

Abstract
Enantioenriched aldehydes are produced through asymmetric hydroformylation of styrene derivatives using BIBOP type ligands. The featured example is enantioselective synthesis of 4-methyl-3,4-dihydroisocoumarin, which was prepared in a 95.1:4.9 enantiomeric ratio from asymmetric hydroformylation of ethyl 2-vinylbenzoate, then in situ lactonization during reduction process. The conditions are compatible with both electron-rich and electron-poor substituents.
3,4-Dihydroisocoumarins and their derivatives are widely present in nature as key intermediates for the synthesis of biologically active molecules.1 They are regarded as highly attractive compounds and have many interesting activities. Naturally occurring 3,4-dihydroisocoumarins rarely have substitution at the 4-position.2 They, nevertheless, have shown activities in many different disease areas such as antifungal,3 antimicrobial,4 antimalarial,5 and insecticidal 6 (Figure 1).
Figure 1.

Representative 4-substituted 3,4-dihydroisocoumarins
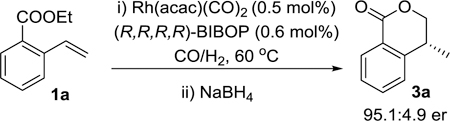
In view of the relevant importance of these molecules, facile and straightforward synthetic routes to 4-substituted 3,4-dihydroisocoumarins are highly desirable. Common methods involve oxidation of methylene groups adjacent to aromatic rings and/or ether oxygen atoms (Scheme 1a),7 which require the use of often toxic oxidation reagents. Most recently, an elegant enantioselective synthesis of 4-methyl-3,4-dihydroisocoumarin was reported by asymmetric hydrogenation of 4-methyl isocoumarin (Scheme 1b).8 Nevertheless, both approaches require pre-construction of the bicyclic ring structure.7,8
Scheme 1.

Synthesis of 4-methyl-3,4-dihydroisocoumarin
Hydroformylation is an industrial process for the production of aldehydes from alkenes via the net addition of a formyl group and a hydrogen atom to a carbon-carbon double bond.9 A number of synthetically useful intermediates can be derived from the resulting aldehydes. Applications to the synthesis of branched chiral aldehydes with a rhodium catalyst have also made much progress in recent years10 with the discovery of effective chiral phosphine ligands including Ph-BPE, Binaphos, Chiraphite, Kelliphite, Yanphos, Duanphos and bis(diazaphospholane) (BDP) ligands.11–16 The emergence of new ligand scaffolds that affect rapid, enantioselective, and regioselective hydroformylation provides new strategies for the efficient synthesis of chiral intermediates which otherwise cannot be accessed readily. Recently we reported that conformationally rigid bisdihydrobenzooxaphosphole ligands (BIBOPs) are highly effective and enantioselective for rhodium-catalyzed asymmetric hydroformylation (AHF) of various mono- and di-substituted alkenes.17 Furthermore, these BIBOP ligands are stable in air and can be isolated as crystalline solid; which make them conveniently synthesized on kilogram scales.18 To apply AHF toward the synthesis of highly functionalized intermediates, we hypothesized that asymmetric hydroformylation of an ortho-ester substituted styrene would produce an in situ cyclized six-membered chiral lactone during reduction of the aldehyde. Herein, we report the direct synthesis of highly enantioenriched α-methyl substituted 4-methyl-3,4-dihydroisocoumarin via asymmetric hydroformylation of ethyl 2-vinylbenzoate, followed by in situ lactonization during a subsequent reduction process.
Commercially available ethyl 2-vinylbenzoate was selected for the study. It can also be accessed through vinylation of aryl bromide.19 The reaction was tested in the presence of 0.5 mol% Rh(acac)(CO)2 and 0.6 mol% of (R,R,R,R)-BIBOP ligand, and syn gas pressure of 15/5 bar of CO/H2 at 60 °C. The starting material fully disappeared as judged by 1H NMR analysis of the reaction mixture after 20 h. A major aldehyde compound was produced in a branched:linear (b/l) ratio of 12:1. The unpurified reaction mixture was then reduced with NaBH4 at 0 °C. The isolated product was confirmed to be 4-methyl 3,4-dihydroisocoumarin 3a with 90% yield and 95.1:4.9 er (Scheme 1c).
