

SUMMARY
Topoisomerase II cleaves DNA at preferred sequences with different efficiencies; however, the mechanism of cleavage site selection is not known. Here we used single-molecule fluorescence assays that monitor several critical steps of DNA-topoisomerase II interactions, including binding/dissociation, bending/straightening, and cleavage/religation, and reveal that the cleavage site is selected mainly during the bending step. Furthermore, despite the sensitivity of the bending rate to the DNA sequence, it is not an intrinsic property of the DNA itself. Rather, it is determined by protein-DNA interactions.
Graphical Abstract
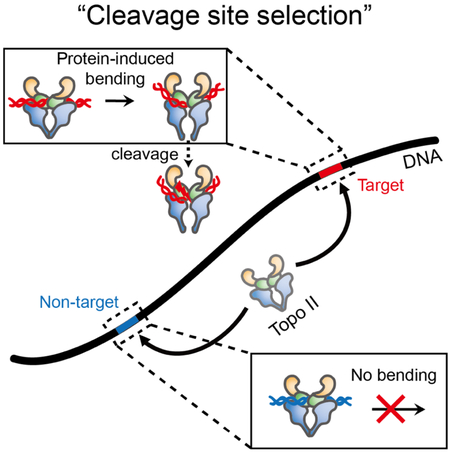
eTOC
Jang et al. describe single-molecule fluorescence experiments that monitor the individual steps of topoisomerase II. Kinetic analysis for six different sequences reveal that the cleavage site is selected in the DNA bending step. It is determined by protein-DNA interactions rather than by an intrinsic property of the sequences.
INTRODUCTION
Topoisomerase II is an indispensable enzyme that resolves topological problems occurring in double-stranded DNA during replication, repair, and transcription(Deweese and Osheroff, 2009; Nitiss, 2009a; Pommier et al., 2016; Vos et al., 2011; Wang, 2002). As part of the essential processes in resolving these problems, the enzyme generates a transient double-strand break in one DNA segment (called the “Gate” or G-segment) and transports another DNA segment (called the “Transport” or T-segment) through the break, resulting in the religation of the cleaved G-segment DNA(Berger et al., 1996; Nitiss, 2009a; Wang, 1985, 2002). Through the double-stranded DNA passage reaction, the enzyme modulates the topological state of DNA. In this reaction, the G-segment breakage is catalyzed by a pair of active-site tyrosine residues that form a covalently linked enzyme–DNA intermediate, termed the cleavage complex(Deweese and Osheroff, 2009).
Although DNA breakage is essential for the cellular function of the enzyme, it also poses a risk to the maintenance of genomic integrity because failure to religate the DNA breaks can trigger cell death pathways(McClendon and Osheroff, 2007) and chromosome rearrangements(Pendleton et al., 2014). Consequently, tight regulation of the DNA cleavage/religation reaction is required to avoid the generation of cytotoxic DNA breaks. Conversely, many clinically important anticancer drugs exploit the potential lethality of the type II enzyme, as discussed above. These drugs lead to an accumulation of the cytotoxic DNA strand breaks by blocking the religation of cleaved DNA ends(Deweese and Osheroff, 2009; Nitiss, 2009b).
Because of the biological and clinical importance of the topoisomerase II-mediated DNA cleavage/religation reaction, its underlying mechanisms have been extensively studied. However, many fundamental questions concerning the DNA cleavage/religation reaction remain unanswered. For the quantitative characterization of this reaction, several studies have focused on uncoupling the cleavage and religation events. For example, the DNA has been biochemically modified(Bromberg et al., 2002; Bromberg et al., 2004; Deweese et al., 2008b), and environmental conditions, such as the reaction temperature or ionic species, have been altered(Bromberg and Osheroff, 2001; Osheroff and Zechiedrich, 1987; Schmidt et al., 2010). Through these studies, it has been suggested that divalent metal ions play critical catalytic and structural roles in the DNA cleavage/religation reaction(Deweese et al., 2008a; Deweese and Osheroff, 2010; Lee et al., 2012; Schmidt et al., 2010). In this regard, it has been demonstrated that topoisomerase II utilizes a non-canonical two-metal-ion mechanism for these catalytic reactions(Deweese et al., 2008a; Deweese and Osheroff, 2010; Schmidt et al., 2010). However, when and how the ions participate during the entire reaction remains incompletely understood. Considering the high activity of topoisomerase II in cell metabolism and its DNA cleavage site preferences, the enzyme might be expected to rapidly scan a large number of locations throughout the genome and then efficiently select its DNA cleavage site. Hence, an important step in elucidating the operational mechanism of the enzyme would be to obtain an improved understanding of the target recognition process. In previous biochemical studies, the sequence-preferential cleavage of DNA by the enzyme has been characterized using a series of DNA substrates with varied sequences(Bromberg et al., 2002; Capranico and Binaschi, 1998; Mueller-Planitz and Herschlag, 2007). However, the mechanistic basis by which topoisomerase II selects its sites of DNA action remains an enigma.
