

Abstract
Usher syndrome (USH) is the leading cause of inherited combined vision and hearing loss. However, mutations in most USH causative genes lead to other diseases, such as hearing loss only or vision loss only. The molecular mechanisms underlying the variable disease manifestations associated with USH gene mutations are unclear. This review focuses on an USH type 2 (USH2) gene encoding whirlin (WHRN; previously known as DFNB31), mutations in which have been found to cause either USH2 subtype USH2D or autosomal recessive non-syndromic deafness type 31 (DFNB31). This review summarizes the current knowledge about different whirlin isoforms encoded by WHRN orthologs in animal models, the interactions of different whirlin isoforms with their partners, and the function of whirlin isoforms in different cellular and subcellular locations. The recent findings regarding the function of whirlin isoforms suggest that disruption of different isoforms may be one of the mechanisms underlying the variable disease manifestations caused by USH gene mutations. This review also presents recent findings about the vestibular defects in Whrn mutant mouse models, which suggests that previous assumptions about the normal vestibular function of USH2 patients need to be re-evaluated. Finally, this review describes recent progress in developing therapeutics for diseases caused by WHRN mutations.
Keywords: stereocilia, hair cell, ankle link, photoreceptor, retinitis pigmentosa, hearing loss, Usher syndrome
Introduction:
Usher syndrome (USH) is an incurable autosomal recessive genetic disease characterized by combined vision and hearing loss. USH is the major cause of inherited deaf-blindness, with a prevalence of 1.6–4.4 per 10,000 individuals worldwide (Boughman et al., 1983; Kimberling et al., 2010). Based on the severity of the phenotypes, USH is subdivided into three categories. USH1 is the most severe, wherein patients have congenital profound hearing loss, early-onset retinal degeneration, and vestibular disorder. USH2 is the most prevalent, characterized by congenital moderate hearing loss, retinal degeneration starting as early as adolescence, and no obvious balance problems. USH3 patients have progressive hearing loss, retinal degeneration with variable onset times, and variable vestibular dysfunction. Thus far, 10 genes have been identified as causative in USH, and more remain to be identified (Vozzi et al., 2011). Among the 10 genes, 6 are USH1 genes; 3 are USH2 genes; and 1 is an USH3 gene.
Sensorineural hearing loss and vestibular dysfunction in USH patients are caused by defective hair cells in the inner ear. In the cochlea, the coil-shaped organ of Corti is positioned on the top of the basilar membrane, which resonates in response to vibrations induced by sound waves. The organ of Corti has three rows of outer hair cells (OHCs) and one row of inner hair cells (IHCs). Each of the OHCs and IHCs contains a hair bundle composed of actin-based stereocilia and a transient microtubule-based kinocilium at its apex. OHC tallest stereociliary tips are embedded into the overlying tectorial membrane. Therefore, vibration of the basilar membrane develops a shearing force between OHC stereocilia and tectorial membrane, which deflects OHC stereocilia and stimulates mechanotransduction at stereociliary tips. The resulting mechanotransduction response induces longitudinal oscillation of the OHC cell body, thereby amplifying the vibration of the basilar membrane. The amplified vibration signals deflect the hair bundle and subsequently induce mechanotransduction in IHCs that synapse with afferent spiral ganglion auditory nerve fibers. Stereocilia in the vestibular hair cells (VHCs) sense movement of the head and function in a similar manner to those of cochlear hair cells. Therefore, the organization and dimension of stereocilia in the cochlea and vestibular system are crucial for normal hearing and balance, respectively.
In the hair bundle, there are various proteinaceous links among the kinocilium and stereocilia, such as tip links, kinociliary links, lateral links, horizontal top connectors, and ankle links (Fig. 1A). Tip links in mature hair cells connect the tip of shorter stereocilia to the lateral wall of their neighboring taller stereocilia and are essential for ion channel opening during mechanotransduction. Kinociliary links connect the kinocilium with its neighboring stereocilia, while lateral links and horizontal top connectors connect adjacent stereocilia to each other. The kinociliary links, lateral links, and horizontal top connectors are thought to play a role in cohesion among kinocilium and stereocilia during hair bundle development (Goodyear et al., 2005). Ankle links are present at the stereociliary base transiently in OHCs and IHCs (e.g., mouse postnatal day [P] 2 – 12) and permanently in VHCs (Grati et al., 2012; Jeffries et al., 1986; McGee et al., 2006). Ankle links are required for stereociliary organization in a cochlear hair cell bundle during development (McGee et al., 2006; Michalski et al., 2007; Zou et al., 2015). USH1 proteins mainly localize to the kinociliary links and lateral links during development and to the tip links and tip link densities in adulthood; USH2 proteins are components of ankle links and ankle link complex; and USH3 protein is present in the hair bundle. Therefore, USH proteins in different regions of hair cell bundles are required directly or indirectly for receiving and processing sound and balance inputs.
Figure 1:
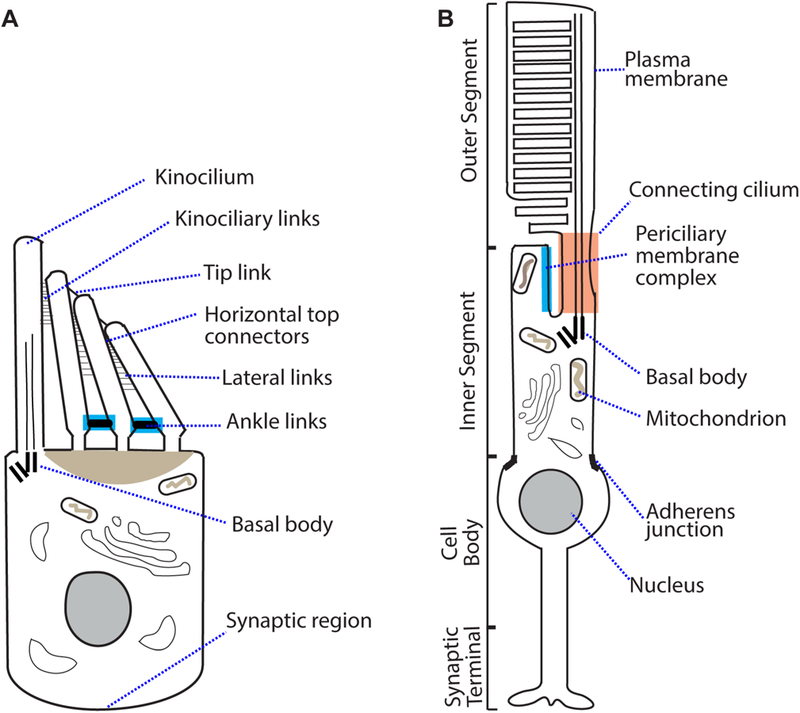
Structure of an inner ear hair cell and a retinal photoreceptor. (A) A developing cochlear hair cell (e.g., mouse P2–P12) has ankle links (highlighted in blue) located at the base of stereocilia. Lateral links and horizontal top connectors are also present. Tip links connect the tip of short stereocilia to the shaft of longer stereocilia in the adjacent row and are required for mechanotransduction. (B) A rod photoreceptor has an outer segment with discs that contain rhodopsin. Rhodopsin detects light in the form of photons. The photoreceptor inner segment contains mainly organelles required for energy metabolism and protein synthesis. The periciliary membrane complex (highlighted in blue) is located at the apical region of inner segment, surrounding the connecting cilium (highlighted in orange). In order to illustrate the periciliary membrane complex and the connecting cilium, the calyceal processes, which are located outside the two subcellular structures, are not shown. The cell body contains the nucleus, while the synaptic region contains ribbon synapses.
Retinal degeneration in USH patients occurs in the form of retinitis pigmentosa (RP), with night-blindness and tunnel vision as typical phenotypes. These phenotypes arise as a result of rod photoreceptor degeneration. In the advanced RP stage, cone photoreceptors degenerate, which leads to complete or legal blindness. Photoreceptors are light-sensitive neurons (Fig. 1B). Photons are detected by rhodopsin present on the discs of the photoreceptor outer segment. The outer segment lacks protein synthesis machinery and relies on the inner segment for supply of proteins. The periciliary membrane complex (PMC) is located at the apex of the inner segment surrounding the connecting cilium and was thought to be involved in transporting protein cargos from the inner to the outer segment (Yang et al., 2010). The calyceal processes containing actin bundles project from the apex of the inner segment and tightly wrap around the base of the outer segment (Mathur et al., 2015a; Sahly et al., 2012). The calyceal processes are thought to maintain the integrity of the connection between the inner and outer segments, probably in response to mechanical stress. While USH2 proteins assemble into the PMC, most USH1 proteins are localized to the calyceal processes (Sahly et al., 2012; Yang et al., 2010). These USH proteins are required for photoreceptor maintenance and function.
