

Summary
Transcription factors (TFs) control gene expression by binding DNA recognition sites in genomic regulatory regions. While most forkhead TFs recognize a canonical forkhead (FKH) motif, RYAAAYA, some forkheads recognize a completely different (FHL) motif, GACGC. Bispecific forkhead proteins recognize both motifs, but the molecular basis for bispecific DNA recognition is not understood. We present co-crystal structures of the FoxN3 DNA binding domain bound to the FKH and FHL sites, respectively. FoxN3 adopts a similar conformation to recognize both motifs, making contacts with different DNA bases using the same amino acids. However, the DNA structure is different in the two complexes. These structures reveal how a single TF binds two unrelated DNA sequences and the importance of DNA shape in the mechanism of bispecific recognition.
Keywords: transcription factors, DNA shape, gene regulation, specificity, forkhead, co-crystal structures, DNA binding sites, protein-DNA interactions
Graphical Abstract
eTOC blurb
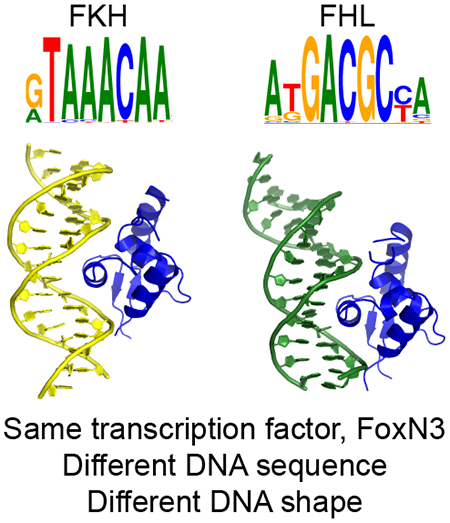
Transcription factors typically bind DNA sites related by sequence similarity. Some forkhead factors bind two classes of DNA sequences, of different length and sequence composition. Rogers et al. show the mechanism of this bispecificity in FoxN3, revealing that DNA conformation differences enable the same protein to bind both sites.
Introduction
Transcription factors (TFs) must accurately distinguish target sites from the rest of the genome in order to properly regulate gene expression. Structural studies have revealed mechanisms used by many TFs to contact specific DNA sequences, largely by hydrogen bonding interactions between amino acid side chains and DNA bases in the major groove (Garvie and Wolberger, 2001; Harrison, 1991). Global structural features of DNA can also be recognized through a shape readout mechanism, involving contacts to the DNA backbone instead of directly to the bases (Otwinowski et al., 1988). TFs can also recognize particular structural features of DNA, such as a narrow minor groove (Rohs et al., 2009).
Forkhead TFs are one of the major TF families in eukaryotes and play prominent roles in development, immunity, metabolism, and cell cycle control (Lam et al., 2013). Within this family, structural studies have revealed how members of this family bind sequences closely matching the canonical forkhead motif, RYAAAYA, and a related lower affinity motif, AHAACA (Boura et al., 2010; Brent et al., 2008; Clark et al., 1993; Li et al., 2017; Littler et al., 2010; Stroud et al., 2006; Tsai et al., 2006, 2007). Forkhead factors share a canonical forkhead DBD, which adopts a winged-helix fold, a modification of the helix-turn-helix DBD, to interact with DNA. In this motif, a three-helix bundle presents the third helix, the recognition helix, into the major groove of DNA. Additionally, two loop structures, known as the wings, make additional DNA contacts, typically to the DNA backbone.
We previously showed that some forkhead proteins can recognize an alternate DNA motif, GACGC, with some individual proteins able to bind both the canonical FKH and alternate FHL motifs (Nakagawa et al., 2013). These two motifs are of different lengths and GC content, and their sequences are divergent enough that there is no clear sequence-based alignment between them; therefore, it is not clear how the documented binding mechanism to the FKH motif could also enable binding to the FHL motif. Previously determined mechanisms of recognition of multiple motifs by a single TF involve binding sequences of the same length that can be aligned (Badis et al., 2009; Gordan et al., 2011; Morgunova et al., 2018). In other cases, dimeric TFs, such as bZIP TFs, can bind motifs comprising very similar or identical DNA half-sites separated by different spacer lengths. Recognition of different DNA sequence motifs can also be achieved by the binding of different sets of fingers, linkers, or flanking regions within a multi-fingered C2H2 zinc finger protein (Siggers et al., 2014). However, the bispecificity observed for forkheads is achieved by a single, monomeric DBD, binding two sites of different lengths and very different sequences, and thus cannot be explained by these mechanisms.
Human FoxN3 was first identified as a suppressor of checkpoint mutations in S. cerevisiae (Pati et al., 1997). It has been shown to act as a transcriptional repressor, and interacts with histone deacetylase complexes involved in the DNA damage response(Busygina et al., 2006; Scott and Plon, 2003, 2005). FoxN3 has also been implicated in craniofacial and eye development, and in regulation of metabolism and the cell cycle (Chang et al., 2005; Huot et al., 2014; Karanth et al., 2016; Markowski et al., 2009; Nagel et al., 2017; Samaan et al., 2010; Schmidt et al., 2011; Schuff et al., 2007; Sun et al., 2016). The molecular mechanisms by which FoxN3 carries out these diverse functions remain unclear.
Here, we show that FoxN3 is a bispecific TF that binds both the FKH and FHL sites in cells. We report the co-crystal structures of the bispecific human protein FoxN3 in complex with both FKH and FHL consensus sequences. The structures reveal that the forkhead DNA binding domain (DBD) adopts remarkably similar structures to contact both motifs, using the same residues to specifically recognize two distinct DNA motifs. However, the shape of the DNA, particularly the bend of the DNA helix, throughout the recognition motif is strikingly different between the structures.
Results
FoxN3 is a bispecific transcription factor
Human FoxN3 is a member of the FoxN forkhead subfamily, which contains bispecific and FHL monospecific TFs (Nakagawa et al., 2013). We assayed the binding specificity of FoxN3 by universal protein binding microarray (PBM), and found that the FoxN3 DBD recognizes both the FKH and FHL motifs (Figure 1a,b). We also measured the binding affinity of FoxN3 to DNA oligonucleotides containing the FKH or FHL sequence, and showed that FoxN3 binds both sequences with mid-nanomolar affinity (Figure 1c). The Kd to the FKH site is 60 ± 20nM, and to the FHL site is 238 ±69nM.
Figure 1:

FoxN3 is a bispecific transcription factor.
The (a) FKH and (b) FHL motifs are bound by FoxN3 in PBM experiments. (c) FoxN3 binds both the FKH and FHL motifs in solution. MicroScale Thermophoresis (MST) measurements of FoxN3 binding to oligonucleotides containing the FKH site (red) or the FHL consensus sequence (blue). Data points show the mean of six measurements, and with error bars show the s.d..