To more fully answer the above questions, a thorough characterization of the fundamental steps in the DNA cleavage/religation reaction is an essential prerequisite. Structural and biochemical studies on topoisomerase II have demonstrated that the ability to bend DNA is critical for the enzyme to cleave the DNA double helix(Dong and Berger, 2007; Hardin et al., 2011; Lee et al., 2013; Schmidt et al., 2010). In our previous single-molecule fluorescence resonance energy transfer (FRET) study using human topoisomerase IIα(Lee et al., 2012), we observed that enzyme-mediated DNA bending preceded DNA cleavage and the enzyme-bent DNA complex was stabilized by the use of a drug or suicide DNA. However, the cleavage reaction could not be directly observed in this FRET study because its efficiency was too low.
It is known that the cleavage efficiency of yeast topoisomerase II (yTopoII) is greatly enhanced in the presence of Ca2+ even though calcium ions have little effect on the selection of cleavage sites(Mueller-Planitz and Herschlag, 2007; Osheroff and Zechiedrich, 1987). Therefore, to overcome the issue of low levels of DNA cleavage, we used yTopoII as a model system to perform single-molecule FRET experiments to monitor individual reaction steps mediated by the enzyme, including DNA binding/unbinding, bending/straightening, and cleavage/religation (Figure 1). In this unique experiment, we focused on the mechanism by which the sequence of DNA alters the cleavage efficiency of topoisomerase II. Results indicate that the cleavage site is selected primarily during the DNA bending step.
Figure 1.

Kinetic Scheme for the Topoisomerase II-mediated DNA Cleavage/Religation Reaction
RESULTS
Single-molecule FRET Assay
A partial DNA duplex (Yclv1) containing a centrally located cleavage site for yTopoII and flanking FRET probes (Cy3 and Cy5) was prepared for the experiments (Figure 2A). To observe the interactions between the duplex and yTopoII in real time, we immobilized the duplexes on a polyethylene glycol (PEG)-coated quartz surface using a biotinylated overhang at one end of the duplex and monitored fluorescence signals from single DNA molecules using a total-internal-reflection fluorescence (TIRF) microscope (Figure 2B). In the present study, for the detection of a substantial number of the DNA cleavage/religation events, we used a reaction buffer containing Ca2+ ions in place of the physiological Mg2+ ions. Under the reaction conditions employed, the DNA cleavage efficiency by yTopoII is significantly increased compared to reactions including Mg2+ ions(Mueller-Planitz and Herschlag, 2007; Osheroff and Zechiedrich, 1987). This enhanced DNA cleavage allowed accurate quantification of the fundamental steps in the DNA cleavage/religation reaction.
Figure 2. Experimental Design for Observing DNA-topoisomerase II Interactions.

(A) DNA construct (Yclv1) used for the experiments. The labeling sites for conjugating dyes, Cy3 and Cy5, are indicated.
(B) Schematic diagram of single-molecule fluorescence resonance energy transfer (FRET) assay.
Direct Observation of Fundamental Steps during the DNA Cleavage and the Religation Reactions of yTopoII
In the absence of yTopoII, we observed fluorescence intensities showing single-state behavior with small fluctuations that were limited by shot noise (Figure S1). However, in the presence of yTopoII, the binding events of single enzyme molecules were clearly identified by large intensity jumps in the Cy3 fluorescence signal (black arrowheads in Figure 3A and C), with appreciable dwell time (~0.25 sec on average) using a single-molecule fluorescence assay termed protein-induced fluorescence enhancement (PIFE) (Hwang et al., 2011; Luo et al., 2007). In this assay, the intensity of a fluorophore is increased when a protein binds in its vicinity due to the enhancement in the quantum yield(Sanborn et al., 2007; Sorokina et al., 2009). In our experiment, the fluorescence enhancement occurred only in Cy3, and almost not in Cy5, upon the enzyme–DNA binding events (Figure S2). As a result of the prominent effect of Cy3-intensity enhancement, a decrease in apparent FRET efficiency was observed simultaneously with a jump in Cy3 intensity during the enzyme binding events (Figure 3G and H).
Figure 3. Real-time Observation of Topoisomerase II-mediated DNA Cleavage/Religation Reaction.

(A,C) Representative time traces showing association/dissociation (black arrowheads) and bending/straightening events (red arrowheads) in the presence of 5 nM yTopoII and 10 mM Ca2+ (A) or Mg2+ (C) ions for Yclv1 DNA construct. In these measurements, a time resolution of 50 ms was used for observing transient interactions. Black arrowheads indicate transient binding of the enzyme to a DNA. Red arrowheads indicate the short-lasting bending events. Cy3 fluorescence, Cy5 fluorescence, and corresponding FRET efficiency are shown in green, red, and blue, respectively. Here, the total intensity is the sum of intensities of Cy3 and Cy5 and FRET efficiency (hereafter FRET) is the ratio of Cy5 intensity to the total intensity. To clearly visualize the enzyme binding events, red and yellow boxes are added as a visual guide. The same color convention is used throughout the paper.
(B,D) Representative time traces showing DNA cleavage/religation (red arrows) events in the presence of 5 nM yTopoII and 10 mM Ca2+ (B) or Mg2+ (D) ions. In (B), experiment was performed under the same reaction condition as in (A) except for the time resolution. In (D), the experimental condition was same as in (C) except for the time resolution. For long-time observation, a time resolution of 500 ms was used to reduce the photobleaching of the fluorophores. The long-lived cleavage events are indicated by red arrows.
