

Abstract
Colloidal drug aggregates have been a nuisance in drug screening, yet, because they inherently comprise drug-rich particles, they may be useful in vivo if issues of stability can be addressed. As the first step toward answering this question, we optimized colloidal drug aggregate formulations using a fluorescence-based assay to study fulvestrant colloidal formation and stability in high (90%) serum conditions in vitro. We show, for the first time, that the critical aggregation concentration of fulvestrant depends on media composition and increases with serum concentration. Excipients, such as polysorbate 80, stabilize fulvestrant colloids in 90% serum in vitro for over 48 h. Using fulvestrant and an investigational pro-drug, pentyloxycarbonyl-(p-aminobenzyl) doxazolidinylcarbamate (PPD), as proof-of-concept colloidal formulations, we demonstrate that the in vivo plasma half-life for stabilized colloids is greater than their respective monomeric forms. These studies demonstrate the potential of turning the nuisance of colloidal drug aggregation into an opportunity for drug-rich formulations.
Graphical Abstract
Colloidal aggregation of small molecules is the leading cause of false hits in early screening and drug discovery.1,2 The formation of these self-assembled colloids is characterized by a critical aggregation concentration (CAC), above which small molecules self-assemble into liquid—liquid phase-separated particles.3,4 Colloidal aggregates cause both false-positives in enzyme- and receptor-based assays and false-negatives in cell-based assays.5–7 The formation of colloidal particles has been reported for thousands of compounds (http://advisor.bkslab.org),8 including those from screening libraries and from clinically used drugs, such as anticancer, cardiovascular, and antiretroviral therapeutics.6,9–13
While these aggregates are a serious nuisance for early drug discovery, they have potentially interesting properties as drug formulations. Colloidal aggregates have many aspects desired for delivery as the self-assembly of these compounds leads to defined nanoparticles composed entirely of the active molecule. To be used for drug delivery, the aggregates must be stable in vivo, however, and only a few studies have investigated how colloidal aggregates behave in biological milieus. Doak et al. found that many biopharmaceutics classification system (BCS) class II and IV drugs form colloidal aggregates in simulated intestinal fluid, suggesting colloid formation could play a role in drug formulation and bioavailability.14 Recently, Wilson et al. demonstrated that colloids formed from amorphous solid dispersions can act as reservoirs and enhance plasma drug exposure after oral delivery.15 Frenkel et al. found that orally administered colloid-forming non-nucleoside reverse transcriptase inhibitors were directed to the lymphatic circulation.16 The presence of proteins can further impact colloidal drug transport. For example, Owen et al. demonstrated that colloids do form in standard cell culture conditions (10% serum) and observed that colloidal chemotherapeutics did not cross into cells, resulting in an apparent loss in drug cytotoxicity.17
Efforts to exploit and study colloidal aggregates in high protein milieus have been hindered by their transient stability. Even in biochemical buffers, most small molecule aggregates are only transiently stable, often flocculating and precipitating over several hours. Recently, strategies to stabilize colloidal particles under physiologically relevant conditions have been developed. Co-aggregation with polymeric surfactants, azodyes, or protein coronas all stabilized drug colloids over many days in buffered and serum-containing media.18–20 Colloids of the estrogen receptor antagonist, fulvestrant, and the investigational anthracycline prodrug, pentyloxycarbonyl-(p-amino- benzyl) doxazolidinylcarbamate (PPD), can be stabilized by coaggregation with ultrapure polysorbate 80 (UP80) and poly(d,l-lactide-co-2-methyl-2-carboxytrimethylene carbonate)-g-poly(ethylene glycol) (PLAC-PEG), respectively (Figure S1).20
While these efforts have yielded drug aggregates that are stable under in vitro conditions, their stability in biomimetic, high serum conditions remains unknown, largely because the methods to even measure such stability have been unavailable. We describe a new method to measure the critical aggregation concentration in high-serum content media and demonstrate that fundamental drug colloid properties, such as those that dictate the onset of aggregation, are significantly changed under in vivo-mimetic conditions. We use fulvestrant and PPD as model colloid-forming compounds to further demonstrate that serum-stable colloidal drug aggregates influence in vivo drug circulation properties and increase the plasma half-life compared to monomeric formulations.
RESULTS AND DISCUSSION
Few techniques are available to probe the integrity of amorphous nanostructures in complex media. In biochemical buffers, drug colloids can readily be defined by dynamic light scattering (DLS); however these techniques are ineffective in the presence of serum due to scattering from serum proteins themselves, which is only further complicated as serum content is increased.21 Alternatively, FRET pairs can be absorbed into the self-assembled colloids, where they can report on their gross structural integrity.20,22,23 Accordingly, we designed such a strategy to study colloidal drug aggregate stability in serum-containing media in vitro. Cholesterol-modified BODIPY dyes can be readily incorporated during colloid formation due to the hydrophobic and amorphous nature of drug aggregates.4,20 These dyes have substantial fluorescence intensity within drug colloids but have very low intensity when not associated with a drug aggregate or when colloids are disrupted with detergents (Figure S2). Thus, we investigated the presence and stability of colloidal aggregates of fulvestrant, in high-serum conditions exploiting the fluorescence intensity changes of a BODIPY FRET pair.