For most USH genes, different mutations of the same genes can cause USH or phenotypes that are characterized as diseases other than USH (see Table 2 in Mathur et al., 2015a). For example, mutations in USH1 gene MYO7A may lead to USH1B subtype (Weil et al., 1995), non-syndromic autosomal recessive deafness (DFNB) DFNB2 subtype, non-syndromic autosomal dominant deafness DFNA11 subtype, or atypical USH (Liu et al., 1997a; Liu et al., 1997b; Liu et al., 1998; Riazuddin et al., 2008; Weil et al., 1997). Understanding the molecular mechanisms underlying these variable disease manifestations is essential not only for early and accurate diagnosis but also for development of therapies. Recent findings with regard to an USH2 gene WHRN, which encodes the whirlin protein, provide novel insights into a potential mechanism underlying the variable disease manifestations caused by USH gene mutations.
The WHRN/Whrn gene yields various mRNA transcripts in humans and mice, because the gene has two predicted promoter regions and undergoes alternative splicing (Belyantseva et al., 2005; Ebrahim et al., 2016; Mburu et al., 2003; Wright et al., 2012). WHRN/Whrn mRNA transcripts are predicted to encode full-length (FL)-, N-, and C-terminal whirlin proteins. The FL-whirlin protein isoform has two harmonin N-like (HNL) domains, three PDZ domains, a proline-rich (PR) region, and a PDZ-binding motif (PBM) (Fig. 2). The transcripts encoding C-whirlin have start sites in either exon 1 or exon 6 and are expected to yield similar C-whirlin protein isoforms with HNL2, PR, PDZ3, and PBM domains (Belyantseva et al., 2005; Mburu et al., 2003; Yang et al., 2010). The transcripts encoding N-whirlin yield isoforms that contain either PDZ1, PDZ2, or both PDZ1 and PDZ2 domains (Ebrahim et al., 2016; Wright et al., 2012). In this review, we will summarize the recent studies on WHRN orthologs in the cochlea, vestibular organs, retina, and other nerve systems in animal models as well as recent exploratory therapeutic studies aiming to treat retinal and inner ear defects caused by Whrn mutations in mice.
Figure 2:
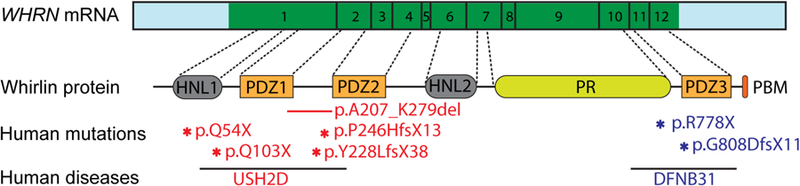
Location and disease manifestation of human WHRN mutations. FL-whirlin mRNA (untranslated region-light blue; coding sequence region-green) is translated into a protein with two harmonin N-like (HNL) domains, three PDZ domains, a proline-rich (PR) region, and a PDZ-binding motif (PBM). Arabic numerals in the coding sequence region denote exon numbers. The asterisks and short line in red denote mutations in the N-terminal region that cause USH2D, except p.Q54X. The patient with p.Q54X mutation has not been tested for hearing. The asterisks in blue denote C-terminal mutations that cause DFNB31.
Mutations in WHRN cause variable disease manifestations:
Mutations in WHRN lead to either USH2D subtype or DFNB31 subtype. USH2D patients have moderate sensorineural hearing loss and RP, while DFNB31 patients have profound sensorineural hearing loss and normal vision. The first WHRN mutation identified in DFNB31 patients is a cysteine to threonine substitution in exon 10, which changes the arginine codon to a stop codon (R778X) (Mburu et al., 2003). Another WHRN mutation in exon 11 (G808DfsX11) was later found to lead to profound deafness (Tlili et al., 2005). On the other hand, patients with compound heterozygous nonsense (G103X) and splice site (A207_K279del) mutations in the 1st and 2nd exons of WHRN, respectively, have USH2D, with congenital moderate hearing loss and RP that begins by age 20 (Ebermann et al., 2007). Patients with either a homozygous one base-pair deletion (P246HfsX13) or compound heterozygous mutations (P246HfsX13;Y228fsX38) in WHRN also show USH2D phenotypes (Audo et al., 2011; Besnard et al., 2012). Recently, a patient with a nonsense mutation (Q54X) in WHRN exon 1 developed relatively late-onset RP, but was unavailable for hearing tests (Nishiguchi et al., 2013). None of these aforementioned patients self-reported any vestibular deficits. Taken together, clinical reports suggest that mutations in the 5′-region of WHRN, corresponding to the N-terminal region of the protein, lead to USH2D, while mutations toward the 3′ end, corresponding to the protein C-terminal region, cause DFNB31 (Fig. 2).
Whrn mouse models recapitulate USH2D and DFNB31 diseases:
The existence of a naturally occurring Whrn mutant mouse model and the generation of other Whrn mutant mouse models provide a variety of excellent tools to study Whrn expression and function in the inner ear and retina, and to understand the pathogenesis of USH2D and DFNB31. The Whrnwi mouse (MGI:1857090) has a spontaneous 592-bp deletion between exons 6–10 in the transcript (Fig. 3A and B). This deletion creates a frameshift leading to a premature translation termination. The Whrnwi mouse is profoundly deaf and shows no retinal degeneration (Holme et al., 2002; Mburu et al., 2003; Yang et al., 2010). Therefore, the Whrnwi mutation reflects the 3′-region mutations found in DFNB31 patients. The Whrntm1Tili mouse (MGI:4462398, hereafter referred to as Whrnneo) was generated by replacing the 5′-region of Whrn exon 1 with a Neor cassette (Yang et al., 2010) (Fig. 3C). This mouse shows moderate hearing loss and progressive retinal degeneration similar to that observed in USH2D patients. The Whrntm1a(EUCOMM)wtsi mouse (MGI:4432119, referred to as Whrntm1a hereafter) carries a gene trap containing a selection cassette between exon 3 and exon 4 (Fig. 3D). By crossing the Whrntm1a mouse with a whole-body Cre mouse line HprtTg(CMV-Cre)Brd, the Whrntm1b mouse was generated with a deletion of exon 4 (Fig. 3E) (Ebrahim et al., 2016). Therefore, both Whrntm1a and Whrntm1b mice have mutations at the 5′ region of Whrn. Furthermore, the Whrnwi+BAC transgenic mouse was generated by inserting a Bacterial Artificial Chromosome (BAC) containing Whrn gDNA from exons 4–13 into the genome of the Whrnwi mouse (Fig. 3F) (Mburu et al., 2003). All Whrntm1a, Whrntm1b, and Whrnwi+BAC mice exhibit moderate hearing loss, similar to the Whrnneo mouse (Ebrahim et al., 2016; Mathur et al., 2015b). Because the mutations in the currently available Whrn mutant mice mimic human WHRN mutations that lead to USH2D and DFNB31, these mouse models have been used to understand the genotype-phenotype correlation found in WHRN-deficient patients.