(E,F) FRET histograms of Yclv1 DNA duplexes with bound enzyme in the presence of 5 nM yTopoII and 10 mM Ca2+ (E) or Mg2+ (F) ions. The cleavage population of Yclv1 (blue line, 83%) in the presence of Ca2+ ions was obtained by fitting the FRET histogram to the sum of three Gaussian functions.
(G,H) Relative distribution of total intensity and FRET efficiency of Yclv1 DNA duplexes in the presence of 5 nM yTopoII and 10 mM Ca2+ (G) or Mg2+ (H) ions. The colors represent the relative frequency of the events. White dashed lines were added for a clear visualization of the three species, including unbound DNA only, enzyme binding, and DNA bending/cleavage.
Interestingly, in the above observations, two types of large (binding mode I, 70%) or small (binding mode II, 30%) intensity jumps existed. Kinetic data and FRET histograms for the two types of binding events revealed that binding mode I had a much longer lifetime and showed greatly increased FRET states during the binding events in comparison with binding mode II (Figure S2). As a subsequent high FRET state (~0.42) only follows after the binding events with the longer lifetimes, we infer that the binding mode I is a prerequisite step for forming a productive enzyme-DNA complex.
In some of the binding events, we observed two kinetically distinct FRET jumps with a lifetime of ~0.1 s (red arrowheads in Figure 3A) or ~100 s (red arrows in Figure 3B) in the presence of Ca2+ ions (Figure S2). This observation is in line with recent structural studies(Bax et al., 2010; Dong and Berger, 2007; Schmidt et al., 2010; Wendorff et al., 2012; Wu et al., 2011) that strongly support the conclusion that two species of DNA deformation, namely bending and cleavage, are stochastically induced by topoisomerase II. The long-lasting FRET jump was much less frequently observed in Mg2+ buffer, which is known to induce less DNA cleavage (Figure 3D), and was completely suppressed in a DNA sequence with very low cleavage efficiency (Figure S1). By combining these results with findings from our previous study (which described a transient DNA bending by topoisomerase II as an intermediate state en route to the cleavage reaction) (Lee et al., 2012), we conclude that the long-lasting FRET jump indicates the cleavage of DNA, whereas the short-lasting FRET jump corresponds to the brief bending of DNA as a pre-cleavage intermediate. Consistently, DNA bending also occurred in the case of nonreactive mutant of yTopo II, Y783F (Figure S1). This conclusion is further supported by the fact that the high FRET population of enzyme-bound DNA (83%, Figure 3E) in the presence of 10 mM Ca2+ ions, which is mostly contributed from long-lasting FRET states, is similar to the fraction of DNA cleavage (76%) with a saturating concentration of enzyme in the previous biochemical study(Mueller-Planitz and Herschlag, 2007). In addition, the FRET histograms observed in Ca2+ or Mg2+ buffer show a significant difference in the high FRET population (Figure 3E and F).
The Ca2+ Ions that are Required for the DNA Bending Step are Maintained during the DNA Cleavage/Religation Reaction
Combining the above observations with findings from the analysis of FRET time traces using hidden Markov modelling (Figure S3), a kinetic scheme for the DNA cleavage/religation reaction is summarized in Figure 1.
By using the unique capability of the FRET technique to identify individual reaction steps, we first investigated the kinetic pathway of divalent ions in the DNA cleavage/religation reaction by topoisomerase II. Previous studies have suggested catalytic and structural roles of the divalent ions(Deweese et al., 2008a; Deweese and Osheroff, 2010; Lee et al., 2012; Schmidt et al., 2010). However, as to when and how the ions play roles during the DNA cleavage/religation reaction remains incompletely understood. To address these questions, we performed Ca2+ titration experiments. The FRET histograms of enzyme-bound DNA are shown in Figure 4A at varying Ca2+ concentrations. As the Ca2+ concentration increased, the population of high FRET state, corresponding to the DNA bending, increased. Since the high FRET population is contributed mostly by the long-lived cleavage events, two important factors responsible for the Ca2+–dependent variance in the high FRET population should be the frequency and/or the duration of the cleavage events.
Figure 4. Kinetic Pathway of Divalent Ions during the DNA Cleavage/Religation Reaction.

(A) Fluorescence resonance energy transfer (FRET) histograms of DNA duplexes with bound enzyme at varying Ca2+ ion concentrations. Cleavage populations at varying Ca2+ ion concentrations were obtained by fitting the FRET histograms to the sum of three Gaussian functions.
(B–G) Comparison of association (B), dissociation (C), bending (D), straightening (E), cleavage (F), and religation rates (G) at varying Ca2+ ion concentrations. Error bars represent standard deviations obtained from three independent experiments.
