

Abstract
The PD-1 immune checkpoint pathway is a highly validated target for cancer immunotherapy. Despite the potential ad-vantages of small molecule inhibitors over antibodies, the discovery of small molecule checkpoint inhibitors has lagged behind. To discover small molecule inhibitors of the PD-1 pathway, we have utilized a fragment-based approach. Small molecules were identified that bind to PD-L1 and crystal structures of these compounds bound to PD-L1 were obtained.
Keywords: Fragment-based screening, Structure-based design, PD-L1 inhibitor, Cancer drug discovery, Immunotherapy
Graphical Abstract
Genomic instability arising from somatic mutations and chromosomal rearrangements is a hallmark of intrinsic carcinogenesis.1 The host immune system can potentially recognize and eliminate highly mutated cells by a process referred to as immune surveillance.2 Cancer cells can adapt to escape this host defense mechanism by exploiting endogenous T cell immune tolerance pathways, termed immune checkpoints. An important mechanism of how cancer can evade the immune response is through the PD-1 / PD-L1 immune checkpoint signaling pathway.3 Programmed Death 1 (PD-1) suppresses T cell cytolytic function when bound to its ligand PD-L1.4, 5 PD-L1 is upregulated in most cancer types via induction of PD-L1 expression by IFNγ (secreted from tumor infiltrating T cells) and by constitutive expression of PD-L1 resulting from oncogene activation.3, 6 Indeed, the presence of PD-L1 in the tumor microenvironment is generally correlated with poor prognosis in multiple cancer types.7
Therapeutic antibodies that target PD-1 and PD-L1 have been successful as single agents in numerous clinical trials and have revolutionized the field of immuno-oncology. To date five antibodies that target the PD-1 pathway are now FDA approved for the treatment of 11 different types of cancer, and their indications are continuously expanding.8 Although current antibody-based therapies can offer substantial benefits, the intrinsic properties of antibodies have negative implications when targeting the PD-1 / PD-L1 signaling axis. These concerns include suboptimal tumor penetration, the expense due to the high cost of manufacturing, and potential immunogenicity.9–13 Most importantly, current PD-1 / PD-L1 blocking antibodies have half-lives on the order of 3 to 4 weeks.14, 15 Long-term inhibition of the PD-1 signaling pathway can result in immune related adverse events (irAEs). The prevalence, severity, and management of various irAEs with checkpoint inhibitors in many cancer types is well documented and has been reviewed extensively.16–19 Moreover, higher toxicity rates are expected when these drugs are combined with chemotherapy and other immunotherapeutic agents.
An alternative therapeutic approach is to use small molecules to block the PD-1 / PD-L1 interaction. Small molecule inhibitors of the PD-1 pathway can address the problems associated with antibody-based therapeutics. A small molecule inhibitor could have improved tumor penetration, oral bioavailability, a longer shelf-life, and lower production costs.10–13, 20 Because the pharmaceutical and pharmacokinetic profile of a small molecule can be easily modulated, inhibitors could be designed to be rapidly cleared from the body to minimize irAEs and allow for more flexible dosing regimens. These advantages are expected to be especially important for combinatorial immunotherapies.