We first investigated the effects of dilution and media composition on the critical aggregation concentration of fulvestrant. In protein-free media, many colloid-forming compounds, including fulvestrant, aggregate at low micromolar concentrations, as measured by light scattering (Figure S3).9 To measure the CAC of fulvestrant in serum-containing media, colloids were formulated with 10 μM of a BODIPY FRET pair in PBS with 0.01% UP80 and subsequently diluted 10-fold into media of varying serum content. We used fluorescence intensity to measure the amount of colloids present following dilution, and hence the CAC (Figures 1A and S4). The CAC of fulvestrant increased with serum content (Figure 1B), perhaps owing to serum proteins themselves sequestering drug monomers, bile acids, and other detergent-like molecules in serum.24 This equilibrium shift requires a higher amount of free drug for colloid formation to occur.
Figure 1.
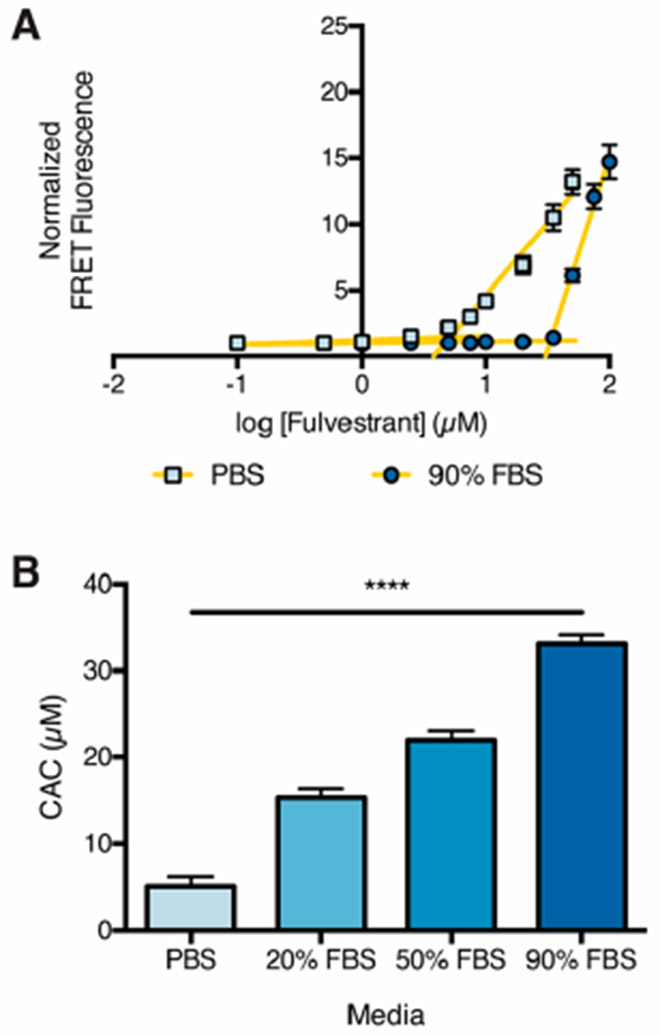
Critical aggregation concentration of fulvestrant in serum-containing media. (A) Fulvestrant colloids with 10 μM BODIPY FRET pair and 0.01% UP80 as a stabilizing excipient were diluted (10-fold) into PBS or 90% FBS. Fluorescence intensity was used to measure the remaining colloids. (B) CAC of fulvestrant in serum-containing media. ****p < 0.0001 between all groups by one-way ANOVA with Tukey’s posthoc (n = 3, mean ± SD).
Drug colloids, which will ordinarily flocculate and precipitate over several hours, require a stabilizing excipient to remain in buffer and serum-containing media for longer times.9 We investigated the role of excipients in stabilizing fulvestrant colloids in high-serum conditions, which mimic the in vivo environment. Here, using the same hydrophobic dyes, which lose fluorescence intensity as they become released when the colloids disassemble or precipitate,20,25 we measured the stability of colloids in complex protein media. Fulvestrant colloids were formulated at 500 μM with 10 μM of the BODIPY FRET pair and a range of UP80 concentrations and subsequently diluted 10-fold into serum-containing media (Figures 2 and S5); this results in a final concentration of 50 μM, which is above the CAC in each of the tested media. Without UP80, colloids quickly disappeared from the liquid phase and were not detectable after 10 h. A low amount of UP80 (0.001% or 0.01%) partially stabilized the colloids, but only a formulation with an initial 0.1% UP80 was stable over the entire 48-h period in the highest amount of serum, with no sign of diminishing signal. At low concentrations of UP80, the surface coverage of the colloid may be insufficient, resulting in flocculation, protein adsorption, colloid destabilization, and eventual precipitation.26,27 These observations are consistent with other studies where nanoparticle PEGylation and PEG surface density influence serum stability.28,29
Figure 2.