The kinetic analysis of the individual reaction steps at varying Ca2+ concentrations revealed that the bending and dissociation rates changed dramatically (Figure 4B–G and Table S1). In particular, the DNA bending rate had the strongest correlation with the Ca2+ concentration (Figure 4D). Although the dissociation rate also showed an appreciable correlation with the Ca2+ concentration (Figure 4C), the dissociation rate was increased at higher Ca2+ concentrations, which produces the opposite effect on Ca2+ concentration dependence of the DNA cleavage efficiency. In addition, we observed the similar salt effect in the dissociation rate under K+ condition in our previous study using human topoisomerase IIα, and found that the dissociation rate was very sensitive to ionic strength(Lee et al., 2012). The subsequent kinetic parameters were largely unaffected, particularly the DNA cleavage and religation rates, which remained constant at varying Ca2+ concentrations (Figure 4B and E–G). The finding that the DNA cleavage rate is at least a 10 times higher than the religation rate is in agreement with a previous report which showed high level of DNA cleavage in the presence of Ca2+ ions(Osheroff and Zechiedrich, 1987). By combining these results with the fact that topoisomerase II requires divalent metal ions for both the DNA cleavage and religation activities(Bromberg et al., 2002; Deweese and Osheroff, 2010; Liu et al., 1983; Osheroff and Zechiedrich, 1987; Sander and Hsieh, 1983), we strongly suggest that Ca2+ ions are first required in the DNA bending step and that those ions are bound to the enzyme–DNA complex during the subsequent cleavage and religation reactions. These suggestions are further supported by the finding that when the Ca2+ buffer in the detection chamber was exchanged by a buffer containing high salts with no divalent ions during data acquisition to remove free Ca2+ ions, 85% of the cleaved DNA was religated by the enzyme, despite the absence of free divalent ions (Figure S4). This finding confirms that the Ca2+ ions bound in the bending step are preserved in the enzyme–DNA complex until religation of the cleaved DNA. Furthermore, the strong correlation between the bending rate and the cleavage population indicates that the bending of G-segment DNA provides a cleavage-competent state for the enzyme. Thus, the frequent bending of DNA at a high Ca2+ concentration increases the occurrence of cleavage events, which is primarily responsible for the high FRET population.
The DNA Bending Rate is Strongly Correlated with the Efficiency of DNA Cleavage
Although topoisomerase II acts at a variety of locations throughout the genome, previous studies using DNA sequences that were identified in plasmid DNA have confirmed that the enzyme cleaves DNA at preferred sites with different efficiencies(Fortune et al., 2002; Mueller-Planitz and Herschlag, 2007; Sander and Hsieh, 1985; Spitzner et al., 1990; Spitzner and Muller, 1988). Because the sequence-preferential cleavage of the DNA is thought to be an underlying strategy for the regulation of overall activity of the enzyme(Burden and Osheroff, 1999; Kas and Laemmli, 1992; Miassod et al., 1997), the mechanism by which the cleavage efficiency is determined poses an important and intriguing question. To address this question, six DNA duplexes with varied sequences (from Yclv1 to Yclv6 in Table S2) were systematically investigated by monitoring the individual steps of the DNA cleavage/religation reaction by the enzyme (Figure 5). The DNA sequences were designed to have different cleavage efficiencies by mutating the central 26 bp region of the duplex that is within van der Waals distance of the protein in the crystal structure of yTopo II-DNA complex(Dong and Berger, 2007; Schmidt et al., 2010; Mueller-Planitz and Herschlag, 2007). We characterized the cleavage efficiency of Yclv1-Yclv6 by the high FRET population of enzyme-bound DNA, representing the fraction of DNA cleavage at equilibrium.
Figure 5. DNA Bending as a Main Determinant of the DNA Cleavage Efficiency.

(A) Fluorescence resonance energy transfer (FRET) histograms of six DNA duplexes with bound enzyme in the presence of 10 mM Ca2+ ions. Cleavage populations of six DNA duplexes were obtained by fitting the FRET histograms to the sum of three Gaussian functions.
(B–G) Comparison of association (B), dissociation (C), bending (D), straightening (E), cleavage (F), and religation rates (G) of DNA duplexes with different DNA base compositions. Error bars indicate standard deviations obtained from three independent experiments. To clearly visualize the corre-lation between each rate constant and the cleavage efficiency, linear fit lines (red line) and average values (blue dashed line) of the entire data set for each rate constant are added.
The high FRET population of a series of enzyme-bound DNA duplexes with different DNA base compositions, representing the fraction of DNA cleavage, was gradually decreased as shown in Figure 5A. This result is in agreement with a previous biochemical study (Figure S5) (Mueller-Planitz and Herschlag, 2007). To identify which reaction step correlated with the cleavage efficiency, all rate constants in our kinetic scheme were obtained for these DNA substrates (Figure 5B–G and Table S1). First, the association and dissociation rates of the enzyme showed no appreciable correlation with the cleavage efficiency (Figure 5B and C). Therefore, we conclude that binding affinity plays no substantial role in determining the efficiency of sequence-preferential cleavage of the DNA, which is consistent with previous ensemble studies(Bromberg et al., 2002; Mueller-Planitz and Herschlag, 2007). In addition, similar straightening rates were observed in all DNA duplexes, showing no correlation with the cleavage efficiency (Figure 5E).
