

Abstract
Fluorescent small molecules are important tools in many aspects of modern biology. A two-stage evaluation process involving fluorescence screening and live-cell imaging was developed to facilitate the identification of new fluorescent probes from extracts housed within the NCI Natural Products Repository. To this end, over 2000 extracts and prefractionated samples were examined, including an extract from the marine crinoid Pterometra venusta. An optically guided evaluation involving stepwise fluorescence screening and live-cell imaging was developed to enable the isolation of fluorescent natural products. These efforts resulted in the isolation of six hydroxyanthraquinone compounds, three of which are new natural products. These purified metabolites were examined for their potential as cellular imaging probes, and they demonstrate that natural product libraries can be a good source of new fluorescent agents.
Graphical Abstract
The use of fluorescent small-molecule probes has had an immense impact on medicine and biomedical research, with applications ranging from biomolecular labels for understanding cellular processes to contrast agents used in surgical procedures.1,2 Fluorescent probes are an essential component of basic and applied cell biology and are widely utilized in a range of chemical biology studies.3 In addition, fluorescent bioconjugates find applications in time-resolved fluorescence measurements,4 as fluorescence resonance energy transfer (FRET) reagents,5 and in fluorescence polarization studies.6 Most of the commercially available fluorescent probes are based on a limited number of structural motifs, including the cyanine dyes (e.g., Cy5, Cy7) and xanthene derivatives such as fluorescein and rhodamine. The optical properties of these chromophores have been tuned to a wide range of applications through, for example, heteroatom substitution or extension of conjugation (e.g., fluorescein to naphthofluorescein) to create families of related fluorophores. These changes to the molecular architecture allow a larger range of emission wavelengths to be obtained. Despite the success and utility of these dyes, there is still a need for new molecular scaffolds that can serve as leads toward fluorescent probe design, specifically those that possess photophysical properties that allow emission in the far-red to near-IR range of the electromagnetic spectrum.
Nature is quite adept at creating molecular architectures that exhibit inherent fluorescent properties. Core secondary metabolite structures such as coumarins, quinolones, azulenes, acridines, and anthraquinones provide the foundation for important classes of fluorescent probes,3 making a strong case to continue to look to natural sources as inspiration for new fluorophores. However, there has been no systematic endeavor to search for new fluorescent scaffolds from natural product extracts. A library of prefractionated samples, generated from extracts obtained from the NCI Natural Products Repository,7,8 provided a unique opportunity to screen for naturally occurring fluorescent compounds that may be useful in the discovery and development of next-generation molecular probes. Here, we describe a high-throughput approach to screen natural product samples and the discovery of a new series of anthraquinone-based fluorescent compounds. Ideal candidate structures are those that exhibit one or more of the following criteria: (i) absorbance/fluorescence in the near-IR range (650–900 nm), (ii) favorable photophysical properties, including high quantum yield (Φ) and molar absorptivity (ε), and (iii) distinct cellular localization, e.g., nuclear, mitochondrial, lysosomal.
RESULTS AND DISCUSSION
An initial test library comprising 2456 prefractionated natural product samples on seven 384-well plates was screened at 100 μg/mL for absorbance and fluorescence properties using a microplate reader. The initial evaluation of these materials consisted of a series of single-point absorbance and fluorescence measurements, spaced 100 nm apart over the range of 350 to 850 nm. Wells with fluorescence values greater than 3-fold background (i.e., of DMSO alone) were then subjected to full absorbance and fluorescence spectral analyses. Samples for which the major chromophoric element was porphyrin-based, indicated by a characteristic absorbance spectrum consisting of an intense Soret band around 400 nm and minor Q bands from approximately 500 to 600 nm, with a sharp fluorescence peak around 700 nm, were excluded at this stage.9 This was based on the likelihood of such extracts being predominantly chlorophyll or other photosynthetic pigments. These criteria for exclusion eliminated a majority of the terrestrial plant, marine algae, and cyanobacterial samples from further analysis.