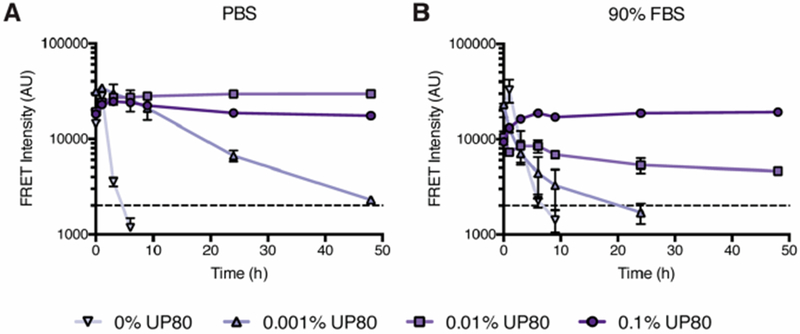
Stability of fulvestrant colloids in buffered solutions and serum-containing media. Fulvestrant (500 μM) colloids with the BODIPY FRET pair (10 μM) and UP80 were diluted (10-fold) into (A) PBS or (B) 90% FBS. Stability was measured over time by monitoring FRET fluorescence. Dashed line indicates baseline fluorescence of dye-only controls (n = 3, mean ± SD).
We hypothesized that serum-stable fulvestrant-UP80 colloids would increase the circulation of fulvestrant compared to a solubilized monomeric form of the drug (Figure 3). As colloid formation can be disrupted by detergents, we used 5% UP80 to yield a fully solubilized, monomeric form of fulvestrant (Figure S6). Tumor-bearing mice were intravenously administered with 6 mg/kg fulvestrant (formulated at 1250 μM) as either stable colloids (with 0.03% UP80) having a diameter of 201 ± 9 nm (PDI = 0.12 ± 0.03, Figure S7) or a monomeric solubilized-drug solution (with 5% UP80). Plasma drug concentrations were quantified at various time points by high performance liquid chromatography with tandem mass spectrometry (HPLC-MS/MS) to obtain a pharmacokinetic profile of fulvestrant (Figure 3A and B). On the basis of a noncompartmental pharmacokinetic analysis, fulvestrant colloids had a plasma half-life (t1/2) that was almost 4 times longer than monomeric fulvestrant (Figure 3C). As only a modest increase in exposure (as seen from the area under the curve, AUC) and a decrease in drug clearance (Cl) was observed, the extended half-life of the colloidal fulvestrant is mainly due to its three-fold increased volume of distribution (Vz). We hypothesize that the UP80-stabilized colloidal fulvestrant likely reduced drug binding to plasma proteins, resulting in increased distribution to other organ tissues. In contrast, in the monomeric form, plasma proteins can sequester free fulvestrant in circulation,30 resulting in a reduced volume of distribution. This is corroborated by the biodistribution data, which show higher tissue accumulation of colloidal fulvestrant (Figure S8).
Figure 3.
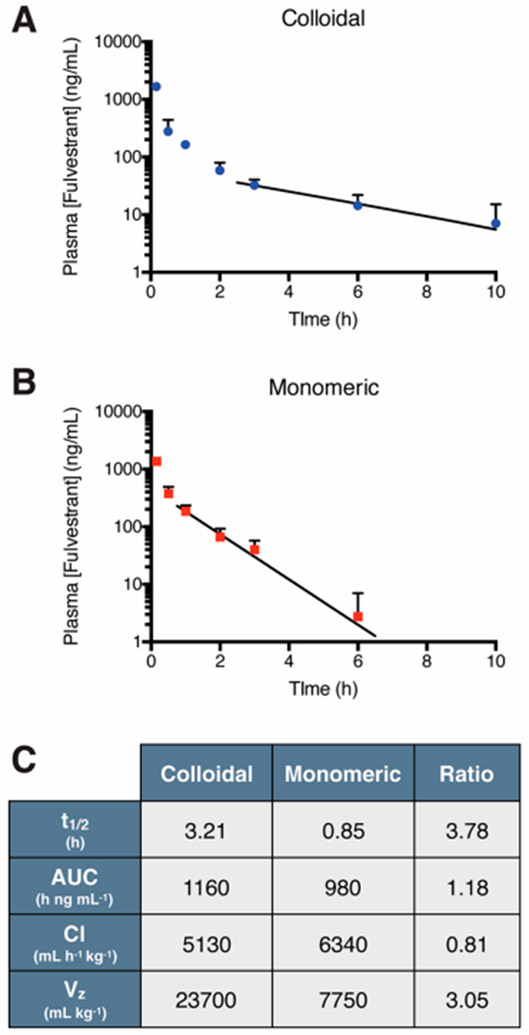
Pharmacokinetic profile of fulvestrant after intravenous administration. Plasma concentration of fulvestrant (initial dose, ID = 6 mg/kg, formulated at 1250 μM) administered as (A) stable colloids (0.03% UP80) or (B) monomer (5% UP80). Trend line denotes exponential decay fitting of lambda elimination phase. (C) Pharmacokinetic parameters of fulvestrant show almost 4-fold increase in drug half-life with colloids compared to monomer (n = 3–6, mean + SD).
