

Abstract
A signature of photo-mediated controlled polymerizations is the ability to modulate the rate of polymerization by turning the light source ‘on’ and ‘off.’ However, in many reported systems, growth can be reproducibly observed during dark periods. In this study, emerging photo-mediated controlled radical polymerizations are evaluated with in situ 1H NMR monitoring to assess their behavior in the dark. Interestingly, it is observed that Cu-mediated systems undergo long-lived, linear growth during dark periods in organic media.
Keywords: photopolymerization, controlled radical polymerization, in-situ monitoring
GRAPHICAL ABSTRACT
The utility and sweeping impact of controlled radical polymerization (CRP) has fundamentally changed the direction of polymer synthesis. By enabling the accurate control of molecular weight, architecture, and dispersity (Ð) for a wide variety of functional monomers, the facile synthesis of complex polymeric materials such as extended multiblocks[1], surface-modified nanoparticles[2,3], and bioconjugates[4–6] is now possible. Recently, the use of external stimuli, such as light[7,8], reducing agents[9], applied voltage[10], and mechanical forces[11] to mediate CRP processes has further increased the usefulness and impact of CRP.[12] Of these stimuli, light is particularly attractive, as it is environmentally benign and highly tunable.[13–15] Numerous examples of photo-mediated controlled radical polymerization (photo-CRP) have recently been developed, including Cu-mediated reversible-deactivation radical polymerization (Cu-mediated RDRP)[16–19], Cu-free atom transfer radical polymerization (Cu-free ATRP)[8,17,20–22], and photo induced electron transfer-reversible addition-fragmentation chain transfer (PET-RAFT).[23,24] These systems operate over a wide variety of wavelengths and employ a variety of catalysts to polymerize different monomer classes,[25–27] with the broad scope of these systems leading to the development of well-defined, functional materials. Notable examples include patterned polymer brushes[28,29], organic light-emitting diodes[30], soft gels[31,32], and complex polymer architectures[33,34].
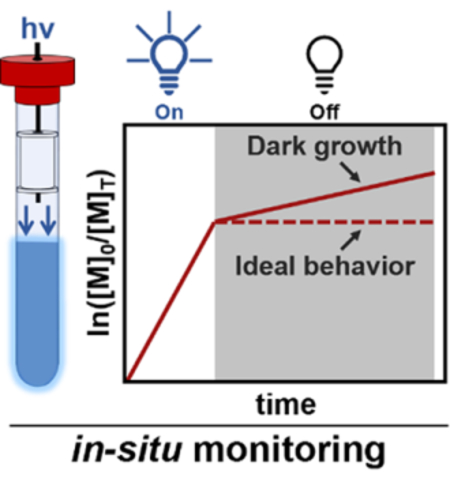
In photo-CRP, temporal control is typically demonstrated through sequential ‘on’ - ‘off’ cycles. This cycling is performed by irradiating the reaction mixture, polymerization then initiates/propagates, followed by a ‘dark’ period where, in an ideal scenario, no additional conversion takes place. However, for many reported photo-CRP systems, a small yet reproducible amount of polymer growth can be observed during the ‘off’ cycles.[22,35–39] This apparent growth has been attributed to several factors, from experimental error to residual active catalyst. While the kinetics of growth and the presence of side reactions has been extensively studied for ‘on’ periods[5,40,41], no systematic examination of the polymerization reaction during the ‘off’ or ‘dark’ periods has been conducted.
To address this challenge and provide insight into photo-CRP processes, a recently developed in situ NMR spectroscopy method is utilized to evaluate temporal control for a selection of widely studied photo-CRP processes (see Figure 1 and Figure S1 for a representative schematic and photograph of the setup).[42] Compared to conventional sampling methods, this approach is uniquely suited for studying temporal control of photo-CRPs, allowing accurate modulation of irradiation intensity and wavelength through the combination of LEDs and fiber optics. In addition, in situ coupling with NMR spectroscopy permits rapid and repeated measurements to be taken without invasive sampling of the polymerization reaction. As a result, accurate polymerization kinetics can be obtained in both the ‘on’ and ‘off’ states.
Figure 1:

Schematic of the in situ fiber-coupled NMR system showing idealized schemes for photo-CRP in active (‘on’) and dormant (‘off’) states.