Despite these potential advantages, the discovery of small molecule inhibitors has greatly lagged behind mABs. This is likely because PD-1 and PD-L1 proteins are predicted to be challenging drug targets for small molecules.21 The PD-1 / PD-L1 interaction is large (1,970 A2) and lacks deep hydrophobic pockets traditionally found in more druggable proteins.22 One approach for targeting challenging protein-protein interactions is to utilize fragment-based methods. Indeed, fragment-based methods have generated high affinity inhibitors to other protein-protein interactions previously thought to be “undruggable”. 23, 24 While many biochemical and biophysical techniques exist to screen fragment libraries, we prefer protein-observed NMR spectroscopy because of the many advantages including direct measurement of weak binding fragments, the ability to measure binding affinity without a secondary assay and the possibility of identifying the binding location on the protein if the resonance assignments are known.25
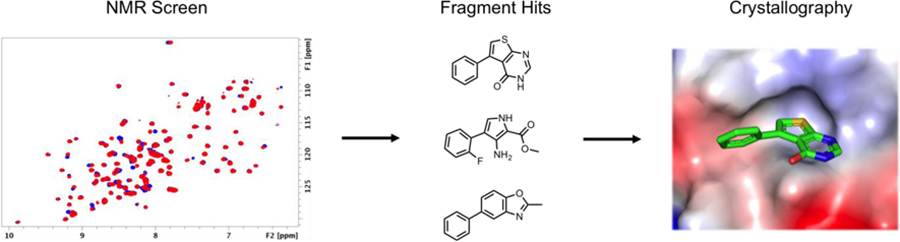
To date there have been no reported attempts to develop small molecule inhibitors of the PD-1 signaling pathway by fragment-based methods. Herein, we report the results of a fragment-based screen of PD-L1 using NMR. From this screen, many novel chemotypes were identified which were subsequently found to displace PD-1. X-ray co-crystal structures of the fragments bound to PD-L1 were obtained to identify their binding site. These results serve as starting points for further optimization of PD-L1 small molecule inhibitors.
PD-L1 is a transmembrane protein that belongs to the Ig superfamily consisting of an extracellular N-terminal V domain (IgV) and one C domain (IgC) connected by a short linker. 1H-15N HMQC spectra containing both domains (18–239) was unsuitable for fragment screening due to numerous unresolved peaks and inconsistent peak intensities. Because the IgV domain of PD-L1 is the sole interaction domain of PD-1, the IgC domain was removed in attempt to improve the HMQC spectrum. However, initial constructs of the IgV domain were unstable at concentrations typically required for generating high quality HMQC spectra (> 15 µM). To obtain a construct that was suitable for protein observed NMR screening, over 100 different PD-L1 IgV constructs were designed and tested for stability. Multiple C-terminal tags were found to stabilize the IgV domain including an 8-Lys tag, S-tag, and a previously reported 6xHis tag.22 These constructs had well resolved HMQC spectra but were unstable when mixed with concentrations of fragments necessary to conduct a fragment-based screen. To further stabilize this construct, H140 was mutated to a glu, H140E, on the C-terminal helix to form additional electrostatic interactions with the IgV domain. Indeed, an X-ray structure obtained of this construct reveals that H140E forms two hydrogen bonds to the backbone amides of Q107 and L106 resulting in a conformational change of Q107 to allow additional hydrogen bonds with S80, R84, and D107 (Figure 1). With exception of these residues, the fold of PD-L1 is unaffected by this mutation with a calculated RMSD of 0.198 Å compared to the wildtype construct (5C3T).22 The H140E mutation was found to stabilize PD-L1 in the presence of fragments and was used to screen our fragment library.
Figure 1:
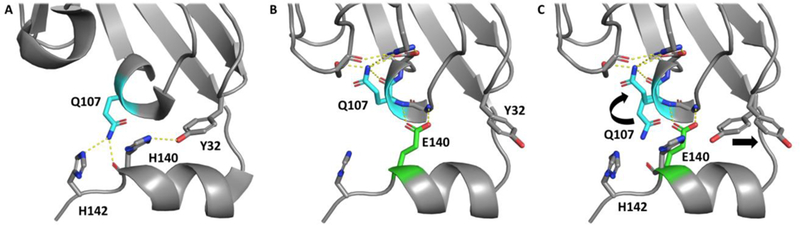
A) PD-L1 IgV-6his X-ray structure with H140, H142, Q107 and Y32 highlighted (sticks) to show stabilizing interactions of the C-terminal 6His helix. B) H140E mutation (green sticks) forms two hydrogen bonds with Q107 and L106 backbone amides. C) Overlay of PD-L1 IgV-6His and PD-L1 IgV-6His-H140E to show conformational change of Q107 to form additional interactions with other residues that stabilize the IgV construct. PD-L1 IgV-6His PDB code: 5C3T and H140E mutant PDB code: 6NP9.