We next investigated whether the in vivo PK trends observed for stable fulvestrant colloids could be extended to another colloid-forming compound. We previously developed stabilized colloids of the investigational prodrug, PPD, using an amphiphilic polymer, PLAC–PEG.20 Colloidal (diameter = 230 ± 10 nm; PDI = 0.20 ± 0.01, Figure S9) or monomeric PPD were intravenously delivered to naive NSG mice, and plasma drug concentrations were measured by HPLC-MS/MS (Figure 4A and B). As with fulvestrant, the colloidal formulation of PPD increased plasma half-life over monomeric PPD, here by 2-fold (Figure 4C). For PPD, the increased half-life could be attributed to a 2-fold decrease in drug clearance and a 2-fold increase in drug exposure (AUC), rather than the volume of distribution effect that was dominant for fulvestrant. PPD is a prodrug that is activated by carboxylesterases overexpressed in some human tumors;31-33 however, rodent plasma also has high levels of these enzymes.34,35 Thus, we postulate that PPD–PLAC–PEG colloids limit access of the prodrug to plasma carboxylesterase activity, preventing degradation and accounting for the lower clearance and greater circulation observed.
Figure 4.
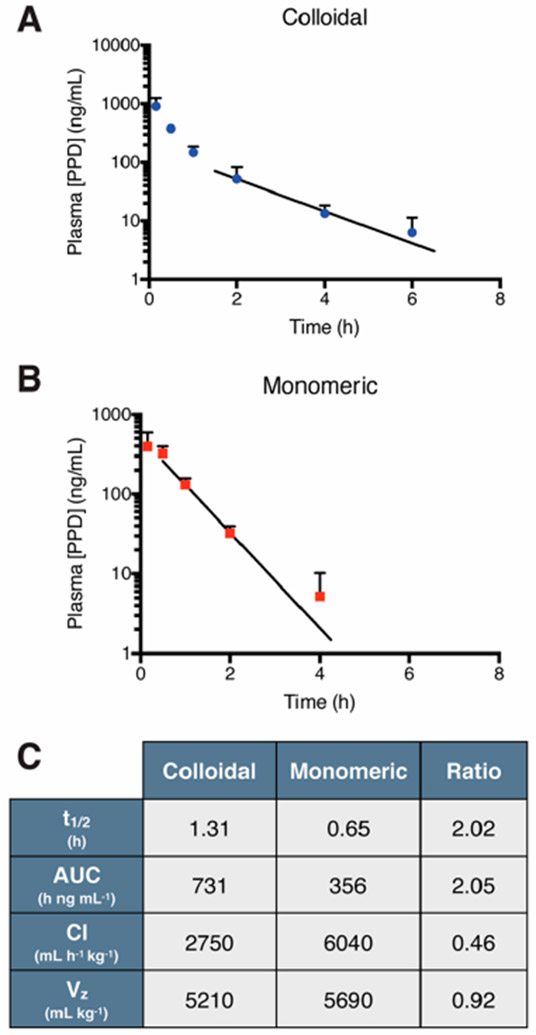
Pharmacokinetic profile of the investigational prodrug PPD. Plasma concentration of PPD (ID = 2 mg/kg, formulated at 500 μM) administered as (A) colloids (0.04% PLAC–PEG) or (B) solubilized monomer (5% UP80). Trend line denotes exponential decay fitting of the lambda elimination phase. (C) Pharmacokinetic parameters of noncompartmental analysis show improvement in colloidal PPD half-life due to decreased clearance, resulting in increased AUC (n = 3–6, mean + SD).
Colloidal drug aggregates have properties that make them intriguing as formulations for drug delivery; they are composed entirely of self-assembled active drug and have nanoscale diameters. To be useful as nanoparticle formulations, however, they must be stable in vivo. Three key observations emerge from this study. First, the critical aggregation concentration of a colloid-forming compound is serum content-dependent. Naturally, for colloids to influence drug fate in vivo, they must be present, and this can only be predicted by CAC measurements in relevant, serum-containing media. To accomplish this, colloidal drug aggregates may be coformulated with excipients, improving their stability in high-serum media from a few hours to several days, likely due to reduced protein adsorption.20 This, in turn, allows them to be injected in vivo where stable colloidal aggregates can extend the circulation half-life over their monomeric counterparts. Second, these studies were enabled by our use of hydrophobic dyes embedded within the colloids to measure colloidal stability. Whereas dynamic light scattering has found wide use in identifying, quantifying, and characterizing colloidal aggregates in buffered solutions,4,21,36 it cannot be used in high-serum media. We observed no difference in the measured CAC of fulvestrant in PBS with or without BODIPY dyes when comparing fluorescence and light scattering methods, indicating minimal impact of the fluorophores on colloid formation. The incorporation of fluorophores may find broad use for characterizing colloidal aggregates in many biological milieus. Third and finally, colloidal aggregation overcomes the major limitation of poor drug loading for many current nanoparticle formulations. While many traditional nanoparticles are limited to loadings of 5–15%,37,38 colloidal drug aggregates offer the opportunity to increase loadings to 50–90%. The stabilized fulvestrant and PPD aggregates, used in this study as proof-of-concept colloidal formulations, have drug loadings of 70% and 50%, respectively. Importantly, these high drug loadings are achieved without chemical modification of the drug. It is the capacity of these drugs to form colloids that results in their high loading. For example, when the PLAC–PEG polymer, used in this study to stabilize PPD, was tested with the noncolloid forming docetaxel,39,40 significantly lower drug loading was achieved. Thus, colloidal aggregation could be combined with conventional micellar and liposomal formulations to increase drug loading while taking advantage of decades of research that has identified many nanocarrier design principles.41,42
Certain caveats bear airing. While the pharmacokinetics demonstrate that colloids can extend the circulation of fulvestrant and PPD, these differences were only observed at the later time points. Thus, there is an opportunity to improve drug pharmacokinetics by enhancing the stability of colloidal drug aggregates earlier in their time course. Our results also suggest that the extent and mechanisms by which stable drug colloids influence pharmacokinetics are dependent on the formulation itself. Drug-dependent processes such as plasma protein adsorption, metabolism, and tissue distribution may contribute to these differences. The two excipients used to stabilize fulvestrant and PPD likely also contribute to the differences observed as they may impact formation of protein coronas, which in turn influence in vivo circulation.28,43 Further studies will be required to determine how the changes to pharmacokinetics observed herein influence drug efficacy and how stable colloidal aggregates of other drug-excipient combinations might behave in vivo.