Figure 3:

Whrn genomic DNA and mRNA variants in wild-type and Whrn mutant mice. (A) FL-, N-, and C-terminal whirlin mRNA variants found in the wild-type mice. For simplicity, only one representative N-whirlin mRNA variant is shown, although several N-whirlin mRNA variants have been reported (Belyantseva et al., 2005; Ebrahim et al., 2016; Mathur et al., 2015b; Wright et al., 2012). In addition, the C-whirlin variants starting from exon 1 are not shown. (B) A deletion from exon 6 to exon 10 (red box) truncates both FL-whirlin mRNA and C-whirlin mRNA variants in Whrnwi mice. A vertical yellow bar indicates a premature stop in truncated Whrnwi mRNA fragments. N-whirlin mRNA variants were not found in our study, although they were found in the Whrnwi inner ear by Ebrahim et al. (Ebrahim et al., 2016; Mathur et al., 2015b; Mathur et al., 2015c). (C) Only a C-terminal whirlin variant is present in Whrnneo mice, which has a Neor cassette (orange box) in the 5′ region of exon 1. (D) A targeting cassette (light blue box) in intron 3 of Whrntm1a genomic DNA (gDNA) disrupts the FL-whirlin variant. However, it is expected that the C-terminal whirlin mRNA variant remains unaffected. L, loxP site. (E) Exon 4 and a part of the targeting cassette (light blue box) in Whrntm1a gDNA are deleted in Whrntm1b mice, which disrupts the FL-whirlin variant. The C-terminal whirlin mRNA variant is expected to be unaffected. (F) Whrnwi+BAC mice are expected to express C-terminal whirlin mRNA variant from the transgenic BAC DNA, in addition to the variants expressed in Whrnwi mice. Brown and light grey in the Whrn gDNA and BAC DNA denote the exons and introns, respectively, with exon numbers in Arabic numerals. White color denotes 5′ and 3′-untranslated regions, and green color denotes the coding sequence region of whirlin mRNA variants.
Whirlin has different spatiotemporal expression patterns in the inner ear and retina:
We identified and compared the Whrn mRNA transcripts expressed in the organ of Corti, vestibular, and retinal tissues of wildtype, Whrnneo, and Whrnwi mice (Mathur et al., 2015b; Mathur et al., 2015c). Our results indicate that wild-type mice have transcripts predicted to encode FL-, N-, and C-whirlin proteins in the retina, but only transcripts predicted to encode FL- and C-whirlin proteins in the inner ear (Table 1, Fig. 3A). We also found that the C-whirlin-encoding transcript has a start site only in exon 6 but not in exon 1 (Mathur et al., 2015b). Whrnneo mice lack all Whrn transcripts except the transcript encoding C-whirlin protein in the inner ear (Table 1, Fig. 3C). Whrnwi mice have truncated FL- and C-whirlin mRNA transcripts in the inner ear and retina (Table 1; Fig. 3B). However, a recent study reported several N-whirlin-encoding transcripts in the wild-type, Whrntm1b, and Whrnwi mouse inner ear, and a C-whirlin-encoding transcript starting in exon 1 in the wild-type mouse inner ear (Ebrahim et al., 2016). The discrepancy between our results and that report may be due to the different mouse genetic backgrounds and ages examined as well as the different PCR primers and experimental conditions used. Notably, some Whrn transcripts may not be robustly translated into proteins in the retina or in the inner ear.
Table 1:
Different Whrn mRNA variants present in the retina and inner ear of WT, Whrnneo, Whrntm1b, and Whrnwi mice (Ebrahim et al., 2016; Mathur et al., 2015b).
| Mouse model | Retina | Cochlea | Vestibular organs |
|---|---|---|---|
| WT | FL-whirlin | FL-whirlin | FL-whirlin |
| C-whirlin | C-whirlin | C-whirlin | |
| N-whirlin | N-whirlin? | N-whirlin? | |
| Whrnneo | None | C-whirlin | C-whirlin |
| Whrnwi | truncated FL-whirlin | truncated FL-whirlin | truncated FL-whirlin |
| truncated C-whirlin | truncated C-whirlin | truncated C-whirlin | |
| Whrntm1b | Not determined | C-whirlin | |
| N-whirlin | |||
: N-whirlin variants in wild-type inner ear were detected only by Ebrahim et al. (Ebrahim et al., 2016), but not by our laboratory.
Studies on whirlin localization in hair cells and photoreceptors have been extensively conducted for more than one decade, although the findings are not exactly consistent. For example, in one study, whirlin was shown to localize only to the stereociliary tip of hair cells (Kikkawa et al., 2005). Another study reported the presence of whirlin at the stereociliary tip and base (Delprat et al., 2005). A third study found whirlin in the OHC synaptic region (van Wijk et al., 2006). We generated antibodies that specifically detect the N- and C-terminal regions of whirlin and utilized Whrn mutant mice as controls for comparison to wild-type mice. In the inner ear of wild-type mice at P4, we found that both FL- and C-whirlin isoforms localize to the IHC and VHC stereociliary tips, whereas only C-whirlin is present at the OHC stereociliary tip. FL-whirlin is the only whirlin isoform at the stereociliary base in all hair cell types (Fig. 4A, B) (Mathur et al., 2015b; Mathur et al., 2015c). In Whrnneo mice, the FL-whirlin isoform at the stereociliary base and tip is missing, but the C-whirlin isoform remains localized to the stereociliary tip of OHCs, IHCs, and VHCs. (Fig. 4E, F) (Mathur et al., 2015b; Mathur et al., 2015c). Localization of whirlin isoforms in Whrntm1a hair cells and a fraction of Whrnwi+BAC hair cells (96% for IHCs and 33% for OHCs) is similar to that of Whrnneo hair cells (Fig. 4E, F and M–O) (Mathur et al., 2015b). Whrnwi mice do not show any whirlin isoforms, either at the stereociliary tip or at the stereociliary base of their inner ear hair cells (Fig. 4I, J) (Mathur et al., 2015b; Mathur et al., 2015c), suggesting that the truncated FL- and C-terminal whirlin transcripts in Whrnwi hair cells are probably degraded by nonsense-mediated mRNA decay. The localization of whirlin isoforms remains unchanged at the stereociliary tip in adult mouse IHCs, while whirlin isoforms are abolished in other regions of adult mouse IHCs and OHCs (Fig. 4C, G and K) (Mathur et al., 2015b).
Figure 4:
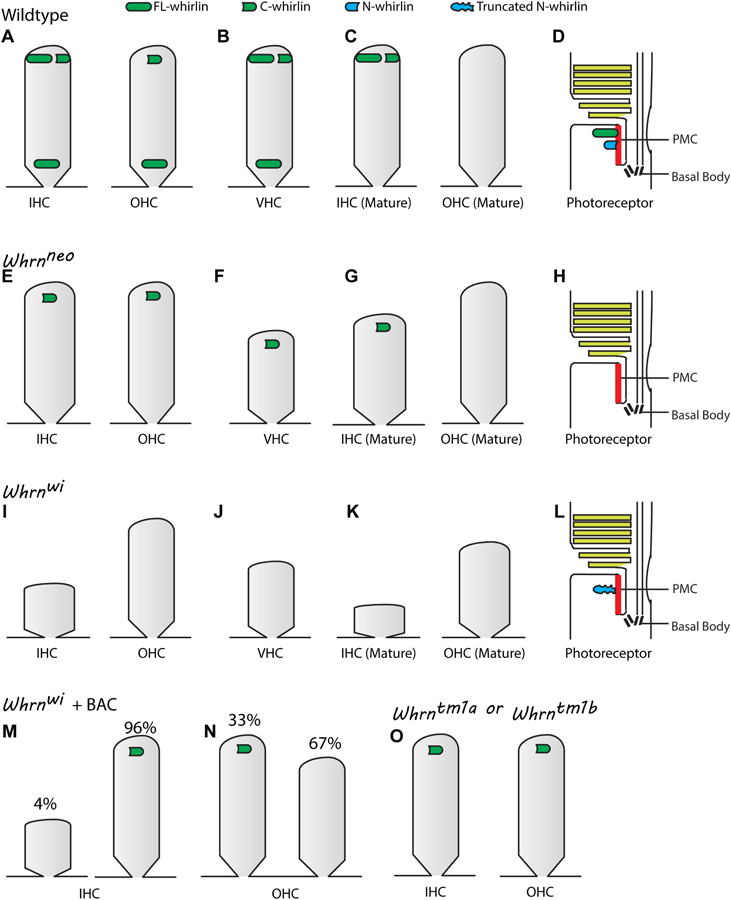
Localization of whirlin isoforms in the wild-type and Whrn mutant inner ear and retina. (A) In the developing wild-type organ of Corti (e.g., during mouse P2–P12), FL-whirlin is localized to the stereociliary base of IHCs and OHCs. Additionally, IHC stereociliary tips contain FL- and C-whirlin, while OHC stereociliary tips contain only C-whirlin. (B) Localization of whirlin isoforms in wild-type VHCs is similar to that of developing IHCs as shown in A. (C) FL- and C-whirlin isoforms remain at the stereociliary tip of mature wild-type IHCs, but no whirlin isoforms are present in mature wild-type OHCs. (D) FL-whirlin and likely N-whirlin are localized to the PMC in wild-type photoreceptor cells. (E-G) C-whirlin is localized to the stereociliary tip of OHCs, IHCs, and VHCs in Whrnneo mice. This localization of C-whirlin in OHCs occurs only during development but not in adulthood. No whirlin isoforms are present at the stereociliary base in any Whrnneo hair cells at any time point. (H) No whirlin isoforms are detected in Whrnneo photoreceptors. (I-K) Whrnwi inner ear hair cells do not show any whirlin expression at any time point. (L) The truncated N-whirlin fragment in Whrnwi retinas localizes to the PMC. (M-N) 96% of IHCs and 33% of OHCs in Whrnwi+BAC mice show localization of C-whirlin to their stereociliary tips during development. (O) Similar to Whrnneo mouse, Whrntm1a and Whrntm1b mice show localization of only C-whirlin to their IHC and OHC stereociliary tips during development. In this figure, the short and thick stereocilia phenotypes in Whrnneo and Whrnwi hair cells are shown.