In marked contrast, the bending and cleavage/religation rates changed dramatically. In particular, the bending rate showed a significant positive correlation with the cleavage efficiency, suggesting that DNA bending is an underlying mechanism determining the cleavage efficiency (Figure 5D). In the subsequent reaction steps, the cleavage rate of Yclv5 and the cleavage/religation rates of Yclv6 could not be obtained since the corresponding events occurred less than 3%. Nonetheless, we were able to assess the relationship between the kinetic rates in these steps and the cleavage efficiency. Although Yclv2 has a much higher cleavage rate than the other DNA duplexes, the cleavage rate showed no correlation with the cleavage efficiency (Figure 5F). On the other hand, the religation rate showed an appreciable correlation, but not as high as that of the bending rate (Figure S5). These observations suggest that although DNA bending acts as the main determinant of cleavage efficiency, the religation step may also play an important role in the selection of the cleavage site.
We next examined the effect of mutations in the specific region of the duplex. Mutations at the isoleucine intercalation and cleavage sites, whose structural roles are already well known, significantly affected DNA bending and cleavage. In addition to these mutations, the alteration at position 9 (C9T) also affected these parameters (Figure 5A–D and Figure S5). The interaction of this position with the αβ fold highly conserved in DNA- and RNA-binding proteins has been expected to act as a buttressing element for the outer ends of bent DNA from the crystal structure of yTopoII-DNA complex(Dong and Berger, 2007). Thus, we infer that the interaction for buttressing the outer DNA ends is also critical for DNA bending and consequently the overall interaction with the central 20 bp region of the duplex plays a significant role in inducing DNA bending. In contrast, mutations outside that region did not substantially influence DNA bending or cleavage (Figure S5). Especially in Yclv3-6, we found that the mutation at the cleavage site (G2T) significantly increased the religation rate (Figure 5G), which is reasonably predictable considering that the site directly interacts with catalytic residues. Interestingly, however, comparing Yclv2 with Yclv3, we found that the mutation at the cleavage site, rather than the isoleucine intercalation site which is structurally known as the kink site of the DNA, most greatly affected the bending rate and consequently cleavage efficiency (Figure 5A–D and Figure S5). This unexpected result can be understood in the same context with our previous finding that divalent ions coordinated at the cleavage site are critical for inducing DNA bending(Lee et al., 2012).
Protein-induced DNA Bending Determines the Cleavage Efficiency of Different DNA Sequences
The strong correlation of the DNA bending rate with the cleavage efficiency raises the question of whether the origin of the correlation is the intrinsic bendability of the DNA sequence or whether it is due to protein-induced effects on the DNA. To address this question, we performed a single molecule cyclization assay(Le and Kim, 2013, 2014; Vafabakhsh and Ha, 2012) to investigate the intrinsic bendability of the DNA sequences (Figure 6A) (Boedicker et al., 2013; Munteanu et al., 1998). This assay directly monitors the looping and unlooping dynamics of single DNA molecules in real time.
Figure 6. Intrinsic Bendability of the DNA Sequences.

(A) Schematic diagram of the single-molecule cyclization assay.
(B) Representative time traces of fluorescence intensity for six DNA duplexes with different DNA base compositions. All cyclization assays are conducted with 40 mM KCl and 1 mM MgCl2. For long-time observations, the excitation laser power was reduced as much as possible in spite of the low signal-to-noise ratio (~ 4) and a time resolution of 0.5 s was used to reduce the photobleaching of fluorophores. In this measurement, a theoretical limit of observation time was estimated to 12~120 minutes. The detailed estimation is provided in STAR Methods.
(C–E) Comparison of looping rates (C), unlooping rates (D), and the ratio of the looping to unlooping rates (E) of DNA duplexes with different DNA base compositions. To clearly visualize the correlation between each rate constant and the cleavage efficiency, average values (blue dashed line) of the entire data set for each rate constant are added.
For this assay, we prepared DNA duplexes containing a central cleavage site and 8 nt single-stranded overhangs on both 5′ ends (Table S2). Probes for FRET (Cy3 and Cy5) were labeled at the 5′ ends of the strands. Single-stranded overhangs of each DNA duplex were complementary so that their hybridization captured the looped state of the DNA duplexes. Therefore, a high FRET signal was observed in the looped state of the duplexes. To observe the looping/unlooping kinetics, we immobilized the duplexes on the surface using a biotin-streptavidin interaction and monitored fluorescence signals from single DNA duplexes. Owing to the steric hindrance by the surface immobilization, in this assay, a considerable reduction in the intrinsic bendability of DNA is expected. Furthermore, it has been reported that the position of biotin in the duplex affects the extent of the reduction (Waters and Kim, 2013). Thus, we tested the extent of DNA looping for two DNA constructs that had a different position of biotin, and found that the influence by the biotin position was not significant (Figure S6). Despite this minor reduction in DNA bendability, we were able to compare the relative bendability of the DNA sequences in this assay.
Figure 6B shows the representative fluorescence intensity-time traces of the DNA duplexes with different DNA base compositions. All traces exhibited a clear two-state transition between the looped and unlooped states. The kinetic analysis of the looping and unlooping transitions in these traces revealed that the looping/unlooping rates show no correlation with the cleavage efficiency (Figure 6C–E). Because the looping/unlooping rates quantify the intrinsic bendability of the DNA duplex, this finding suggested that the strong correlation of the DNA bending rate with the DNA cleavage efficiency of yTopoII was not due to the intrinsic bendability of DNA sequences. Furthermore, potential difference in the intrinsic bendability of DNA in the presence of Mg2+ and Ca2+ ions is insignificant (Figure S6). Consequently, the sequence-preferential cleavage of DNA duplexes appears to result from the DNA bendability determined by protein-DNA interactions.