Screening conditions were optimized by adjusting the number of fragments and their concentration per mixture. The quality of 1H-15N HMQC spectra for each sample were monitored at various time points. Optimal screening conditions were found to be 30 µM of PD-L1 with mixtures of 6 fragments at 400 µM each. Using these conditions, our fragment library which consists of 13,800 fragments was screened against PD-L1 by recording 1H-15N SOFAST-HMQC NMR spectra. Hits in mixture samples were noted by monitoring chemical shift changes compared to HMQC spectra of PD-L1 in the absence of fragments (Figure 2B). Mixture samples containing chemical shifts greater than 1 standard deviation above the average chemical shift were designated as hits for deconvolution. Mixtures were deconvoluted by testing each fragment individually at 800 μM concentration. In total, a diverse set of 226 fragments (1.6% hit rate) were found to bind to PD-L1, and these were clustered into 18 different chemotypes (Figure 2A). A 400-fragment analog screen designed by substructure similarity searches generated an additional 100 fragments that bound to PD-L1. The binding affinities of hits were determined by NMR titration experiments to range from 1.2 mM to > 3 mM with ligand efficiencies less than 0.24 (Figure 2A). However, the binding affinities of many fragments were not able to be measured accurately due to limited compound solubility and/or peak broadening at higher concentrations.
Figure 2:
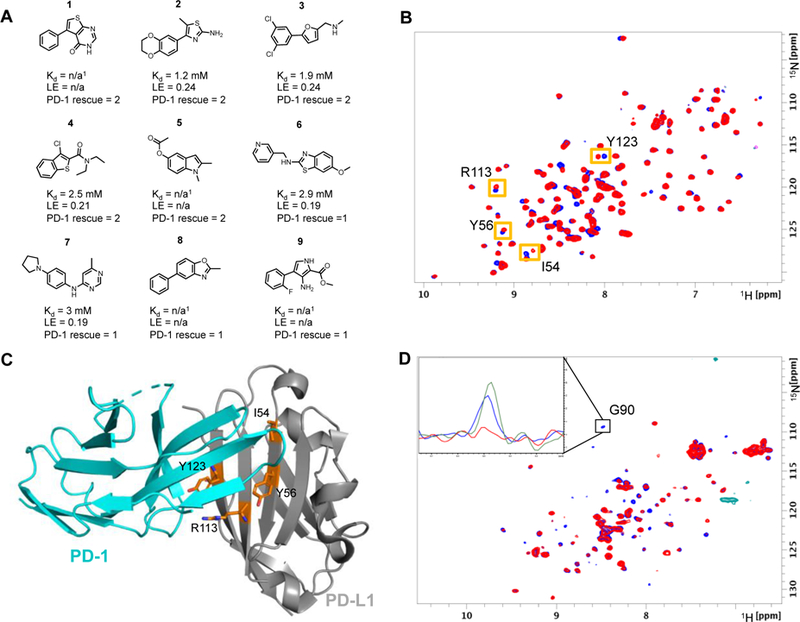
A) Selected fragment hits identified in the fragment screen that displace PD-1. PD-1 rescue score is based on the percent of G90 signal rescued of 15N labeled PD-1 at 800 µM fragment concentration. Score 1 indicates 1 – 15% signal rescue and score 2 indicates > 15% signal rescue. 1Kd values could not be determined due to solubility limits and/or peak broadening. B) SOFAST HMQC spectra of PD-L1-6His-H140E in the absence (blue) and presence of 800 µM fragment #2 (red). Shifted peaks and corresponding amino acid are indicated by orange boxes. C) X-ray structure of PD-1 bound to PD-L1 with highlighted residues (orange sticks) that have resonance perturbations from HMQC spectra suggest fragments bind to PD-L1 at the PD-1 binding site (PDB Code: 4ZQK). D) NMR based ADIA assay showing fragment displace PD-1 from PD-L1. HMQC PD-1 spectrum is broadened by the addition of unlabeled PD-L1 (red). Rescue of signal intensity by addition of 2 mM of fragment 1 (blue) suggests fragment 1 displaces PD-1 from PD-L1. Inset 1D slice of the HMQC spectra shows rescue of signal intensity from G90 of PD-1. The peak intensity is broadened by the addition of 14N PD-L1 (red) compared to 15N PD-1 alone (green). Addition of 2 mM of fragment 1 rescues approximately 75% of the signal intensity (blue).
