

SUMMARY
Genetically encoded biosensors are useful tools for detecting the presence and levels of diverse biomolecules in living cells. However, low-abundance targets are difficult to detect because they are often unable to bind and activate enough biosensors to detect using standard microscopic imaging approaches. Here we describe a type of RNA-based biosensor, an RNA integrator, which enables detection of low-abundance targets in vitro and in living cells. The RNA integrator is an RNA sequence comprising a ribozyme and an unfolded form of the fluorogenic aptamer Broccoli. Upon binding its target, the ribozyme undergoes cleavage and releases Broccoli, which subsequently folds and becomes fluorescent. Importantly, each target molecule can bind and induce cleavage of multiple copies of the integrator sensor, resulting in an amplified signal. We show that this approach can be generalized to numerous different ribozyme types for the detection of various small molecules.
Graphical Abstract
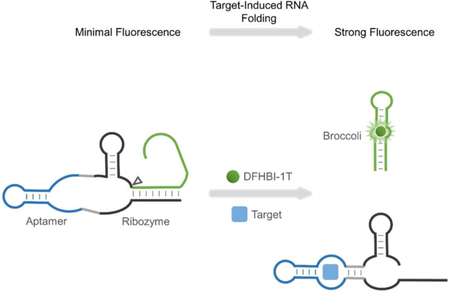
In Brief
You et al. describe a novel type of genetically encoded biosensor, an RNA integrator, which enables detection of low-abundance targets in vitro and in living cells. Expression of RNA integrators in live bacterial cells allows metabolite levels to be imaged with high sensitivity and large dynamic range.
INTRODUCTION
Detecting the levels of cellular metabolites and signaling molecules is important in understanding metabolism and signal transduction networks. A number of fluorescent chemical dyes and genetically encoded sensors have been developed for metabolite imaging in live cells (Okumoto, 2010; Specht et al., 2017; Zhang et al., 2002). In particular, genetically encoded fluorescent sensors can be expressed within specific cell types and particular intracellular locations. They can be incorporated for long-term imaging without interfering with cellular functions. As a result, these genetically encoded sensors, based on either fluorescent proteins or RNA molecules, have been powerful tools for the live-cell measurement of metabolites and signaling molecules (Frommer et al., 2009; Ni et al., 2018; Strack and Jaffrey, 2013).
Fluorescent protein-based sensors normally comprises a target-binding protein domain flanked by two fluorescent proteins (Frommer et al., 2009). These sensors require target molecule to bind and induce a conformational change. Sensors undergo a conformational change that repositions the fluorescent proteins, which resulting in changes in Förster resonance energy transfer (FRET) between two fluorescent proteins. However, for many physiologically important metabolites and signaling molecules, protein domains that selectively bind these target analytes and undergo an induced conformational change are not readily available (Specht et al., 2017). In addition, the signal-to-noise ratio and sensitivity of fluorescent protein-based sensors is normally quite limited (Miyawaki, 2011; Palmer et al., 2011). As a result, sensors for many critical metabolites and signaling molecules have not been created.
Metabolite sensors have also been developed based on RNA riboswitches that can regulate gene expression levels. These metabolite-binding riboswitches have been inserted into transcripts encoding fluorescent proteins (Wachter et al., 2007; Borujeni et al., 2016; Zhou et al., 2016). As a result, changes in metabolite levels lead to changes in cellular fluorescence. However, the temporal resolution of the sensors may be limited because of the time required for nascent fluorescent proteins to mature to a fluorescent form.
We and others recently developed an alternative class of RNA-based genetically encoded sensors. These sensors are based on fluorogenic RNA aptamers, named Spinach and Broccoli (Filonov et al., 2014; Paige et al., 2011). These RNA aptamers can bind and switch on otherwise nonfluorescent fluorophores, such as 4-(3,5-difluoro-4-hydroxybenzylidene)-2-methyl-1-(2,2,2-trifluoroethyl) imidazolinone (DFHBI-1T). Unbound DFHBI-1T is cell-membrane permeable and exhibits nearly undetectable fluorescence under microscope unless bound to a Spinach or Broccoli aptamer (Song et al., 2014). As a result, Spinach and Broccoli has been used as an imaging tag to monitor specific RNAs in living cells.
There are currently two approaches for converting the constitutively fluorescent RNA/DFHBI-1T complex into metabolite sensors, termed Spinach riboswitches (You et al., 2015) and allosteric sensors (Paige et al., 2012). Both types of sensors comprise three domains: fluorogenic Spinach/Broccoli, a transducer, and a target-binding aptamer. Both sensors operate using the same general principle: when the target binds to the target-binding aptamer, there is a conformational change that allows Spinach/Broccoli to fold and bind DFHBI-1T, which leads to fluorescence that can be detected in vitro or in cells. Since aptamers can be generated that selectively bind to a wide spectrum of molecules, including ions, nucleotides, cofactors, small peptides, and large proteins (Stoltenburg et al., 2007), RNA-based sensors could be readily generated against these various biomolecules.
Using these two approaches, RNA-based sensors have been created for the intracellular imaging of S-adenosyl-methionine (SAM), adenosine diphosphate (ADP), thiamine pyrophosphate (TPP), cyclic di-guanylate (c-di-GMP) and several other metabolites (Kellenberger et al., 2013; Paige et al., 2012; Porter et al., 2017; Su et al., 2016; You et al., 2015).
Even though selective RNA-based sensors can be developed in a relatively straightforward manner, this method is limited to target molecules that are present in the micromolar to millimolar range in cells. For these sensors, one target molecule can only activate one RNA sensor, and exhibit maximally one equivalent fluorescence signal. Thus, the target concentration places a limit on the total possible fluorescence that can be achieved. For fluorescence microscopy, micromolar concentrations of folded Spinach/ Broccoli are normally needed for visualization (Zhang et al., 2015). RNA-based biosensors are typically expressed in the low micromolar concentration in E. coli (Paige et al., 2012). In the case of the RNA-based sensor for S-adenosylmethionine, which is present at >100 μM in bacterial cells, enough sensor-SAM complexes form to detect by standard epifluorescence microscopy (Paige et al., 2012). However, for other metabolites and signaling molecules, concentrations are often in the nanomolar to low micromolar range. For example, metabolites such as c-di-GMP, c-GMP-AMP, and homoserine lactones, are found in the cytosol in the low nanomolar range (Hengge, 2009). Thus, even if all of the target molecules in the cytosol were bound to an RNA sensor, there may not be enough fluorescence to detect. Indeed, in the case of c-di-GMP, this metabolite was mainly detected by artificially increasing its concentration by expressing diguanylate cyclase (Kellenberger et al., 2013; Wang et al., 2016). Importantly, this problem also occurs with protein-based biosensors (Christen et al., 2010).
Here we describe RNA-based sensors that detect metabolites that are present in the cell at low concentration. In this approach, RNA-based sensors are expressed as nonfluorescent Broccoli aptamers that are inhibited by intramolecular hybridization that prevents Broccoli folding. The sensor contains a target-activated ribozyme that triggers release of the inhibitory sequence, leading to production of a functional Broccoli aptamer. Target molecules can perform this reaction on multiple sensors, leading to time-dependent accumulation of Broccoli. These RNA-based sensors thus functions as “integrators,” reporting on both target concentration and time. We show that this class of RNA-based biosensor enables detection and quantification of specific target molecules in vitro and in live cells.
RESULTS
Design and Optimization of Ribozyme-activated Broccoli
Ribozymes are catalytic RNAs that are capable of self-cleavage or trans-cleaving complementary RNA strands at specific positions (Ferre-D’Amare and Scott, 2010). We asked if the self-cleavage mechanisms of ribozymes could be used to switch Broccoli from non-fluorescent to fluorescent states. We chose to use Broccoli because, compared to Spinach, it has increased cellular fluorescence and improved folding at the low free magnesium concentrations found in the cytosol of the cell (Filonov et al., 2014).