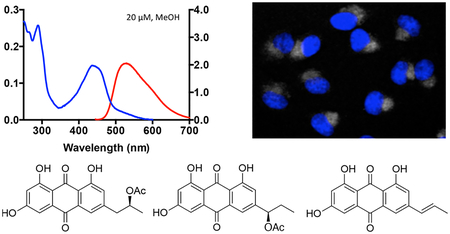
A prefractionated sample (fraction C, 100% EtOAc)7 from an extract of the crinoid Pterometra venusta (Clark 1912, family Asterometridae) was subjected to full spectral analysis, and it showed a strong absorbance band with a maximum at approximately 450 nm and a broad fluorescence emission centered at approximately 550 nm (Figure 1A). The crinoid extract then progressed to the next level of evaluation–cellular imaging. HeLa cells were treated with the extract for 24 h and then imaged over the full 350 to 850 nm wavelength range indicated above. These studies showed that the chromophoric components of this extract localize intracellularly and give rise to a measurable fluorescence signal upon addition to cells (Figure 1B). Given its fluorogenic properties and potential utility as a cellular stain, we set out to isolate the individual compounds in the extract responsible for these characteristics. The organic solvent (1:1 MeOH−CH2Cl2) extract of P.venusta was partitioned across DIOL bonded-phase silica to replicate the prefractionation procedure and validate the initial fluorescence screen. Chromatographic fractions that exhibited the same absorbance and fluorescence profile shown in Figure 1A were progressively purified using C18 HPLC to afford three new natural products (1–3) and three known compounds (4–6).
Figure 1.

Initial spectroscopic and cellular imaging results for the prefractionated P. venusta sample. (A) Absorbance and fluorescence spectra at a concentration of 100 μg/mL in DMSO. Excitation for the fluorescence measurements was at the absorbance maximum (λabs). (B) Fluorescence images of cells treated with 20 μg/mL of the sample for 24 h and costained with 1 μM of the nuclear tracer Hoechst 33342 for 15 min. Fluorescent emission from Hoechst 33342 is shown on the DAPI channel (left) and the P. venusta sample on the RFP channel (right).
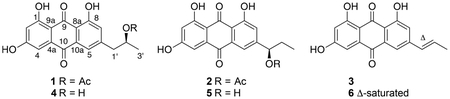
Compound 1 was isolated as an optically active orange film, [α]25D +40, and its molecular formula was assigned as C19H16O7 through HRESIMS measurements and NMR analyses. The 12 degrees of unsaturation and low hydrogen count were indicative of a highly aromatic structure. All 19 carbon signals (Table 1) were apparent in the 13C NMR spectrum, and 15 corresponded to sp2-hybridized carbons (δC 107.9–189.2). A multiplicity-edited HSQC experiment revealed four sp2-methines, one oxymethine, one aliphatic methylene, and two methyl groups. The three additional hydrogens required by the molecular formula were assigned as exchangeable OH protons. Of the 15 sp2-hybridized carbons, two (δC 189.2, 181.5) were typical of α,β-unsaturated carbonyl groups, one suggested a carboxylic acid derivative (δC 169.7), and the remaining 12 centers were consistent with two polarized aromatic rings. The 1H NMR and COSY data revealed three simple spin systems: two sets of meta-coupled pairs (δH 6.54/7.09 and δH 7.54/7.19) and a 1,2-substituted propyl group. HMBC correlation data from the aromatic protons were used to construct the anthracene-9,10-dione core (Figure 2). Key 4J correlations from H-2 and H-7 to C-9 (δC 189.2) along with 3J correlations from H-4 and H-5 to C-10 (δC 181.5) helped define the central dione ring. Reciprocal HMBC correlations between CH-5, CH-7, and CH2-1′ were used to affix the propyl chain to C-6. An acetoxy group was linked to this unit by an HMBC correlation between H-2′ (δH 5.07) and C-4′ (δC 169.7). The absolute configuration of 1 was determined to be 2′S by base hydrolysis of the acetyl group to give the secondary alcohol, followed by application of the advanced Mosher’s ester analysis (Figure 3).10,11 The structure, chiroptical properties, and NMR data for the hydrolysis product of 1 were virtually identical with those recently reported for (+)-2′ S-isorhodoptilometrin by the Oberlies group.12
Table 1.