The ability of a forkhead factor to recognize both the FKH and FHL sites in the same cells in vivo has not been reported in prior studies. Therefore, we performed ChIP-Seq experiments on FoxN3 and found that FoxN3 also binds both motifs in HepG2 cells (Figure 2a,b, Figure S1, Table S1). The FKH (P=1×10−21) and FHL (P=1×10−8) motifs are both enriched among the top 2,000 ChIP-seq peaks (Figure 2c), with centralization of both motifs (Figure 2d, e), supporting direct DNA binding of FoxN3 to these motifs within the peaks. 78 of these peaks contained an FHL match and 365 contained an FKH match (1 peak had matches to both motifs).
Figure 2:

FoxN3 recognizes both motifs in cells.
(a,b) Browser shot for a ~40 kb region of chromosome 3 flanking a peak with an FKH binding site (a) or a ~40 kb region of chromosome 15 flanking a peak with an FHL binding site (b). An additional track for ChromHMM genome segmentations is displayed (red regions are annotated as promoters, yellow regions are annotated as enhancers, and green regions are annotated as regions of transcription) (Ernst and Kellis, 2012a). (c) A table summarizing enrichment P-values for FKH or FHL motifs in individual and pooled replicate samples, as well as the percent of the top 2,000 peaks from the pooled sample that contain matches to each motif. (d,e) Composite profile of binding site location within the top 2,000 peaks after scanning with either the FKH (d) or FHL (e) motif. LOESS fits of the data are displayed in black. (See also Figure S1 and Table S1).
FoxN3 adopts the canonical winged helix fold to bind both the FKH and FHL DNA sequences
In order to understand how the forkhead domain contacts these two sites, we determined co-crystal structures of the DBD of FoxN3 in complex with the FKH and, separately, the FHL consensus sequence (Figure 3a,b, Table 1). In both structures, FoxN3 adopts the same overall forkhead winged-helix fold that has been observed for other forkhead proteins (Boura et al., 2010; Brent et al., 2008; Clark et al., 1993; Li et al., 2017; Littler et al., 2010; Stroud et al., 2006; Tsai et al., 2006, 2007). In the FoxN3:FHL structure, two DBDs bind the same DNA in the crystallographic asymmetric unit (Figure S2a). Molecule A directly contacts the FHL motif within the DNA, while molecule B contacts the end of the crystallized DNA sequence, making contacts with a weak match to an FKH site created by formation of a pseudo-continuous DNA helix in the crystal (Figure S2b). Therefore, for the rest of the analysis of the FHL site presented here, we will discuss molecule A of this structure. The root mean square deviation (RMSD) between the forkhead domain in the FKH-bound structure and that in the FHL-bound structure is 0.533 Å. In both structures, there is no observed electron density for wing 1 for amino acid (a.a.) positions 178 to 185, indicating that this wing may be flexible when FoxN3 binds DNA. Wing 2 is partially alpha-helical, with electron density extending to a.a. position 207 in the FKH structure and position 210 in the FHL structure.
Figure 3:

FKH and FHL DNA adopt different structures
(a) FoxN3 (magenta) in complex with DNA containing the FKH motif (yellow): GTAAACA. (b) FoxN3 (blue), in complex with DNA containing the FHL motif (green): GACGC. Alpha helices (α1-α4), wing 1 (w1), and wing 2 (w2) are labeled. The helical axis calculated by Curves+ (Lavery et al., 2009) is shown for both structures. (c) The minor groove of the FHL DNA (green) is much narrower than that of the FKH DNA (yellow) through the core binding site. The DNA molecules from other published structures with FKH sites (PDB codes 1VTN, 2C6Y, 2A07, 3CO6, 3CO7, 3COA, 2UZK, 3L2C, 3G73, and 5X07 (Boura et al., 2010; Brent et al., 2008; Clark et al., 1993; Li et al., 2017; Littler et al., 2010; Stroud et al., 2006; Tsai et al., 2006, 2007)) were aligned with respect to the FoxN3 DBD, and are shown in transparent grey. (d) The bases in the FHL site (green) are positioned differently from those in the FKH (yellow for FoxN3, transparent grey for other published crystal structures) site, enabling fewer bases to cover the same amount of space. (e) Major groove width and (f) helical rise are shown for the FKH (red) and FHL sites (blue). Shape parameters were calculated by Curves+ (Lavery et al., 2009) (See also Figures S2-S3).
Table 1:
Crystallographic Data
| FoxN3:FKH | FoxN3:FHL | |
|---|---|---|
| Data collection | ||
| Space Group | C 1 2 1 | P 21 21 21 |
| Cell dimensions | ||
| a,b,c (Å) | 94.94 102.08 34.45 | 42.54 72.31 102.63 |
| α, β, γ (°) | 90 102.648 90 | 90 90 90 |
| Rmerge | 0.04832 (1.328) | 0.09034 (1.949) |
| <I/σ(I)> | 12.60 (1.09) | 11.66 (0.85) |
| CC1/2 | 0.996 (0.563) | 0.999 (0.34) |
| Completeness (%) | 96.62 (94.47) | 99.16 (98.87) |
| Redundancy | 3.5 (3.5) | 6.5 (6.6) |
| Refinement | ||
| Resolution (Å) | 30.57-2.598 | 39.3 - 2.704 |
| Number of Reflections | 33381 (3307) | 59615(5791) |
| Wilson B-factor (Å) | 85.51 | 81.03 |
| Rwork/Rfree (%) | 23.46/27.06 | 23.37/27.55 |
| Number of non-H atoms | ||
| Macromolecules | 1381 | 2125 |
| Water | 19 | 13 |
| Ligands | 8 | 2 |
| Average B factors (Å2) | ||
| Macromolecules | 136.50 | 76.11 |
| Water | 105.83 | 66.50 |
| Ligands | 134.30 | 73.06 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.004 | 0.003 |
| Bond angles (°) | 0.54 | 0.49 |
| Ramachandran favored (%) | 98.82 | 98.24 |
| Ramachandran allowed (%) | 1.18 | 1.76 |
| Ramachandran outliers (%) | 0.00 | 0.00 |
Each data set was collected from a single crystal. The values in parentheses show the values for the highest resolution shell.
FKH and FHL DNA adopt different shapes in complex with FoxN3
Although the overall protein conformation is similar between the FKH-contacting and FHL-contacting structures, the DNA molecules in the two structures adopt different conformations (Figure 3a,b). The DNA is bent away from the FoxN3 DBD by 22.2° in the FHL structure, but towards the DBD in the FKH structure by 13.5°. This bend towards the protein in the FKH structure is consistent with the DNA conformation in other co-crystal structures of forkhead domains with the FKH motif, but the FHL DNA has a very different conformation from that observed in any other published forkhead co-crystal structure (Boura et al., 2010; Brent et al., 2008; Clark et al., 1993; Li et al., 2017; Littler et al., 2010; Stroud et al., 2006; Tsai et al., 2006, 2007) (Figure 3c, d).