Our findings in the mouse cochlea during development are generally consistent with those of a recent report on wild-type, Whrntm1b, and Whrnwi mice by Ebrahim et al. (Ebrahim et al., 2016), except the following differences. First, although the whirlin immunofluorescent confocal signal pattern in cochlear hair cells is similar between our study and the study by Ebrahim et al., the latter used structured illumination microscopy and high-resolution spinning disc confocal microscopy to localize whirlin to the neighboring shorter stereocilia close to the stereociliary tip (Ebrahim et al., 2016). In our study, we concluded that whirlin is present at the stereociliary base, because whirlin colocalizes with ADGRV1 in that region (Grati et al., 2012; Zou et al., 2011) and ADGRV1 is a major component of ankle links at the stereociliary base (McGee et al., 2006). It is possible that ankle links end close to the tip at the shorter stereocilia and close to the base at the longer stereocilia (McGee et al., 2006). To resolve this discrepancy, it will be crucial to examine whirlin subcellular localization in hair bundles using immunoelectron microscopy. Second, Ebrahim et al. did not detect FL-whirlin at the stereociliary tip in IHCs (Ebrahim et al., 2016), likely because they used an antibody and sample treatment procedure different from ours. In general, both our study and the study from Ebrahim et al. showed that the 5′-region mutations in Whrnneo, Whrntm1a, and Whrntm1b mice retain the C-whirlin isoform, which is located at the stereociliary tip, while the 3′-region mutation in Whrnwi mice leads to loss of both FL- and C-whirlin isoforms in the inner ear hair cells (Fig. 3 and Fig. 4).
Whirlin was previously found at the PMC in photoreceptors (Yang et al., 2010); however, whirlin isoform-specific information at the PMC was unknown. We found that FL- and likely N-whirlin isoforms are expressed and localize to the PMC in wild-type photoreceptors (Fig. 4D), while no whirlin isoforms are expressed in Whrnneo photoreceptors (Fig. 4H) (Mathur et al., 2015b). This result indicates that, despite the existence of C-whirlin transcript, the C-whirlin protein isoform is not expressed in photoreceptors. In Whrnwi mice, the truncated FL-whirlin mRNA transcript (Fig. 3B) is translated into a truncated N-whirlin protein fragment, although its expression level is low (Mathur et al., 2015b). The truncated N-whirlin protein localizes normally to the PMC (Fig. 4L). Together, these findings demonstrate that whirlin isoforms have distinct spatiotemporal expression patterns in the inner ear and retina.
Whirlin forms different multiprotein complexes in the inner ear and retina:
Whirlin is a scaffold protein with multiple protein-protein interaction domains, such as HNL domains, PDZ domains, a PR region, and PBM. Whirlin thus is thought to aid in assembling large multiprotein complexes. Whirlin isoforms likely form different complexes at the stereociliary tip and base in inner ear hair cells and at the PMC in photoreceptors. To understand the function of whirlin isoforms at different subcellular and cellular locations and the mechanisms underlying the different phenotypes observed in Whrn mutants, it is important to identify the proteins that interact with whirlin and to determine how whirlin isoforms mediate these interactions.
Proteins interacting with whirlin at the stereociliary tip in hair cells:
Actin regulatory protein EPS8, actin motor protein myosin XVa, and MAGUK scaffold proteins p55 and CASK bind to and co-localize with whirlin at the stereociliary tip (Fig. 5A) (Manor et al., 2011; Mburu et al., 2006). Loss of EPS8, myosin XVa, and whirlin leads to short stereociliary length, as observed in Eps8 knockout (Eps8−/−), shaker2 (Myo15ash2), Whrnwi, and Whrnneo mice (Holme et al., 2002; Mathur et al., 2015b; Mustapha et al., 2007; Zampini et al., 2011), indicating that the multiprotein complex containing these three proteins plays a role in maintaining stereocilia length. The N-terminal region of EPS8 binds to the PDZ1, PDZ2, and PR domains of whirlin, while its C-terminal region binds to myosin XVa (Manor et al., 2011). The C-terminal PBM of myosin XVa interacts with the PDZ3 domain of whirlin and delivers whirlin to the tip of stereocilia (Belyantseva et al., 2005). By contrast, another study showed that the SH3-MyTH4 domain of myosin XVa binds to whirlin PDZ3 domain, and that the MyTH4-FERM domain of myosin XVa also interacts with whirlin PDZ1 and PDZ2 domains (Delprat et al., 2005). However, our findings suggest that only the C-terminal half of whirlin is required for the interaction between myosin XVa and whirlin, and that both the whirlin N- and C-terminal halves are involved in the interaction between EPS8 and whirlin (Mathur et al., 2015b). The GUK domain of p55 interacts with whirlin PDZ3 (Mburu et al., 2006; Mburu et al., 2010), and the GUK domain of CASK interacts with the whirlin C-terminal region including PDZ3 (Yap et al., 2003). In addition, p55 and CASK were shown to localize to the OHC stereociliary tip. The localization of P55 is sporadic at the OHC stereociliary tip on embryonic day 17.5 (E17.5), and its signal is completely missing in Whrnwi mice at P5 (Mburu et al., 2006). Because only C-whirlin is localized to the stereociliary tip of OHCs (Mathur et al., 2015b), it is highly likely that p55 and CASK interact with the PDZ3 domain of C-whirlin in vivo. However, the exact function of interactions of whirlin with P55 and CASK in hair cells remains unknown.
Figure 5:
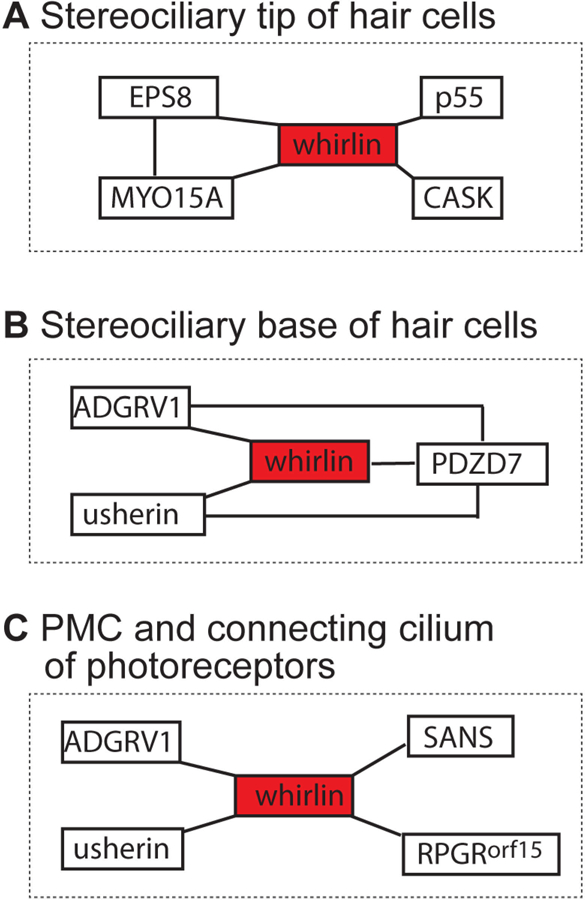
Interacting partners of whirlin at various subcellular locations in the inner ear and retina. (A) Whirlin interacts with EPS8, MYO15A, p55, and CASK proteins at the stereociliary tip of inner ear hair cells, where MYO15A and EPS8 also interact with each other. (B) Whirlin interacts with ADGRV1, usherin, and PDZD7 at the stereociliary base in inner ear hair cells, where the four proteins interact among one another and form the ALC. (C) Whirlin interacts with ADGRV1 and usherin at the PMC and with SANS and RPGRorf15 at the connecting cilium in photoreceptors.