The AHF conditions were also applied to other ortho-substituted styrene derivatives (Table 1). Both electron rich and electron deficient styrene derivatives are compatible under the reaction conditions. 2-Fluoro styrene produced a high branched:linear ratio of 21.3:1 with 93.1:6.9 er for compound 2b.20 2-Chloro and 2-methoxy styrenes gave similar selectivity at enantiomeric ratios of 93.9:6.1 and 94.2:5.8, respectively. For CH3 or CF3 substituted styrenes, same enantioselectivity of the aldehydes was obtained at 93:7 er, but with slightly decreased b/l ratios at 4.7:1 and 2.2:1 respectively. The highest b/l ratio was obtained with bis-ortho fluoro-substituted styrene, where >100:1 of b/l ratio was obtained for 2g. The reduced alcohol product 3g was isolated in 94% yield and 94.5:5.5 er. 1-Vinylnaphthalene was hydroformylated in 5.1:1 of b/l ratio (2h) and an enantiomeric ratio of 89.5:10.5.
Table 1.
AHF of the styrene derivatives
 |
The mechanisms of rhodium catalyzed hydroformylation reactions are complex due to potentially reversible steps, steps disproportionately affected by CO or H2 pressure, off-cycle intermediates, and the possibility of multiple selectivity and TOF determining steps.21 A simplified version of the likely pathway is outlined in Scheme 2a. In related systems,21,22 the most catalytically relevant steps are alkene coordination, alkene insertion, and CO insertion. Using a BP86 functional based on a published benchmarking study for related processes,23 alkene insertion was found to be a high energy barrier, whereas the barriers for alkene coordination and CO insertion were considerably lower. Even so, the reaction of styrene with D2/CO gave rise to products incorporating hydrogen instead of deuterium and recovered styrene containing deuterium (see SI), which is consistent with reversibility in the alkene insertion step. Varying the CO pressure (Scheme 2b) revealed a slight increase in enantioselectivity consistent with the barriers to alkene insertion becoming higher, and reducing the reversibility.21d In such a scenario, an energy span analysis is the most accurate protocol to estimate enantioselectivity or regioselectivity (b/l) by taking into account reversibility.21a However, a much simpler analysis using the relative transition state energies for the alkene insertion step has been found to provide a good approximation of these values.21a,23,24
Scheme 2.
Mechanism of enantiodetermining olefin insertion in the asymmetric hydroformylation of styrenea


a Structures in red are calculated. Remaining structures are presented based on reported studies (see refs 21–24). For clarity, only one structure is shown per intermediate/transition state with the exception of the alkene insertion where the lowest energy transition states leading to the three possible isomers are shown.
Thus, the alkene adducts and the alkene insertion step were exhaustively explored with a total of 16 isomers of the latter being possible, eight that could lead to the chiral branched aldehyde and eight to linear the aldehyde. Ultimately, eight unique transition states were found that give the branched aldehyde (Scheme 2). Notably, the ligand was most stable in an axial/equatorial coordination mode and the lowest energy transition states positioned the hydride axial which, in turn, dictates approach of the styrene. The amount of (R)-2-phenylpropanal relative to that of (S)-2-phenylpropanal was calculated via a Boltzmann distribution analysis of these transition states. The results indicate that the model predicts the stereochemical outcome in good to excellent agreement with experiment for three substrates (Table 2). The lowest energy transition state leading to the linear isomer was also located for styrene and when the free energy was compared to that of the lowest energy branched isomer provided a fair estimate of the b/l ratios (expt = 10.2:1, computed = 4.3:1).
Table 2.
Enantioselectivity predictions for the asymmetric hydroformylation of styrene derivatives
 |
Our model and a distortion/interaction analysis25 suggests that steric factors are the dominant control element in the hydroformylation. As shown in the lowest energy transition states leading to each enantiomer (Figure 2), the (R,R,R,R)-BIBOP ligand promotes asymmetric hydroformylation by sterically crowding the substrate in transition states leading to the (S)-enantiomer more significantly than in those leading to the (R)-enantiomer. In Panel A, the (S)-enantiomer approach places the phenyl group of styrene close to the large tert-butyl group on the ligand. Conversely, in Panel B, the (S)-enantiomer approach tilts the phenyl group of styrene away from the tert-butyl group on the ligand, and this structure is consequently more stable by 1.38 kcal/mol. This model also indicates that the position ortho to the phosphorous on the aromatic rings of the ligand comes in close proximity to the substrate and may be used to modulate the selectivity. With methoxy groups in these positions (Table 2, entry 1), slightly higher enantioselectivities were observed, illustrating the potential for exploration of further ligand variants along these lines.