In this study, PET-RAFT, Cu-free ATRP, and Cu-mediated RDRP systems were selected as representative examples of photo-CRP methods. To facilitate an unbiased comparison across techniques, irradiation conditions were held constant (equivalent photon flux) and polymerization conditions, such as monomer concentration and targeted degree of polymerization, were fixed at 33 wt% and DP=150. Temporal control experiments were also carried out with equal ‘on’ and ‘off’ times targeting conversions of ~40% with an initial ‘off’ period conducted to establish a baseline before exposure to light. To show the general trends of a given technique, a representative catalyst/ligand combination will be discussed, however full data for all catalysts studied is available in the Supporting Information.
As they both utilize the photocatalyst as an electron transfer agent, the initial systems chosen for study were PET-RAFT and Cu-free ATRP, Scheme 1a.[43] Under traditional PET-RAFT conditions, the polymerization of methyl acrylate (MA) in DMSO was examined, (Figure 2a) and after an inhibition period attributed to residual oxygen being consumed,[24,44,45] the polymerization demonstrated linear kinetics with significant deviation from linearity only observed at high monomer conversions (Figure 2b). As expected, the polymerization of methyl methacrylate (MMA) under Cu-free ATRP conditions (Figure 3a) was slower than the polymerization of MA by PET-RAFT due to the increased kp values. However, in both the PET-RAFT and Cu-free ATRP experiments, linear kinetics with little to no deviation were observed up to conversions of 30–40%. To simplify comparison, this conversion range was thereby targeted in the temporal control studies (Figure 2c, 3c).
Scheme 1:

Simplified mechanisms reported for a) PET-RAFT / Cu-free ATRP and b) Cu-mediated RDRP.
See review by Johnson and co-workers[13] for in-depth discussions of the above photo-CRP mechanisms.
Figure 2:

a) PET-RAFT conditions for the polymerization of methyl acrylate (MA) using 470 nm light and tris(2,2′-bipyridyl)dichlororuthenium(II) hexahydrate. b) Kinetic plots of the polymerizations at a fixed photon flux. c) Temporal control experiments for the PET-RAFT demonstrate ideal temporal control.
Figure 3:
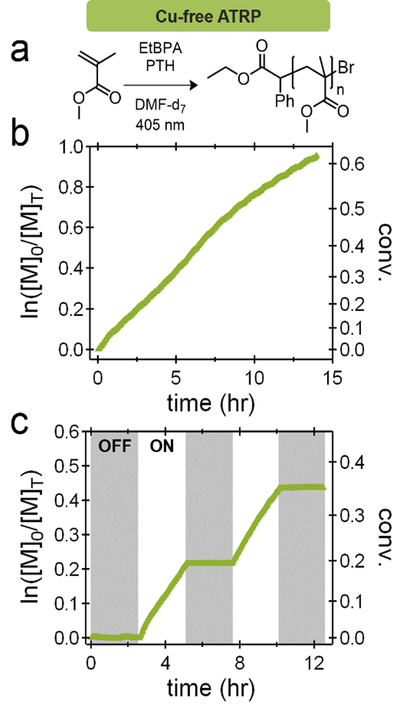
a) Cu-free ATRP conditions for the polymerization of methyl methacrylate (MMA) using 405 nm light and 10-phenylphenothiazine. b) Kinetic plots of the polymerizations at a fixed photon flux. c) Temporal control experiments for the Cu-free ATRP reactions demonstrate ideal temporal control.
Significantly, for all PET-RAFT and Cu-free ATRP systems studied, high fidelity is observed with no observable conversion being measured during the ‘dark’ period.
Unlike PET-RAFT and Cu-free ATRP, which directly drive polymerization through light-driven electron transfer events, Cu-mediated RDRP in a secondary fashion, generating active Cu(I) from inactive Cu(II), potentially leaving residual catalytic Cu(I) in solution after irradiation has stopped, Scheme 1b. To examine this behavior and compare Cu-mediated RDRP to both PET-RAFT and Cu-free ATRP, Me6TREN and CuBr2 were employed in the polymerization of both MA and MMA (Figure 4a). Although there are differences in the overall behavior of the polymerization of MA and MMA compared to the PET-RAFT and Cu-free ATRP examples, namely a lack of inhibition for MA and evidence of severe termination for MMA, both systems show linear kinetics up to monomer conversions of ~30–40% (Figure 4b).