NMR Spectroscopic Data (1H 600 MHz, 13C 150 MHz) for Compounds 1–3 in DMSO-d6
| 1 | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| pos. | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) |
| 1 | 164.6, C | 164.8, C | n.o.a | |||
| 2 | 107.9, CH | 6.54, s | 108.0, CH | 6.47, s | 107.9, CH | 6.38, s |
| 3 | 166.7, C | 168.1, C | n.o.a | |||
| 4 | 109.5, CH | 7.09, s | 110.6, CH | 7.06, s | 111.8, CH | 7.00, s |
| 4a | 135.1, C | 135.0, C | 134.9, C | |||
| 5 | 120.4, CH | 7.54, s | 116.5, CH | 7.60, s | 116.5, CH | 7.66, s |
| 6 | 147.7, C | 149.6, C | 145.0, C | |||
| 7 | 124.6, CH | 7.19, s | 121.2, CH | 7.27, s | 119.8, CH | 7.29, s |
| 8 | 161.2, C | 161.3, C | 161.7, C | |||
| 8a | 114.2, C | 115.2, C | 114.6, C | |||
| 9 | 189.2, C | 188.3, C | 187.6, C | |||
| 9a | 108.5, C | 107.9, C | 107.6, C | |||
| 10 | 181.5, C | 181.7, C | 182.1, C | |||
| 10a | 132.9, C | 133.4, C | 133.4, C | |||
| 1′ | 41.1, CH2 | 2.92, dd (13.6, 7.5) 2.96, dd (13.8, 5.3) | 75.5, CH | 5.67, t (6.5) | 129.5, CH | 6.54, d (16.1) |
| 2′ | 70.2, CH | 5.07, sext (6.3) | 28.5, CH2 | 1.83, m | 131.8, CH | 6.65, dq (16.1, 6.5) |
| 3′ | 19.5, CH3 | 1.21, d (6.3), m | 9.5, CH3 | 0.86, t (7.3) | 18.6, CH3 | 1.91, d (6.5) |
| 4′ | 169.7, C | 169.9, C | ||||
| 5′ | 20.9, CH3 | 1.93, s | 20.8, CH3 | 2.11, s | ||
Not observed.
Figure 2.

Selected HMBC correlations for compound 1.
Figure 3.

Mosher’s ester analysis of the base hydrolysis product of compounds 1 and 2.
Compound 2 had a molecular formula of C19H16O7 that was isomeric with 1, and comprehensive NMR analyses showed it possessed the same connectivity as 1, except for placement of the acetoxy group at C-1′. HMBC correlations from H-5, H-7, and H-3′ to C-1′ and from H-1′ to C-4′ confirmed the acetoxy location in 2. The absolute configuration of C-1′ was assigned as R by Mosher’s ester analysis of the base hydrolysis product (Figure 3).