In brief, in our RNA integrator design, the target molecule induces folding of its cognate aptamer, which then results in the catalytic self-cleavage of ribozyme. By fusing a folding-inhibited form of Broccoli RNA in proximity to the cleavage site, the activation of ribozyme leads to release of Broccoli, allowing it to fold and activate the fluorescence of DFHBI-1T (Figure 1).
Figure 1. Secondary structure and design of RNA integrators.

(A) Schematic of RNA integrators based on the minimal catalytic domain of hammerhead ribozyme (HHR). Shown are the sequences from the Inhibitor 3 complex.
(B) Secondary structure and design of RNA integrators that function in low magnesium levels. The Inhibitor 3 complex (sequences shown) was incorporated as the reporter.
We first examined if hammerhead ribozyme (HHR) (Birikh et al., 1997), a well-characterized natural catalytic RNA, can be used to develop RNA integrator. Allosterically regulated hammerhead ribozymes have been developed previously for in vitro measurements of metabolites (Gu et al., 2012; Soukup and Breaker, 2000; Rueda and Walter, 2006; Soukup et al., 2000) and intracellular gene regulation (Beilstein et al., 2015; Chang et al., 2012; Win and Smolke, 2008). These allosteric ribozyme (Breaker, 2002) or aptazyme sensors (Sefah et al., 2009) have laid the foundation for our current study. The minimal catalytic domain of HHR comprises three duplexed stem regions, H1 – H3 (Figure 1A). The proper folding of all three stem regions is required for the catalytic self-cleavage of HHR. By fusing target-binding aptamers into the H2 stem of HHR via a transducer module, target-induced allosteric activation of HHR can be achieved (Soukup and Breaker, 2000; Soukup et al., 2000).
We wanted to modify this design so that HHR activation would generate a fluorescence signal. Since Broccoli needs to be nonfluorescent prior to HHR activation, we designed the H1 stem to form a duplex that uses sequences that are required for stem formation in Broccoli. This sequence, designated A, can hybridize to the complementary A’ sequence in the H1 stem and can also hybridize with the Broccoli A” sequence, which is a component of a stem that is required for Broccoli folding (Figure 1A). Critically, we designed the A stem sequence so that it is more stable when hybridized to A’ rather than A”. In this way, Broccoli would be non-fluorescent since the A sequence preferentially hybridize with the A’ sequence.
The sensor would be activated if the target binds its cognate aptamer, causing allosteric self-cleavage of HHR. This would release the A strand from its hybridization to A’, which would allow A to form a duplex with A”, and thus reconstitute a critical stem in Broccoli and subsequently bind and activate fluorescence of DFHBI-1T. It should be noted that the A-A’ hybrid is not intrinsically stable since there are relatively few base pairs in this duplex. Instead the hybrid is stable because A and A’ are placed in proximity to each other due to the overall structure of the RNA (Yin and Zhao, 2011). Thus, when the A strand is no longer part of the overall RNA structure due to ribozyme cleavage, the A-A’ strand will readily dissociate due to its thermal instability.
To test this design principle, we first developed an RNA-based integrator sensor for detecting theophylline. RNA aptamers that selectively bind theophylline has been identified previously (Jenison et al., 1994). Additionally, theophylline-regulated HHRs were previously described which allows theophylline to allosterically induce HHR activity (Ketzer et al., 2014; Soukup et al., 2000; Win and Smolke, 2008).
We therefore modified this previous design by adding Broccoli such that the A strand of Broccoli was hybridized to the A’ stem in the HHR (Figure 1A). We synthesized the RNA by in vitro transcription, and then asked if this modified allosteric HHR is regulated by theophylline. Indeed, addition of theophylline (200 μM) induced self-cleavage of HHR, as confirmed using gel electrophoresis (Figure S1).
We next wanted to optimize the performance of the RNA integrator by testing different A’ inhibitor sequences (Figure 2A). We modified the number and composition of nucleotides in the A strand and the complementary A’ strand. In general, we designed the A’ inhibitor strands to be 8- or 9-nucleotides long, which would favor a stable A-A’ duplex prior to cleavage. However, this 8 – 9 bp duplex would no longer form a stable duplex after cleavage since the A stand would no longer be tethered in proximity to the A’ strand by the residual HHR structure. In addition to base composition, we also tested different lengths of the A-A’ duplex relative to the competing A-A” duplex. We tested A-A’ duplex regions that were between 2 – 4 base pairs longer than the corresponding A-A” duplex.
Figure 2. Optimization of the RNA integrator performance.

(A) Optimization of inhibitor sequences. In each case, the A’ inhibitor strand (yellow) forms a duplex with the A strand region (green) in Broccoli. After the target-induced cleavage of HHR, the A strands can instead form duplexes with an A” sequence (grey), which is a component of Broccoli stem required for the fluorescence activation. Shown are mean and SEM values of three independent replicates. The predicted Gibbs free energy change (ΔG) during hybridization switch between the A-A’ duplex and A-A’’ duplex was calculated using mfold online software (Zuker, 2003).
(B and C) Optimization of (B) cyclic di-guanylate-targeting and (c) cyclic adenosine monophosphate-targeting RNA integrators. Sequences of Inhibitor 1 – 5 are indicated in Figure 2A. Shown are mean and SEM values of three independent replicates.
We next asked if the released Broccoli fragment was capable of folding and binding DFHBI-1T. Here, we used Inhibitors 1 – 8 to represent each tested A-A’-A’’ composition (Figure 2A). Indeed, screening results indicated that the various Inhibitors exhibited between 2.5- and 9.9-fold increases in fluorescence at 2 h after the addition of 200 μM theophylline (Figure 2A). Notably, the fold of fluorescence enhancement from the RNA sensor was directly proportional to the various amounts of added theophylline and the percentage of HHR cleavage as indicated by gel analysis (Figure S1). The optimal sensor was seen with Inhibitor 3, which formed eight A-A’ and five A-A’’ Watson-Crick base pairs. These data indicate that the integrator signal can be activated through target-induced self-cleavage of the HHR construct.
Modular Generation of RNA Integrator for Metabolite Detection
We next asked if this RNA integrator design can be used to detect other small molecules. We focused on developing sensors for c-di-GMP and cyclic adenosine monophosphate (cAMP), two second messenger molecules involved in several cell signaling processes. Aptamers that can selectively target these two molecules have been identified and, in the case of c-di-GMP, have been used to make allosteric Spinach sensors (Kellenberger et al., 2013; Wang et al., 2016). To convert these aptamers into RNA integrators, similar to the theophylline sensor, we fused these aptamers into the H2 stem of HHR, and inserted the Broccoli A’ and the A’ inhibitor strand into the H1 region. Here, to test the modularity of the integrator design, we chose the same Inhibitors 1 – 5 which provided reasonable fluorescence enhancement in theophylline detection.
In our test, following the addition of 2 μM c-di-GMP or 1 mM cAMP, a 4.3 – 7.4 fold and 3.4 – 4.9 fold increase in fluorescence was observed with the corresponding RNA integrator (Figure 2B, C). Interestingly, in both cases, similar to the theophylline sensor, the incorporation of the Inhibitor 3 resulted in markedly improved performance (i.e., signal-to-noise ratio) of the c-di-GMP and cAMP integrator sensors. This suggests that sensor performance may be independent of the target-binding aptamer in the H2 stem.