Only one set of resonance perturbations were identified in the screen. These resonances corresponded to the PD-1 binding site of PD-L1 based on the partially assigned 1H and 15N resonances (Figure 2B-C). To further validate these hits, a PD-1 / PD-L1 NMR-based antagonist induced dissociation assay (AIDA) was utilized.26, 27 The AIDA assay was performed by the addition of a slight molar excess of 14N PD-L1 to 15N PD-1 causing HMQC signals to broaden corresponding to complex formation. The addition of fragments that displace 15N PD-1 from 14N PD-L1 result in a rescue of the 15N PD-1 signal. In total, 36 of the 104 tested fragments were able to displace PD-1 at fragment concentrations of 800 µM. The rescue percentages of fragments 1 – 9 are shown in Figure 2A. Higher concentrations of fragments resulted in higher percent rescue values, with Fragment 1 resulting in 75% rescue at 2 mM concentration (Figure 2D). In total, 36 of the 104 tested fragments were able to displace PD-1. These hits belonged to multiple hit clusters yielding a diverse set of chemotypes that displace PD-1. Certain chemotypes overrepresented from this assay are shown in Figure 2A. In general, fragments with higher binding affinity displaced PD-1, suggesting the ability to inhibit PD-1 binding is based on affinity to PD-L1. Unique chemotypes identified contain 5,6 fused bicycles (fragments 1, 4–6, 8). 5,6 linked biaryl ring systems (fragments 1–3, 9), and 6,6 linked biphenyl ring systems (fragment 8). Prevalent moieties included mono and di substituted phenyl rings (fragments 2, 3, 5–7, 9) and sulfur containing heterocycles (fragments 1, 2, 4, 6). These chemotypes were prioritized for generating X-ray co-crystal structures and further SAR development.
The PD-1 binding site of PD-L1 is blocked by crystal packing in the IgV6His-H140E construct. To force crystal packing into a new space group with an accessible PD-1 binding site, V76 was mutated to threonine. Using a V76T construct, 14 PD-L1 fragment co-crystal structures were obtained. All fragments co-crystalized as a PD-L1 homodimer with a fragment at the interface of two monomers (2:1 stoichiometric ratio of PD-L1 to fragment), similar to previously reported structures (Figure 3A).26, 28, 29 While the V76T mutation allowed for successful co-crystallization of fragments to PD-L1, this mutation did not alter the protein compared to the wildtype construct (5C3T) with a calculated RSMD of 0.38 Å. Fragments all share a similar binding pose inside a cylindrical shaped hydrophobic pocket formed by the residues I54, Y56, M115, Y123, and A121 of each PD-L1 monomer (Figure 3A). These residues are also critical for the hydrophobic interactions between PD-1 and PD-L1 (Figure 3B) and thus rationalize the displacement of PD-1 using the AIDA NMR assay. Interactions of the fragment with PD-L1 are mostly hydrophobic with only a few examples forming additional hydrogen bonds. For example, fragment 1 forms multiple interactions with monomer A including: face-to-edge π stacking interactions with AY56, π-alkyl interactions with AM115 and AA121 and water mediated hydrogen bonding with AD122 and with the amide of AY123 (Figure 3C). Additional interactions with monomer B include: π-alkyl interactions with BA121, BM115 and BY56 and a σ-hole interaction with the amide carbonyl of BM115. (Figure 3D).