This difference in the DNA shape explains how the same DBD can recognize two motifs of different lengths: the bend in the FHL DNA reduces the number of DNA base pairs that contact the DBD (Figure 4a). The guanines at the 5’ end of both the FKH (G7) and FHL (G10) motifs are in the same position with respect to the protein, as are G12’ in the FKH motif and G14’ in the FHL motif. These guanines can be thought of as ‘registration positions’ that orient the DNA with respect to the protein. Between these two positions, the FKH DNA is bent 6.5° towards the protein, while FHL is bent 9.5° away. In the FKH motif, there are four base pairs between these two positions, while there are three in the FHL motif. In order to accommodate one fewer base in the FoxN3-FHL structure, many aspects of DNA shape are different between this structure and the FoxN3-FKH structure. The minor groove width in the FHL motif is much narrower than that in the FKH motif, averaging 5.90 Å over the FHL motif compared to 7.05 Å over the FKH motif (Figure 3c,e). Additionally, the helical rise (the distance between adjacent base pairs in the DNA helix) is much larger in the FHL structure than in the FKH structure (average of 3.38 Å over the FHL motif compared to 3.17 Å over the FKH motif) (Figure 3d,f). The helical twist is lower in the FKH structure (average 31.4°) than in the FHL structure (average 33.8°), while roll is higher in FKH (average 2.7°) than in FHL (average −0.5°) (Figure S3a,b).
Figure 4:

Contacts between FoxN3 and the FKH and FHL sites
(a) The guanine bases at the end of each DNA strand in both motifs are in the same position with respect to the protein. The helical axis calculated by Curves+ (Lavery et al., 2009) is shown for both structures. (b)Specific contacts between FoxN3 (magenta) and the FKH DNA (yellow) are shown. (c) Contacts between these same amino acids in FoxN3 (blue) and the FHL DNA (green) are shown. (d) The FKH structure (magenta protein, yellow DNA) and the FHL structure (blue protein, green DNA) are aligned to highlight the differences in positions of wing 2. (e) In the FHL structure, wing 2 contacts the DNA backbone through His209. (See also Figure S4).
In the two DNA conformations, the sequence of the intervening bases between the registration positions is very different; these sequences may be partially specified by their propensity to form the required DNA structure, as well as by specific interactions with base-contacting protein side chains. The predicted DNA shape is different from that observed in the crystal structure for both the FKH and FHL sequences, so some deformation from inherent shape must occur to accommodate protein binding (Figure S3c-h). Specific DNA base contacts are largely made through the recognition helix, helix 3, in both structures (Figure 4b,c). In both structures, Arg163 makes bidentate hydrogen bonds to one of the guanine registration positions: G12’ in the FKH structure and G14’ in the FHL structure. The other conserved base-contacting positions in the recognition helix (His164 and Asn160) contact different bases in the two complexes (Figure 4b,c, Figure S4a,b). In both structures, contacts to the DNA backbone are made by the bases of wing 1 and helix 1 (Figure S4c-f).
In contrast, wing 2 adopts different positions in the two structures (Figure 4d). In the FKH structure, wing 2 is angled closer to helix 1, not making any direct ordered contacts to the DNA backbone. In the FHL structure, wing 2 extends towards the DNA backbone and contacts the backbone of G10 through His209 (Figure 4e).
Chimeric proteins reveal the importance of the wings in FHL binding
These observed differences between the structures suggest that the wings may contribute to DNA binding specificity. Therefore, we created chimeric proteins in which we swapped segments of the forkhead DBD between FoxN3 and the FKH monospecific FoxJ3 (Table S2). Replacing the wings of FoxN3 with the wings of FoxJ3 significantly reduced binding to the FHL site, while maintaining binding to the FKH site (Figure 5, Figure S5). Performing these swaps in the context of a different bispecific forkhead protein, FoxN2, also showed the requirement of the wings for FHL binding, indicating that this effect is not limited to FoxN3 (Figure S5). A shorter stretch of 6 amino acids in wing 2, L199-K204, recapitulated the effect of swapping the entire wing 2 sequence. This stretch of amino acids is conserved between the bispecific proteins FoxN2 and FoxN3 (Figure S6), and does not directly contact DNA in either structure. Interestingly, swapping the sequence of the FoxN3 wings into FoxJ3 did not increase FHL binding. These findings reveal that features in the wings of the proteins that bind the FHL motif are necessary, but not sufficient, for binding this motif, indicating allosteric interactions with other regions of the protein.
Figure 5:

Subdomain swap experiments show the involvement of the wings in FHL recognition
(a) Chimeric proteins were designed to test the importance of parts of the forkhead DBD for binding specificity. Positions are numbered with respect to the full-length FoxN3 protein. (b) The location of the swaps on the forkhead structure are shown. (c) Boxplots show PBM E-scores for 8-mers containing the FKH site (GTAAACA) (top panel) or the FHL site (GACGC) (bottom panel) for each chimeric protein. FoxN3 and chimeras of it are shown in blue, and FoxJ3 is shown in red. The FoxN3 boxplot represents the average of three replicate PBM experiments, and N2(J3-wing) and N3(J3-6aa) represent the average of two replicates. Boxplots for individual replicates are shown in Figure S5. (* indicates P<9×10−16, one-sided Mann-Whitney test) (See also Figures S5 and S6 and Table S2).
Given that the repositioned portion of wing 2 is important for DNA binding specificity, we also tested other portions of the domain that adopted different positions in the two structures: helix 4, the short helix preceding the recognition helix, and the N-terminal loop. Neither of these regions affected binding to either the FKH or FHL sequence (Figure S5). The N-terminal loop contacts wing 2 and helix 4, suggesting that combining these swaps may be sufficient to confer FHL binding to FoxJ3 as the wing swap was not sufficient on its own. However, none of the tested combinations of swaps could increase FHL binding, indicating that a more distributed set of amino acids throughout the forkhead domain is required for FHL binding.