Proteins interacting with whirlin at the stereociliary base in hair cells:
Three USH2-associated proteins, ADGRV1, usherin, and PDZD7, bind to one another as well as to whirlin to form the ankle link complex (ALC) at the stereociliary base (Fig. 5B). This complex is required for hair bundle organization. Several studies have confirmed the existence and function of this USH2 protein complex in vivo (Grati et al., 2012; Michalski et al., 2007; van Wijk et al., 2006; Yang et al., 2010; Zou et al., 2015). The PDZ1 domain of whirlin binds to the PDZ2 domain of PDZD7 and the cytoplasmic C-termini of usherin and ADGRV1. The whirlin PDZ2 domain also binds to the C-terminus of usherin, but not ADGRV1 (Chen et al., 2014). We found that loss of whirlin at the stereociliary base leads to partial mislocalization of ADGRV1 to the stereociliary tip, while usherin and PDZD7 remain relatively unaffected (Zou et al., 2015). ADGRV1, probably together with other proteins, constitutes and stabilizes ankle links of developing hair cell stereocilia (McGee et al., 2006). Ankle links are essential for the typical ‘V’-shaped stereociliary organization of the OHC hair bundle (McGee et al., 2006; Michalski et al., 2007). These findings suggest that loss of whirlin at the stereociliary base causes partial destabilization of ankle links. ADGRV1 is also implicated in G protein-coupled receptor (GPCR) signaling pathways (Hu et al., 2014; Weston et al., 2004). Therefore, it is likely that formation of the ALC is required for efficient and proper GPCR signaling. Whether or not loss of whirlin from the ALC affects GPCR signaling remains to be studied.
Proteins interacting with whirlin at the PMC in photoreceptors:
ADGRV1 and usherin are co-localized with whirlin at the PMC in photoreceptors (Yang et al., 2010) (Fig. 5C). Our in vitro studies demonstrated that the formation of a stable USH2 complex inside cells requires PDZD7 protein, apart from whirlin, usherin, and ADGRV1 (Chen et al., 2014). Interestingly, the absence of PDZD7 protein in the retina (Zou et al., 2014) suggests that usherin and ADGRV1 may interact along their extracellular regions. The role of this USH2 protein complex at the PMC in photoreceptors is currently unknown, although it was originally proposed to be involved in protein trafficking from the inner segment to the outer segment (Yang et al., 2010). Additionally, it was reported that the Retinitis Pigmentosa GTPase Regulator RPGRorf15 isoform and USH1-associated scaffold protein SANS co-localize and interact with whirlin at the connecting cilium but not the PMC in photoreceptors (Sorusch et al., 2017; Wright et al., 2012). Specifically, PDZ1 and PDZ2 domains of whirlin interact with the C-terminal region of RPGRorf15 (Wright et al., 2012) and the PBM of SANS (Sorusch et al., 2017). The functional significance of the interactions of whirlin with RPGRorf15 or SANS has not been explored.
Correlation of genotypes with phenotypes reveals the function of different whirlin isoforms:
Phenotypes caused by the absence of whirlin isoforms at a specific subcellular location in the retina or the inner ear provide insights into the function of whirlin isoforms. For example, whirlin isoforms are completely missing in Whrnneo mouse photoreceptors, which causes mislocalization of ADGRV1 and usherin and subsequent retinal degeneration (Yang et al., 2010). In contrast, Whrnwi retinas retain a truncated N-terminal whirlin fragment containing PDZ1 and PDZ2 domains, which partially rescues the normal localization of ADGRV1 and usherin to the PMC (Mathur et al., 2015b). This partial rescue of the USH2 complex at the PMC is sufficient to prevent or delay retinal degeneration in Whrnwi mice (Yang et al., 2010). Therefore, the whirlin isoforms containing the N-terminal two PDZ domains are critical for USH2 complex formation at the PMC in photoreceptors. Additionally, a 2014 study reported that Whrnwi mice showed a delay in transducin translocation from the outer segment to the inner segment upon light stimulation and that light exposure could induce photoreceptor degeneration (Tian et al.,2014), suggesting a potential role of whirlin in protein translocation in response to light in photoreceptors. However, this study did not include Whrnneo or other Whrn mutant mice. It would be interesting to see whether other Whrn mutant mice have a similar or more severe transducin translocation phenotype and photoreceptor degeneration upon light stimulation, compared with Whrnwi mice.
In the cochlea, localization of both FL- and C-whirlin to the IHC stereociliary tip is essential for stereociliary elongation, whereas C-whirlin at the OHC stereociliary tip, which is the only whirlin isoform at this position, is required for the same stereociliary function. Whrnneo, Whrntm1a, Whrntm1b, and Whrnwi+BAC mice harbor C-whirlin isoforms or fragments at their hair cell stereociliary tips (Fig. 4E, G, M–O) (Ebrahim et al., 2016; Mathur et al., 2015b). In Whrnneo mice and perhaps also in Whrntm1a, Whrntm1b and Whrnwi+BAC mice, loss of FL-whirlin, but not C-whirlin, at the IHC stereociliary tip leads to relatively short stereocilia in adult IHCs, while C-whirlin at the OHC stereociliary tip maintains a normal stereocilia length in adult OHCs (Fig. 4E and G) (Mathur et al., 2015b). Whrnwi mice lack both FL- and C-whirlin at their IHC and OHC stereociliary tips and have significantly shorter IHC and OHC stereocilia compared with those in Whrnneo and wild-type mice (Fig. 4I and K) (Ebrahim et al., 2016; Mathur et al., 2015b). As a result, Whrnwi mice have a more severe hearing loss than Whrnneo, Whrntm1a, Whrntm1b, and Whrnwi+BAC mice. At the ALC of the stereociliary base, Whrnneo, Whrntm1a, Whrntm1b, Whrnwi+BAC, and Whrnwi mice all lack the FL-whirlin isoform, and their phenotype of OHC stereociliary disorganization is similar. These mice have similar reductions of distortion product otoacoustic emission (DPOAE) responses, indicating similar dysfunction of OHCs (Ebrahim et al., 2016; Mathur et al., 2015b). Considering the transient existence of the ALC in the developing cochlea, these observations suggest that FL-whirlin is required at the OHC ALC for hair bundle organization during development.
In the vestibular organs, the subcellular localization of whirlin isoforms is similar to that in IHCs. The function of FL- and C-whirlin in stereociliary elongation at the stereociliary tip is also similar to that in the cochlea. Whrnneo stereocilia are taller than Whrnwi stereocilia, but shorter than wild-type stereocilia (Fig. 4F and J) (Mathur et al., 2015c). Consistently, Whrnwi mice have overt abnormal vestibular behaviors (Holme et al., 2002), whereas Whrnneo mice do not (Mathur et al., 2015c). However, both Whrnneo and Whrnwi mice show similar severe to profound loss of linear vestibular evoked potential (VsEP) responses. One reason for the similar reduction in the linear VsEP response between Whrnneo and Whrnwi mice could be that tall VHC stereocilia are present in the peripheral region of Whrnneo cristae and linear VsEP only tests otolith organs. Another possibility could be that some uncharacterized functional role of C-whirlin in the central nervous system (CNS) compensates in Whrnneo mice.