Figure 3:
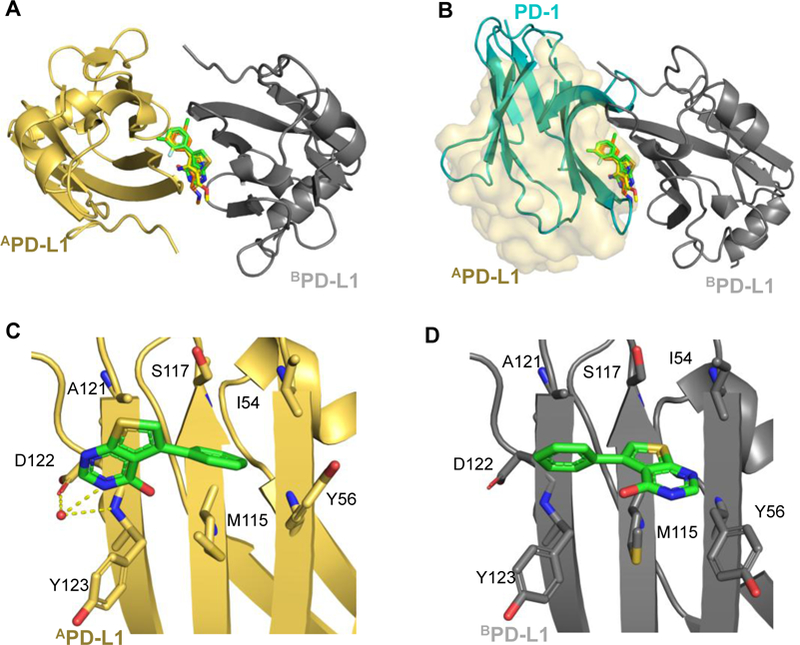
A) Overlay of fragments 1, 3 and 9 PD-L1 co-crystal structures. B) Fragments occupy the PD-1 binding site of PD-L1. PD-1 (blue cartoon PDB: 4ZQK) clashes with fragments shown on monomer A. Monomer B of the PD-L1 dimer is indicated by transparent surface. C and D) Highlighted binding pose of fragment 1 on monomer B (gold) and monomer A (gray). Residues in contact with fragment 1 are shown as sticks. PBD codes: Fragment 1: 6NM7, fragment 3 6NOJ: fragment 9: 6NOS.
In addition to the fragment co-crystal structures, structural information of other PD-L1 inhibitors including macrocyclic peptides and small molecule inhibitors patented by Bristol-Myers Squibb (BMS) have been obtained by us and others.20, 22, 28–30 BMS small molecule inhibitors also crystallize as PD-L1 homodimers. Overlay of co-crystal structures of the reported BMS small molecule ligands with our fragment containing structures show a similar binding pose to PD-L1 (Figure 4, green and yellow sticks). The structural information on how this diverse set of fragment chemotypes bind to PD-L1 can be used to design novel PD-L1 small molecule inhibitors.
Figure 4:
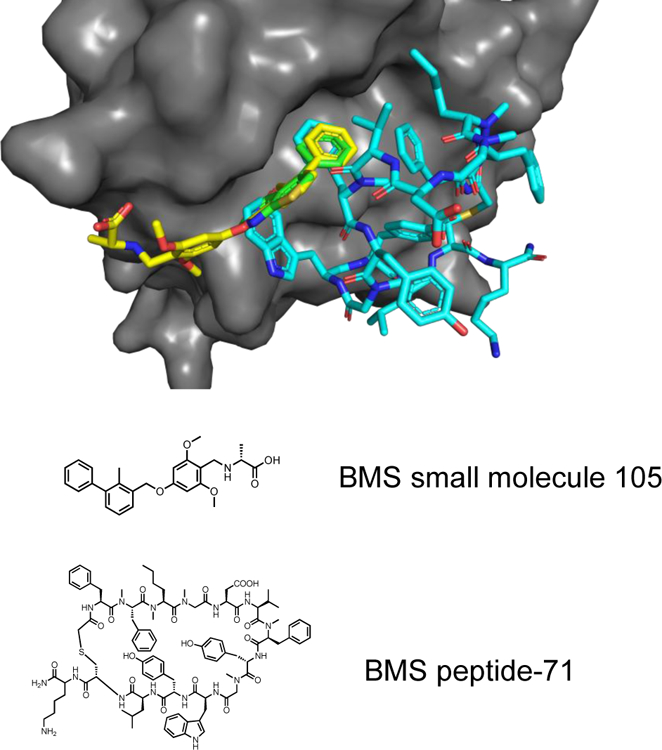
Overlay of Fragment 1 (green) with BMS small molecule inhibitor (yellow) and BMS macrocyclic peptide inhibitor (teal) of PD-L1. PDB codes: Fragment 1: 6NM7, BMS small molecule 105: 6NM8, BMS peptide-71: 6NNV.