DISCUSSION
We have developed a single-molecule FRET assay that allows the direct monitoring of the fundamental steps leading up to and including the topoisomerase II-mediated DNA cleavage/religation reaction. The independent characterization of two tightly coupled reactions (DNA cleavage and religation) has long been a topic of interest; however, a deeper understanding of these reactions has been hindered by population averaging that is inherent in ensemble measurements. Recently, we made a direct observation of the DNA cleavage reaction by human topoisomerase IIα using the single-molecule FRET method(Lee et al., 2012). However, due to the highly transient and infrequent nature of the cleavage events, an external agent such as etoposide or a 3′-bridging phosphorothiolate modification in the DNA was adapted for artificially trapping of the cleavage state (resulting in the loss of the subsequent religation step). The single-molecule FRET assays described in the current study overcome these limitations by using a yTopoII-Ca2+ combination, which induces long-lived and frequent cleavage events separately from short-lived DNA bending. Consequently, this approach enables the identification and characterization of the religation step after the cleavage reaction in real time.
Based on this unique capability, we have determined the kinetic pathway of divalent ions during the DNA cleavage/religation reaction. Previously, we have demonstrated that divalent ions are used for the DNA bending step, resulting in the cleavage-competent state(Lee et al., 2012). The current observations additionally revealed that the divalent ions first used for DNA bending are preserved in the cleavage complex during the DNA cleavage/religation reaction. From a biological perspective, DNA breaks caused by external assaults are potentially dangerous. Therefore, it is imperative that the ions preserved in the cleavage complex allow immediate religation during the unexpected perturbation to prevent permanent breaks in the genome.
It has long been known that topoisomerase II has significant sequence preferences in the DNA cleavage reaction(Burden and Osheroff, 1999; Miassod et al., 1997; Mueller-Planitz and Herschlag, 2007; Sander and Hsieh, 1985; Spitzner and Muller, 1988). On the basis of the existence of enzyme-induced DNA bending, an indirect readout mechanism(Fuxreiter et al., 2011; Garvie and Wolberger, 2001; Martin et al., 1999; Otwinowski et al., 1988; Rohs et al., 2010) has been proposed for G-segment selection by topoisomerase II in which DNA bending plays an important role in determining the cleavage efficiency(Deweese and Osheroff, 2010; Lee et al., 2013; Velez-Cruz et al., 2005). We consistently found that the bending rate had the strongest correlation with the cleavage efficiencies among six different DNA sequences. In this G-segment DNA selection step, the question of whether the enzyme plays an active or passive role in the DNA bending as a cleavage-competent state is also intriguing. Our observation regarding the intrinsic bendability of the DNA sequences revealed that the intrinsic bendability does not correlate with the efficiency of DNA cleavage. This finding suggests that the enzyme induces a sequence-preferential DNA bending for G-segment selection by active protein-DNA interactions rather than by the intrinsic bendability of the sequences. This conclusion is further supported by the fact that the sequence variances within the central 20 bp region including the intercalation and cleavage sites, which is structurally important for the protein-DNA interaction, greatly affected the DNA bending and consequently cleavage efficiency (Figure 5A-D).
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents and resources should be directed to and will be fulfilled by the corresponding author Sanghwa Lee (sanglee@gist.ac.kr).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Line
S. cerevisiae topoisomerase protein was purified from S. cerevisiae DBY745 strain as described in the Key resource table and Method detail (Worland and Wang, 1989).
METHOD DETAILS
Protein Preparation
Yeast topoisomerase II was overexpressed using an inducible promoter of GAL1 gene, PGAL1 in S. Cerevisiae DBY745. The cells transformed with YEpTOP2-PGAL1 plasmid were grown until OD600 reached 0.75 and then 2% (w/v) galactose was added. 12h-grown cells were harvested and frozen rapidly in liquid nitrogen.
After cell debris was removed by centrifugation, polyethyleneimine solution (10%) was added to the supernatant with a final concentration of 0.1%. After stirring in ice for 30 min, Celite was added (80 g/L) and then the whole solution was loaded into 4.9 cm2 × 30 cm column. The column was washed with KCl and saturated ammonium sulfate and the precipitate was collected by centrifugation. The precipitate dissolved in buffer I (50 mM Tris –HCl(pH 7.7), 1 mM EDTA, 1 mM EGTA, 10% glycerol (v/v), 1 mM phenylmethylsulfonyl fluoride, and 1 mM 2-mercaptoethanol) was applied to a phosphocellulose column. The yeast topoisomerase II was eluted by a linear gradient of 250 mM, 1 M KCl in buffer I. The peak fraction were pooled and dialyzed against buffer I plus 150 mM KCl.