Whirlin isoforms have potential functions outside the inner ear and retina:
Several recent studies have reported the expression and role of whirlin in the nervous system. Loss of whirlin disrupts the axonal domain organization and causes paranodal abnormalities in both mouse central and peripheral nervous systems during development (Green et al., 2013). Another study revealed that Whrntm1a mice have elevated nociceptive thresholds (White et al., 2013). Subsequently, it was found that whirlin is selectively expressed in mouse proprioceptive sensory neurons, where it functions in afferent firing in response to static muscle stretch (de Nooij et al., 2015). Recently, both FL- and C-whirlin isoforms were found to associate with TRPV1, a thermosensory channel, and increase TRPV1 stability and clustering at the cell membrane of rodent nociceptive neurons (Ciardo et al., 2016). dysc (dyschronic), the closest homolog of whirlin in Drosophila, is required for locomotor behavior and circadian rhythm (Jepson et al., 2012). dysc is expressed at the presynaptic region of larval neuromuscular junctions and is essential in synaptic development and output. dysc mutants show increased evoked and spontaneous synaptic transmission (Jepson et al., 2014).
Together, these findings suggest that whirlin isoforms have multiple functions in the nervous system beyond the inner ear and retina. Expression and function of whirlin in the murine and Drosophila nervous systems suggest that patients with WHRN mutations might have abnormalities, to some extent, in the nervous system.
Attempts to rescue Whrn mutant phenotypes have been partially successful:
Gene therapy is a promising therapeutic approach to treat inherited diseases. To test its feasibility for WHRN-deficient patients, several attempts have been made to rescue the phenotypes of Whrn mutant mice using a gene replacement approach. Adeno-Associated Virus (AAV) carrying FL-whirlin cDNA successfully restores the USH2 complex at the PMC in Whrnneo retinas (Zou et al., 2011). However, because of the very late onset and weak features of retinal degeneration in this mutant mouse, the rescue effect of AAV-FL-whirlin on retinal degeneration in the Whrnneo mouse has not been evaluated. The N-terminal truncated whirlin in Whrnwi retinas is able to stabilize and maintain the normal localization of a sufficient amount of usherin and ADGRV1 (Mathur et al., 2015b), suggesting that AAV carrying an N-terminal whirlin fragment may potentially prevent retinal degeneration in the Whrnneo mouse and WHRN-deficient patients.
Whrn restoration in the inner ear (Belyantseva et al., 2005; Chien et al., 2015; Isgrig et al., 2017; Mburu et al., 2003) has been explored in several studies; however, only partial rescue of hair bundle morphology, hearing, and vestibular function has been achieved. The Whrnwi+BAC mouse, which carries a mouse genome fragment containing Whrn exons 4–12, does not circle and has longer stereocilia and better hearing compared with the Whrnwi mouse (Mburu et al., 2003). However, the Whrnwi+BAC mouse still has an abnormal ‘U’-shaped OHC stereociliary arrangement and impaired hearing function comparable to that seen in the Whrnneo mouse (Mathur et al., 2015b). This phenotype probably results from the absence of FL-whirlin, which is required for ALC stabilization and IHC stereociliary elongation.
Gene gun transfection of VHCs with GFP-tagged FL-whirlin cDNA is able to elongate stereocilia and rescue the missing staircase pattern of the hair bundle to some extent in Whrnwi mice (Belyantseva et al., 2005). However, whether the rescued hair cells have a stereociliary length similar to that of wild-type hair cells has not been reported. Our studies showed that C-whirlin is sufficient to partially restore the missing staircase pattern and increase the stereociliary length in Whrnneo VHCs and that both FL- and C-whirlin are required for VHC stereociliary elongation (Mathur et al., 2015c). Therefore, it is possible that replacement of both FL- and C-whirlin in Whrnwi mice is necessary to fully restore VHC stereociliary staircase pattern and length.
FL-whirlin cDNA has also been packaged into an AAV8 vector and delivered into the inner ear of Whrnwi mice through two different routes (Chien et al., 2015; Isgrig et al., 2017). The cochlear round window delivery partially increases the stereocilia length in IHCs but not OHCs, and hearing loss in Whrnwi mice is not restored (Chien et al., 2015). On the other hand, the posterior semicircular canal delivery enables AAV-FL-whirlin to infect VHCs as well as cochlear hair cells (Isgrig et al., 2017). The infection efficiency of cochlear hair cells through posterior semicircular canal injection is significantly higher than that through cochlear round window injection (~36% vs ~15%, respectively). Posterior semicircular canal delivery of AAV-FL-whirlin was found to increase the stereociliary length in utricular hair cells as well as cochlear inner hair cells, and partially improve both the vestibular and auditory function of Whrnwi mice (Isgrig et al., 2017). The partial functional rescue in that study is probably due to an uncertain infection efficiency of AAV-FL-whirlin into OHCs (which needs to be examined during postnatal development), abnormal FL-whirlin expression in mature OHCs, and the absence of C-whirlin at the VHC and IHC stereociliary tips.
Conclusions and future perspectives:
The high similarity between human and mouse gene sequences supports the notion that the findings regarding Whrn gene transcripts, isoforms, protein localization, and protein-protein interactions in mice may translate to human WHRN. Analysis of human WHRN cDNA sequences (AB040959, AL11028, AK022854, AL110228, and etc.) predicts that human WHRN can be expressed as FL-, N-, and C-terminal protein isoforms, although the information about the promoter regions that lead to expression of multiple WHRN/Whrn variants is still missing. With 88% amino acid sequence identity between human and mouse whirlin proteins, which rises to 94.4% in the PDZ domains (Mburu et al., 2003), it is likely that the function of whirlin isoforms in humans and mice is similar. Therefore, our findings in mice provide valuable information that can be applied to the early differential diagnosis of USH2D and DFNB31, genetic counselling of patients and their families, and educational planning of the disabilities occurring at a late age. This is important because retinal degeneration in USH2D patients does not occur until about age 20, and early diagnosis, before the onset of retinal degeneration, will allow ample time for early intervention and perhaps cure. Furthermore, eight out of eleven currently known USH causative genes, not including WHRN, show variable disease manifestations when mutated (Mathur et al., 2015a). Similar to WHRN, these USH genes express multiple splicing variants. Therefore, disruption of different protein isoforms by mutations in these USH genes may be one of the mechanisms underlying the variable disease manifestations, which can be investigated and verified in future studies.
Abnormal vestibular responses in the Whrnneo mouse (Mathur et al., 2015c), which is an animal model for USH2D patients, suggest that USH2 patients may have an occult vestibular dysfunction. Whether USH2 patients have abnormal vestibular phenotypes is currently under debate. One study reported defective vestibular responses in eight out of eleven USH2 patients examined (Magliulo et al., 2015). However, the lack of a clear genetic diagnosis of the patients in that study weakened the authors’ conclusion (Hartel et al., 2016). Therefore, vestibular tests of other USH2 mouse models and USH2 patients in clinics are imperative. The role of other USH2 proteins in VHCs and the role of the ALC in vestibular function need to be further investigated. In addition, because of the localization of whirlin to the nervous system other than the retina and inner ear, studies on whirlin and perhaps other USH2 proteins in the nervous system are warranted. Finally, experimental designs that test the therapeutic efficacy of specific whirlin isoforms in various Whrn mutant mouse models at different time points including postnatal development period are needed in order to move the field toward treatment of WHRN-deficient diseases.
Highlights.
Whirlin isoforms have different spatiotemporal expression patterns in the inner ear and retina.
Whirlin isoforms form specific multiprotein complexes at different subcellular and cellular positions in the inner ear and retina.
Disruption of different whirlin isoform expression causes different phenotypes in mice and is the potential mechanism underlying variable disease manifestation in WHRN-deficient patients.
Whirlin has a function in the nervous systems other than the inner ear and retina.
Understanding whirlin isoform expression and function is critical for designing effective treatments for WHRN-associated diseases.
Acknowledgements:
National Eye Institute (EY020853, EY026521, and EY014800), Research to Prevent Blindness, Foundation Fighting Blindness, E. Matilda Ziegler Foundation for the Blind, Inc.