Figure 4:

a) Cu-mediated RDRP conditions for the polymerization of MA and MMA using CuBr2 and tris[2-(dimethylamino)ethyl]amine (Me6TREN). b) Kinetic plots of the polymerizations at a fixed photon flux. c) Temporal control experiments wherein distinct linear growth during dark periods after initial irradiation are observed for both polymerizations (~10–15% of the ‘on’ rate).
For both MA and MMA, significant differences were observed during ‘off’ periods (Figure 4c). While the initial ‘dark’ periods did not result in any monomer conversion, the Cu-mediated systems exhibited substantial polymer growth during the subsequent ‘dark’ periods (~5–10% of the ‘on’ rate in both the MA and MMA systems). Interestingly, upon extending the dark window from ~10 minutes to ~5 hours, linear polymerization kinetics in the ‘off’ state are still observed. Even at high conversions, the Cu catalyst was active with linear kinetics being observed (86 to 91%) despite being in an ‘off’ or ‘dark’ period for 3.5 hours (Figure S24). These results suggest that during the ‘off’ periods a significant amount of Cu(I) (initially produced by reduction of Cu(II)) remains in solution and is responsible for polymer growth through a conventional ATRP mechanism, rather than a photo-mediated ATRP process. To further investigate the temporal control of Cu-mediated RDRP systems, the dark periods were extended for different Cu/ligand pairs (Me6TREN and TPMA). The equilibrium constants for Me6TREN and TPMA are reported in the literature, and it has been shown that TPMA has a KATRP value approximately an order of magnitude lower than Me6TREN.[46] After initial irradiation to similar conversions, both systems did show growth during the ‘dark’ period. However, Me6TREN displays a considerably higher rate of conversion (approximately an order of magnitude) when compared to the corresponding TPMA system (Figure 5). This result illustrates that Cu-mediated RDRP in organic media does not exhibit ideal temporal control for any of the conditions/ligands studied due to the unwanted presence and extended lifetime of CuBr during ‘dark’ periods.
Figure 5:

Kinetics of MMA polymerizations using Me6TREN/CuBr2 (DMF-d7) and TPMA/CuBr2 (DMSO-d6) photosystems at equal loadings. Me6TREN undergoes more growth in the dark period due to its higher activity (KATRP).
To improve the temporal control of Cu-mediated polymerizations, we envisage that a system must exhibit rapid consumption of residual Cu(I) catalyst during the ‘off’ cycles. Aqueous systems are subject to high equilibrium constants,[47,48] and the concentration of Cu(I) should therefore decrease rapidly during ‘dark’ periods, translating to increased temporal control relative to the corresponding organic systems. As a control, Cu-mediated RDRP of poly(ethylene glycol) methyl ether acrylate (PEGA, Mn = 480) using Me6TREN/CuBr2 was conducted in organic and aqueous media (Figure 6). In analogy with the Cu-mediated polymerization of MA in DMSO, a linear increase in conversion for PEGA occurs during an extended ‘dark’ period of ~5 hours after initial irradiation (Figure 6b). To achieve a comparable controlled polymerization of PEGA in water, the copper loading was increased 5x relative to that used in DMSO.[48] Interestingly, after irradiation with 365 nm light, rapid polymerization continued for 2 hours in the dark (though in a non-linear fashion) before decreasing to undetectable levels. Importantly, the polymerization continued upon further irradiation, highlighting that the end groups were still active and implying that the active Cu(I) was consumed during the ‘off’ period, presumably by conversion to Cu(II). While this aqueous system demonstrated the potential for improved temporal control compared to a similar polymerization in organic media, significant monomer conversion does occur in the ‘dark’ after turning off the light source. In an attempt to increase fidelity further, it was hypothesized that a lower amount of initial CuBr2 would generate less residual catalyst, which could then be deactivated more rapidly in the absence of light. In order to maintain control with a reduced amount of CuBr2, NaBr was therefore added to the polymerization mixture.[48] Indeed, under these conditions, nearly immediate cessation of the polymerization was observed upon switching the light ‘off,’ leading to a high degree of temporal control. These results highlight the importance of mechanistic understanding in the development of strategies for temporal control of Cu-mediated CRP processes.