Compound 3, with a molecular formula of C17H12O5, shared the same anthraquinone core as compounds 1 and 2, but had a prop-1-enyl substituent attached to C-6. NMR signals for a benzylic disubstituted olefin (δH 6.54/δC 129.5 and δH 6.65/δC 131.8) and allylic methyl group (δH 1.91/δC 18.6) were apparent. HMBC correlations from H-1′ and H-2′ to C-6 confirmed the point of substitution. The configuration of the alkene was established as E based on the olefinic protons vicinal coupling of 16.1 Hz. The isolation of 3 represents the first natural occurrence of this compound; however it has previously been reported as a synthetic intermediate toward the synthesis of (±)-5.13
The planar structures of compounds 4 and 5 were identified as the (iso)rhodoptilometrins based on spectroscopic comparisons with literature values12 and with 1 and 2, respectively (Supporting Information). The absolute configuration of 4 in this study was determined to be 2′S by preparation and analysis of the Mosher’s ester derivatives. However, our sample of 4 had a levorotary optical rotation ([α]25D −24), which is antipodal to the specific rotation reported for (+)-2′S-isorhodoptilometrin ([α]25D +30) for which the absolute configuration was verified by Mosher’s ester analysis.12 This discrepancy likely resulted from co-occurrence of a minor constituent or metal ion complex with a large negative optical rotation, but even after rigorous and repeated purification efforts the sample of 4 we isolated still showed a negative rotation. Attempts to determine the absolute configuration of naturally isolated 5 by Mosher’s ester derivatization revealed two reaction products for each preparation from the chiral acid chlorides. Each of these ester derivatives was spectroscopically identical to one or the other diastereomers prepared by treating the hydrolysis product of 2 with R-MTPA-Cl or S-MTPA-Cl. As racemization at C-1′ was not observed following hydrolysis of 2, this suggested the existence of a partial racemate of 5 in the crinoid extract. As isolated from the extract, compound 5 showed a large negative specific rotation ([α]25D −39), even though it clearly was a mixture of R and S enantiomers. This was in contrast to the positive rotation ([α]25D +29) measured for alcohol 5 obtained from hydrolysis of 2 (see Figure 3). This difference in rotation is also suggestive of sample contamination or interference in the specific rotation measurements. Crinemodin (6), a well-known anthraquinone metabolite that has been isolated from several echinoderm species, was also identified from the P. venusta extract, and its spectroscopic data closely matched the reported literature values.14
Each of the purified compounds was analyzed for a range of photophysical properties (Table 2). Compounds 1–6 (20 μM in MeOH) showed λabs between 436 and 452 nm. Excitation of these compounds at the respective λabs afforded fluorescence emission spectra with maxima (λem) ranging between 526 and 534 nm. Quantum yields (ΦF) in MeOH ranged between 0.6% and 2%. Notably, absorbance and fluorescence emission spectra for 1–6 (Supporting Information) closely resemble those obtained with the original prefractionated crinoid sample (Figure 1).
Table 2.
Optical Properties for Compounds 1–6 (in MeOH)
| compound | λabs (nm) | λem (nm) | ε (×103 M−1 cm−1) | ΦF (%) |
|---|---|---|---|---|
| 1 | 438 | 528 | 7.7 | 0.7 |
| 2 | 438 | 531 | 3.2 | 0.6 |
| 3 | 452 | 534 | 2.3 | 2 |
| 4 | 436 | 526 | 9.0 | 0.9 |
| 5 | 437 | 526 | 13 | 0.8 |
| 6 | 437 | 527 | 7.4 | 1 |
Each compound was then profiled in cellular imaging studies. HeLa cells were treated with 1–6 at 5 μM and incubated for 24 h, followed by treatment with 1 μM Hoechst 33342. Representative cellular images are shown in Figure 4 for compound 3. Imaging results for the other anthraquinones (1, 2, 4–6) were all very similar (Supporting Information), and their localization pattern in cells suggested endosomal/lysosomal accumulation of these compounds.15
Figure 4.

Representative cellular fluorescence imaging of HeLa cells treated with 5 μM compound 3 and 1 μM Hoechst 33342. Fluorescence emission of 3 on the RFP channel (left) and overlay with Hoechst 33342 (right), on the DAPI channel, are shown.
CONCLUSION
The tiered screening approach developed here enabled rapid identification of natural product samples that contain fluorescent materials. Using the UV absorbance and fluorescence emission spectra, the initial screening hits could be readily refined to eliminate those containing chlorophyll and other photosynthetic pigments. Cellular imaging then identified samples that exhibited a particular optical property and/or cellular localization pattern (in our case, detectable fluorescence staining of the intracellular environment). UV and fluorescence monitoring during the fractionation of complex natural product samples can then guide the isolation and purification of the desired chromophoric compounds. Of note, the majority of the analyses were performed on relatively small amounts of the extract or prefractionated samples, making this an attractive approach for mining natural product libraries for new fluorescent agents with desired photophysical properties and/or cellular applications.