Discussion
The structures presented in this work demonstrate the ability of the forkhead DBD to recognize vastly different DNA sequences using the same overall fold and DNA-contacting residues. The FKH and FHL sites are much more dissimilar than other examples of sequences bound by the same DBD (Badis et al., 2009; Gordan et al., 2011). Structures of FoxO1 in complex with the FKH primary site and a related lower affinity motif (AHAACA) showed that this protein also rearranges the interactions of the amino acids in the recognition helix to bind different sequences (Brent et al., 2008). The FoxN3 structures presented here reveal even more flexibility in the interactions of these amino acids, as they can not only tolerate substitutions leading to DNA sequences still related to the FKH motif, but also interact with the highly divergent FHL motif of different sequence length. The interactions made by FoxN3 with the registration positions in both the FKH and FHL motifs are enabled by the difference in DNA shape, orienting the FHL DNA in a strikingly different conformation than the FKH DNA. The structures presented here highlight that DNA motif recognition cannot be cleanly partitioned into sequence versus shape recognition, as TFs recognize the totality of the DNA structure. The role of DNA shape in motif recognition represents a different mechanism of binding to distinct DNA sequences than others previously reported. For example, dimeric TFs can bind DNA half sites with different spacer lengths (Badis et al., 2009; Gordan et al., 2011). Alternatively, the monomers of a dimeric TF can adopt different protein conformations to recognize distinct half site sequences(Kalodimos et al., 2002). Monomeric HoxB13 can recognize two sequences (CCAATAAA and CTCGTAAA) using different contributions of enthalpy and entropy to binding (Morgunova et al., 2018). FoxN3 uses a different strategy, recognizing two DNA sequences without major structural differences within the DBD, via differences in DNA shape.
Co-crystal structures of other forkhead TFs in complex with the FKH site show that these wings can adopt vastly different positions and conformations, and so may be a wider source of functional divergence within this family (Boura et al., 2010; Brent et al., 2008; Clark et al., 1993; Littler et al., 2010; Stroud et al., 2006; Tsai et al., 2006, 2007). Previous studies of FKH-binding forkhead proteins showed that swapping wings between two forkhead proteins was sufficient to switch their preferences for the flanking sequences surrounding the core FKH motif (Pierrou et al., 1994). Our study reveals that the protein sequence of the wings can exert a much more dramatic effect in determining specificity for the core recognition sequence.
Lastly, the differences in DNA recognition between the two complexes might have gene regulatory consequences. For example, different co-factors might interact with FoxN3 when bound to an FKH versus FHL site; this mode of sequence readout by TFs affecting co-factor recruitment has been observed for single-nucleotide differences in NF-κB binding sites, suggesting that the much more dramatic differences over the lengths of the FKH versus FHL motifs might also contribute to different regulatory output (Leung et al., 2004).
Overall, the work presented here reveals the ability of the FoxN3 DBD to recognize two DNA bind sites of vastly different DNA sequences and structures, highlighting the surprising plasticity of DNA recognition within the forkhead family. Given the prevalence of diverse DNA binding motif preferences within numerous TF families, such flexibility in TF-DNA recognition may be a more universal principle and might play an important role in the evolution of gene regulatory networks (Badis et al., 2009; Gordan et al., 2011; Nakagawa et al., 2013).
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Martha L. Bulyk (mlbulyk@genetics.med.harvard.edu).
EXPERIMENT MODEL AND SUBJECT DETAILS
Cell culture
HepG2 cells were purchased from ATCC (HB-8065). Cells were cultured in DMEM with High Glucose and 4.0 mM L-Glutamine, without Sodium Pyruvate (HyClone SH30022.01), and supplemented with 10% heat inactivated fetal bovine serum (Gibco 10082139) and 1% penicillin/streptomycin.
Protein Production
One Shot® BL21(DE3) Chemically Competent E. coli cells were purchased from Thermo Fisher Scientific.
METHOD DETAILS
Preparation of whole cell lysates
Whole cell lysates were prepared by placing a 15-cm culture dish on ice, aspirating culture media, and washing once in 15 mL cold PBS. Two mL of ice cold RIPA buffer (150 mM NaCl, 1% NP-40 substitute, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris pH 8.0) was then added. Cells were scraped in RIPA buffer and transferred to a cold microcentrifuge tube. The tube was then placed on a shaker platform at 100 rpm for 30 minutes at 4°C. After lysis, cell debris was pelleted by spinning at 14,000 rcf for 20 minutes at 4°C. The supernatant was removed, aliquoted into 300-μL aliquots, flash frozen in liquid nitrogen and stored at −80°C. One cOmplete ULTRA mini protease-inhibitor tablet was used per 10 mL of buffer (RIPA or PBS).
Western blot
Anti-FoxN3 antibody (Abgent AP19255B) was first evaluated for specificity via western blot against HepG2 whole cell lysate (Figure S1a). Ten to fifteen μL of whole cell lysate was run on a 4-12% Criterion Bis-Tris acrylamide gel (Bio-Rad 3450125), and was blotted with a 1:100 dilution of primary antibody, followed by 1:2,000 dilution of an HRP-conjugated goat anti-rabbit secondary (Thermo Fisher #31460).
Cross-linking and harvest of cells for ChIP
To prepare material for ChIP-seq, cells were grown on 15-cm culture dishes. Two independent passages were maintained simultaneously, harvested independently, and processed in parallel, yielding two biological replicate datasets. Plates were removed from the incubator and placed at room temperature. Formaldehyde (Sigma F87750) was added directly to the culture medium to a final concentration of 1%. Plates were incubated at room temperature for 10 minutes, with swirling every 2.5 minutes. Crosslinking was quenched by adding 2.5 M stock glycine to a final concentration of 0.125 M and plates were swirled to mix. Media was then aspirated and cells were washed once in 1X PBS. Eight milliliters of cold lysis buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% NP-40 substitute, prepared fresh and filter sterilized) was then added directly to cells on the dish. Cells were then scraped, transferred to a 15-cm conical tube, and spun at 2000 rpm for 5 minutes at 4° C. The supernatant was then removed and flash frozen in liquid nitrogen at a concentration of 2×107 cells/mL for storage at −80° C.
Sonication of cross-linked material for ChIP
A 1-mL aliquot of cells was thawed and gently resuspended before being passed through a 20-gauge needle 20 times. Crude nuclear prep was then collected by spinning the lysate at 2000 rpm for 5 minutes at 4°C. The resulting pellet was resuspended in 300 μL RIPA buffer and then processed in a Biorupter Twin circulating bath sonicator for 50 cycles of 30 seconds on and 30 seconds off at the high setting in a 4°C environmental chamber. Following sonication, samples were spun at 16,000 rcf for 15 minutes at 4°C and the supernatant was either snap frozen and stored at −80°C or used as input for immunoprecipitation. Prior to immunoprecipitation, the total protein concentration of each sample was measured by Bradford assay, and all sample concentrations were normalized to 1.75 mg/mL with 1X RIPA buffer and split into 1-mL aliquots.