Figure 6:

a) General reaction scheme for polymerization of poly(ethylene glycol) methyl ether acrylate (PEGA) macromonomers. b) Temporal experiments for PEGA polymerized in DMSO (top), water at an elevated Cu concentration (middle), and water with a reduced Cu content and NaBr (bottom). Only the reduced Cu-concentration aqueous polymerization shows ideal temporal behavior.
In summary, a modular in situ NMR technique was utilized to investigate monomer conversion during the ‘on’ and ‘off’ cycles for a selection of photo-CRP procedures. Temporal control during metal-free ATRP and PET-RAFT was demonstrated to have high fidelity and little to no conversion during ‘dark’ periods. In direct contrast, Cu-mediated polymerizations conducted in DMSO showed significant growth during ‘off’ cycles, which is attributed to the increased lifetime of residual Cu(I) catalyst after initial photoactivation. The use of aqueous conditions (low Cu(II) concentration and added NaBr) quickly consumes the residual catalytic species and alleviates this problem. This allows well-controlled polymers with no observable ‘dark’ growth to be obtained. However, it should be noted that these conditions cannot currently be broadly generalized with understanding and improving the temporal control in Cu-mediated polymerizations in organic media being an area of future focus. The findings of non-ideal temporal behaviour herein also illustrate the necessity for employing long ‘off’ periods when studying temporal control to ensure measurement fidelity and accuracy.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the MRSEC Program of the National Science Foundation (DMR 1720256), the National Institute of General Medical Sciences (R35GM119702) of the National Institutes of Health, and the Institute for Collaborative Biotechnologies (W911NF-09–0001) from the U.S. Army Research Office for support. The content of the information presented does not necessarily reflect the position or policy of the funding entities, and no official endorsement should be inferred. C.B. acknowledges Australian Research Council (ARC) for his Future Fellowship (FT12010096). E.H.D. and B.G.M. are grateful for support from an NSF GRFP. G.R.J. and R.W. thank Lubrizol and Syngenta for support.
Footnotes
EXPERIMENTAL
See Supporting Information for full experimental details.
REFERENCES AND NOTES
- [1].a) Chuang YM, Ethirajan A, Junkers T, ACS Macro Lett. 2014, 3, 732–737; [DOI] [PubMed] [Google Scholar]; b) Stehling UM, Malmstrom EE, Waymouth RM, Hawker CJ, Macromolecules, 1998, 31, 4396–4398; [Google Scholar]; c) Beck JB, Killops KL, Kang T, Sivanandan K, Bayles A, Mackay ME, Wooley KL, Hawker CJ, Macromolecules, 2009, 42, 5629–5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Skaff H, Emrick T, Angew. Chemie - Int. Ed 2004, 43, 5383–5386. [DOI] [PubMed] [Google Scholar]
- [3].Babin J, Lepage M, Zhao Y, Macromolecules 2008, 41, 1246–1253. [Google Scholar]
- [4].Niu J, Lunn DJ, Pusuluri A, Yoo JI, O’Malley MA, Mitragotri S, Soh HT, Hawker CJ, Nat. Chem 2017, 9, 537–545. [DOI] [PubMed] [Google Scholar]
- [5].Niu J, Page ZA, Dolinski ND, Anastasaki A, Hsueh AT, Soh HT, Hawker CJ, ACS Macro Lett. 2017, 6, 1109–1113. [DOI] [PubMed] [Google Scholar]