Figure 2.

Three-dimensional representations of the computed lowest energy transition states leading to (S)-2-phenylpropanal (A) and (R)-2-phenylpropanal (B). The red double-headed arrows represent steric interactions. Both structures were calculated using BP86/(cc-pVTZ; Rh: SDD)-IEFPCM-Toluene // BP86/(6–31G*; Rh: LANL2DZ).
In summary, asymmetric hydroformylation is an atom-economical homogenous catalytic process that constructs optically active aldehydes in one step from alkenes in the presence of syn gas with minimal waste generation. BIBOP ligands have been found as effective in the enantioselective Rh-catalyzed asymmetric hydroformylation of styrene derivatives. Enantioenriched 4-methyl-3,4-dihydrocumarine was effectively synthesized from 2-vinylbenzoate using this method followed by reduction. Styrenes with different substituents underwent similar transformations. Calculations reveal a very rigid ligand architecture that reduces conformational freedom and positions the stereogenic tert-butyl groups directly into the reactive space. The stereochemistry was largely controlled by steric effects between the approaching styrene and these tert-butyl groups. Interaction with the arene backone of the ligand, indicate an opportunity for further catalyst engineering to improve outcomes.
EXPERIMENTAL SECTION
General Information.
All reactions were carried out under an atmosphere of argon or nitrogen in dry glassware with magnetic stirring. All commercially available reagents and solvents were used without further purification. 1H and 13C NMR spectra were recorded on a Bruker DRX400 MHz or DRX500 MHz spectrometer with TMS as internal reference. 1H NMR data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, hex = hexet, m = multiplet, br = broad), coupling constant, and integration. High resolution mass spectra (HRMS) were obtained on a Thermo LTQ FT Ultra Mass Spectrometer with heated electrospray ionization (positive), and using linear ion trap with a Fourier Transform ion cyclotron resonance (FTICR) MS detector. Chiral analyses were performed on an Agilent HP 1200 Series HPLC system or on SFC systems.
General Procedure for the Asymmetric Hydroformylation.
In a glovebox filled with nitrogen, ligand (0.012 mmol) and [Rh(acac)(CO)2] (0.01 mmol in 0.4 mL toluene) were added to a 2 mL vial. After stirring for 10 minutes, the substrate (1.0 mmol) and additional solvent were added to bring the total volume of the reaction mixture to 0.5 mL. The vial was transferred into an autoclave and taken out of the glovebox. Hydrogen and carbon monoxide were added sequentially. The reaction mixture was stirred at 60 °C for 20 hours under CO/H2 pressure of 15/5 bar. The reaction was then cooled and the pressure was carefully released in a well-ventilated fume hood. The conversion and branched/linear ratio of this reaction were determined by 1H NMR spectroscopy from the crude reaction mixture. The reaction vial was then placed into an ice-water bath, NaBH4 and MeOH were added, after stirring for 30 min, water was added to quench the reaction. The mixture was extracted with EtOAc 3 times, organic phases were combined and dried with anhydrous Na2SO4 and concentrated under reduced pressure, the residue was chromatographed on silica gel to give the pure alcohol products 3a-h. The enantiomeric excess was determined by HPLC or SFC analysis. For deuterioformylation of styrene, deuterium (5 bar) and carbon monoxide (15 bar) was added sequentially. The reaction mixture was stirred at 60 °C for 5 hours under CO/D2 pressure of 15/5 bar.
Experimental Data of Products.
(R)-4-methylisochroman-1-one (3a).