Footnotes
Declarations of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Audo I, Bujakowska K, Mohand-Said S, Tronche S, Lancelot ME, Antonio A, Germain A, Lonjou C, Carpentier W, Sahel JA, Bhattacharya S, Zeitz C 2011. A novel DFNB31 mutation associated with Usher type 2 syndrome showing variable degrees of auditory loss in a consanguineous Portuguese family. Mol. Vis 17, 1598–606. [PMC free article] [PubMed] [Google Scholar]
- Belyantseva IA, Boger ET, Naz S, Frolenkov GI, Sellers JR, Ahmed ZM, Griffith AJ, Friedman TB 2005. Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat Cell Biol 7, 148–56. [DOI] [PubMed] [Google Scholar]
- Besnard T, Vache C, Baux D, Larrieu L, Abadie C, Blanchet C, Odent S, Blanchet P, Calvas P, Hamel C, Dollfus H, Lina-Granade G, Lespinasse J, David A, Isidor B, Morin G, Malcolm S, Tuffery-Giraud S, Claustres M, Roux AF 2012. Non-USH2A mutations in USH2 patients. Hum. Mutat 33, 504–10. [DOI] [PubMed] [Google Scholar]
- Boughman JA, Vernon M, Shaver KA 1983. Usher syndrome: definition and estimate of prevalence from two high-risk populations. J. Chronic Dis 36, 595–603. [DOI] [PubMed] [Google Scholar]
- Chen Q, Zou J, Shen Z, Zhang W, Yang J 2014. Whirlin and PDZ domain containing 7 (PDZD7) proteins are both required to form the quaternary protein complex associated with Usher syndrome type 2. J. Biol. Chem 289, 36070–36088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien WW, Isgrig K, Roy S, Belyantseva IA, Drummond MC, May LA, Fitzgerald TS, Friedman TB, Cunningham LL 2015. Gene Therapy Restores Hair Cell Stereocilia Morphology in Inner Ears of Deaf Whirler Mice. Molecular therapy : the journal of the American Society of Gene Therapy [DOI] [PMC free article] [PubMed]
- Ciardo MG, Andres-Borderia A, Cuesta N, Valente P, Camprubi-Robles M, Yang J, Planells-Cases R, Ferrer-Montiel A 2016. Whirlin increases TRPV1 channel expression and cellular stability. Biochim. Biophys. Acta 1863, 115–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Nooij JC, Simon CM, Simon A, Doobar S, Steel KP, Banks RW, Mentis GZ, Bewick GS, Jessell TM 2015. The PDZ-domain protein Whirlin facilitates mechanosensory signaling in mammalian proprioceptors. J. Neurosci 35, 3073–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delprat B, Michel V, Goodyear R, Yamasaki Y, Michalski N, El-Amraoui A, Perfettini I, Legrain P, Richardson G, Hardelin JP, Petit C 2005. Myosin XVa and whirlin, two deafness gene products required for hair bundle growth, are located at the stereocilia tips and interact directly. Hum. Mol. Genet 14, 401–10. [DOI] [PubMed] [Google Scholar]
- Ebermann I, Scholl HP, Charbel Issa P, Becirovic E, Lamprecht J, Jurklies B, Millan JM, Aller E, Mitter D, Bolz H 2007. A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum. Genet 121, 203–11. [DOI] [PubMed] [Google Scholar]
- Ebrahim S, Ingham NJ, Lewis MA, Rogers MJC, Cui R, Kachar B, Pass JC, Steel KP 2016. Alternative Splice Forms Influence Functions of Whirlin in Mechanosensory Hair Cell Stereocilia. Cell Rep 15, 935–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyear RJ, Marcotti W, Kros CJ, Richardson GP 2005. Development and properties of stereociliary link types in hair cells of the mouse cochlea. J. Comp. Neurol 485, 75–85. [DOI] [PubMed] [Google Scholar]
- Grati M, Shin JB, Weston MD, Green J, Bhat MA, Gillespie PG, Kachar B 2012. Localization of PDZD7 to the stereocilia ankle-link associates this scaffolding protein with the Usher syndrome protein network. J. Neurosci 32, 14288–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JA, Yang J, Grati M, Kachar B, Bhat MA 2013. Whirlin, a cytoskeletal scaffolding protein, stabilizes the paranodal region and axonal cytoskeleton in myelinated axons. BMC Neurosci 14, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartel BP, Pennings RJ, van Wijk E 2016. Comment on “Usher’s Syndrome: Evaluation of the Vestibular System with Cervical and Ocular Vestibular Evoked Myogenic Potentials and the Video Head Impulse Test”. Otol Neurotol 37, 608. [DOI] [PubMed] [Google Scholar]
- Holme RH, Kiernan BW, Brown SD, Steel KP 2002. Elongation of hair cell stereocilia is defective in the mouse mutant whirler. J. Comp. Neurol 450, 94–102. [DOI] [PubMed] [Google Scholar]
- Hu QX, Dong JH, Du HB, Zhang DL, Ren HZ, Ma ML, Cai Y, Zhao TC, Yin XL, Yu X, Xue T, Xu ZG, Sun JP 2014. Constitutive Galphai Coupling Activity of VLGR1 and its Regulation by PDZD7. J. Biol. Chem 289, 24215–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isgrig K, Shteamer JW, Belyantseva IA, Drummond MC, Fitzgerald TS, Vijayakumar S, Jones SM, Griffith AJ, Friedman TB, Cunningham LL, Chien WW 2017. Gene Therapy Restores Balance and Auditory Functions in a Mouse Model of Usher Syndrome. Molecular therapy : the journal of the American Society of Gene Therapy 25, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffries DJ, Pickles JO, Osborne MP, Rhys-Evans PH, Comis SD 1986. Crosslinks between stereocilia in hair cells of the human and guinea pig vestibular labyrinth. J. Laryngol. Otol 100, 1367–74. [DOI] [PubMed] [Google Scholar]
- Jepson JE, Shahidullah M, Lamaze A, Peterson D, Pan H, Koh K 2012. dyschronic, a Drosophila homolog of a deaf-blindness gene, regulates circadian output and Slowpoke channels. PLoS Genet 8, e1002671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepson JE, Shahidullah M, Liu D, le Marchand SJ, Liu S, Wu MN, Levitan IB, Dalva MB, Koh K 2014. Regulation of synaptic development and function by the Drosophila PDZ protein Dyschronic. Development 141, 4548–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkawa Y, Mburu P, Morse S, Kominami R, Townsend S, Brown SD 2005. Mutant analysis reveals whirlin as a dynamic organizer in the growing hair cell stereocilium. Hum. Mol. Genet 14, 391–400. [DOI] [PubMed] [Google Scholar]
- Kimberling WJ, Hildebrand MS, Shearer AE, Jensen ML, Halder JA, Trzupek K, Cohn ES, Weleber RG, Stone EM, Smith RJ 2010. Frequency of Usher syndrome in two pediatric populations: Implications for genetic screening of deaf and hard of hearing children. Genetics in medicine : official journal of the American College of Medical Genetics 12, 512–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XZ, Walsh J, Tamagawa Y, Kitamura K, Nishizawa M, Steel KP, Brown SD 1997a. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat. Genet 17, 268–9. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, Brown SD 1997b. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat. Genet 16, 188–90. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Hope C, Walsh J, Newton V, Ke XM, Liang CY, Xu LR, Zhou JM, Trump D, Steel KP, Bundey S, Brown SD 1998. Mutations in the myosin VIIA gene cause a wide phenotypic spectrum, including atypical Usher syndrome. Am. J. Hum. Genet 63, 909–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magliulo G, Iannella G, Gagliardi S, Iozzo N, Plateroti R, Plateroti P, Re M, Vingolo EM 2015. Usher’s Syndrome: Evaluation of the Vestibular System with Cervical and Ocular Vestibular Evoked Myogenic Potentials and the Video Head Impulse Test. Otol Neurotol 36, 1421–7. [DOI] [PubMed] [Google Scholar]