Applying this screening protocol to a subset of the NCI Natural Product Repository has yielded three new anthraqui-none-based fluorophores with potential application for cellular imaging. While these compounds exhibit modest fluorescence quantum yields, they bear significant structural similarity to the commercial dye DRAQ5, a nuclear DNA stain.16 Because compounds 1–6 accumulate in cytoplasmic organelles rather than the nucleus, these scaffolds provide unique starting points for structural modification that could afford improved imaging agents for these other cellular compartments. Additionally, these efforts would help define the photophysical SAR of anthraquinone-based fluorophores. Continued screening of the NCI Repository for new fluorescent natural products, particularly those active in the red to near-IR range, would certainly seem warranted.
EXPERIMENTAL SECTION
General Experimental Procedures.
Optical rotations were measured on a PerkinElmer 241 polarimeter in a 100 × 2 mm cell. UV absorption and fluorescence emission spectra were obtained using a Synergy Mx multimode microplate reader operated by Gen5 software (BioTek Instruments). Molar absorption coefficients were measured using 1 cm quartz cuvettes (Hellma Analytics) on a UV-2550 spectrophotometer operated by UVProbe 2.32 software (Shimadzu). Fluorescence quantum yields were similarly measured using a QuantaMaster steady-state spectrofluorimeter operated by FelixGX 4.0.3 software (PTI), with 5 nm excitation and emission slit widths and a 0.1 s integration rate. Cellular imaging was performed on an Evos FL auto cell imaging system (ThermoFisher Scientific) at 20× magnification operating with DAPI (λabs 357 ± 22 nm, λem 447 ±30 nm) and RFP (λabs 531 ± 20 nm, λem 593 ± 20 nm). Images were processed using ImageJ. IR spectra were recorded with a Bruker ALPHA II FT-IR spectrometer. 1H, 13C, COSY, HSQC, HMBC (optimized for nJHC = 8 Hz), and other NMR experiments were performed in DMSO-d6 using a Bruker Avance III spectrometer operating at 600 MHz for 1H and 150 MHz for 13C and equipped with a 3 mm cryogenically cooled probe. Spectra were calibrated to residual solvent signals at δH 2.50 and δC 39.5. ESIMS studies were carried out on an Agilent 6130 Quadrapole LC/MS system, and (+)HRESIMS data were acquired on an Agilent Technology 6530 Accurate-mass Q-TOF LC/MS. Flash chromatography was per formed using an automated system (CombiFlash, Teledyne Isco). Reversed-phase HPLC was carried out using a Varian PrepStar solvent delivery system. Water used for chromatography was distilled prior to use; all other solvents were HPLC-grade.
Animal Material.
Samples of the marine crinoid Pterometra venusta were collected by scuba at a depth of 5–23 feet, in the Philippines in 1996, and kept frozen until extraction. The collection was carried out by the Coral Reef Research Foundation under contract with the Natural Products Branch, U.S. National Cancer Institute. A voucher specimen (voucher ID # 0CDN3466) was deposited at the Smithsonian Institution, Washington, DC. The animal material (301.5 g wet wt) was ground and processed using the standard NCI method for marine samples to provide 7.38 g of organic solvent extract (NSC C015855).17
Isolation.