DNA Preparation for Single-molecule FRET Experiment
High performance liquid chromatography (HPLC)-purified DNAs (IDT, USA) were labeled with either Cy3 or Cy5 NHS-ester dyes (GE Healthcare, USA) at the amine group of an internal amino modifier (dTC6) (Table S2). DNA duplexes for single-molecule cyclization assays were designed based on a previous single molecule looping assay (Vafabakhsh and Ha, 2012). Nucleotides totaling 12 and 3 were added at the 5’ and 3’ ends of each sequence used for the single-molecule enzyme assay, respectively (Table S2). To monitor looping events, Cy3 and Cy5 were labeled at the 5’ end of each strand. The DNA strands were annealed by slowly cooling down the mixture of the biotinylated strand labeled with Cy5 and the non-biotinylated strand labeled with Cy3 (2:3 molar ratio) in a buffer containing 10 mM Tris-HCl (pH 8.0) and 50 mM NaCl.
Single-molecule FRET Measurement
To prevent the nonspecific adsorption of enzymes, the quartz slide and coverslip were thoroughly cleaned and coated with polyethylene glycol (Laysan Bio Inc., USA) and biotinylated PEG (Laysan Bio, Inc., USA) in a 40:1 ratio (Roy et al., 2008). DNA duplexes were immobilized on the PEG-coated surface via a streptavidin-biotin interaction. Single-molecule fluorescence images were taken in an imaging buffer (10 mM Tris-HCl (pH 8.0) with 0.4 % (w/v) glucose (Sigma, USA), 1 % (v/v) Trolox (Sigma, USA), 1 mg/mL glucose oxidase (Sigma, USA), 0.04 mg/mL catalase (Sigma, USA), 150 mM KCl, and MgCl2 or CaCl2 with varying concentrations) in a home-built prism-type total-internal-reflection fluorescence microscope (Rasnik et al., 2006; Roy et al., 2008). The enzymes contained in a storage buffer (50 mM Tris, 1 mM ethylenediaminetetraacetic acid (EDTA), 260 mM NaCl, 10 % (v/v) glycerol) were diluted 100-fold in the imaging buffer for each experiment (final concentration, 5 nM).
As an excitation source, a green laser (532 nm, Compass215M; Coherent) was used. Fluorescence signals from Cy3 and Cy5 were collected by a water immersion objective lens (UPlanSApo 60x; Olympus), filtered through a 540 nm long-pass filter (LP03-532RU-25; Semrock), separated with a dichroic mirror (635dcxr; Chroma Technology), and imaged onto an electron multiplying (EM)-charge coupled device (CCD) camera (Ixon DV887, Andor). During the buffer exchange experiments, a new buffer was infused in real time into the detection chamber by using a syringe pump (PHD 22/2000; Harvard Apparatus) while single-molecule images were being taken. For stable long time FRET measurement in the single molecule cyclization assay, a home-built autofocusing system was employed (Hwang et al., 2012).
Fluorescence Image Processing
The time course of FRET efficiency traces, which is defined as the ratio of acceptor intensity to the sum of donor and acceptor intensities, were obtained using IDL (Exelis, USA) and Matlab (Mathworks, USA) scripts. Background subtraction and bleed-through correction of the donor signal to the acceptor channel were considered.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data fitting for histograms and rate constants were performed using OriginPro 9.1 (OriginLabs, USA).
Determination of Kinetic Rates
Association/dissociation, bending/straightening, and religation events were determined by the simple threshold method. Occasionally, we used an eye inspection of fluorescence intensity and FRET time traces to fix errors. Dwell-time histograms of each state were fitted by an exponential decay function to obtain corresponding kinetic rates (Table S1). Since DNA duplexes have two competing reaction pathways of bending and dissociation, the dwell-time histogram of the enzyme-bound state without bending provides the sum of bending and dissociation rates, which is multiplied by the relative frequency of each event to obtain individual rates of bending and dissociation (Table S3) (Lee et al., 2012; Nahas et al., 2004). The cleavage time τ3 = 1/k3 was approximately calculated as the total time to stay in the bent state before occurring a cleavage event. For this calculation, we used the following equation:
where T is the total time required for the transition from unbound to cleaved states, which was characterized by the average time interval between cleavage events. Next, k1 is approximately the frequency of binding event. Thus, T × k1 represents total number of binding trials before occurring a cleavage events. Finally, k2/k−1 is the relative frequency of bending event to binding event, 1/k−2 is the average lifetime of single bending events. Consequently, τ3 represents the total time that the DNA stays in the bent state before occurring a cleavage event. For facilitating the measurement of T, the concentration of enzyme was raised (final concentration 44 nM) and rescaled in the above calculation. In the measurement of cleavage and religation rates, the long-lived high FRET state continuing for more than 5 s was considered as a cleavage event (Figure S2). It is possible that multiple cleavage/religation events occur during the single long-lived state. However, we assumed that the long-lived bent state is a single cleavage/religation event based on the fact that the transition probability of straightening was much higher than the probability of cleavage as the other competing reaction pathway under our experimental conditions (Table S4).
Looping/unlooping rates for each construct were estimated by fitting high/low FRET dwell time histograms to single exponential decay functions constructed by collecting looping and unlooping time from individual time traces. To collect the dwell times without bias, only the initial five consecutive looping and unlooping dwell times were collected from each time trace as a few time traces showed faster kinetics containing more looping/unlooping events as compared to other traces.
Estimation for a Theroretical Limit of Observation Time in Cyclization Assay
In our single-molecule cyclization assay, surface-immobilized DNA molecules were imaged. The image was taken with exposure time 0.5 sec, with 1000x EM gain. First, we converted the signal intensity shown in Figure 6 to the number of real incident photons by following the calculation method presented by the manufacturer of our EMCCD. As a result, we found that the average number of photons collected from single fluorophore during single exposure time (0.5 s) was about 70 photons under our experimental condition as described in Calculation 1 below.