- Manor U, Disanza A, Grati M, Andrade L, Lin H, Di Fiore PP, Scita G, Kachar B 2011. Regulation of stereocilia length by myosin XVa and whirlin depends on the actin-regulatory protein Eps8. Curr. Biol 21, 167–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur P, Yang J 2015a. Usher syndrome: hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 1852, 406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur P, Zou J, Zheng T, Almishaal A, Wang Y, Chen Q, Wang L, Vashist D, Brown S, Park A, Yang J 2015b. Distinct expression and function of whirlin isoforms in the inner ear and retina: an insight into pathogenesis of USH2D and DFNB31. Hum. Mol. Genet 24, 6213–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur PD, Vijayakumar S, Vashist D, Jones SM, Jones TA, Yang J 2015c. A study of whirlin isoforms in the mouse vestibular system suggests potential vestibular dysfunction in DFNB31-deficient patients. Hum. Mol. Genet 24, 7017–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mburu P, Kikkawa Y, Townsend S, Romero R, Yonekawa H, Brown SD 2006. Whirlin complexes with p55 at the stereocilia tip during hair cell development. Proc. Natl. Acad. Sci. U. S. A 103, 10973–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mburu P, Romero MR, Hilton H, Parker A, Townsend S, Kikkawa Y, Brown SD 2010. Gelsolin plays a role in the actin polymerization complex of hair cell stereocilia. PLoS One 5, e11627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mburu P, Mustapha M, Varela A, Weil D, El-Amraoui A, Holme RH, Rump A, Hardisty RE, Blanchard S, Coimbra RS, Perfettini I, Parkinson N, Mallon AM, Glenister P, Rogers MJ, Paige AJ, Moir L, Clay J, Rosenthal A, Liu XZ, Blanco G, Steel KP, Petit C, Brown SD 2003. Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat. Genet 34, 421–8. [DOI] [PubMed] [Google Scholar]
- McGee J, Goodyear RJ, McMillan DR, Stauffer EA, Holt JR, Locke KG, Birch DG, Legan PK, White PC, Walsh EJ, Richardson GP 2006. The very large G-protein-coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J. Neurosci 26, 6543–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski N, Michel V, Bahloul A, Lefevre G, Barral J, Yagi H, Chardenoux S, Weil D, Martin P, Hardelin JP, Sato M, Petit C 2007. Molecular characterization of the ankle-link complex in cochlear hair cells and its role in the hair bundle functioning. J. Neurosci 27, 6478–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustapha M, Beyer LA, Izumikawa M, Swiderski DL, Dolan DF, Raphael Y, Camper SA 2007. Whirler mutant hair cells have less severe pathology than shaker 2 or double mutants. J Assoc Res Otolaryngol 8, 329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiguchi KM, Tearle RG, Liu YP, Oh EC, Miyake N, Benaglio P, Harper S, Koskiniemi-Kuendig H, Venturini G, Sharon D, Koenekoop RK, Nakamura M, Kondo M, Ueno S, Yasuma TR, Beckmann JS, Ikegawa S, Matsumoto N, Terasaki H, Berson EL, Katsanis N, Rivolta C 2013. Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc. Natl. Acad. Sci. U. S. A 110, 16139–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazuddin S, Nazli S, Ahmed ZM, Yang Y, Zulfiqar F, Shaikh RS, Zafar AU, Khan SN, Sabar F, Javid FT, Wilcox ER, Tsilou E, Boger ET, Sellers JR, Belyantseva IA, Riazuddin S, Friedman TB 2008. Mutation spectrum of MYO7A and evaluation of a novel nonsyndromic deafness DFNB2 allele with residual function. Hum. Mutat 29, 502–11. [DOI] [PubMed] [Google Scholar]
- Sahly I, Dufour E, Schietroma C, Michel V, Bahloul A, Perfettini I, Pepermans E, Estivalet A, Carette D, Aghaie A, Ebermann I, Lelli A, Iribarne M, Hardelin JP, Weil D, Sahel JA, El-Amraoui A, Petit C 2012. Localization of Usher 1 proteins to the photoreceptor calyceal processes, which are absent from mice. J. Cell Biol 199, 381–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorusch N, Bauss K, Plutniok J, Samanta A, Knapp B, Nagel-Wolfrum K, Wolfrum U 2017. Characterization of the ternary Usher syndrome SANS/ush2a/whirlin protein complex. Hum. Mol. Genet 26, 1157–1172. [DOI] [PubMed] [Google Scholar]
- Tian M, Wang W, Delimont D, Cheung L, Zallocchi M, Cosgrove D, Peng YW 2014. Photoreceptors in whirler mice show defective transducin translocation and are susceptible to short-term light/dark changes-induced degeneration. Exp. Eye Res 118, 145–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlili A, Charfedine I, Lahmar I, Benzina Z, Mohamed BA, Weil D, Idriss N, Drira M, Masmoudi S, Ayadi H 2005. Identification of a novel frameshift mutation in the DFNB31/WHRN gene in a Tunisian consanguineous family with hereditary non-syndromic recessive hearing loss. Hum. Mutat 25, 503–507. [DOI] [PubMed] [Google Scholar]
- van Wijk E, van der Zwaag B, Peters T, Zimmermann U, Te Brinke H, Kersten FF, Marker T, Aller E, Hoefsloot LH, Cremers CW, Cremers FP, Wolfrum U, Knipper M, Roepman R, Kremer H 2006. The DFNB31 gene product whirlin connects to the Usher protein network in the cochlea and retina by direct association with USH2A and VLGR1. Hum. Mol. Genet 15, 751–65. [DOI] [PubMed] [Google Scholar]
- Vozzi D, Aaspollu A, Athanasakis E, Berto A, Fabretto A, Licastro D, Kulm M, Testa F, Trevisi P, Vahter M, Ziviello C, Martini A, Simonelli F, Banfi S, Gasparini P 2011. Molecular epidemiology of Usher syndrome in Italy. Mol. Vis 17, 1662–8. [PMC free article] [PubMed] [Google Scholar]
- Weil D, Kussel P, Blanchard S, Levy G, Levi-Acobas F, Drira M, Ayadi H, Petit C 1997. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat. Genet 16, 191–3. [DOI] [PubMed] [Google Scholar]
- Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Mburu P, Varela A, Levilliers J, Weston MD, et al. 1995. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 374, 60–1. [DOI] [PubMed] [Google Scholar]
- Weston MD, Luijendijk MW, Humphrey KD, Moller C, Kimberling WJ 2004. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am. J. Hum. Genet 74, 357–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JK, Gerdin AK, Karp NA, Ryder E, Buljan M, Bussell JN, Salisbury J, Clare S, Ingham NJ, Podrini C, Houghton R, Estabel J, Bottomley JR, Melvin DG, Sunter D, Adams NC, Tannahill D, Logan DW, Macarthur DG, Flint J, Mahajan VB, Tsang SH, Smyth I, Watt FM, Skarnes WC, Dougan G, Adams DJ, Ramirez-Solis R, Bradley A, Steel KP 2013. Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell 154, 452–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright RN, Hong DH, Perkins B 2012. RpgrORF15 connects to the usher protein network through direct interactions with multiple whirlin isoforms. Invest. Ophthalmol. Vis. Sci 53, 1519–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu X, Zhao Y, Adamian M, Pawlyk B, Sun X, McMillan DR, Liberman MC, Li T 2010. Ablation of whirlin long isoform disrupts the USH2 protein complex and causes vision and hearing loss. PLoS Genet 6, e1000955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap CC, Liang F, Yamazaki Y, Muto Y, Kishida H, Hayashida T, Hashikawa T, Yano R 2003. CIP98, a novel PDZ domain protein, is expressed in the central nervous system and interacts with calmodulin-dependent serine kinase. J. Neurochem 85, 123–34. [DOI] [PubMed] [Google Scholar]
- Zampini V, Ruttiger L, Johnson SL, Franz C, Furness DN, Waldhaus J, Xiong H, Hackney CM, Holley MC, Offenhauser N, Di Fiore PP, Knipper M, Masetto S, Marcotti W 2011. Eps8 regulates hair bundle length and functional maturation of Mammalian auditory hair cells. PLoS Biol 9, e1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Luo L, Shen Z, Chiodo VA, Ambati BK, Hauswirth WW, Yang J 2011. Whirlin replacement restores the formation of the USH2 protein complex in whirlin knockout photoreceptors. Invest. Ophthalmol. Vis. Sci 52, 2343–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Mathur PD, Zheng T, Wang Y, Almishaal A, Park AH, Yang J 2015. Individual USH2 proteins make distinct contributions to the ankle link complex during development of the mouse cochlear stereociliary bundle. Hum. Mol. Genet 24, 6944–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Zheng T, Ren C, Askew C, Liu XP, Pan B, Holt JR, Wang Y, Yang J 2014. Deletion of PDZD7 disrupts the Usher syndrome type 2 protein complex in cochlear hair cells and causes hearing loss in mice. Hum. Mol. Genet 23, 2374–90. [DOI] [PMC free article] [PubMed] [Google Scholar]