These caveats should not obscure the principal observations from this work. While the in vitro implications of colloidal aggregates have been intensely studied,4,5,17,44 investigations into the in vivo consequences of small molecule aggregates have been rare. This study suggests that serum-stable colloidal drug aggregates do indeed influence drug fate and furthermore that they may be intentionally designed to do so. It is conceivable that such a colloidal formulation strategy may be adapted to benefit the many drugs that aggregate at relevant concentrations.
MATERIALS AND METHODS
Materials.
Fulvestrant was purchased from SelleckChem, and PPD was synthesized as previously described.32 Ultrapure polysorbate 80 (H2X, UP80) was purchased from NOF America Corporation. Poly(d,l-lactide-co-2-methyl-2-carboxy-trimethylene carbonate)-graft-poly(ethylene glycol) (PLAC–PEG) was synthesized as previously described.29 CholEsteryl BODIPY FL C12 and 543/563 C11 were purchased from Thermo Fisher Scientific. Charcoal-stripped fetal bovine serum was purchased from Wisent Bio Products. RPMI and DMEM-F12 media were purchased from Sigma-Aldrich. Growth factor-reduced Matrigel was purchased from Corning.
Colloid Formation and Stability Studies.
Colloids of fulvestrant and PPD were formed as previously described.20 Briefly, a typical formulation comprised 880 μL of double-distilled water (ddH2O) and 20 μL of drug stock solution (in DMSO) followed by the addition of 10× PBS (100 μL). Drug stock solutions were prepared to obtain the desired final concentration. Excipients were incorporated into ddH2O prior to colloid formation. Fluorescent colloids were prepared by including the FRET pair of CholEsteryl BODIPY dyes (FL C12 and 542/563 C11) in the drug stock solution. On the basis of previous experiments, a final dye concentration of 10 μM and a donor-to-acceptor ratio of 0.875:0.125 was used.20 For CAC measurement and stability experiments, colloids were formulated in PBS, as above, and then diluted 10-fold into various media. Colloids were incubated at 37 °C for the duration of the stability studies.
Colloid Characterization.
Diameter, polydispersity, and normalized scattering intensity were measured using dynamic light scattering (DLS). A DynaPro Plate Reader II (Wyatt Technologies), with a laser width optimized for colloidal aggregate detection by the manufacturer, was used with a 60 mW laser at 830 nm wavelength and a detector angle of 158°. Fluorescence intensity of colloids coformulated with the BOPDIY dye FRET pair was measured using the Tecan Infinite 200 Pro plate reader. The FRET pair was excited at 490 nm, and the acceptor emission was measured at 575 nm.
Morphology of colloids was assessed by transmission electron microscopy where 5 μL of colloid solution was deposited on glow-discharged 400 mesh carbon-coated copper grids (Ted Pella Inc.) and allowed to adhere for 3 min. Excess liquid was removed and grids washed with 5 μL of double-distilled water. Grids were stained with 1% ammonium molybdate (pH 7, 5 μL) for 30 s. After excess stain removal, samples were imaged using a Talos L120C transmission electron microscope operating at 80 kV. Images were captured using a CETA CMOS camera and analyzed using ImageJ software.
Cell Maintenance and Preparation for Xenografts.
Cells were maintained at 37 °C in 5% CO2 in media supplemented with 10% FBS, 10 units/mL penicillin, and 10 μg/mL streptomycin. MCF-7 cells were purchased from ATCC and cultured in DMEM-F12 media. To prepare cells for injection, cells were detached using trypsin-ethylenediamine tetraacetic acid (trypsin-EDTA) followed by pelleting and washing with PBS (three times). MCF-7 cells were resuspended at 108 cells/mL in 50% Matrigel in PBS.
Orthotopic Breast Tumor Model.
Animal study protocols were approved by the University Health Network Animal Care Committee and performed in accordance with current institutional and national regulations. Mice were housed in a 12 h light/dark cycle with free access to food and water. NOD-scid-Il2rgnull (NSG) female mice were bred in-house and received tumor xeno-transplantation at 9-weeks old. Slow release 17β-estradiol pellets (0.72 mg/pellet, 60-day release) were subcutaneously implanted in mice 4 days prior to tumor cell transplantation. For orthotopic mammary fat pad surgeries, mice were anaesthetized with isoflurane-oxygen, and the surgical area was depilated and cleaned with betadine. An incision in the skin of the lower abdomen to the right of the midline was made to reveal the mammary fat pad. Cells were injected into the right inguinal region (50 μL injection, 5 × 106 cells/mouse). The incision was then sutured, and lactate Ringer’s solution and buprenorphine were postoperatively administered for pain management and recovery.