The title compound was prepared according to the general procedure and purified by column chromatography (hexane/ethyl acetate = 4:1) to give the product as colorless oil: 146.0 mg, 90% yield, 95.1:4.9 er; HPLC Chiralpak AD-3, isocratic: heptane/2-propanol = 85/15, flow rate = 0.5 mL/min; λ = 254 nm, retention time: 6.1 min (major), 7.0 min (minor). NMR data match those reported in the literature.8 1H NMR (500 MHz, CDCl3) δ 8.09 (d, J = 7.8 Hz, 1H), 7.58 (t, J = 7.6 Hz, 1H), 7.39 (t, J = 7.6 Hz, 1H), 7.31 (d, J = 7.7 Hz, 1H), 4.51 (dd, J = 11.0, 4.1 Hz, 1H), 4.24 (dd, J = 10.9, 6.6 Hz, 1H), 3.21 – 3.12 (m, 1H), 1.37 (d, J = 7.2 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 165.1, 144.6, 133.9, 130.4, 127.5, 125.7, 124.4, 72.5, 31.7, 16.7. HRMS (ESI) [M+H]+ m/z calcd for [C10H11O2]+ is 163.0754, found 163.0754.
(R)-2-(2-fluorophenyl)propan-1-ol (3b)
The title compound was prepared according to the general procedure and purified by column chromatography (hexane/ethyl acetate = 4:1) to give the product as colorless oil: 143.4 mg, 93% yield, 93.1:6.9 er; [α]D22 = +13.97 (c = 1.335 in CHCl3) ([α]D25 = +13.5, c = 0.85 in DCM, 95% ee);26 SFC Chiralpak IC-3, 4.6 mm x 150 mm, temperature: 10 °C, A: CO2, B: isopropanol, isocratic: A/B: 98.5/1.5, v/v, flow rate = 2.5 mL/min; λ = 254 nm, retention time: 4.7 min (major), 5.0 min (minor). NMR data match those reported in the literature.26 1H NMR (400 MHz, CDCl3) δ 7.26 – 7.13 (m, 2H), 7.08 (td, J = 7.5, 1.2 Hz, 1H), 7.00 (ddd, J = 10.4, 8.1, 1.2 Hz, 1H), 3.69 (ddd, J = 17.1, 10.6, 6.9 Hz, 2H), 3.26 (hex, J = 6.9 Hz, 1H), 2.06 (br s, 1H), 1.27 (d, J = 7.1 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 161.1 (d, J = 245.0 Hz), 130.6 (d, J = 14.5 Hz), 128.5 (d, J = 5.1 Hz), 127.9 (d, J = 8.4 Hz), 124.2 (d, J = 3.4 Hz), 115.5 (d, J = 22.9 Hz), 67.2, 35.6, 16.6.
(R)-2-(2-chlorophenyl)propan-1-ol (3c).
The title compound was prepared according to the general procedure and purified by column chromatography (hexane/ethyl acetate = 4:1) to give the product as colorless oil: 146.8 mg, 86% yield, 93.9:6.1 er; SFC Chiralpak IC-3, 4.6 mm x 150 mm, temperature: 10 °C, A: CO2, B: isopropanol, isocratic: A/B: 98.5/1.5, v/v, flow rate = 2.5 mL/min; λ = 220 nm, retention time: 7.2 min (major), 7.9 min (minor). NMR data match those reported in the literature.27 1H NMR (400 MHz, CDCl3) δ 7.36 (dd, J = 7.9, 1.2 Hz, 1H), 7.30 – 7.20 (m, 2H), 7.18 – 7.10 (m, 1H), 3.73 (dddd, J = 39.9, 10.8, 6.4, 1.5 Hz, 2H), 3.52 (hex, J = 6.7 Hz, 1H), 1.71 (br s, 1H), 1.28 (d, J = 7.0 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 140.9, 134.3, 129.7, 127.6, 127.6, 127.1, 67.1, 38.1, 16.8.
(R)-2-(2-methoxyphenyl)propan-1-ol (3d).
The title compound was prepared according to the general procedure and purified by column chromatography (hexane/ethyl acetate = 4:1) to give the product as colorless oil: 141.3 mg, 85% yield, 94.2:5.8 er; SFC Chiralpak IC-3, 4.6 × 150 mm: 10 °C, A: CO2, B: isopropanol; isocratic: A/B: 99/1, v/v, flow rate 2.5 mL/min, λ = 220 nm, retention time: 13.0 min (major), 14.5 min (minor). NMR data match those reported in the literature.28 1H NMR (400 MHz, CDCl3) δ 7.25 – 7.16 (m, 2H), 6.98 – 6.90 (m, 1H), 6.87 (d, J = 8.4 Hz, 1H), 3.81 (s, 3H), 3.77 – 3.61 (m, 2H), 3.47 – 3.36 (m, 1H), 1.74 (br s, 1H), 1.25 (d, J = 7.1 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 157.3, 131.8, 127.4, 127.4, 120.8, 110.6, 67.8, 55.4, 35.2, 16.6.