Development of RNA Integrator based on Different Ribozymes
We next wanted to test additional design strategies for RNA integrators. A number of cis-acting self-cleavage RNA ribozymes have been discovered in nature (Ferre-D’Amare and Scott, 2010; Jimenez et al., 2015). The ability to choose different ribozymes can be useful for several reasons. Some aptamers may allosterically activate one ribozyme better than another. Additionally, speed of detection may be important for certain applications, and this may be different if another ribozyme is used. For example, the cleavage rate of twister ribozyme is estimated to be ~1,000/min (Roth et al., 2014), markedly faster than most existing ribozymes, including HHR (~1 – 10/min) (Breaker et al., 2003; Emilsson et al., 2003). Therefore, we wondered if the performance of the RNA integrator can be further optimized by using ribozymes other than the HHR. We tested eight additional classes of natural small cis-acting ribozymes, i.e., glmS, hairpin, hatchet, hepatitis delta virus, pistol, twister, twister sister, and Varkud satellite ribozymes, as potential platforms for engineering RNA integrators. Broccoli A strands and A’ inhibitor strands were fused at the 3’- and 5’-end of each ribozyme, respectively (Figure 3A-E). In most cases, these insertion sites are close to the catalytic cleavage position of the ribozyme. We expect that the activation of ribozyme cleavage will separate Broccoli from the inhibitor strand and induce an enhanced fluorescence signal.
Figure 3. Generation of RNA integrators with diverse natural cis-acting ribozymes.

(A – E) Secondary structure and design of the optimal (A) twister ribozyme integrator, (B) pistol ribozyme integrator, (C) Varkud satellite ribozyme integrator, (D) twister sister ribozyme integrator, and (E) hairpin ribozyme integrator. The integrator is generated by swapping Broccoli-inhibitor A-A’-A’’ complex (green-yellow-grey) into one domain of the ribozyme. Shown are the sequences from the Inhibitor 3 complex. In experiments to test the performance of theophylline-regulated integrator, a theophylline-binding aptamer module (dotted blue) was inserted into the corresponding domain of the ribozyme.
(F) Performance of optimal RNA integrators using each of the six ribozymes. Shown are mean and SEM values of three independent replicates. The fluorescence signal was normalized to that of Broccoli.
To experimentally test this idea, we first studied the constitutive ribozymes without any target-binding aptamer. We wanted to determine if ribozyme cleavage could release Broccoli and induce fluorescence. Since no target-binding aptamer was incorporated into the ribozyme, we added metal ions (i.e., Mg2+ or Co(NH3)63+) to activate cleavage of the ribozyme. In these experiments we again tested the above-mentioned Inhibitor 1 – 4. After in vitro transcription of each of these ribozyme-Broccoli fusions, we measured fluorescence before and after addition of metal ions. In all these cases, an optimal cleavage-induced fluorescence signal was again observed with Inhibitor 3 (Figure S2). Five classes of ribozymes were associated with a 4.4-fold to 9.8-fold enhancement of Broccoli fluorescence relative to background. These ribozymes were the twister ribozyme (Roth et al., 2014), pistol ribozyme (Weinberg et al., 2015), Varkud satellite ribozyme (Saville and Collins, 1990), twister sister ribozyme (Weinberg et al., 2015), and hairpin ribozyme (Walter and Burke, 1998) in the order of smallest to largest fold of signal enhancement.
In our preliminary test, the Broccoli-inhibitor complex could not be constructed from a hatchet ribozyme (Weinberg et al., 2015), hepatitis delta virus ribozyme (Kuo et al., 1988), or glmS ribozyme (Winkler et al., 2004) (Figure S2). Based on the biochemical structural studies and crystal structures (Ferre-D’Amare et al., 1998; Klein and Ferre-D’Amare, 2006; Li et al., 2015), no natural 5’- and 3’-end hybridization-based H1-like stems exist in these three ribozymes. Therefore, an artificially introduced A-A’ duplex may not be functional or could prevent the activity of these ribozymes. To engineer fluorescent reporters for these classes of ribozymes, it might be necessary to perform circular permutation (Su et al., 2016) and place Broccoli and the inhibitor strand in other stem regions of these ribozymes.
Among the five Broccoli-compatible ribozyme classes, we next asked whether they could be converted into theophylline-regulated RNA integrators by inserting a theophylline-binding aptamer module. After analyzing the crystal structure of each ribozyme, a theophylline-targeting aptamer module was fused to the H3 domain of the twister ribozyme, the H4 domain of the pistol ribozyme, the H5 domain of the Varkud satellite ribozyme, the H5 domain of the twister sister ribozyme, and the H4 domain of hairpin ribozyme (Figure 3F). Although the design of the allosteric twister ribozyme follows a previous report (Felletti et al., 2016), the pistol ribozyme has also been recently engineered into riboswitch-like devices (Kobori et al., 2017), the Varkud satellite ribozyme, twister sister ribozyme, and hairpin ribozyme have not previously been made into an allosteric sensor. Using the optimal Broccoli-Inhibitor 3 complex as the reporter, the addition of 200 μM theophylline induced a 2.9-fold to 6.1-fold increase in the fluorescence signal of each in vitro transcribed ribozyme sensor. Taken together, these data indicate that the modular design principle of RNA integrator can be achieved using various ribozyme scaffolds.
Based on its optimal signal-to-noise ratio, subsequent experiments focused on the HHR-based integrator.
In Vitro Characterization of RNA Integrator
In order to determine whether a HHR-based RNA integrator can be used to detect target molecules in cells, we wanted to assess the activation kinetics of the sensor. Based on the gel electrophoresis result, the theophylline-regulated RNA integrator exhibited a rapid apparent cleavage rate constant of ~1.4/min, comparable with known cleavage rate constant of HHR (~1 – 10/min) under similar physiological conditions (Figure 4A and Figure S1). In contrast, the activation rate of fluorescence signal (~0.5/min) appeared to be slower than the self-cleavage rate of HHR (Figure 4A). This delay in the fluorescence activation is likely due to the slow folding of Broccoli (You et al., 2015), while the time needed for the release of the RNA from the A-A’ duplex is likely to also be important. Overall, after adding 200 μM theophylline, half-maximal fluorescence signal of the RNA integrator was reached in ~55 min (Figure 4B). Similarly, 50% of the maximal fluorescence was exhibited in ~69 min and ~85 min upon the addition of 2 μM c-di-GMP or 1 mM cAMP when using the corresponding RNA integrators, respectively (Figure S1). For all these sensors, minimal background fluorescence was observed, as measured by adding vehicle rather than the target molecule, throughout the whole detection period (Figure 4B and Figure S1).
Figure 4. In vitro characterization of the HHR-based RNA integrator.

(A) The apparent cleavage rate constant and fluorescence activation rate constant of theophylline-regulated RNA integrator. The apparent cleavage rate constant was measured in a 6% TBE-urea gel using either RNA integrator or previously reported allosteric theophylline-regulated HHR structure (HHR-theo) (Soukup et al., 2000). Shown are the measured rate constants after adding either 0 μM (open grey circle) or 200 μM theophylline (blue star).
(B) Measurement of the rate of RNA integrator activation. As can be seen, addition of 200 μM theophylline led to rapid signal acquisition (blue), with more than half of the total fluorescence signal appearing in ~55 min. Shown are mean and SEM values of three independent replicates.
(C) Fluorescence accumulation of the RNA integrator during repeated addition-and-removal of the target. A rapid signal increase was observed during the 30 min incubation with 200 μM theophylline (blue star). A gel filtration spin column was then used to remove the theophylline (open grey circle). After 30 min, 200 μM fresh theophylline was added again (blue star). This cycle was repeated three times.
(D – F) Dose-response curve for fluorescence activation of the (D) theophylline, (E) cyclic di-guanylate, and (F) cAMP RNA integrator by target or analogs. Shown are mean and SEM values of three independent replicates. Chemical structures of analogs are drawn with differences from the target indicated in red.