Intravenous Injection.
Tumors were allowed to grow for 3 weeks until palpable (100 mm3). Formulations were injected intravenously using a BD324702 insulin syringe. The volume of injection was based on the weight of the mouse in order to administer the intended dose and no more than 10% of the blood volume.
Pharmacokinetics and Biodistribution Study.
In each study, mice received either a colloidal formulation (as described above) or a monomeric formulation, comprising the same dose of drug solubilized with 5% polysorbate 80. Formulations were administered at the intended dose via tail vein injection. At time points after the injection, blood was drawn via the saphenous vein (<30 μL) into EDTA-coated microcuvette tubes (Sarstead 16.444.100) such that each mouse was not sampled more than three times. At terminal time points, blood was collected by cardiac puncture after CO2 asphyxiation. After collection of blood, tubes were immediately centrifuged, and isolated plasma was flash-frozen. For biodistribution, organ tissues (tumor, heart, lung, liver, kidney, spleen, brain) at terminal time points of PK studies were collected in preweighed vials after rinsing with PBS and flash frozen.
Drug Extraction and Protein Precipitation.
Drug concentrations were determined by HPLC-MS/MS following drug extraction and protein precipitation. Plasma samples (10 μL) were diluted with 10 μL of 1% formic acid solution and 10 μL of internal standard at 10× the final concentration in acetonitrile (ACN) followed by vortexing (10 s, 2 times). Samples were further diluted with 70 μL of ACN followed by a second round of vortexing and then centrifuged at 16 000g for 15 min at 4 °C. The resulting supernatant was collected for analysis by HPLC-MS/MS. Standard curves were prepared in a similar manner with 10 μL of a 10×-concentrated standard solution spiked into blank plasma prior to the first round of vortexing.
To prepare organ tissue, samples were thawed and weighed followed by the addition of 20 zirconia beads (1.0 mm diameter) to facilitate homogenization. To each vial were added 100 μL of 1% formic acid solution and 100 μL of 10×-concentration internal standard solution. Samples were then homogenized for 1 min (2 times) using a bead beater with 1 min on ice between each run. Cold ACN was then added (800 μL) followed by an additional round of homogenization. Samples were centrifuged at 16 000g for 15 min at 4 °C and supernatant collected for analysis. Standard curves were prepared by spiking blank liver tissue with 100 μL of 10×-concentration drug solution.
Drug Quantification by HPLC-MS/MS.
Fulvestrant and PPD concentrations were determined by HPLC-MS/MS. Chromatographic separation was performed using a Waters XTerra C8 column (5 μm) on an Agilent 1100 HPLC equipped with an AB Sciex API 4000 triple quadrupole mass spectrometer with electrospray ionization source detector. Mobile phases of 0.1% formic acid in water (solvent A) and methanol (solvent B) were used. Fulvestrant and PPD standard curves were prepared in blank plasma or liver as described above (0.5–500 ng/mL) using norethindrone or docetaxel (50 ng/mL) as an internal standard, respectively. If necessary, samples were diluted to be within the standard curve with blank plasma or liver that had been spiked with IS solution.
PK and Statistical Analysis.
PK parameters were determined using Phoenix WinNonlin software. Statistical analysis was performed using GraphPad Prism 5.0 software.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Canadian Cancer Society Research Institute (to M.S.S. and B.K.S.), the U.S. National Institutes of General Medical Sciences (GM71630 to B.K.S. and M.S.S.), and the U.S. National Cancer Institute (CA143549 to T.H.K.). A.N.G. and J.L. were supported, in part, by the Natural Sciences and Engineering Research Council Postgraduate Research Scholarships. The authors wish to thank P. Poon for assistance with animal experiments, A. Keating (Princess Margaret Hospital) for help in establishing the mouse tumor model, S. Doyle and B. Calvieri (University of Toronto Microscopy Imaging Laboratory) for assistance with TEM imaging, and members of both Shoichet laboratories for thoughtful discussions.
Footnotes
The authors declare no competing financial interest
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.9b00032.