Immunoprecipitation
Sixty μL of Protein G Sepharose beads (Sigma P3296) were used per sample. Beads were washed twice in 1X PBS and resuspended in 1X PBS to 60 μL. Thirty μL of washed beads were blocked by adding 9 μL of 0.3 mg/mL salmon sperm DNA and 12 μL 1 mg/mL BSA and incubating at 4° C for 1 hour on a rotisserie. The remaining 30 μL was added directly to a 1-mL sonicated cell aliquot and incubated for 1 hour at 4° C on a rotisserie to pre-clear the lysate. Following clearing, the sample was spun at 2500 rcf for 1 minute and 950 μL of supernatant was transferred to a new tube. Ten micrograms of the anti-FoxN3 antibody (Abgent AP19255B) and 30 μL of blocked bead slurry was then added and samples were incubated at 4° C overnight with rotation on a rotisserie. A ‘no antibody’ negative control sample was also prepared from a paired aliquot by incubating overnight with 30 μL of blocked bead slurry, without addition of the primary antibody. Approximately 18 hours later, samples were washed at 4° C by spinning at 2,500 rcf for 1 minute, adding 1 mL of the following buffers, incubating for 1 minute, and repeating. Washes began with 2 1X PBS washes, 4 washes with IP wash buffer (100 mM Tris pH 7.5, 500 mM LiCl, 1% NP-40 substitute, and 1% sodium deoxycholate), and 1 wash with PBS-RIPA buffer (1X PBS, 1% NP-40 substitute, 0.5% sodium deoxycholate, and 0.1% SDS). Immunoprecipitated material was eluted off the beads by adding 200 μL elution buffer (70 mM Tris-Cl pH 8.0, 1 mM EDTA, 1.5% SDS), and incubating at 65° C for 10 minutes with vortexing every 2 minutes. Samples were then spun at 2500 rcf for 2 minutes and the supernatants were transferred to fresh tubes.
Reversal of crosslinks
To each sample (antibody or no antibody), 13 μL of 4 M NaCl (200 mM final) was added, and samples were incubated at 65° C for 18 hours. Subsequently, 20 μg of Proteinase K was added to each sample and incubated at 45° C for 60 minutes. Five volumes of Qiagen Buffer PB (QIAquick PCR purification kit) was then added to one volume of sample. This material was then processed according to the manufacturer’s instructions for the QIAquick PCR purification kit, eluting twice with 30 μL of pre-warmed Qiagen Buffer EB.
Preparation of sequencing libraries and sequencing
Libraries for sequencing with Illumina high-throughput sequencing chemistry were prepared using the NEBNext Ultra II DNA Library Prep Kit (NEB E7645) with adaptors from the NEBNext Singleplex Oligos for Illumina Kit (NEB E7350) according to manufacturer’s instructions. A final cycle number of 12 cycles of amplification was required during the PCR step. Libraries were multiplexed at equal concentrations based on integration under TapeStation D1000 tape traces and sequenced on an Illumina NextSeq instrument at the Bauer Core Facility at Harvard University.
ChIP-seq data processing and analysis
Read quality was evaluated using FastQC v0.11.5. Reads were trimmed to 36 nt to remove lower quality 3’ end base calls using fastx-trimmer v0.0.13. Reads were then aligned to hg38 using bowtie v1.1.1 (bowtie -n 2 -m 1) (Langmead et al., 2009). Aligned reads from replicate sequencing runs were pooled either within biological replicates or across all replicates. Peaks were then called on pooled aligned reads using MACS2 v2.1.1.20160309 (macs2 callpeak -g hs -bw 400) (Feng et al., 2012; Zhang et al., 2008). Following peak calls, enriched motifs in the top 2,000 peaks, as ranked by the -log10(q-value) for each peak, were detected using HOMER v4.9 (findMotifsGenome.pl -size 50 -len 6,8,10 with hg38) (Heinz et al., 2010). Motif enrichment p-values were calculated by the HOMER findMotifsGenome.pl script against a binomial null distribution.
The top 2,000 peaks were partitioned according to the presence of a match to the FoxN3 PBM-derived FKH or FHL motifs using an empirically-derived log odds detection threshold in HOMER (annotatePeaks.pl -size 50 or -size 200 with hg38). Composite profiles were generated using the HOMER annotatePeaks.pl function (-m -hist 10 -size 2000 with hg38). Plots were produced using R v3.2.4. The background data set used for motif centralization was generated using the GENRE utility, matching on dinucleotide content and promoter enrichment (Mariani et al., 2017). Genome browser shots were depicted using Integrative Genomics Viewer (IGV), and chromatin state was analyzed using ChromHMM (Ernst and Kellis, 2012b; IGV (Integrative Genomic Viewer), 2013). The top 2,000 peaks, annotated for FKH and FHL motif occurrence, are provide in Supplemental Table 1.
FoxA1 ChIP-seq data from HepG2 cells were downloaded from the ENCODE repository (ENCFF396NXZ and ENCFF988UCQ) with corresponding control data (ENCFF190EPQ) (Dunham et al., 2012). Peaks were called using MACS2, as above, and the top 2,000 peaks as ranked by −log10(q-value) were analyzed for the presence of matches to either the FKH or the FHL motif, as above. Composite profiles were generated using the HOMER annotatePeaks.pl function (-m -hist 5 -size 2000 with hg38).
Cloning
Forkhead DBDs were generated through gene synthesis, flanked by Gateway attB recombination sites (GenScript USA, Inc.; IDT). For protein expression, the FoxN3 construct was transferred into the pDEST17 vector, which confers an N-terminal 6xHis tag, using the Gateway cloning system (Invitrogen). The sequence encoding the TEV protease cleavage site (ENLYFQG) was inserted between the pDEST17 recombination site and the beginning of the FoxN3 sequence, in order to enable tag cleavage. For use in PBM experiments, constructs were transferred into pDEST15, which confers an N-terminal Glutathione S-transferase (GST) tag, using the Gateway system (Invitrogen). All cloned protein sequences are provided in Supplemental Table 2.
Protein Purification
To produce protein for crystallization, BL21(DE3) cells were transformed with pDEST17 containing the FoxN3 DBD, grown at 37°C to OD600 of 0.6, and protein production was induced with 0.5 mM IPTG overnight at 16°C. Cells were harvested, resuspended in lysis buffer (20 mM Tris HCl pH7.7, 300 mM NaCl, 0.03% TritonX-100, 5 mM beta-mercaptoethanol, and protease inhibitor tablets (Roche)), lysed by sonication and clarified. Protein was bound to Ni-NTA beads (Qiagen), washed in wash buffer (20 mM Tris HCl pH 7.7, 300 mM NaCl, 0.03% Triton X-100, 10 mM imidazole, 5 mM 1,4-dithiothreitol (DTT)), and eluted in elution buffer (20 mM Tris HCl pH 7.7, 300 mM NaCl, 250 mM imidazole (500 mM in final elution), 5 mM DTT). The 6xHis tag was removed by overnight digestion with Tobacco Etch Virus (TEV) protease at 4°C. The cleaved protein solution was re-bound to Ni-NTA beads to capture the cleaved tag, and the flow-through was collected and loaded on to a Mono-S 10/100 GL column (GE) and eluted in 20 mM Tris HCl pH 7.7, 5 mM DTT with a 0 to 1 M NaCl gradient. Peak fractions were pooled and further purified by size exclusion chromatography on a Superdex 75 10/300GL column (GE) in 20 mM Tris HCl pH 7.7, 150 mM NaCl, 2 mM tris(2-carboxyethyl)phosphine (TCEP). The protein was either used fresh, or concentrated to 1 mg/mL and flash frozen in 20% glycerol for later use.