We next wanted to test if the integrator fluorescence is maintained after removing the target. In this experiment, the RNA integrator was incubated with 200 μM theophylline for 1 h, and then the theophylline was removed by a rapid gel filtration step to separate the RNA from the small molecule. Indeed, >95% of the Broccoli signal was retained for the 1 h measurement period after the gel filtration step (Figure S1). In contrast to the previously described Spinach riboswitches (You et al., 2015) or allosteric Spinach sensors (Paige et al., 2012), irreversible self-cleavage of HHR allows the sensor to function as an integrator that can allow a single target molecule to produce multiple fluorescent Broccoli-DFHBI-1T complexes.
To test if each target metabolite can bind and induce cleavage of multiple copies of the integrator sensor, we next wanted to characterize the turnover efficiency of the c-di-GMP RNA integrator. In our study, the initial cleavage rate was measured after mixing 200 nM c-di-GMP with 0.5 – 5 μM RNA integrator in a gel electrophoresis assay (Figure S1). The turnover number, k’cat, can then be calculated based on an Eadie-Hofstee diagram (Hofstee, 1959) by considering c-di-GMP as the catalyst and the RNA integrator as the substrate. Our results indicated that c-di-GMP turned over at a rate of 0.22 min−1 in activating the RNA integrator. Half of the maximum reaction rate, KM’, was exhibited at 0.97 μM RNA integrator. These results indicate that each target molecule can indeed activate multiple RNA integrators, resulting in an amplified signal due to the generation of multiple copies of Broccoli per target molecule.
As a mimic of the cellular cycles of generation-and-degradation of metabolites, we next asked if this sensor can integrate fluorescence during the repeated addition-and-removal of a target theophylline. In our test, the RNA integrator was incubated with 200 μM theophylline for 30 min, and then theophylline was removed by rapid gel filtration. After 30 min of target-free incubation, 200 μM fresh theophylline was added again. The whole process was repeated three times. As shown in Figure 4C, the re-activation of fluorescence signal was indeed observed following the repeated addition of theophylline. The overall level of fluorescence increase is similar as that during the continuous 90 min incubation with the same concentration of theophylline (Figure 4B). Thus, the RNA integrator can record the dynamic changes in the levels of small molecules by accumulating fluorescence during periods when the small molecule is present.
To further study the sensitivity of this approach, we next measured the dose-response curve for each target molecule with the optimized RNA integrators. After 2 h incubation, the measured half-maximal fluorescence was reached at 12 μM, 150 nM, and 58 μM for theophylline, c-di-GMP, and cAMP, respectively (Figure 4D-F). In addition, if we define the dynamic range as the target concentration that can induce 10% – 90% of maximal fluorescence, a large dynamic range of three orders of magnitude was observed for these RNA integrators. 0.3 – 250 μM theophylline, 6 – 3,500 nM c-di-GMP, and 2 – 850 μM cAMP can be measured with the corresponding integrator (Figure 4D-F). Traditional genetically encoded RNA or fluorescent protein sensors usually exhibit a dynamic range of two orders of magnitude (Miyawaki, 2011; Paige et al., 2012; Palmer et al., 2011; You et al., 2015). RNA integrators can therefore be used to detect a broader concentration range of the target analytes, including those at relatively low levels.
Lastly, we asked if the target specificity of aptamers has been conserved after their incorporation into the RNA integrator. To test this, we incubated these RNA sensors with analogs that are structurally similar to the target analytes. Indeed as shown in Figure 4D-F, these non-target molecules show minimal binding to the aptamers and induce nearly undetectable levels of fluorescence. The RNA integrator retains the high target selectivity that is present in the aptamer domain (Jenison et al., 1994; Koizumi et al., 1999; Sudarsan et al., 2008). In sum, these different experiments suggest that RNA integrators can selectively and sensitively detect target analytes.
An HHR-based RNA Integrator that Detects Target Molecules in Low Magnesium
We next wanted to develop an HHR-based integrator sensor to detect target molecules in the cytoplasm of bacterial cells. The minimal conserved domain of HHR requires at least 10 mM Mg2+ for active cleavage (Dahm and Uhlenbeck, 1991). As a result, the integrators described above could have limited performance under physiological free Mg2+ levels (0.25 – 5 mM) (Grubbs, 2002; Romani, 2013). We therefore took advantage of a modification to the minimal HHR which allows it to function at lower Mg2+ levels (Khvorova et al., 2003). These studies showed that introducing an internal loop into the H1 stem to form a pseudoknot with the H2 loop allows efficient self-cleavage of HHR to occur at low Mg2+ levels (Figure 1B).
To test if the Mg2+ dependence of RNA integrator can be reduced using this modified sequence, we engineered a pseudoknot-containing RNA integrator. In this design, the target-binding aptamer was inserted into the H3 region of extended HHR to avoid the disruption of the critical H1-H2 loop-loop interactions. Again, Broccoli, together with the five tested inhibitor strands (Inhibitor 1 – 5), was incorporated into the extended H1 stem region.
We developed three pseudoknot-containing RNA integrators to target theophylline, c-di-GMP, and thiamine 5’-pyrophosphate (TPP). After the addition of each target, fluorescence increase was readily observed in solutions containing 1 mM Mg2+ (Figure S3). In contrast, a similar fluorescence increase was only observed when ≥10 mM Mg2+ was added to RNA integrators that utilized the minimal HHR domain (Figure 4). The optimal signal-to-noise ratio was again seen using Inhibitor 3. With this optimal Broccoli-inhibitor complex, after adding 200 μM theophylline, 2 μM c-di-GMP, and 100 μM TPP, the corresponding RNA integrators exhibited a 3.9-fold, 3.5-fold, and 5.4-fold increases in fluorescence during the 2 h incubation time (Figure S3). The target-selectivity of the original aptamer in these integrators were also retained (Figure S3). Overall, these data indicate that the modified pseudoknot-containing HHR-based RNA integrators function efficiently at the lower magnesium concentrations expected to occur in physiological settings.
Live-Cell Imaging of Small Molecules with RNA Integrators
We next used the pseudoknot-containing HHR-based RNA integrator to image analytes in living E. coli cells. Using theophylline as an example, plasmids expressing these RNA integrators were transformed into BL21 (DE3)* cells. Theophylline is permeable through bacterial cell membranes (Ketzer et al., 2014; Win and Smolke, 2008). After adding 0.2 – 2,000 μM theophylline to the RNA integrator-expressing E. coli and incubating for 3 h, the fluorescence of individual cells was quantified by flow cytometry. Under these conditions, we observed a theophylline concentration-dependent 1.3-fold to 13.3-fold increase in the mean cellular fluorescence compared to that in the absence of theophylline (Figure 5A, B). During 3 h incubation, minimal fluorescence signal was observed in these cells without adding theophylline (Figure S4). The measured half-maximal cellular fluorescence was reached after adding 52 μM theophylline. The addition of a wide range of theophylline (2.7 – 680 μM) can be detected based on fluorescence levels that range from 10% – 90% of maximal fluorescence (Figure 5B). The fluorescence increase sigmoidally correlates with the amount of theophylline added to the solution. Our results indicated that the intracellular turnover rate of theophylline-targeted integrator is around 0.018 min−1. Target concentration at which the reaction rate is half of maximum, KM’, is measured to be 160 nM for theophylline (Figure S4). As another control, we constructed the pseudoknot-containing HHR-Broccoli (Table S1) and transformed the plasmid into BL21 (DE3)* cells. In these E. coli cells, the addition of theophylline will not influence the cellular fluorescence activation of HHR-Broccoli (Figure S4). These data indicate that the RNA integrator can be used to measure the cellular levels of target molecules with a large dynamic range.
Figure 5. Metabolite detection in living cells using the RNA integrator.

(A) Theophylline-induced fluorescence activation in E. coli. Cells expressing the theophylline integrator were incubated with 0, 2, 20, or 200 μM theophylline (up to bottom) for 3 h and then fluorescence signal from 5,000 individual cells was quantified in each case by flow cytometry.
(B) Dose-response curve for cellular fluorescence activation of the theophylline integrator. At each concentration, shown are the individual fluorescence values of 1,000 representative cells as measured by flow cytometry after 3 h incubation with theophylline.