A 1.11 g aliquot of the P. venusta organic solvent extract was fractionated on DIOL-derivatized silica eluted with (i) 9:1 hexane−CH2Cl2, (ii) 20:1 CH2Cl2−EtOAc, (iii) 100% EtOAc, (iv) 5:1 EtOAc−MeOH, and (v) 100% MeOH, to give fractions A–E. Fractions C and D were combined (197.0 mg total) and further fractionated on octadecyl-derivatized silica (C18), eluting with 75–100% MeOH−H2O in a step-gradient fashion to give fractions S–X. Fraction S afforded compound 1 (12.3 mg). A 5 mg portion of fraction T (25.6 mg total) was purified by C18 HPLC to give additional 1 (1.4 mg, tR = 5.6 min) and 2 (1.0 mg, tR = 6.1 min). Fraction W was a 114.5 mg mixture of two compounds, and a 5 mg portion was purified using C18 HPLC (250 × 10 mm, 5 μ, 85% MeCN−H2O−0.1% HCOOH, 4 mL/min) to afford 3 (0.6 mg, tR = 7.7 min) and 6 (1.6 mg, tR = 8.2 min).
Fraction F from the DIOL column (117.3 mg) was fractionated on C18 (75–100% MeOH−H2O) to give fractions Y–AG, from which AA–AC were combined (54.3 mg total) and chromatographed on silica gel (10–100% EtOAc−CH2Cl2) to afford fractions AH–AO. An 8 mg portion of fraction AH (16.5 mg total) was purified by C18 HPLC (250 × 10 mm, 5 μ, 50% MeCN−H2O−0.1% HCOOH, 4 mL/min) to give 4 (1.6 mg, tR = 12.1 min) and 5 (4.4 mg, tR = 16.4 min).
1,3,8-Trihydroxy-6-(2′-acetoxypropyl)anthracene-9,10-dione (1):
orange film; [α]25D +40 (c 0.05, MeOH); UV data, see Table 2; IR (neat) νmax 2921, 1732, 1626, 1477, 1373, 1262, 1024, 764 cm−1; NMR data, see Table 1; HRESIMS m/z 357.0971 [M + H]+ (calcd for C19H17O7, 357.0974).
1,3,8-Trihydroxy-6-(1′-acetoxypropyl)anthracene-9,10-dione (2):
orange film; [α]25D −33 (c 0.05, MeOH); UV data, see Table 2; IR (neat) νmax 2924, 1736, 1626, 1372, 1256, 1020, 766 cm−1; NMR data see, Table 1; HRESIMS m/z 357.0964 [M + H]+ (calcd for C19H17O7, 357.0974).
(E)-1,3,8-Trihydroxy-6-(prop-1′-enyl)anthracene-9,10-dione (3):
orange film; UV data, see Table 2; IR (neat) νmax 2922, 2852, 1626, 1388, 1263, 1221, 766 cm−1; NMR data, see Table 1; HRESIMS m/z 297.0756 [M + H]+ (calcd for C17H13O5, 297.0763).
1,3,8-Trihydroxy-6-(2′-hydroxypropyl)-anthracene-9,10-dione (isorhodoptilometrin, 4):
orange film; [α]25D −24 (c 0.05, MeOH), cf. [α]24D +30 (c 0.13, MeOH) from Figueroa et al.;12 UV data, see Table 2; NMR data, see Table S31; HRESIMS m/z 315.0860 [M + H]+ (calcd for C17H15O6, 315.0869).
1,3,8-Trihydroxy-6-(1′-hydroxypropyl)anthracene-9,10-dione (rhodoptilometrin, 5):
orange film; [α]25D −39 (c 0.05, MeOH), cf. [α]20D −22 (c 0.1, MeOH) from Wright et al.;18 UV data, see Table 2; NMR data, see Table S31; HRESIMS, m/z 315.0869 [M + H]+ (calcd for C17H15O6, 315.0869).
1,3,8-Trihydroxy-6-propylanthracene-9,10-dione (crinemodin, 6):
orange film; UV data, see Table 2; NMR data, see Table S31; HRESIMS, m/z 299.0915 [M + H]+ (calcd for C17H15O5, 299.0920).
Hydrolysis and Mosher’s Analysis of 1 and 2.