Macrocyclic peptides (MCP) also developed by BMS bind to monomeric PD-L1 with low nanomolar affinity.31 Co-crystal structures reveal a phenylalanine side chain of the MCP overlays with the phenyl moiety present in most fragment hits (Figure 4, green and blue sticks). This structural information suggests these fragments could be incorporated into an MCP to make additional interactions with PD-L1. Indeed, this strategy has been used previously in our lab to significantly improve the binding affinity of a peptide to replication protein A (RPA).32
In conclusion, small molecule inhibitors of immune checkpoint proteins are highly sought after due to the potential advantageous pharmaceutical properties compared to currently used mABs. However, despite the exciting potential of small molecule inhibitors, there are limited reports of efforts to develop such molecules. This is likely due to the notion that the checkpoint proteins are predicted to be challenging targets for small molecules due to the relatively flat binding surfaces of these proteins.33 Indeed, fragment screens of both CTLA-4 and PD-1 against our fragment library resulted in low hit rates and suboptimal starting points for further development. Based on these screening results, we concluded that PD-1 and CTLA-4 are unlikely to be druggable by small molecules. However, a fragment screen of PD-L1 resulted in a much higher hit rate (1.6%) and appears to be druggable.
Herein, we report the results of our fragment-based screen on the PD-L1 IgV domain. In total, 226 hits were identified from a screen of 13,800 fragments. Of these hits, 36 were able to displace PD-1 in solution using an NMR-based AIDA assay. Co-crystal structures were obtained of 14 fragments as PD-L1 homodimers as seen in previously characterized BMS PD-L1 inhibitors.26, 28, 29 Using structure-based design and fragment merging approaches, these fragments could be rapidly incorporated into existing PD-L1 inhibitors to generate novel inhibitors with higher binding affinity and/or improved pharmaceutical properties. Although the PD-1 / PD-L1 interaction is thought to be difficult to target by small molecules, the fragment screening results reported here yielded novel chemotypes that could serve as starting points for the further development of PD-L1 inhibitors.