(C) Sensitivity of the RNA integrator for the cellular imaging of TPP biosynthesis. We used a previously reported Spinach riboswitch (You et al., 2015) as the control. In this experiment, after culturing in thiamine-free media for 2 h, we added 0, 0.1 μM, or 10 μM thiamine and imaged the cellular fluorescence level after another 3 h incubation. Images are pseudocolored to show the fold increase in fluorescence at each time point relative to 0 min without adding thiamine. Color scale represents 0- to 15.0-fold changes (black to yellow) in fluorescence signal. Scale bar, 5 μm. In addition, flow cytometry was used to quantify the fluorescence of 100,000 individual cells in these cases.
We next asked if the pseudoknot-containing HHR-based RNA integrator design can be used to detect endogenously generated metabolites. We focused on sensors to detect cellular TPP levels. TPP is normally synthesized in bacterial cells from thiamine, a vitamin molecule that is actively transported into cells (Du et al., 2011). In our experiments, RNA integrator-expressing E. coli were first cultured in thiamine-free media for 2 h, and then 10 μM thiamine was added followed by cellular fluorescence measurement with a flow cytometer every 30 min over 3 h (Figure S4A). A time-dependent increase in the mean cellular fluorescence was observed at each time point (Figure S4).
Next, we wanted to determine how the increase in RNA integrator fluorescence corresponds to the actual TPP concentrations in cells. An HPLC assay was used to measure the intracellular TPP level changes over time (Gerrits et al., 1997). By correlating the increase in cellular fluorescence with the measured cellular TPP concentrations, our data indicated that the RNA integrator signal is indeed directly correlated to the TPP accumulation in E. coli (Figure S4).
Lastly we asked if the RNA integrator exhibits improved sensitivity than existing RNA-based sensors. We chose to use a previously reported TPP-targeting Spinach riboswitch (You et al., 2015) as the control. BL21 (DE3)* cells were transformed with the RNA integrator or Spinach riboswitch, respectively, using a pET28c-based vector (Paige et al., 2011). After culturing in thiamine-free media for 2 h, we added 0.1 – 10 μM thiamine and imaged the cellular fluorescence level after another 3 h incubation (Figure 5C). Under these conditions, we observed an enhanced fluorescence signal in cells expressing RNA integrators compared to those with the Spinach riboswitch. Using flow cytometry to quantify the fluorescence of individual cells, the RNA integrator exhibited a 4.7 – 13.2-fold increase in the mean cellular fluorescence. In contrast, a 1.2 – 9.7-fold signal enhancement was observed in cells expressing the Spinach riboswitch. By comparing the mean cellular fluorescence under the same experimental conditions, the RNA integrator is between 6.0-fold and 17.4-fold higher than that of the Spinach riboswitch. Based on an HPLC assay to measure the intracellular TPP levels in these experiments, the RNA integrator can reliably image ~1.0 μM TPP after 3 h incubation, which is more than 8-fold sensitive than the Spinach riboswitch (Figure S4). These data suggest that the RNA integrator can be used to image metabolites at lower cellular concentrations than the non-integrating Spinach riboswitch.
DISCUSSION
Synthetic riboswitches have been engineered to detect metabolites by regulating the cellular expression of fluorescent proteins (Wachter et al., 2007; Borujeni et al., 2016; Zhou et al., 2016). The RNA integrator is an alternative class of purely RNA-based sensor system with target-induced signal amplifications. Without the requirement of sequential transcription, translation, and the maturation of nascent fluorescent proteins, RNA integrators may potentially provide better temporal resolution than protein-based sensors. In addition, RNA integrators are substantially shorter in sequence than riboswitch-based fluorescent protein sensors, and as a result, may be more suitable for viruses or other expression systems where there are limits in the amount of encoded material that can be added to the genome.
RNA integrators are expected to be highly versatile because of the ease with which aptamers can be generated that can bind to target molecules (Stoltenburg et al., 2007). One limitation of this approach is the background fluorescence that is generated by spontaneous ribozyme cleavage in the absence of target molecule. We have identified several sensor designs that reduce this overall background fluorescence. However, it is important to establish background fluorescence in cells lacking the target molecule and to use HPLC or other assays to independently generate a standard curve to interpret how the fluorescence levels correlate to intracellular target concentrations.
Importantly, the limit of sensitivity applies to both RNA-based sensors and protein-based sensors. The integrator approach described here provides a relatively straightforward method to detect intracellular molecules. Allosteric hammerhead ribozymes have been inserted into transcripts encoding fluorescent proteins, thus allowing changes in metabolite levels to result in differences in levels of these transcripts (Beilstein et al., 2015; Chang et al., 2012; Ketzer et al., 2014; Win and Smolke, 2008). RNA integrators have potential advantages over these ribozyme-controlled transcripts. The temporal resolution of these protein sensors can be limited because of the time required for translation and for nascent GFP to mature to a fluorescent form (Beilstein et al., 2015; Chang et al., 2012; Ketzer et al., 2014; Win and Smolke, 2008).
It should be noted that there is a trade-off between high sensitivity and the ease of observing fluctuations in target levels with these integrator sensors. In contrast to the integrator sensors, the previously described Spinach riboswitches (You et al., 2015) and allosteric Spinach sensors (Paige et al., 2012) are reversible, such that a drop in intracellular target levels leads to a drop in fluorescence levels. Integrator sensors instead show a cessation in the rate of signal accumulation when the target levels drop. Thus, to infer the dynamics of intracellular metabolite concentrations, a continuous measurement of fluorescence over time would be needed followed by analysis of the slope in order to infer concentration levels at different time points.
Our results indicate that the various ribozyme-based integrator sensors are also useful for in vitro quantification of metabolites. The lower limit of detection is improved with integrator sensors using allosteric Spinach sensors or Spinach riboswitch sensors. Quantifications can either involve direct measurement of fluorescence or gel staining to detect cleaved products.
Although our approach primarily relied on the hammerhead ribozyme, other ribozymes could be useful for these integrator sensors as well. Indeed, we show that six different classes of ribozymes are capable of producing target-dependent fluorescence signals. Among these, the Varkud satellite ribozyme, twister sister ribozyme, and hairpin ribozyme have not been made into an allosteric sensor. However, the exceptionally high activity of some ribozymes, such as twister (Roth et al., 2014), might make them difficult to use because of their high background fluorescence due to spontaneous cleavage even in the absence of the target molecule.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Mingxu You (mingxuyou@umass.edu).
EXPERIMENTAL MODEL DETAILS
Cell Culture
BL21 (DE3)* E. coli cells were obtained from Thermo Fisher. RNA integrators were expressed in these cells from pET28c vector under the control of a T7 promoter as previously described (Paige et al., 2012). Cells were plated and grown overnight on LB Agar plate containing 50 μg/ml kanamycin, and afterwards single colonies were picked for inoculation overnight in LB medium supplemented with 50 μg/ml kanamycin. All the LB Agar plates were freshly made and stored at 4°C for less than one month.
METHOD DETAILS
Reagents and Equipment
Commercially available reagents were used without further purification. Absorbance spectra were recorded with a Thermo Scientific NanoDrop 2000 spectrophotometer with cuvette capability. Fluorescence emission and kinetics spectra were measured with a Horiba Fluoromax-4C fluorescence spectrometer. HPLC assays were performed on an Agilent 1100 series HPLC with a Zorbax C8 column (4.6 × 150 mm, 5 μm particle size). In-gel fluorescence imaging was recorded using a ChemiDoc MP imager (Bio-Rad). FACS experiments were performed using FACSAria III instrument (BD Biosciences).