Additional characterization of formulations in 20% and 50% serum, those used for in vivo studies, and in vivo biodistribution data (PDF)
REFERENCES
- (1).Shoichet BK (2006) Screening in a spirit haunted world. Drug Discovery Today 11, 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Aldrich C, Bertozzi C, Georg GI, Kiessling L, Lindsley C, Liotta D, Merz KMJ, Schepartz A, and Wang S (2017) The Ecstasy and Agony of Assay Interference Compounds. J. Med. Chem. 60, 2165–2168. [DOI] [PubMed] [Google Scholar]
- (3).Coan KE, and Shoichet BK (2008) Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc. 130, 9606–9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ilevbare GA, and Taylor LS (2013) Liquid-Liquid Phase Separation in Highly Supersaturated Aqueous Solutions of Poorly Water-Soluble Drugs: Implications for Solubility Enhancing Formulations. Cryst. Growth Des. 13, 1497–1509. [Google Scholar]
- (5).McGovern SL, Helfand BT, Feng B, and Shoichet BK (2003) A specific mechanism of nonspecific inhibition. J. Med. Chem. 46, 4265–4272. [DOI] [PubMed] [Google Scholar]
- (6).Owen SC, Doak AK, Wassam P, Shoichet MS, and Shoichet BK (2012) Colloidal aggregation affects the efficacy of anticancer drugs in cell culture. ACS Chem. Biol. 7, 1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sassano MF, Doak AK, Roth BL, and Shoichet BK (2013) Colloidal aggregation causes inhibition of G protein-coupled receptors. J. Med. Chem. 56, 2406–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Irwin JJ, Duan D, Torosyan H, Doak AK, Ziebart KT, Sterling T, Tumanian G, and Shoichet BK (2015) An Aggregation Advisor for Ligand Discovery. J. Med. Chem. 58, 7076–7087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ganesh AN, Donders EN, Shoichet BK, and Shoichet MS (2018) Colloidal aggregation: From screening nuisance to formulation nuance. Nano Today 19, 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).McGovern SL, and Shoichet B (2003) Kinase inhibitors: not just for kinases anymore. J. Med. Chem. 46, 1478–1483. [DOI] [PubMed] [Google Scholar]
- (11).Duan D, Doak AK, Nedyalkova L, and Shoichet BK (2015) Colloidal aggregation and the in vitro activity of traditional Chinese medicines. ACS Chem. Biol. 10, 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Almeida e Sousa L, Reutzel-Edens SM, Stephenson GA, and Taylor LS (2015) Assessment of the amorphous “solubility” of a group of diverse drugs using new experimental and theoretical approaches. Mol. Pharmaceutics 12, 484–495. [DOI] [PubMed] [Google Scholar]
- (13).Seidler J, McGovern SL, Doman TN, and Shoichet BK (2003) Indentification and prediction of promiscuous aggregating inhibitors among known drugs. J. Med. Chem. 46, 4477–4486. [DOI] [PubMed] [Google Scholar]
- (14).Doak AK, Wille H, Prusiner SB, and Shoichet BK (2010) Colloid formation by drugs in simulated intestinal fluid. J. Med. Chem. 53, 4259–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wilson V, Lou X, Osterling DJ, Stolarik DF, Jenkins G, Gao W, Zhang GGZ, and Taylor LS (2018) Relationship between amorphous solid dispersion in vivo absorption and in vitro dissolution: phase behavior during dissolution, speciation, and membrane mass transport. J. Controlled Release 292, 172–182. [DOI] [PubMed] [Google Scholar]
- (16).Frenkel YV, Clark AD, Das K, Wang YH, Lewi PJ, Janssen PA, and Arnold E (2005) Concentration and pH dependent aggregation of hydrophobic drug molecules and relevance to oral bioavailability. J. Med. Chem. 48, 1974–1983. [DOI] [PubMed] [Google Scholar]
- (17).Owen SC, Doak AK, Ganesh AN, Nedyalkova L, McLaughlin CK, Shoichet BK, and Shoichet MS (2014) Colloidal drug formulations can explain “bell-shaped” concentration-response curves. ACS Chem. Biol. 9, 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).McLaughlin CK, Duan D, Ganesh AN, Torosyan H, Shoichet BK, and Shoichet MS (2016) Stable Colloidal Drug Aggregates Catch and Release Active Enzymes. ACS Chem. Biol. 11, 992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ganesh AN, McLaughlin CK, Duan D, Shoichet BK, and Shoichet MS (2017) A New Spin on Antibody-Drug Conjugates: Trastuzumab-Fulvestrant Colloidal Drug Aggregates Target HER2-Positive Cells. ACS Appl. Mater. Interfaces 9, 12195–12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ganesh AN, Logie J, McLaughlin CK, Barthel BL, Koch TH, Shoichet BK, and Shoichet MS (2017) Leveraging Colloidal Aggregation for Drug-Rich Nanoparticle Formulations. Mol. Pharmaceutics 14, 1852–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Fischer K, and Schmidt M (2016) Pitfalls and novel applications of particle sizing by dynamic light scattering. Biomaterials 98, 79–91. [DOI] [PubMed] [Google Scholar]
- (22).Gaudin A, Tagit O, Sobot D, Lepetre-Mouelhi S, Mougin J, Martens TF, Braeckmans K, Nicolas V, Desmaele D, de Smedt SC, Hildebrandt N, Couvreur P, and Andrieux K (2015) Transport Mechanisms of Squalenoyl-Adenosine Nanoparticles Across the Blood-Brain Barrier. Chem. Mater. 27, 3636–3647. [Google Scholar]