(R)-2-(2-(trifluoromethyl)phenyl)propan-1-ol (3e).
The title compound was prepared according to the general procedure and purified by column chromatography (hexane/ethyl acetate = 4:1) to give the product as colorless oil: 155.2 mg, 76% yield, 93.2:6.8 er; SFC Chiralpak IC-3, 4.6 × 150 mm: 10 ºC, A: CO2, B: isopropanol; isocratic: A/B: 98.5/1.5, v/v, flow rate 2.5 mL/min, λ = 220 nm, retention time: 2.6 min (major), 2.8 min (minor). 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 7.9 Hz, 1H), 7.53 (t, J = 7.5 Hz, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.31 (t, J = 7.6 Hz, 1H), 3.84 – 3.65 (m, 2H), 3.41 (hex, J = 7.0 Hz, 1H), 1.59 (br s, 1H), 1.29 (d, J = 6.9 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 143.2, 132.0, 128.7 (d, J = 6.0 Hz), 127.7, 126.3, 125.9 (d, J = 6.0 Hz), 123.2 (q, J = 271.8 Hz), 67.9, 37.4 (d, J = 1.5 Hz), 18.5. HRMS (ESI) [M+NH4]+ m/z calcd for [C10H15ONF3]+ is 222.1100, found 222.1101.
(R)-2-(o-tolyl)propan-1-ol (3f).
The title compound was prepared according to the general procedure and purified by column chromatography (hexane/ethyl acetate = 4:1) to give the product as pale brown oil: 82.6 mg, 55% yield, 93.0:7.0 er; Aquity UPLC Lux 3µ Amylose-1, 3.0 × 150 mm, 10 °C, A: CO2, B: isopropanol; Isocratic: A/B: 99.7/0.3, v/v, flow rate 2.5 mL/min, λ = 220 nm, retention time: 3.7 min (minor), 4.1 min (major). NMR data match those reported in the literature.26,28 1H NMR (400 MHz, CDCl3) δ 7.20 (m, 1H), 7.18 (d, J = 7.4 Hz, 1H), 7.13 (m, 2H), 3.72 (m, 2H), 3.28 (m, 1H), 2.35 (s, 3H), 1.58 (br s, 1H), 1.24 (d, J = 8.0 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 141.8, 136.4, 130.5, 126.4, 126.3, 125.5, 68.0, 37.2, 19.6, 17.6.
(R)-2-(2,6-difluorophenyl)propan-1-ol (3g).
The title compound was prepared according to the general procedure and purified by column chromatography (hexane/ethyl acetate = 4:1) to give the product as colorless oil: 161.8 mg, 94% yield, 94.5:5.5 er; SFC Chiralpak OD-3, 4.6 × 150 mm: 10 °C, A: CO2, B: isopropanol; isocratic: A/B: 99/1, v/v, flow rate 2.5 mL/min, λ = 220 nm, retention time: 4.0 min (major), 4.6 min (minor). 1H NMR (500 MHz, CDCl3) δ 7.16 – 7.11 (m, 1H), 6.85 – 6.82 (m, 2H), 3.85 – 3.77 (m, 2H), 3.45 (hex, J = 7.2 Hz, 1H), 2.05 (br s, 1H), 1.32 (d, J = 7.2 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 162.8 (d, J = 9.4 Hz), 160.9 (d, J = 9.3 Hz), 127.9 (t, J = 10.6 Hz), 119.0 (t, J = 18.2 Hz), 111.6 (d, J = 5.8 Hz), 111.5 (d, J = 5.8 Hz), 66.0 (d, J = 2.5 Hz), 33.1, 15.7 (t, J = 2.5 Hz). HRMS (ESI) [M+NH4]+ m/z calcd for [C9H14ONF2]+ is 190.1038, found 190.1039.
(R)-2-(naphthalen-1-yl)propan-1-ol (3h).