Preparation of RNA Sensors and Mutants
Secondary structure prediction was performed using mfold online software (Zuker, 2003). RNA sensors and mutants were prepared by in vitro transcription using single-stranded DNA templates (Oligo Synthesis Resource, Yale School of Medicine) containing the desired sequences or mutations. Templates were PCR-amplified to create double-stranded DNA templates using primers (Integrated DNA Technologies) which included a 5′-T7 promoter sequence as described previously (Paige et al., 2011). Purified PCR products were used as templates for in vitro T7 transcription (Epicentre). To prevent the spontaneous RNA self-cleavage during the in vitro transcription, a concentrated blocker DNA strand (25-fold concentration of the PCR template) that can hybridize with the catalytic core of each ribozyme was added during the process.
In Vitro Fluorescence Measurements
All in vitro RNA properties were measured in 40 mM HEPES buffer at pH 7.4 with 125 mM KCl unless specified. 10 mM MgCl2 was added for integrator measurement with minimal conserved domain of hammerhead ribozyme (HHR). 1 mM MgCl2 was used for pseudoknot-containing HHR-based integrator. All values presented are the average of at least three independent experiments, and error bars indicate SEM values. The dose-response curve for each sensor were determined by measuring the increase in fluorescence as a function of increasing target concentration in the presence of a fixed concentration of RNA sensor (1 μM) and a fixed concentration of DFHBI-1T fluorophore (20 μM).
Fluorescence emission was recorded using the following instrument parameters: excitation wavelength, 470 nm; emission wavelength, 503 nm; slit width, 3 nm. The fluorescence increase was then plotted against target concentration or incubation time and normalized to the maximum intensity. Dose-response curves were determined using a nonlinear regression analysis in GraphPad Prism software and matched by least squares fitting to a standard dose-response model for 1:1 complex.
To measure integrator fluorescence during the repeated addition-and-removal of target theophylline, a solution of RNA sensor (1 μM) and DFHBI-1T (20 μM) was incubated with theophylline (200 μM) for 30 min, and then the solution was transferred to an RNase-free Micro Bio-Spin column (Bio-Rad) containing Bio-Gel P-30. This column was buffer-exchanged with the above buffer without theophylline, and spun at 1,000 × g for 3 min. The flowthrough after gel filtration contains the sensor in a theophylline-free buffer. The fluorescence emission kinetics of the flowthrough was then immediately recorded over a 30-min period. Afterwards, 200 μM fresh theophylline was added to the solution. The whole process was repeated three times. The control experiment (buffer-exchanged columns with buffer containing theophylline) was performed similarly. The fluorescence of the control sample was similar as Figure 4A, not significantly affected by the spin and incubation process.
In Gel Analysis of RNA Integrators
Typically 10 – 200 ng of in vitro transcribed RNA was loaded into a well of precast 6% TBE-Urea Gel (Life Technologies) and ran at 270 V in 1x TBE buffer. RiboRuler low range RNA ladder (Thermo Scientific) was used as a molecular weight size standard. After the gel was run to completion, the gel was washed 3× 5 min with water and then stained for 30 min with SYBR Gold dye (Life Technologies) diluted 1/10,000 in TBE buffer. Then gel was imaged using a ChemiDoc MP (Bio-Rad) with 302 nm excitation and 590/110 nm emission. Gel bands intensities were quantified in Image Lab 5.0 software (Bio-Rad).
HPLC Measurement of TPP Aggregation
Cells transformed with TPP-targeting RNA integrators were grown overnight in LB-Kan. Cultures were then diluted to OD600 = 0.1 in 100 mL LB-Kan and grown to OD600 = 0.4. Cells were then induced for 2 hours with 1 mM IPTG at 37°C with constant shaking. The entire culture was centrifuged for 2 min at 5,000 × g, pellets were resuspended and washed once with M9 media and then resuspended in 300 mL of M9 media containing 200 μM DFHBI-1T and divided into three samples for replicates. After 45 min incubation at 37°C and 1 h incubation at 25°C, 10 μM thiamine or vehicle alone was then added to each sample and cultures were incubated at 25°C. 15 ml aliquots were removed from replicated sample at each time point, centrifuged for 2 min at 5,000 × g, and flash frozen in liquid nitrogen. Cells were thawed on ice and lysed by resuspending the pellet in 500 μl of cold 4% perchloric acid. Insoluble material was then pelleted by spinning at 5,000 × g for 2 min and the supernatant was added with final concentration of 2 mM K3[Fe(CN)6] in 0.56 M NaOH solution for oxidation. The solution was transferred to a dialysis spin column with a 10,000 molecular weight cut off (Amicon) and spun at 10,000 × g for 10 min at 4°C. A 50 μl portion of the flow through was then injected on a Zorbax C8 column (Agilent) on an Agilent 1100 HPLC. Samples were loaded and eluted for 15 min with an aqueous buffer A containing 12% methanol, 1.5% N,N-dimethylformamide, 140 mM K2HPO4, and 0.3 mM tetrabutylammonium hydroxide, which was brought to pH 7.0 with phosphoric acid. After each sample was eluted, the column was washed with a gradient of 0 – 20% buffer B (70% methanol in water) for 10 min prior to the next samples being injected. Fluorescence emission signal was monitored at 454 nm with excitation at 360 nm. TPP standards were used to calibrate TPP levels measured using this assay.
Cloning Sensors for Expression in E. coli
RNA sensors were expressed in E. coli using a pET28c vector (EMD Biosciences). The RNA integrator was PCR amplified with primers containing either BglII or XhoI restriction sites on the 5’- or 3’-end of the sensor sequence, respectively. This PCR product was then cloned into a pET28c-based vector (Filonov et al., 2014) directly upstream of the T7 terminator sequence with BglII and XhoI restriction sites. This expression condition was based on our previous experience and optimization experiments (Filonov et al., 2014).
Live-Cell Imaging of TPP Synthesis
BL21 (DE3)* E. coli cells (Thermo Fisher) were transformed to express the RNA integrator from pET28c under the control of a T7 promoter as previously described (Paige et al., 2012). Control cells were transformed with pET28c. Cells were plated and grown overnight, and afterwards single colonies were picked for inoculation overnight in LB medium supplemented with 50 μg/ml kanamycin. At OD600 = 0.4, 1 mM IPTG was added to the culture and shaking was continued at 37°C for another 2 h. After induction, 100 μl culture was removed, spun down to pellet the culture and then resuspended in 1 ml of pH 6.0 M9 minimal medium. A 200 μl aliquot of resuspended culture was then plated on poly-D-lysine-coated 24-well glass-bottom dishes (MatTek) and incubated for 45 min at 37°C. Adheren t cells were washed twice and then incubated with 200 μM DFHBI-1T in pH 6.0 M9 minimal medium or PBS for 1 h at 25°C. Cells were then treated with various concentrations of thiamine or theophylline. Live fluorescence images were taken with a CoolSnap HQ2 CCD camera through a 60× oil objective mounted on a Nikon TE2000 microscope and analyzed with a NIS-Elements software, as described previously (Paige et al., 2012). The filter set was a sputter-coated filter cube with excitation filter 470/40 nm, dichroic mirror 495 nm (long pass) and emission filter 525/50 nm (Chroma Technology). The background signal for DFHBI-1T alone was determined by imaging cells expressing an empty control vector under identical conditions, and subtracted using the NIS-Elements software.
Flow Cytometry Analysis of RNA Integrator
Cells expressing pET28c-RNA integrator were incubated with 200 μM of DFHBI-1T dye and 0.2 – 2,000 μM theophylline or 0.1 – 10 μM thiamine for 3 h, and then analyzed on FACSAria III instrument (BD Biosciences). In the kinetic study of TPP synthesis, after adding 10 μM thiamine, cellular fluorescence was measured every 30 min for 3 h. The cellular signal was excited with 488 nm laser and their emission was collected using 525/50 nm emission filter. 5,000 to 100,000 events were collected for each sample at each time point. A FlowJo software was used for the data analysis and quantification.