A 7 mg sample of fraction T containing a mixture of 1 and 2 (see Isolation) was dissolved in a solution of MeOH (8 mL) and 5 M NaOH (1 mL) and stirred at reflux for 3 h. To the cooled reaction mixture was added concentrated HCl (1 mL), and after dilution with H2O (5 mL), the solution was passed through a 5 mL column of HP20ss preequilibrated in MeOH. The adsorbed material was washed with H2O (50 mL) and eluted with acetone (15 mL). The acetone eluent was evaporated to dryness to yield a mixture of (+)-4 and (+)-5, verified by LCMS. The reaction products were reconstituted in MeOH and purified by C18 HPLC (250 × 10 mm, 5 μ, 50% MeCN−H2O−0.1% HCOOH, 4 mL/min) to give (+)-4 (1.5 mg, tR = 12.3 min) and (+)-5 (1.5 mg, tR = 16.5 min). A 0.5 mg aliquot of hydrolysis product (+)-4 dissolved in 200 μL of pyridine-d5 was treated with 10 μL of R-MTPA-Cl at rt for 16 h. The reaction was quenched by addition of MeOH, and the S-MTPA ester product 1a was purified by C18 HPLC eluted with 100% MeOH−0.1% HCOOH. An additional 0.5 mg of (+)-4 was reacted and purified in the same manner using S-MTPA-Cl to generate the R-MTPA ester derivative 1b. The location of the unreacted phenolic OH group at C-3 in these products was established by diagnostic HMBC correlations from the OH proton (δH 11.68, DMSO-d6) to C-2 (δC 112.8), C-3 (δC 163.6), and C-4 (δC 116.0). Hydrolysis product (+)-5 was derivatized, purified, and characterized in an identical manner to that described above to afford 2a and 2b, respectively.
Derivatization product 1a:
1H NMR (600 MHz) δ 8.12 (H-5), 7.44 (H-7), 5.46 (H-2′), 3.25 (H-1′b), 3.15 (H-1′a), 1.31 (H3-3′); MS m/z 961.1930 [M − H]− (calcd for C47H34F9 O12, 961.1907).
Derivatization product 1b:
1H NMR (600 MHz) δ 7.89 (H-5), 7.30 (H-7), 5.46 (H-2′), 3.19 (H-1′b), 3.05 (H-1′a), 1.43 (H3-3′); HRESIMS m/z 961.1935 [M − H]− (calcd for C47H34F9 O12, 961.1907).
Derivatization product 2a:
1H NMR (600 MHz) δ 8.13 (H-5), 7.32 (H-7), 6.16 (H-1′), 1.98 (H-2′b), 1.93 (H-2′a), 0.90 (H3-3′); HRESIMS m/z 961.1926 [M − H]− (calcd for C47H34F9 O12, 961.1907).
Derivatization product 2b:
1H NMR (600 MHz) δ 8.23 (H-5), 7.42 (H-7), 6.21 (H-1′), 1.93 (H-2′b), 1.89 (H-2′a), 0.77 (H3-3′); HRESIMS m/z 961.1929 [M − H]− (calcd for C47H34F9 O12, 961.1907).
Optical Screening and Characterization.
Extracts were plated into 384-well plates at a concentration of 100 μg/mL in DMSO (80 μL/well). Single-point absorbance measurements were recorded using a microplate reader, at 350, 450, 550, 650, and 750 nm. Single-point fluorescence was similarly measured by exciting at the wavelengths above and recording emission at 450, 550, 650, 750, and 850 nm (emission bandwidth = 9 nm). For wells giving greater than 3-fold the background value (i.e., of DMSO alone), full absorbance and fluorescence emission curves were obtained (excitation at λabs).
Optical Properties and Cellular Imaging of 1–6.