The title compound was prepared according to the general procedure and purified by column chromatography with 100% dicholoromethane to give the product as colorless oil: 134 mg, 72% yield, 89.5:10.5 er; SFC Chiralpak IC-3, 4.6 × 150 mm: 10 ºC, A: CO2, B: isopropanol; isocratic: A/B: 97/3, v/v, flow rate 2.5 mL/min, λ = 220 nm, retention time: 14.5 min (minor), 15.9 min (major). NMR data matches to those reported.26,28 1H NMR (500 MHz, CDCl3) 1H NMR: (500 MHz, CDCl3): 8.16 (d, J = 8.5 Hz, 1H), 7.88 (d, J = 8.1 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.50 (m, 4 H), 3.95 (m, 1H), 3.89 (m, 2 H), 1.45 (d, J = 6.7 Hz, 3H), 1.37 (br s, 1H). 13C{1H} NMR: (126 MHz, CDCl3): 139.5, 134.0, 131.9, 129.0, 127.1, 126.0, 125.5, 123.05, 123.04, 68.1, 36.4, 17.8.
Supplementary Material
Acknowledgement
We thank the NIH (GM087605 M.C.K.) and Boehringer Ingelheim Pharmaceuticals for financial support. Computational support was provided by XSEDE (TG-CHE120052).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publication website HPLC, NMR spectra and calculation data (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Braca A; Bader A; De Tommasi N Plant and Fungi 3,4-Dihydroisocoumarins: Structures, Biological Activity, and Taxonomic Relationship. Stud. Nat. Prod. Chem 2012, 37, 191–215. [Google Scholar]
- (2).Qi J; Shao C-L; Li Z-Y; Gan L-S; Fu X-M; Bian W-T; Zhao H-Y; Wang C-Y Isocoumarin Derivatives and Benzofurans from a Sponge-Derived Penicillium sp. Fungus. J. Nat. Prod 2013, 76, 571–579. [DOI] [PubMed] [Google Scholar]
- (3).Nozawa K; Yamada M; Tsuda Y; Kawai K; Nakajima S Synthesis of Antifungal Isocoumarins. III. Synthesis and Antifungal Activity of 3-Aryl-3,4-dihydro-4-substituted-isocoumarins. Chem. Pharm. Bull 1981, 29, 3486–3493. [DOI] [PubMed] [Google Scholar]
- (4).Bogdanov MG; Kandinska MI; Dimitrova DB; Gocheva BT; Palamareva MD Preliminary Evaluation of Antimicrobial Activity of Diastereomeric cis/trans-3-Aryl(Heteroaryl)-3,4-dihydroisocoumarin-4-carboxylic Acids. Z. Naturforsch 2007, 62c, 477–482. [DOI] [PubMed] [Google Scholar]
- (5).Cedrón JC; Gutiérrez D; Flores N; Ravelo ÁG; Estévez-Braun A Preparation and antimalarial activity of semisynthetic lycorenine derivatives. Eur. J. Med. Chem 2013, 63, 722–730. [DOI] [PubMed] [Google Scholar]
- (6).Ozoe Y; Kuriyama T; Tachibana Y; Harimaya K; Takahashi N; Yaguchi T; Suzuki E; Imamura K; Oyama K Isocoumarin Derivative as a Novel GABA Receptor Ligand from Neosartorya quadricincta. J. Pestic. Sci, 2004, 29, 328–331. [Google Scholar]
- (7).(a) Zhang Z; Gao Y; Liu Y; Li J; Xie H; Li H; Wang W Organocatalytic Aerobic Oxidation of Benzylic sp3 C–H Bonds of Ethers and Alkylarenes Promoted by a Recyclable TEMPO Catalyst. Org. Lett 2015, 17, 5492–5495. [DOI] [PubMed] [Google Scholar]; (b) Gonzalez-de-Castro A; Robertson CM; Xiao J Dehydrogenative α-Oxygenation of Ethers with an Iron Catalyst. J. Am. Chem. Soc 2014, 136, 8350–8360. (b) [DOI] [PubMed] [Google Scholar]
- (8).Li W; Wiesenfeldt MP; Glorius F Ruthenium−NHC−Diamine Catalyzed Enantioselective Hydrogenation of Isocoumarins. J. Am. Chem. Soc 2017, 139, 2585–2588. [DOI] [PubMed] [Google Scholar]
- (9).Bohnen H-W; Cornils B Hydroformylation of Alkenes: An Industrial View of the Status and Importance. Adv. Catal 2002, 47, 1–64. [Google Scholar]
- (10).For representative reviews, see(a) Klosin J; Landis CR Ligands for Practical Rhodium-Catalyzed Asymmetric Hydroformylation. Acc. Chem. Res 2007, 40, 1251–1259. [DOI] [PubMed] [Google Scholar]; (b) Franke R; Selent D; Börner A Applied Asymmetric Hydroformylation. Chem. Rev 2012, 112, 5675–5732. [DOI] [PubMed] [Google Scholar]; (c) Brezny AC; Landis CR Recent Developments in the Scope, Practicality, and Mechanistic Understanding of Enantioselective Hydroformylation Recent Developments in the Scope, Practicality, and Mechanistic Understanding of Enantioselective Hydroformylation. Acc. Chem. Res 2018, 51, 2344–2354, and references cited therein [DOI] [PubMed] [Google Scholar]
- (11).(a) Whiteker GT; Briggs JR; Babin JE; Barner BA Asymmetric Catalysis Using Bisphosphite Ligands, in Chemical Industries Marcel Dekker: New York, 2003; Vol. 89, 359–367. [Google Scholar]; (b) Babin JE; Whiteker GT Asymmetric Synthesis WO 9303839, 1993.