Protein for PBM experiments was produced through in vitro transcription and translation using the PURExpress in vitro protein synthesis kit (New England BioLabs), according to the manufacturer’s instructions. Protein quality and concentration was assessed by anti-GST Western blot with a dilution series of a recombinant GST standard (Sigma G5663). The western blot was performed with 20 ng/mL anti-GST primary antibody (Sigma G7781) and a 1:2000 dilution of HRP-conjugated goat anti-rabbit secondary antibody (Thermo Fisher #31460).
Crystallization and Data Collection
DNA oligonucleotides (IDT) for crystallization were resuspended to 100 μΜ in DNA hybridization buffer (10 mM Tris HCl pH 8, 50 mM NaCl), and annealed by mixing the forward and reverse sequences at a 1:1 ratio, heating to 95°C for 5 minutes, and slowly cooling to room temperature overnight. The DNA sequences are: FKH-F (5’-TCTTAAGTAAACAATG-3’), FKH-R (5’-ACATTGTTTACTTAAG-3’), FHL-F (5’-TCATGCTAAGACGCTA-3’), and FHL-R (5’ ATAGCGTCTTAGCATG-3’). These sequences were selected from among PBM probes that were highly bound by bispecific forkhead proteins in our previous study (Nakagawa et al., 2013).
FoxN3 was mixed with the annealed DNA at a molar ratio of 1:1.2, and the mixture was incubated on ice for 5 min. The complexes were concentrated to a final protein concentration of 4 mg/mL for the FKH complex, and 5 mg/mL for the FHL complex. Initial crystallization tests with the Natrix, Peg-Ion, and Peg-Rx screens (Hampton) were performed using the NT8 liquid handling robot, in sitting drop format. Crystals were optimized in hanging drops in 24-well format at room temperature. The FoxN3:FKH crystals formed in 0.1 M BisTris pH 5.4, 0.2 M MgCl2, 22% PEG 3350. The FoxN3:FHL crystals formed in 0.1 M BisTris pH 5.5, 0.2 M NaCl, 22% PEG 3350.
Crystals were harvested, moved to cryoprotectant solution (for FKH crystal: 0.1 M BisTris pH 5.4, 0.2 M MgCl2, 25% PEG 3350, 5% glycerol; for FHL crystal: 0.1 M BisTris pH 5.5, 0.2 M NaCl, 22% PEG 3350, 10% glycerol), and flash frozen with liquid nitrogen. Diffraction data was collected at the Advanced Photon Source, beam-line 24-ID-C for the FoxN3-FKH structure, and 24-ID-E for the FoxN3-FHL structure (NE-CAT).
Structure Determination
Diffraction images were indexed using XDS (Kabsch, 2010). Phases were produced by molecular replacement in Phenix (Adams et al., 2010). For the FoxN3:FKH structure, the structure of FoxK1a (PDB 2C6Y, chains A,C,D) was used as a search model (Tsai et al., 2006). The DNA and protein structures from the FoxN3:FKH structure were used as search models for the FoxN3:FHL structure. Model building was done in COOT (Emsley and Cowtan, 2004), and structures were refined with Phenix (Adams et al., 2010) using reciprocal space optimization of xyz coordinates, individual atomic B factors, optimization of X-ray/stereochemistry weights and optimization of ADP weights for both structures. TLS was used in the refinement of the FoxN3:FKH structure. The crystallographic data table (Table 1) was generated using Phenix. Figures were created using PyMOL (Schrödinger).
DNA Shape Analysis
Protein-DNA contacts were identified using the DNAproDB tool(Sagendorf et al., 2017). Superpositions of the FKH and FHL structures were performed using the align function in PyMOL, aligning the FoxN3 protein in the two structures. DNA shape parameters were determined from the structures using Curves+ (Lavery et al., 2009). For MGW for C14 at the end of the FHL DNA, which Curves+ could not compute, the measurement tool in PyMOL was used to determine the distance between the phosphate group of the +2 and −2’ nucleotides. 5.8 Å was subtracted from the measurement, for concordance with the Curves+ results. Shape parameters for unbound DNA were predicted using the DNAshape webserver (Zhou et al., 2013). Average shape parameter values were calculated over the GTAAAC or GACGC bases for the FKH and FHL motifs, respectively.
Protein Binding Microarrays
PBM assays were carried out essentially as described, using our 8 × 60K “all 10-mer” universal array design (Agilent Technologies, AMADID #030236) (Berger and Bulyk, 2009; Berger et al., 2006). Briefly, GST-tagged forkhead DBDs, synthesized by IVT, were applied to the double-stranded DNA array, and detected with a fluorescently conjugated anti-GST antibody (Thermo Fisher #A-11131). Full experimental conditions (protein concentration and buffers used for each protein) are provided in Supplemental Table 2. Microarray data were quantified as described previously (Berger and Bulyk, 2009; Berger et al., 2006).
MicroScale Thermophoresis
Fluorescein-labeled oligonucleotides (IDT) were resuspended to 100 μM in DNA hybridization buffer (10 mM Tris HCl pH 8.0, 50 mM NaCl), and annealed by mixing the forward and reverse sequences at a 1:1 ratio, heating to 95°C for 5 minutes, and slowly cooling to room temperature overnight. Concentrations of the labeled, annealed oligonucleotidess were determined by measuring A280 and A495, and using the following formula, with ε260, fluorescein = 20960, ε495, fluorescein = 75000, ε260, FKH = 252,998, and ε260, FHL = 264,337.
MST reaction buffer was 12mM Tris pH7.5, 200mM KCl, 1mM DTT, 5mM MgCl2, 0.05% Tween-20. A 1:2 dilution series of FoxN3 from 7.5 μM to 228pM was made in MST reaction buffer, and incubated with 20nM DNA for at least 30 minutes before measuring binding. Binding was measured by MicroScale Thermophoresis using the NanoTemper Monolith NT. 115pico. MST measurements were made at 22°C, at 30% excitation power for the FKH sequence, 50% excitation power for the FHL sequence, and medium MST power (40%) for both. 3 dilution series of FoxN3 were performed for each DNA, and each dilution series was read twice by MST, resulting in six replicates.
QUANTIFICATION AND STATISTICAL ANALYSIS
Protein Binding Microarray Analysis
Boxplots were generated by filtering all 8-bp sequences for those that match the indicated sequence (FKH: GTAAACA, FHL: GACGC). The PBM E-scores of those 8-mers for each protein were plotted using the boxplot function in R. The negative control GST PBM data are from Barrera et al. (Barrera et al., 2016). P-values were computed using the one-sided Mann-Whitney test function in R, and were corrected for multiple hypothesis testing using the Bonferroni correction.
