickets among children in the United States: review of cases reported between 1986 and 2003. Am J Clin Nutr. 2004. Dec; 80(6 Suppl): 1697S–705S. PMID: 15585790. [DOI] [PubMed] [Google Scholar]
- 76).Blok B.H., Grant C.C., McNeil A.R., Reid I.R. Characteristics of children with florid vitamin D deficient rickets in the Auckland region in 1998. N Z Med J. 2000. Sep 8; 113(1117): 374–6. PMID: 11050902. [PubMed] [Google Scholar]
- 77).Shaw N.J., Pal B.R. Vitamin D deficiency in UK Asian families: activating a new concern. Arch Dis Child. 2002. Mar; 86(3): 147–9. PMID: 11861227. PMCID: PMC1719105. 10.1136/adc.86.3.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78).Karrar Z.A. Vitamin D deficiency rickets in developing countries. Ann Trop Paediatr. 1998. Sep; 18 Suppl: S89–92. PMID: 9876274. 10.1080/02724936.1998.11833490. [DOI] [PubMed] [Google Scholar]
- 79).Al-Mustafa Z.H., Al-Madan M., Al-Majid H.J. et al. Vitamin D deficiency and rickets in the Eastern Province of Saudi Arabia. Ann Trop Paediatr. 2007. Mar; 27(1): 63–7. PMID: 17469734. 10.1179/146532807X170529. [DOI] [PubMed] [Google Scholar]