- (12).Cobley CJ; Gardner K; Klosin J; Praquin C; Hill C; Whiteker GT; Zanotti-Gerosa A; Petersen JL; Abboud KA Synthesis and Application of a New Bisphosphite Ligand Collection for Asymmetric Hydroformylation of Allyl Cyanide. J. Org. Chem 2004, 69, 4031–4040. [DOI] [PubMed] [Google Scholar]
- (13).Yan Y; Zhang X A Hybrid Phosphorus Ligand for Highly Enantioselective Asymmetric Hydroformylation. J. Am. Chem. Soc 2006, 128, 7198–7202. [DOI] [PubMed] [Google Scholar]
- (14).(a) Abrams ML; Foarta F; Landis CR Asymmetric Hydroformylation of Z-Enamides and Enol Esters with Rhodium-Bisdiazaphos Catalysts. J. Am. Chem. Soc 2014, 136, 14583–14588. [DOI] [PubMed] [Google Scholar]; (b) Clark TP; Landis CR; Freed SL; Klosin J; Abboud KA Highly Active, Regioselective, and Enantioselective Hydroformylation with Rh Catalysts Ligated by Bis-3,4-diazaphospholane. J. Am. Chem. Soc 2005, 127, 5040–5042. [DOI] [PubMed] [Google Scholar]
- (15).Wang X; Buchwald SL Synthesis of Optically Pure 2-Trifluoromethyl Lactic Acid by Asymmetric Hydroformylation. J. Org. Chem 2013, 78, 3429–3433. [DOI] [PubMed] [Google Scholar]
- (16).Yu Z; Eno MS; Annis AH; Morken JP Enantioselective Hydroformylation of 1-Alkenes with Commercial Ph-BPE Ligand. Org. Lett 2015, 17, 3264–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tan R; Zheng X; Qu B; Sader CA; Fandrick KR; Senanayake CH; Zhang X Tunable P-Chiral Bisdihydrobenzooxaphosphole Ligands for Enantioselective Hydroformylation. Org. Lett 2016, 18, 3346–3349. [DOI] [PubMed] [Google Scholar]
- (18).(a) Li W; Rodriguez S; Duran A; Sun X; Tang W; Premasiri A; Wang J; Sidhu K; Patel ND; Savoie J; Qu B; Lee H; Haddad N; Lorenz JC; Nummy L; Hossain A; Yee N; Lu B; Senanayake CH The P-Chiral Phosphane Ligand (MeO-BIBOP) for Efficient and Practical Large-Scale Rh-Catalyzed Asymmetric Hydrogenation of N-Acetyl Enamides with High TONs. Org. Process Res. Dev 2013, 17, 1061–1065. [Google Scholar]; (b) Tang W; Qu B; Capacci AG; Rodriguez S; Wei X; Haddad N; Narayanan B; Ma S; Grinberg N; Yee NK; Krishnamurthy D; Senanayake CH Novel, Tunable, and Efficient Chiral Bisdihydrobenzooxaphosphole Ligands for Asymmetric Hydrogenation. Org. Lett 2010, 12, 176–179. [DOI] [PubMed] [Google Scholar]
- (19).Denmark SE; Butler CR Vinylation of Aryl Bromides Using an Inexpensive Vinylpolysiloxane. Org. Lett 2006, 8, 63–66. [DOI] [PubMed] [Google Scholar]
- (20). Asymmetric hydroformylation of o-fluoro styrene using Yanphos ligand was reported to produce 2b in 99:1 er and 10.1:1 b/l ratio in benzene: see ref 13.