Supplementary Material
Acknowledgments
E.P. gratefully acknowledges support from the NIH sponsored chemistry-biology interface training grant # 5T32GM065086. The biomolecular NMR facility at Vanderbilt University is supported in part by grants for NMR instrumentation from the NSF (0922862), NIH (S10 RR025677) and Vanderbilt University matching funds. Vanderbilt robotic crystallization facility is supported by NIH grant S10 RR026915. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02–06CH11357. The authors also thank Corbin Whitwell and co-workers at the Vanderbilt High-Throughput Screening Core facility for compound management.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Experimental details for protein expression and purification, NMR spectroscopy, and protein crystallization are included in the supplementary. Supplementary data to this article can be found online at https://doi.org/
References and notes
- 1.Hanahan D, and Weinberg RA (2011) Hallmarks of cancer: the next generation, Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 2.Swann JB, and Smyth MJ (2007) Immune surveillance of tumors, J Clin Invest 117, 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy, Nat Rev Cancer 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, and Honjo T (2000) Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation, J Exp Med 192, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Francisco LM, Sage PT, and Sharpe AH (2010) The PD-1 pathway in tolerance and autoimmunity, Immunol Rev 236, 219–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, and Minato N (2002) Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade, Proc Natl Acad Sci U S A 99, 12293–12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, Rost S, Leabman M, Xiao Y, Mokatrin A, Koeppen H, Hegde PS, Mellman I, Chen DS, and Hodi FS (2014) Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients, Nature 515, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gong J, Chehrazi-Raffle A, Reddi S, and Salgia R (2018) Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations, J Immunother Cancer 6, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maute RL, Gordon SR, Mayer AT, McCracken MN, Natarajan A, Ring NG, Kimura R, Tsai JM, Manglik A, Kruse AC, Gambhir SS, Weissman IL, and Ring AM (2015) Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging, Proc Natl Acad Sci U S A 112, E6506–6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee CM, and Tannock IF (2010) The distribution of the therapeutic monoclonal antibodies cetuximab and trastuzumab within solid tumors, BMC Cancer 10, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott AM, Wolchok JD, and Old LJ (2012) Antibody therapy of cancer, Nat Rev Cancer 12, 278–287. [DOI] [PubMed] [Google Scholar]
- 12.Imai K, and Takaoka A (2006) Comparing antibody and small-molecule therapies for cancer, Nat Rev Cancer 6, 714–727. [DOI] [PubMed] [Google Scholar]
- 13.Beasley D (2017) The cost of cancer: new drugs show success at a steep price, Reuters.com.
- 14.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, Gilson MM, Wang C, Selby M, Taube JM, Anders R, Chen L, Korman AJ, Pardoll DM, Lowy I, and Topalian SL (2010) Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates, J Clin Oncol 28, 3167–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patnaik A, Kang SP, Rasco D, Papadopoulos KP, Elassaiss-Schaap J, Beeram M, Drengler R, Chen C, Smith L, Espino G, Gergich K, Delgado L, Daud A, Lindia JA, Li XN, Pierce RH, Yearley JH, Wu D, Laterza O, Lehnert M, Iannone R, and Tolcher AW (2015) Phase I Study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors, Clin Cancer Res 21, 4286–4293. [DOI] [PubMed] [Google Scholar]
- 16.Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, Berdelou A, Varga A, Bahleda R, Hollebecque A, Massard C, Fuerea A, Ribrag V, Gazzah A, Armand JP, Amellal N, Angevin E, Noel N, Boutros C, Mateus C, Robert C, Soria JC, Marabelle A, and Lambotte O (2016) Immune-related adverse events with immune checkpoint blockade: a comprehensive review, In Eur J Cancer 2016/01/15 ed., pp 139–148. [DOI] [PubMed]