- (23).Owen SC, Chan DPY, and Shoichet MS (2012) Polymeric micelle stability. Nano Today 7, 53–65. [Google Scholar]
- (24).Krebs H (1950) Chemical composition of blood plasma and serum. Annu. Rev. Biochem. 19, 409–430. [DOI] [PubMed] [Google Scholar]
- (25).Ilevbare GA, Liu H, Pereira J, Edgar KJ, and Taylor LS (2013) Influence of additives on the properties of nanodroplets formed in highly supersaturated aqueous solutions of ritonavir. Mol. Pharmaceutics 10, 3392–3403. [DOI] [PubMed] [Google Scholar]
- (26).Gref R, Luck M, Quellec P, Marchand M, Dellacherie E, Harnisch S, Blunk T, and Muller RH (2000) Stealth” corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf., B 18, 301–313. [DOI] [PubMed] [Google Scholar]
- (27).Riley T, Govender T, Stolnik S, Xiong CD, Garnett MC, Illum L, and Davis SS (1999) Colloidal stability and drug incorporation aspects of micellar-like PLA–PEG nanoparticles. Colloids Surf., B 16, 147–159. [Google Scholar]
- (28).Bertrand N, Grenier P, Mahmoudi M, Lima EM, Appel EA, Dormont F, Lim J-M, Karnik R, Langer R, and Farokhzad OC (2017) Mechanistic understanding of in vivo protein corona formation on polymeric nanoparticles and impact on pharmacokinetics. Nat. Commun. 8, 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Logie J, Owen SC, McLaughlin CK, and Shoichet MS (2014) PEG-Graft Density Controls Polymeric Nanoparticle Micelle Stability. Chem. Mater. 26, 2847–2855. [Google Scholar]
- (30).Zhang F, Xue J, Shao J, and Jia L (2012) Compilation of 222 drugs’ plasma protein binding data and guidance for study designs. Drug Discovery Today 17, 475–485. [DOI] [PubMed] [Google Scholar]
- (31).Barthel BL, Torres RC, Hyatt JL, Edwards CC, Hatfield MJ, Potter PM, and Koch TH (2008) Identification of Human Intestinal Carboxylesterase as the Primary Enzyme for Activation of a Doxazolidine Carbamate Prodrug. J. Med. Chem. 51, 298–304. [DOI] [PubMed] [Google Scholar]
- (32).Barthel BL, Zhang Z, Rudnicki DL, Coldren CD, Polinkovsky M, Sun H, Koch GG, Chan DCF, and Koch TH (2009) Preclinical Efficacy of a Carboxylesterase 2-Activated Prodrug of Doxazolidine. J. Med. Chem. 52, 7678–7688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Barthel BL, Rudnicki DL, Kirby TP, Colvin SM, Burkhart DJ, and Koch TH (2012) Synthesis and Biological Characterization of Protease-Activated Prodrugs of Doxazolidine. J. Med. Chem. 55, 6595–6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Rudakova EV, Boltneva NP, and Makhaeva GF (2011) Comparative Analysis of Esterase Activities of Human, Mouse, and Rat Blood. Bull. Exp. Biol. Med. 152, 73–75. [DOI] [PubMed] [Google Scholar]
- (35).Li B, Sedlacek M, Manoharan I, Boopathy R, Duysen EG, Masson P, and Lockridge O (2005) Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma. Biochem. Pharmacol. 70, 1673–1684. [DOI] [PubMed] [Google Scholar]
- (36).McGovern SL, Caselli E, Grigorieff N, and Shoichet BK (2002) A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 45, 1712–1722. [DOI] [PubMed] [Google Scholar]
- (37).Kumar V, and Prud’homme RK (2008) Thermodynamic limits on drug loading in nanoparticle cores. J. Pharm. Sci. 97, 4904–4914. [DOI] [PubMed] [Google Scholar]
- (38).Chu KS, Schorzman AN, Finniss MC, Bowerman CJ, Peng L, Luft JC, Madden AJ, Wang AZ, Zamboni WC, and DeSimone JM (2013) Nanoparticle drug loading as a design parameter to improve docetaxel pharmacokinetics and efficacy. Biomaterials 34, 8424–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Logie J, Ganesh AN, Aman AM, Al-Awar RS, and Shoichet MS (2017) Preclinical evaluation of taxane-binding peptide-modified polymeric micelles loaded with docetaxel in an orthotopic breast cancer mouse model. Biomaterials 123, 39–47. [DOI] [PubMed] [Google Scholar]
- (40).Ho KS, Aman AM, Al-awar RS, and Shoichet MS (2012) Amphiphilic micelles of poly(2-methyl-2-carboxytrimethylene carbonate-co-D,L-lactide)-graft-poly(ethylene glycol) for anti-cancer drug delivery to solid tumours. Biomaterials 33, 2223–2229. [DOI] [PubMed] [Google Scholar]
- (41).Torchilin VP (2014) Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat. Rev. Drug Discovery 13, 813–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Dawidczyk CM, Kim C, Park JH, Russell LM, Lee KH, Pomper MG, and Searson PC (2014) State-of-the-art in design rules for drug delivery platforms: lessons learned from FDA-approved nanomedicines. J. Controlled Release 187, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Perry JL, Reuter KG, Kai MP, Herlihy KP, Jones SW, Luft JC, Napier M, Bear JE, and DeSimone JM (2012) PEGylated PRINT Nanoparticles: The Impact of PEG Density on Protein Binding, Macrophage Association, Biodistribution, and Pharmacokinetics. Nano Lett. 12, 5304–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Raina SA, Zhang GGZ, Alonzo DE, Wu J, Zhu D, Catron ND, Gao Y, and Taylor LS (2014) Enhancements and limits in drug membrane transport using supersaturated solutions of poorly water soluble drugs. J. Pharm. Sci. 103, 2736–2748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.