- [6].Xu J, Jung K, Corrigan NA, Boyer C, Chem. Sci 2014, 5, 3568–3575. [Google Scholar]
- [7].Shi Y, Liu G, Gao H, Lu L, Cai Y, Macromolecules 2009, 42, 3917–3926. [Google Scholar]
- [8].Fors BP, Hawker CJ, Angew. Chemie - Int. Ed 2012, 51, 8850–8853. [DOI] [PubMed] [Google Scholar]
- [9].Jakubowski W, Matyjaszewski K, Macromolecules 2005, 38, 4139–4146. [Google Scholar]
- [10].Magenau AJD, Strandwitz NC, Gennaro A, Matyjaszewski K, Science 2011, 332, 81–84. [DOI] [PubMed] [Google Scholar]
- [11].Mohapatra H, Kleiman M, Esser-Kahn AP, Nat. Chem 2017, 9, 135–139. [Google Scholar]
- [12].Pan X, Fantin M, Yuan F, Matyjaszewski K, Chem. Soc. Rev 2018, 47, 5457–5490. [DOI] [PubMed] [Google Scholar]
- [13].Chen M, Zhong M, Johnson JA, Chem. Rev 2016, 116, 10167–10211. [DOI] [PubMed] [Google Scholar]
- [14].Pan X, Tasdelen MA, Laun J, Junkers T, Yagci Y, Matyjaszewski K, Prog. Polym. Sci 2016, 62, 73–125. [Google Scholar]
- [15].Corrigan N, Shanmugam S, Xu J, Boyer C, Chem. Soc. Rev 2016, 45, 6165–6212. [DOI] [PubMed] [Google Scholar]
- [16].Konkolewicz D, Schröder K, Buback J, Bernhard S, Matyjaszewski K, ACS Macro Lett. 2012, 1, 1219–1223. [DOI] [PubMed] [Google Scholar]
- [17].Kork S, Ciftci M, Tasdelen MA, Yagci Y, Macromol. Chem. Phys 2016, 217, 812–817. [Google Scholar]
- [18].Frick E, Anastasaki A, Haddleton DM, Barner-Kowollik C, J. Am. Chem. Soc 2015, 137, 6889–6896. [DOI] [PubMed] [Google Scholar]
- [19].Nikolaou V, Anastasaki A, Brandford-Adams F, Whitfield R, Jones GR, Nurumbetov G, Haddleton DM, Polym. Chem 2016, 7, 191–197. [Google Scholar]
- [20].Treat NJ, Sprafke H, Kramer JW, Clark PG, Barton BE, Read De Alaniz J, Fors BP, Hawker CJ, J. Am. Chem. Soc 2014, 136, 16096–16101. [DOI] [PubMed] [Google Scholar]
- [21].Theriot JC, Lim C-H, Yang H, Ryan MD, Musgrave CB, Miyake GM, Science 2016, 352, 1082–1086. [DOI] [PubMed] [Google Scholar]
- [22].a) Pearson RM, Lim CH, McCarthy BG, Musgrave CB, Miyake GM, J. Am. Chem. Soc 2016, 138, 11399–11407; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ryan MD, Theriot JC, Lim C-H, Yang H, Lockwood AG, Garrison NG, Lincoln SR, Musgrave CB, Miyake GM, J. Polym. Sci., Part A: Polym. Chem, 2017, 55, 3017–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shanmugam S, Xu J, Boyer C, Macromolecules 2014, 47, 4930–4942. [Google Scholar]
- [24].a) Fu Q, Xie K, McKenzie TG, Qiao GG, Polym. Chem 2017, 8, 1519–1526; [Google Scholar]; b) Watanabe A, Niu J, Lunn DJ, Lawrence J, Knight AS, Zhang M, Hawker CJ, J. Polym. Sci., Part A: Polym. Chem 2018, 56, 1259–1268; [Google Scholar]; c) Yang Q, Zhang X, Ma W, Ma Y, Chen D, Wang L, Zhao C, Yang W, J. Polym. Sci., Part A: Polym. Chem 2018, 56, 229–236. [Google Scholar]
- [25].Kottisch V, Michaudel Q, Fors BP, J. Am. Chem. Soc 2016, 138, 15535–15538. [DOI] [PubMed] [Google Scholar]
- [26].Michaudel Q, Kottisch V, Fors BP, Angew. Chemie - Int. Ed 2017, 56, 9670–9679. [DOI] [PubMed] [Google Scholar]
- [27].Michaudel Q, Chauviré T, Kottisch V, Supej MJ, Stawiasz KJ, Shen L, Zipfel WR, Abruña HD, Freed JH, Fors BP, J. Am. Chem. Soc 2017, 139, 15530–15538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Poelma JE, Fors BP, Meyers GF, Kramer JW, Hawker CJ, Angew. Chemie - Int. Ed 2013, 52, 6844–6848. [DOI] [PubMed] [Google Scholar]