MST Affinity Analysis
Fnorm was calculated from the MST traces, with the ‘cold region’ defined as −1s to 0s, and the ‘hot region’ defined as 4s to 5s. Individual capillary traces were visualized to remove fluorescence outliers outside 20% of the average fluorescence. Data were fit to a one-to-one binding model accounting for ligand depletion in Prism to determine Kd: Y=Unbound+ (Bound-Unbound)*((Ro + X + Kd)-sqrt((Ro + X + Kd)^2 - 4*Ro*X))/(2*Ro), where Ro = the total fluorescent oligo concentration, fixed at 20nM, X is the protein concentration, Bound and Unbound are the Maximum and Minimum binding response measurements, and Y is the binding measurement (here, Fnorm). For plotting, data were normalized to represent fraction bound: fraction = (unbound-Fnorm)/(unbound-bound).
DATA AND SOFTWARE AVAILABILITY
ChIP data has been deposited in NCBI GEO under series ID GSE112672. PBM data have been deposited in UniPROBE under accession code ROG18A, at http://the_brain.bwh.harvard.edu/uniprobe/. Structures are deposited in PDB, with accession codes 6NCE and 6NCM.
Supplementary Material
Table S1, Related to Figure 2
ChIP-Seq peak locations, annotated by presence of FKH or FHL motifs.
Table S2, Related to Figure 5
Forkhead DBD sequences and PBM experimental conditions.
Highlights.
FoxN3 recognizes two different DNA motifs, both in vitro and in cells.
Co-crystal structures of FoxN3 with both sites were determined.
Differences in DNA shape enable FoxN3 to bind the two sites of different length.
Parts of the forkhead domain that do not contact DNA influence binding specificity.
Acknowledgments
We thank Jesse V. Kurland for performing a PBM experiment, and Katy Weinand for assistance in ChIP-seq analysis. We thank Steve Gisselbrecht, Kelly Arnett, Karen Adelman, Remo Rohs, and Kevin Struhl for helpful discussion. We thank The Bauer Core Facility at Harvard University for high-throughput sequencing services. We thank the Snyder lab and the ENCODE Consortium for generating and hosting FOXA1 ChIP-seq data. MST experiments were performed at the Center for Macromolecular Interactions at Harvard Medical School. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Pilatus 6M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant (S10 RR029205). The Eiger 16M detector on 24-ID-E beam line is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. A.N.H. was supported by NSF DBI-1263215 and NIH R25HL121029. This work was supported by National Institutes of Health Grant R01 HG003985 to MLB, R35 CA220340 to S.C.B., and by National Science Foundation Graduate Research Fellowships to JMR and CTW.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
S.C.B. is a grantee of the Novartis/Dana-Farber Cancer Institute Drug development translational research program, and a consultant for IFM Therapeutics. M.L.B. is a co-inventor on issued U.S. patents on protein binding microarray technology.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. (2009). Diversity and complexity in DNA recognition by transcription factors. Science 324, 1720–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera LA, Vedenko A, Kurland JV, Rogers JM, Gisselbrecht SS, Rossin EJ, Woodard J, Mariani L, Kock KH, Inukai S, et al. (2016). Survey of variation in human transcription factors reveals prevalent DNA binding changes. Science (80-. ). 351, 1450–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, and Bulyk ML (2009). Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat. Protoc 4, 393–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Philippakis AA, Qureshi AM, Fangxue HS, Estep PW, and Bulyk ML (2006). Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol 24, 1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boura E, Rezabkova L, Brynda J, Obsilova V, and Obsil T (2010). Structure of the human FOXO4-DBD-DNA complex at 1.9 Å resolution reveals new details of FOXO binding to the DNA. Acta Crystallogr. D. Biol. Crystallogr. 66, 1351–1357. [DOI] [PubMed] [Google Scholar]
- Brent MM, Anand R, and Marmorstein R (2008). Structural basis for DNA recognition by FoxO1 and its regulation by posttranslational modification. Structure 16, 1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busygina V, Kottemann MC, Scott KL, Plon SE, and Bale AE (2006). Multiple endocrine neoplasia type 1 interacts with forkhead transcription factor CHES1 in DNA damage response. Cancer Res. 66, 8397–8403. [DOI] [PubMed] [Google Scholar]
- Chang JT, Wang HM, Chang KW, Chen WH, Wen MC, Hsu YM, Yung BYM, Chen IH, Liao CT, Hsieh LL, et al. (2005). Identification of differentially expressed genes in oral squamous cell carcinoma (OSCC): Overexpression of NPM, CDK1 and NDRG1 and underexpression of CHES1. Int. J. Cancer 114, 942–949. [DOI] [PubMed] [Google Scholar]
- Clark KL, Halay ED, Lai E, and Burley SK (1993). Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature. [DOI] [PubMed] [Google Scholar]
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis C. a., Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2012a). ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2012b). ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, and Liu XS (2012). Identifying ChIP-seq enrichment using MACS. Nat. Protoc 7, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvie CW, and Wolberger C (2001). Recognition of specific DNA sequences. Mol. Cell 8, 937–946. [DOI] [PubMed] [Google Scholar]
- Gordan R, Murphy K, McCord R, Zhu C, Vedenko A, and Bulyk M (2011). Curated collection of yeast transcription factor DNA binding specificity data reveals novel structural and gene regulatory insights. Genome Biol. 12, R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SC (1991). A structural taxonomy of DNA-binding domains. Nature 353, 715–719. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huot G, Vernier M, Bourdeau V, Doucet L, Saint-Germain E, Gaumont-Leclerc M-F, Moro A, and Ferbeyre G (2014). CHES1/FOXN3 regulates cell proliferation by repressing PIM2 and protein biosynthesis. Mol. Biol. Cell 25, 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IGV (Integrative Genomic Viewer) (2013). Integrative Genomics Viewer. Broad Inst. 29, 24–26. [Google Scholar]
- Kabsch W (2010). Xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalodimos CG, Bonvin AMJJ, Salinas RK, Wechselberger R, Boelens R, and Kaptein R (2002). Plasticity in protein-DNA recognition: lac repressor interacts with its natural operator O1 through alternative conformations of its DNA-binding domain. EMBO J. 21, 2866–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanth S, Zinkhan EK, Hill JT, Yost HJ, and Schlegel A (2016). FOXN3 Regulates Hepatic Glucose Utilization. Cell Rep. 15, 2745–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam EW, Brosens JJ, Gomes AR, and Koo CY (2013). Forkhead box proteins: tuning forks for transcriptional harmony. Nat. Rev. Cancer 13, 482–495. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavery R, Moakher M, Maddocks JH, Petkeviciute D, and Zakrzewska K (2009). Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 37, 5917–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung TH, Hoffmann A, and Baltimore D (2004). One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell 118, 453–464. [DOI] [PubMed] [Google Scholar]