Absorbance and fluorescence emission curves were measured at 20 μM in MeOH. Molar absorption coefficients (ε) were measured in MeOH using Beer’s law. Fluorescence quantum yields (ΦF) were measured in MeOH using a relative method,15 with quinine (ΦF 54% in 0.1 M H2SO4) as the standard.19 HeLa cells (human cervical adenocarcinoma) were obtained from ATCC and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B. HeLa cells (5000 cells/well) were seeded into #1.5 coverglass-bottomed 96-well plates (Cellvis) and allowed to adhere overnight. The media was exchanged with that containing 5 μM of 1–6 and incubated for 24 h. The media was exchanged with that containing 1 μM Hoechst 33342 and incubated for 15 min. The media was again exchanged, and cells were imaged using an Evos FL Auto Cell Imaging System (ThermoFisher Scientific) at 20× magnification. Fluorescence of 1–6 was collected on the RFP channel and of Hoechst 33342 on the DAPI channel (see General Experimental Procedures). False coloring and contrast adjustment were applied uniformly to all images.
Supplementary Material
ACKNOWLEDGMENTS
Grateful acknowledgement goes to the Natural Products Support Group (NCI at Frederick) for extract preparation and R. Rao (CBL) for assistance with optical experiments. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnat-prod.8b00761.
Experimental procedures, additional figures, and full spectroscopic data for all new compounds (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Lavis LD; Raines RT ACS Chem. Biol 2008, 3, 142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Nguyen QT; Tsien RY Nat. Rev. Cancer 2013, 13, 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Duval R; Duplais C Nat. Prod. Rep 2017, 34, 161–193. [DOI] [PubMed] [Google Scholar]
- (4).Bright FV; Munson CA Anal. Chim. Acta 2003, 500, 71–104. [Google Scholar]
- (5).Sapford KE; Berti L; Medintz IL Angew. Chem., Int. Ed 2006, 45, 4562–4588. [DOI] [PubMed] [Google Scholar]
- (6).Owicki JC J. Biomol. Screening 2000, 5, 297–306. [DOI] [PubMed] [Google Scholar]
- (7).Henrich CJ; Cartner LK; Wilson JA; Fuller RW; Rizzo AE; Reilly KM; McMahon JB; Gustafson KR J. Nat. Prod 2015, 78, 2776–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Thornburg CC; Britt JR; Evans JR; Akee RK; Whitt JA; Trinh SK; Harris MJ; Thompson JR; Ewing TL; Shipley SM; Grothaus PG; Newman DJ; Schneider JP; Grkovic T; O’Keefe BR ACS Chem. Biol 2018, 13, 2484–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Giovannetti R In Macro to Nano Spectroscopy, 1st ed.; Uddin J, Ed.; InTech Europe: Rijeka, Croatia, 2012; Vol. 1, pp 87–108. [Google Scholar]
- (10).Ohtani I; Kusumi T; Kashman Y; Kakisawa H J. Am. Chem. Soc 1991, 113, 4092–4096. [Google Scholar]
- (11).Kusumi T; Ohtani I In The Biology Chemistry Interface; Cooper R, Snyder JK, Eds.; Marcel Dekker, Inc.: New York, NY, 1999; pp 103–137. [Google Scholar]
- (12).Figueroa M; Jarmusch AK; Raja HA; El-Elimat T; Kavanaugh JS; Horswill AR; Cooks RG; Cech NB; Oberlies NH J. Nat. Prod 2014, 77, 1351–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Banville J; Brassard P J. Chem. Soc., Perkin Trans. 1 1976, 613–619. [Google Scholar]
- (14).Powell VH; Sutherland MD Aust. J. Chem 1967, 20, 541–553. [Google Scholar]
- (15).Gorka AP; Nani RR; Zhu J; Mackem S; Schnermann MJ J. Am. Chem. Soc 2014, 136, 14153–14159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Smith PJ; Blunt N; Wiltshire M; Hoy T; Teesdale-Spittle P; Craven MR; Watson JV; Amos WB; Errington RJ; Patterson LH Cytometry 2000, 40, 280–291. [DOI] [PubMed] [Google Scholar]
- (17).McCloud TG Molecules 2010, 15, 4526–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wright AD; Nielson JL; Tapiolas DM; Motti CA; Ovendon SPB; Kearns PS; Liptrot CH Mar. Drugs 2009, 7, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Melhuish WH J. Phys. Chem 1961, 65, 229–235. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.