- (21).(a) Dingwall P; Fuentes JA; Crawford L; Slawin AMZ; Bühl M; Clarke ML Understanding a Hydroformylation Catalyst that Produces Branched Aldehydes from Alkyl Alkenes. J. Am. Chem. Soc 2017, 139, 15921–15932. [DOI] [PubMed] [Google Scholar]; (b) Brezny AC; Landis CR Unexpected CO Dependencies, Catalyst Speciation, and Single Turnover Hydrogenolysis Studies of Hydroformylation via High Pressure NMR Spectroscopy. J. Am. Chem. Soc 2017, 139, 2778–2785. [DOI] [PubMed] [Google Scholar]; (c) Nelsen ER; Brezny AC; Landis CR Interception and Characterization of Catalyst Species in Rhodium Bis(diazaphospholane)-Catalyzed Hydroformylation of Octene, Vinyl Acetate, Allyl Cyanide, and 1-Phenyl-1,3-butadiene. J. Am. Chem. Soc 2015, 137 (44), 14208–14219. [DOI] [PubMed] [Google Scholar]; (d) Watkins AL; Landis CR Origin of Pressure Effects on Regioselectivity and Enantioselectivity in the Rhodium-Catalyzed Hydroformylation of Styrene with (S,S,S)-BisDiazaphos. J. Am. Chem. Soc 2010, 132, 10306–10317. [DOI] [PubMed] [Google Scholar]
- (22).Gellrich U; Seiche W; Keller M; Breit B Mechanistic Insights into a Supramolecular Self-Assembling Catalyst System: Evidence for Hydrogen Bonding during Rhodium-Catalyzed Hydroformylation. Angew. Chem. Int. Ed 2012, 51, 11033–11038. [DOI] [PubMed] [Google Scholar]
- (23).Silva VD; Dias RP; Rocha WR Insertion reaction of ethylene into the Rh-H bond: A comparative theoretical study. Chem. Phys. Lett 2007, 439, 69–75. [Google Scholar]
- (24).(a) Lei M; Wang Z; Du X; Zhang X; Tang Y Asymmetric Hydroformylation Catalyzed by RhH(CO)2[(R,S)-Yanphos]: Mechanism and Origin of Enantioselectivity. J. Phys. Chem. A 2014, 118, 8960–8970. [DOI] [PubMed] [Google Scholar]; (b) Wassenaar J; de Bruin B; Reek JNH Rhodium-Catalyzed Asymmetric Hydroformylation with Taddol-Based IndolPhos Ligands. Organometalics 2010, 29, 2767–2776. [Google Scholar]; (c) Gleich D; Schmid R; Herrmann WA A Molecular Model To Explain and Predict the Stereoselectivity in Rhodium-Catalyzed Hydroformylation. Organometallics 1998, 17, 2141–2143. [Google Scholar]
- (25).Bickelhaupt FM; Houk KN Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem. Int. Ed 2017, 56, 10070–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Allmendinger S; Hirotaka K; Bernhard B Easily Accessible TADDOL-Derived Bisphosphonite Ligands: Synthesis and Application in the Asymmetric Hydroformylation of Vinylarenes. Adv. Synth. Catal 2015, 357, 41–45. [Google Scholar]
- (27).Cheung LLW; Vasapollo G; Howard A Synthesis of Alcohols via a Rhodium-Catalyzed Hydroformylation - Reduction Sequence using Tertiary Bidentate Amine Ligands. Adv. Synth. Catal 2012, 354, 2019–2022. [Google Scholar]
- (28).Denmark SE; Werner NS On the Stereochemical Course of the Palladium-Catalyzed Cross-Coupling of Allylic Silanolate Salts with Aromatic Bromides. J. Am. Chem. Soc 2010, 132, 3612–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.