Real-Time PCR Quantification of RNA Integrators
BL21 (DE3)* cells expressing the RNA integrators were grown overnight in LB media at 37°C with 50 μg/ml kanamycin. At OD600 = 0.4, 1 mM IPTG was added to the culture and shaking was continued at 37°C for another 0.5 – 2 h. Total cellular RNAs were then isolated using the TRIzol Reagent (Invitrogen). Meanwhile, standard RNA samples were synthesized by in vitro T7 transcription (Epicentre). After reverse transcribing these RNAs using a ProtoScript® II first strand cDNA synthesis kit by following the manufacturer’s protocol (New England Biolabs), cDNA products were further used for the real-time PCR analysis. The real-time PCR was performed using PowerUp SYBR™ Green PCR Master Mix (Thermo Fisher) in a Bio-Rad C1000 Touch Thermal Cycler. In the PCR cycle, samples were first held at 95 °C for 10 min, followed by 40 cycles of 95°C for 15 s and 58°C for 1 min.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical and quantification details of experiments can be found in the figure legends. Data are presented as of the mean and SEM values from at least three independent experiments. FlowJo Version 10.4.2 was used to determine the distributions of sensor fluorescence in each individual cells.
Supplementary Material
SIGNIFICANCE.
Imaging low-abundance metabolites and signaling molecules in living cells is particularly challenging. Here we describe RNA integrators, a class of genetically encoded metabolite sensors based on ribozymes, a group of naturally occurring catalytic RNAs that are capable of self-cleavage at specific positions. RNA integrators utilize a target-activated ribozyme that triggers the production of a fluorogenic Broccoli aptamer. Broccoli is an RNA sequence that binds and activates the fluorescence of an otherwise nonfluorescent small molecule fluorophore, DFHBI-1T. RNA integrators can produce higher fluorescence signals than conventional RNA-based biosensors since a target molecule can bind multiple RNA integrators to generate multiple Broccoli aptamers. Drawing upon structural insights into the mechanism of self-cleavage in different ribozymes, we show that this approach can be used with various ribozyme classes, revealing different potential RNA integrator designs. Expression of RNA integrators in cells allows metabolite levels to be imaged with high sensitivity and large dynamic range in live bacterial cells. RNA integrators thus provide an alternative approach to detect cellular metabolites in vitro and in living bacterial cells.
Highlights.
A genetically encoded RNA-based sensor is developed for metabolite imaging
Catalytic activation of fluorogenic RNA enables detection of low-abundance target
This approach can be generalized to different ribozyme types
The modular design principle allows cellular detection of various small molecules
ACKNOWLEDGEMENTS
We thank members of the Jaffrey lab for helpful comments and suggestions. This work was supported by NIH grants to S.R.J. (R01 NS064516 and R01 EB010249) and to M.Y. (R01 AI136789).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
S.R.J. is a co-founder of Lucerna Technologies and has equity in this company. Lucerna has licensed commercialization of technology related to Spinach and other RNA–fluorophore complexes.
REFERENCES
- Beilstein K, Wittmann A, Grez M, and Suess B (2015). Conditional control of mammalian gene expression by tetracycline-dependent hammerhead ribozymes. ACS Synth. Biol 4, 526–534. [DOI] [PubMed] [Google Scholar]
- Birikh KR, Heaton PA, and Eckstein F (1997). The structure, function and application of the hammerhead ribozyme. Eur. J. Biochem 245, 1–16. [DOI] [PubMed] [Google Scholar]
- Borujeni AE, Mishler DM, Wang J, Huso W, and Salis HM (2016). Automated physics-based design of synthetic riboswitches from diverse RNA aptamers. Nucleic Acids Res 44, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breaker RR (2002). Engineered allosteric ribozymes as a biosensor components. Curr. Opin. Biotechnol 13, 31–39. [DOI] [PubMed] [Google Scholar]
- Breaker RR, Emilsson GM, Lazarev D, Nakamura S, Puskarz IJ, Roth A, and Sudarsan N (2003). A common speed limit for RNA-cleaving ribozymes and deoxyribozymes. RNA 9, 949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AL, Wolf JJ, and Smolke CD (2012). Synthetic RNA switches as a tool for temporal and spatial control over gene expression. Curr. Opin. Biotechnol 23, 679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen M, Kulasekara HD, Christen B, Kulasekara BR, Hoffman LR, and Miller SI (2010). Asymmetrical distribution of the second messenger c-di-GMP upon bacterial cell division. Science 328, 1295–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahm SC, and Uhlenbeck OC (1991). Role of divalent metal ions in the hammerhead RNA cleavage reaction. Biochem 30, 9464–9469. [DOI] [PubMed] [Google Scholar]
- Du Q, Wang H, and Xie J (2011). Thiamin (vitamin B1) biosynthesis and regulation: a rich source of antimicrobial drug targets? Int. J. Biol. Sci 7, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emilsson GM, Nakamura S, Roth A, and Breaker RR (2003). Ribozyme speed limits. RNA 9, 907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felletti M, Stifel J, Wurmthaler LA, Geiger S, and Hartig JS (2016). Twister ribozymes as highly versatile expression platforms for artificial riboswitches. Nat. Commun 7, 12834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre-D’Amare AR, and Scott WG (2010). Small self-cleaving ribozymes. Cold Spring Harb Perspect. Biol 2, a003574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre-D’Amare AR, Zhou K, and Doudna JA (1998). Crystal structure of a hepatitis delta virus ribozyme. Nature 395, 567–574. [DOI] [PubMed] [Google Scholar]
- Filonov GS, Moon JD, Svensen N, and Jaffrey SR (2014). Broccoli: rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J. Am. Chem. Soc 136, 16299–16308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommer WB, Davidson MW, and Campbell RE (2009). Genetically encoded biosensors based on engineered fluorescent proteins. Chem. Soc. Rev 38, 2833–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrits J, Eidhof H, Brunnekreeft JW, and Hessels J (1997). Determination of thiamin and thiamin phosphates in whole blood by reversed-phase liquid chromatography with precolumn derivatization. Methods Enzymol 279, 74–82. [DOI] [PubMed] [Google Scholar]
- Grubbs RD (2002). Intracellular magnesium and magnesium buffering. Biometals 15, 251–259. [DOI] [PubMed] [Google Scholar]
- Gu H, Furukawa K, and Breaker RR (2012). Engineered allosteric ribozymes that sense the bacterial second messenger cyclic diguanosyl 5’-monophosphate. Anal. Chem 84, 4935–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofstee BHJ (1959). Non-inverted versus inverted plots in enzyme kinetics. Nature 184, 1296–1298. [DOI] [PubMed] [Google Scholar]
- Hengge R (2009). Principles of c-di-GMP signaling in bacteria. Nat. Rev. Microbiol 7, 263–273. [DOI] [PubMed] [Google Scholar]
- Jenison RD, Gill SC, Pardi A, and Polisky B (1994). High-resolution molecular discrimination by RNA. Science 263, 1425–1429. [DOI] [PubMed] [Google Scholar]
- Jimenez RM, Polanco JA, and Luptak A (2015). Chemistry and Biology of Self-Cleaving Ribozymes. Trends Biochem. Sci 40, 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger CA, Wilson SC, Sales-Lee J, and Hammond MC (2013). RNA-based fluorescent biosensors for live cell imaging of second messengers cyclic di-GMP and cyclic AMP-GMP. J. Am. Chem. Soc 135, 4906–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketzer P, Kaufmann JK, Engelhardt S, Bossow S, von Kalle C, Hartig JS, Ungerechts G, and Nettelbeck DM (2014). Artificial riboswitches for gene expression and replication control of DNA and RNA viruses. Proc. Natl. Acad. Sci. U.S.A 111, E554–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khvorova A, Lescoute A, Westhof E, and Jayasena SD (2003). Sequence elements outside the hammerhead ribozyme catalytic core enable intracellular activity. Nat. Struct. Biol 10, 708–712. [DOI] [PubMed] [Google Scholar]