- 17.Alatrash G, Jakher H, Stafford PD, and Mittendorf EA (2013) Cancer immunotherapies, their safety and toxicity, In Expert Opin Drug Saf 2013/05/15 ed., pp 631–645. [DOI] [PubMed]
- 18.Kumar V, Chaudhary N, Garg M, Floudas CS, Soni P, and Chandra AB (2017) Current Diagnosis and Management of Immune Related Adverse Events (irAEs) Induced by Immune Checkpoint Inhibitor Therapy, In Front Pharmacol 2017/02/24 ed., p 49. [DOI] [PMC free article] [PubMed]
- 19.Day D, and Hansen AR (2016) Immune-Related Adverse Events Associated with Immune Checkpoint Inhibitors, In BioDrugs 2016/11/17 ed., pp 571–584. [DOI] [PubMed]
- 20.Magiera-Mularz K, Skalniak L, Zak KM, Musielak B, Rudzinska-Szostak E, Berlicki Ł, Kocik J, Grudnik P, Sala D, Zarganes-Tzitzikas T, Shaabani S, Dömling A, Dubin G, and Holak TA (2017) Bioactive Macrocyclic Inhibitors of the PD-1/PD-L1 Immune Checkpoint, Angew Chem Int Ed Engl 56, 13732–13735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zarganes-Tzitzikas T, Konstantinidou M, Gao Y, Krzemien D, Zak K, Dubin G, Holak TA, and Domling A (2016) Inhibitors of programmed cell death 1 (PD-1): a patent review (2010–2015), Expert Opin Ther Pat 26, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, Dömling A, Dubin G, and Holak TA (2015) Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1, Structure 23, 2341–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shuker SB, Hajduk PJ, Meadows RP, and Fesik SW (1996) Discovering high-affinity ligands for proteins: SAR by NMR, Science 274, 1531–1534. [DOI] [PubMed] [Google Scholar]
- 24.Hajduk PJ, Meadows RP, and Fesik SW (1997) Discovering high-affinity ligands for proteins, Science 278, 497,499. [DOI] [PubMed] [Google Scholar]
- 25.Harner MJ, Frank AO, and Fesik SW (2013) Fragment-based drug discovery using NMR spectroscopy, J Biomol NMR 56, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zak KM, Grudnik P, Guzik K, Zieba BJ, Musielak B, Dömling A, Dubin G, and Holak TA (2016) Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1), Oncotarget 7, 30323–30335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krajewski M, Rothweiler U, D’Silva L, Majumdar S, Klein C, and Holak TA (2007) An NMR-based antagonist induced dissociation assay for targeting the ligand-protein and protein-protein interactions in competition binding experiments, J Med Chem 50, 4382–4387. [DOI] [PubMed] [Google Scholar]
- 28.Guzik K, Zak KM, Grudnik P, Magiera K, Musielak B, Törner R, Skalniak L, Dömling A, Dubin G, and Holak TA (2017) Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1, J Med Chem 60, 5857–5867. [DOI] [PubMed] [Google Scholar]
- 29.Skalniak L, Zak KM, Guzik K, Magiera K, Musielak B, Pachota M, Szelazek B, Kocik J, Grudnik P, Tomala M, Krzanik S, Pyrc K, Dömling A, Dubin G, and Holak TA (2017) Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells, Oncotarget 8, 72167–72181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chupak LS, Zheng X, and Company B-MS (2015) Compounds useful as immunomodulators
- 31.Miller MMP, NJ, US), Mapelli Claudio (Langhorne, PA, US), Allen Martin Patrick (Flemington, NJ, US), Bowsher Michael S. (Prospect, CT, US), Boy Kenneth M. (Durham, CT, US), Gillis Eric P. (Cheshire, CT, US), Langley David R. (Meriden, CT, US), Mull Eric(Guilford, CT, US), Poirier Maude A. (Pennington, NJ, US), Sanghvi Nishith (Franklin Park, NJ, US), Sun Li-qiang (Glastonbury, CT, US), Tenney Daniel J. (Madison, CT, US), Yeung Kap-sun (Madison, CT, US), Zhu Juliang (North Haven, CT, US), Reid Patrick C. (Tokyo, JP), Scola Paul Michael (Glastonbury, CT, US). (2014) MACROCYCLIC INHIBITORS OF THE PD-1/PD-L1 AND CD80(B7–1)/PD-L1 PROTEIN/PROTEIN INTERACTIONS, BRISTOL-MYERS SQUIBB COMPANY, United States. [Google Scholar]
- 32.Frank AO, Vangamudi B, Feldkamp MD, Souza-Fagundes EM, Luzwick JW, Cortez D, Olejniczak ET, Waterson AG, Rossanese OW, Chazin WJ, and Fesik SW (2014) Discovery of a potent stapled helix peptide that binds to the 70N domain of replication protein A, J Med Chem 57, 2455–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zak KM, Grudnik P, Magiera K, Dömling A, Dubin G, and Holak TA (2017) Structural Biology of the Immune Checkpoint Receptor PD-1 and Its Ligands PD-L1/PD-L2, Structure 25, 1163–1174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.