- [29].Discekici EH, Pester CW, Treat NJ, Lawrence J, Mattson KM, Narupai B, Toumayan EP, Luo Y, McGrath AJ, Clark PG, et al. , ACS Macro Lett. 2016, 5, 258–262. [DOI] [PubMed] [Google Scholar]
- [30].Page ZA, Narupai B, Pester CW, Bou Zerdan R, Sokolov A, Laitar DS, Mukhopadhyay S, Sprague S, McGrath AJ, Kramer JW, et al. , ACS Cent. Sci 2017, 3, 654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chen M, Gu Y, Singh A, Zhong M, Jordan AM, Biswas S, Korley LTJ, Balazs AC, Johnson JA, ACS Cent. Sci 2017, 3, 124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Daniel WFM, Burdyńska J, Vatankhah-varnoosfaderani M, Matyjaszewski K, Paturej J, Rubinstein M, V Dobrynin A, Sheiko SS, 2016, 15, 183–190. [DOI] [PubMed] [Google Scholar]
- [33].Chen M, MacLeod MJ, Johnson JA, ACS Macro Lett 2015, 4, 566–569. [DOI] [PubMed] [Google Scholar]
- [34].a) Wenn B, Martens AC, Chuang Y-M, Gruber J, Junkers T, Polym. Chem 2016, 2720–2727; [Google Scholar]; b) Narupai B, Poelma JE, Pester CW, McGrath AJ, Toumayan EP, Luo Y, Kramer JW, Clark PG, Ray PC, Hawker CJ, J. Polym. Sci., Part A: Polym. Chem 2016, 54, 2276–2284. [Google Scholar]
- [35].Tasdelen MA, Uygun M, Yagci Y, Macromol. Chem. Phys 2010, 211, 2271–2275. [Google Scholar]
- [36].Reeves JA, Allegrezza ML, Konkolewicz D, Macromol. Rapid Commun 2017, 38, 1–5. [DOI] [PubMed] [Google Scholar]
- [37].Anastasaki A, Nikolaou V, Zhang Q, Burns J, Samanta SR, Waldron C, Haddleton AJ, McHale R, Fox D, Percec V, Wilson P, Haddleton DM, J. Am. Chem. Soc 2014, 136, 1141–1149. [DOI] [PubMed] [Google Scholar]
- [38].Anastasaki A, Nikolaou V, Brandford-Adams F, Nurumbetov G, Zhang Q, Clarkson GJ, Fox DJ, Wilson P, Kempe K, Haddleton DM, Chem. Commun 2015, 51, 5626–5629. [DOI] [PubMed] [Google Scholar]
- [39].Discekici EH, Anastasaki A, Kaminker R, Willenbacher J, Truong NP, Fleischmann C, Oschmann B, Lunn DJ, Read De Alaniz J, Davis TP, Bates CM, Hawker CJ, J. Am. Chem. Soc 2017, 139, 5939–5945. [DOI] [PubMed] [Google Scholar]
- [40].McKenzie TG, da M. Costa LP, Fu Q, Dunstan DE, Qiao GG, Polym. Chem 2016, 7, 4246–4253. [Google Scholar]
- [41].Ryan MD, Pearson RM, French TA, Miyake GM, Macromolecules 2017, 50, 4616–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dolinski ND, Page ZA, Eisenreich F, Niu J, Hecht S, Read de Alaniz J, Hawker CJ, ChemPhotoChem 2017, 1, 125–131. [Google Scholar]
- [43].Theriot JC, Miyake GM, Boyer CA, ACS Macro Lett. 2018, 7, 662–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Xu J, Jung K, Atme A, Shanmugam S, Boyer C, J. Am. Chem. Soc 2014, 136, 5508–5519. [DOI] [PubMed] [Google Scholar]
- [45].Gormley AJ, Yeow J, Ng G, Conway Ó, Boyer C, Chapman R, Angew. Chemie - Int. Ed 2018, 57, 1557–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang Y, Kwak Y, Buback J, Buback M, Matyjaszewski K, ACS Macro Lett. 2012, 1, 1367–1370. [DOI] [PubMed] [Google Scholar]
- [47].Simakova A, Averick SE, Konkolewicz D, Matyjaszewski K, Macromolecules 2012, 45, 6371–6379. [Google Scholar]
- [48].Jones GR, Whitfield R, Anastasaki A, Haddleton DM, J. Am. Chem. Soc 2016, 138, 7346–7352. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.