- Klein DJ, and Ferre-D’Amare AR (2006). Structural basis of glmS ribozyme activation by glucosamine-6-phosphate. Science 313, 1752–1756. [DOI] [PubMed] [Google Scholar]
- Kobori S, Takahashi K, and Yokobayashi Y (2017). Deep sequencing analysis of aptazyme variants based on a pistol ribozyme. ACS Synth. Biol 6, 1283–1288. [DOI] [PubMed] [Google Scholar]
- Koizumi M, Soukup GA, Kerr JN, and Breaker RR (1999). Allosteric selection of ribozymes that respond to the second messengers cGMP and cAMP. Nat. Struct. Biol 6, 1062–1071. [DOI] [PubMed] [Google Scholar]
- Kuo MY, Sharmeen L, Dinter-Gottlieb G, and Taylor J (1988). Characterization of self-cleaving RNA sequences on the genome and antigenome of human hepatitis delta virus. J. Virol 62, 4439–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Lunse CE, Harris KA, and Breaker RR (2015). Biochemical analysis of hatchet self-cleaving ribozymes. RNA 21, 1845–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litke JL, You M, and Jaffrey SR (2016). Developing Fluorogenic Riboswitches for Imaging Metabolite Concentration Dynamics in Bacterial Cells. Methods Enzymol 572, 315–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A (2011). Development of probes for cellular functions using fluorescent proteins and fluorescence resonance energy transfer. Annu. Rev. Biochem 80, 357–373. [DOI] [PubMed] [Google Scholar]
- Ni Q, Mehta S, and Zhang J (2018). Live-cell imaging of cell signaling using genetically encoded fluorescent reporters. FEBS J 285, 203–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura Y, Zhou L, Miu A, and Yokobayashi Y (2013). Controlling mammalian gene expression by allosteric hepatitis delta virus ribozymes. ACS Synth. Biol 2, 684–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumoto S (2010). Imaging approach for monitoring cellular metabolites and ions using genetically encoded biosensors. Curr. Opin. Biotechnol 21, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige JS, Nguyen-Duc T, Song W, and Jaffrey SR (2012). Fluorescence imaging of cellular metabolites with RNA. Science 335, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige JS, Wu KY, and Jaffrey SR (2011). RNA mimics of green fluorescent protein. Science 333, 642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AE, Qin Y, Park JG, and McCombs JE (2011). Design and application of genetically encoded biosensors. Trends Biotechnol 29, 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter EB, Polaski JT, Morck MM, and Batey RT (2017). Recurrent RNA motifs as scaffolds for genetically encodable small-molecule biosensors. Nat. Chem. Biol 13, 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani AM (2013). Magnesium homeostasis in Mammalian cells. Met. Ions Life Sci 12, 69–118. [DOI] [PubMed] [Google Scholar]
- Roth A, Weinberg Z, Chen AG, Kim PB, Ames TD, and Breaker RR (2014). A widespread self-cleaving ribozyme class is revealed by bioinformatics. Nat. Chem. Biol 10, 56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda D, and Walter NG (2006). Fluorescent energy transfer readout of an aptazyme-based biosensor. Methods Mol. Biol 335, 289–310. [DOI] [PubMed] [Google Scholar]
- Saville BJ, and Collins RA (1990). A site-specific self-cleavage reaction performed by a novel RNA in Neurospora mitochondria. Cell 61, 685–696. [DOI] [PubMed] [Google Scholar]
- Sefah K, Phillips JA, Xiong X, Meng L, Van Simaeys D, Chen H, Martin J, and Tan W (2009). Nucleic acid aptamers for biosensors and bio-analytical applications. Analyst 134, 1765–1775. [DOI] [PubMed] [Google Scholar]
- Song W, Strack RL, Svensen N, and Jaffrey SR (2014). Plug-and-play fluorophores extend the spectral properties of Spinach. J. Am. Chem. Soc 136, 1198–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukup GA, and Breaker RR (2000). Allosteric nucleic acid catalysts. Curr. Opin. Struct. Biol 10, 318–325. [DOI] [PubMed] [Google Scholar]
- Soukup GA, Emilsson GA, and Breaker RR (2000). Altering molecular recognition of RNA aptamers by allosteric selection. J. Mol. Biol 298, 623–632. [DOI] [PubMed] [Google Scholar]
- Specht EA, Braselmann E, and Palmer AE (2017). A Critical and Comparative Review of Fluorescent Tools for Live-Cell Imaging. Annu. Rev. Physiol 79, 93–117. [DOI] [PubMed] [Google Scholar]
- Stoltenburg R, Reinemann C, and Strehlitz B (2007). SELEX--a (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng 24, 381–403. [DOI] [PubMed] [Google Scholar]
- Strack RL, and Jaffrey SR (2013). New approaches for sensing metabolites and proteins in live cells using RNA. Curr. Opin. Chem. Biol 17, 651–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Hickey SF, Keyser SG, and Hammond MC (2016). In Vitro and In Vivo Enzyme Activity Screening via RNA-Based Fluorescent Biosensors for S-Adenosyl-l-homocysteine (SAH). J. Am. Chem. Soc 138, 7040–7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudarsan N, Lee ER, Weinberg Z, Moy RH, Kim JN, Link KH, and Breaker RR (2008). Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science 321, 411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachter A, Tunc-Ozdemir M, Grove BC, Green PJ, Shintani DK, and Breaker RR (2007). Riboswitch control of gene expression in plants by splicing and alternative 3’ end processing of mRNAs. Plant Cell 19, 3437–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter NG, and Burke JM (1998). The hairpin ribozyme: structure, assembly and catalysis. Curr. Opin. Chem. Biol 2, 24–30. [DOI] [PubMed] [Google Scholar]
- Wang XC, Wilson SC, and Hammond MC (2016). Next-generation RNA-based fluorescent biosensors enable anaerobic detection of cyclic di-GMP. Nucleic Acids Res 44, e139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg Z, Kim PB, Chen TH, Li S, Harris KA, Lunse CE, and Breaker RR (2015). New classes of self-cleaving ribozymes revealed by comparative genomics analysis. Nat. Chem. Biol 11, 606–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Win MN, and Smolke CD (2008). Higher-order cellular information processing with synthetic RNA devices. Science 322, 456–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler W, Nahvi A, and Breaker RR (2002). Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature 419, 952–956. [DOI] [PubMed] [Google Scholar]
- Winkler WC, Nahvi A, Roth A, Collins JA, and Breaker RR (2004). Control of gene expression by a natural metabolite-responsive ribozyme. Nature 428, 281–286. [DOI] [PubMed] [Google Scholar]
- Yin Y, and Zhao X (2011). Kinetics of dynamics of DNA hybridization. Acc. Chem. Res 44, 1172–1181 [DOI] [PubMed] [Google Scholar]
- You M, Litke JL, and Jaffrey SR (2015). Imaging metabolite dynamics in living cells using a Spinach-based riboswitch. Proc. Natl. Acad. Sci. U.S.A 112, E2756–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Campbell RE, Ting AY, and Tsien RY (2002). Creating new fluorescent probes for cell biology. Nat. Rev. Mol. Cell. Biol 3, 906–918. [DOI] [PubMed] [Google Scholar]
- Zhang J, Fei J, Leslie BJ, Han K, Kuhlman TE, and Ha T (2015). Tandem Spinach array for mRNA imaging in living bacterial cells. Sci. Rep 5, 17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Zheng C, Su J, Chen B, Fu Y, Xie Y, Tang Q, Chou SH, and He J (2016). Characterization of a natural triple-tandem c-di-GMP riboswitch and application of the riboswitch-based dual-fluorescence reporter. Sci. Rep 6, 20871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.