- Li J, Dantas Machado AC, Guo M, Sagendorf JM, Zhou Z, Jiang L, Chen X, Wu D, Qu L, Chen Z, et al. (2017). Structure of the Forkhead Domain of FOXA2 Bound to a Complete DNA Consensus Site. Biochemistry 56, 3745–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littler DR, Alvarez-Fernandez M, Stein A, Hibbert RG, Heidebrecht T, Aloy P, Medema RH, and Perrakis A (2010). Structure of the FoxM1 DNA-recognition domain bound to a promoter sequence. Nucleic Acids Res. 38, 4527–4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani L, Weinand K, Vedenko A, Barrera LA, and Bulyk ML (2017). Identification of Human Lineage-Specific Transcriptional Coregulators Enabled by a Glossary of Binding Modules and Tunable Genomic Backgrounds. Cell Syst. 5, 187–201.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowski J, Tyszkiewicz T, Jarzab M, Oczko-Wojciechowska M, Gierek T, Witkowska M, Paluch J, Kowalska M, Wygoda Z, Lange D, et al. (2009). Metal-proteinase ADAM12, kinesin 14 and checkpoint suppressor 1 as new molecular markers of laryngeal carcinoma. Eur. Arch. Oto-Rhino-Laryngology 266, 1501–1507. [DOI] [PubMed] [Google Scholar]
- Morgunova E, Yin Y, Das PK, Jolma A, Zhu F, Popov A, Xu Y, Nilsson L, and Taipale J (2018). Two distinct DNA sequences recognized by transcription factors represent enthalpy and entropy optima. Elife 7, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel S, Pommerenke C, Meyer C, Kaufmann M, MacLeod RAF, and Drexler HG (2017). Identification of a tumor suppressor network in T-cell leukemia. Leuk. Lymphoma 0, 1–15. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Gisselbrecht SS, Rogers JM, Hartl DL, and Bulyk ML (2013). DNA-binding specificity changes in the evolution of forkhead transcription factors. Proc. Natl. Acad. Sci. U. S. A 110, 12349–12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Schevitz RW, Zhang RG, Lawson CL, Joachimiak A, Marmorstein RQ, Luisi BF., and Sigler PB. (1988). Crystal structure of trp repressor/operator complex at atomic resolution. Nature. 335, 321–329. [DOI] [PubMed] [Google Scholar]
- Pati D., Keller C., Groudine M, and Plon SE. (1997). Reconstitution of a MEC1-independent checkpoint in yeast by expression of a novel human fork head cDNA. Mol. Cell. Biol 17, 3037–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierrou S, Hellqvist M, Samuelsson L, Enerback S, and Carlsson P (1994). Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J. 13, 5002–5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohs R, West SM, Sosinsky A, Liu P, Mann RS, and Honig B (2009). The role of DNA shape in protein-DNA recognition. Nature 461, 1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagendorf JM, Berman HM, and Rohs R (2017). DNAproDB: An interactive tool for structural analysis of DNA-protein complexes. Nucleic Acids Res. 45, W89–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaan G, Yugo D, Rajagopalan S, Wall J, Donnell R, Goldowitz D, Gopalakrishnan R, and Venkatachalam S. (2010). Foxn3 is essential for craniofacial development in mice and a putative candidate involved in human congenital craniofacial defects. Biochem. Biophys. Res. Commun 400, 60–65. [DOI] [PubMed] [Google Scholar]
- Schmidt J, Schuff M, and Olsson L (2011). A role for FoxN3 in the development of cranial cartilages and muscles in Xenopus laevis (Amphibia: Anura: Pipidae) with special emphasis on the novel rostral cartilages. J. Anat 218, 226–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger LLC. The PyMOL Molecular Graphics System, Version 1.8.
- Schuff M, Rossner A, Wacker S. a., Donow C, Gessert S, and Knochel W (2007). FoxN3 is required for craniofacial and eye development of Xenopus laevis. Dev. Dyn 236, 226–239. [DOI] [PubMed] [Google Scholar]
- Scott KL, and Plon SE (2003). Loss of Sin3 / Rpd3 Histone Deacetylase Restores the DNA Damage Response in Checkpoint-Deficient Strains of Saccharomyces cerevisiae Loss of Sin3 / Rpd3 Histone Deacetylase Restores the DNA Damage Response in Checkpoint-Deficient Strains of Saccharomyces. Mol. Cell. Biol 23, 4522–4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott KL, and Plon SE (2005). CHES1/FOXN3 interacts with Ski-interacting protein and acts as a transcriptional repressor. Gene 359, 119–126. [DOI] [PubMed] [Google Scholar]
- Siggers T, Reddy J, Barron B, and Bulyk ML (2014). Diversification of transcription factor paralogs via noncanonical modularity in C2H2 Zinc finger DNA binding. Mol. Cell 55, 640–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud JC, Wu Y, Bates DL, Han A, Nowick K, Paabo S, Tong H, and Chen L (2006). Structure of the Forkhead Domain of FOXP2 Bound to DNA. Structure 14, 159–166. [DOI] [PubMed] [Google Scholar]
- Sun J, Li H, Huo Q, Cui M, Ge C, Zhao F, Tian H, Chen T, Yao M, and Li J (2016). The transcription factor FOXN3 inhibits cell proliferation by downregulating E2F5 expression in hepatocellular carcinoma cells. Oncotarget 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai K-L, Huang C-Y, Chang C-H, Sun Y-J, Chuang W-J, and Hsiao C-D (2006). Crystal structure of the human FOXK1a-DNA complex and its implications on the diverse binding specificity of winged helix/forkhead proteins. J. Biol. Chem 281, 17400–17409. [DOI] [PubMed] [Google Scholar]
- Tsai K-L, Sun Y-J, Huang C-Y, Yang J-Y, Hung M-C, and Hsiao C-D (2007) Crystal structure of the human FOXO3a-DBD/DNA complex suggests the effects of post-translational modification. Nucleic Acids Res. 35, 6984–6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Yang L, Lu Y, Dror I, Dantas Machado AC, Ghane T, Di Felice R, and Rohs R (2013). DNAshape: a method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res. 41, 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, Related to Figure 2
ChIP-Seq peak locations, annotated by presence of FKH or FHL motifs.
Table S2, Related to Figure 5
Forkhead DBD sequences and PBM experimental conditions.
Data Availability Statement
ChIP data has been deposited in NCBI GEO under series ID GSE112672. PBM data have been deposited in UniPROBE under accession code ROG18A, at http://the_brain.bwh.harvard.edu/uniprobe/. Structures are deposited in PDB, with accession codes 6NCE and 6NCM.