Calculation 1: # of real photons collected in a measurement = [Signal intensity in a measurement (~650 count)] × [pre-amplifier gain (4)] × [data scaler (25)] ÷ [EM gain (1000)] ÷ [Quantum Efficiency @ 550-nm (~0.9)] ≈70 photons from single fluorophore per a measurement (during 0.5 s exposure time)
By combining this result with the fact that a robust organic fluorophore such as Cy3 and Cy5 could emit 105~106 photons before photobleaching, we estimated a theoretical limit of observation time for our single-molecule cyclization assay and found that the observation time would be expected to 12~120 minutes as described in Calculation 2 below. As expected from the theoretical estimation, most of the time traces were photobleached within 90 minutes.
Calculation 2: the expected observation time = [total # of emitted photons from organic dye before photobleaching (105~106 photons)] ÷ [# of photons collected in a measurement (70 photons)] × time resolution (0.5-s) ÷ [second/minute (60)] ≈ 12~120 minutes
DATA AND SOFTWARE AVAILABILITY
IDL and Matlab scripts were used for analyzing fluorescence images from EM-CCD. Custom scripts are available upon request to the corresponding author Sanghwa Lee (sanglee@gist.ac.kr).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial Strains | ||
| Saccharomyces cerevisiae DBY745 | Worland and Wang, 1989 | N/A |
| Chemicals and Recombinant Proteins | ||
| mPEG-Succinimidyl Valerate, MW5000 | Laysan Bio Inc. | mPEG-SVA-5000 |
| Biotin-PEG-SVA, MW5000 | Laysan Bio Inc. | Biotin-PEG-SVA-5000 |
| Streptavidin | Thermo Fisher | S888 |
| Cy3/Cy5 NHS Ester | GE Healthcare | PA13101 / PA15101 |
| D-(+)-Glucose | Sigma | G7528-250G |
| Trolox | Sigma | 238813-1G |
| Glucose oxidase | Sigma | G2133-50KU |
| Catalase | Sigma | C100-50MG |
| Oligonucleotides | ||
| Yclv1 for enzyme assay AGCGGTATCAGCAGGATGACGATGCGCGCATCGTCATCCTACAGAATCAGTTTTTTTTTTTTTTTT |
Integrated DNA Technology (IDT) | N/A (custom) |
| Yclv2 for enzyme assay AGCGGTATCAGCAATCTAACAATGCGCGCATTGTTAGATTACAGAATCAGTTTTTTTTTTTTTTTT |
Integrated DNA Technology (IDT) | N/A (custom) |
| Yclv3 for enzyme assay AGCGGTATCAGCAGGATGACGATGAGCTCATCGTCATCCTACAGAATCAGTTTTTTTTTTTTTTTT |
Integrated DNA Technology (IDT) | N/A (custom) |
| Yclv4 for enzyme assay AGCGGTATCAGCAGGATAACGATGAGCTCATCGTTATCCTACAGAATCAGTTTTTTTTTTTTTTTT |
Integrated DNA Technology (IDT) | N/A (custom) |
| Yclv5 for enzyme assay AGCGGTATCAGCAGGATAACAATGAGCTCATTGTTATCCTACAGAATCAGTTTTTTTTTTTTTTTT |
Integrated DNA Technology (IDT) | N/A (custom) |
| Yclv6 for enzyme assay CCCTTTCCTTCATCGATAAGCTTTAATTAAAGCTTATCGATGAAAAGGAATTTTTTTTTTTTTTTT |
Integrated DNA Technology (IDT) | N/A (custom) |
| Complementary oligonucleotides for sequences above, See Table S2 | Integrated DNA Technology (IDT) | N/A (custom) |
| Oligonucleotides for cyclization assay, See Table S2 | Integrated DNA Technology (IDT) | N/A (custom) |
| Software and Algorithms | ||
| IDL | Exelis Visual Information Solutions | https://www.harrisgeospatial.com/Software-Technology/IDL |
| Matlab R2017b | Mathworks | https://www.mathworks.com/products/matlab.html |
| OriginPro 9.1 | OriginLabs | https://www.originlab.com/Origin |
| smFRET data acquisition program | Taekjip Ha (Johns Hopkins Univ.) | http://ha.med.jhmi.edu/resources |
| IDL scripts (custom) | This work | N/A |
| Matlab scripts (custom) | This work | N/A |
SIGNIFICANCE.
The regulation of the DNA cleavage reaction by topoisomerase II is an essential for cell survival and is required for proper chromosomal structure and segregation. Beyond its critical physiological functions, the DNA cleavage reaction is also an important target for anticancer drugs. Because of this biological and clinical importance of the enzyme-mediated cleavage reaction, its underlying mechanisms have been extensively studied. Especially, decades of biochemical studies have revealed that the cleavage reactions occur only at preferred DNA sequences. Although this sequence-preferential cleavage of the DNA is well characterized, population averaging inherent in ensemble measurements makes it difficult to understand the mechanism of the cleavage site selection. Here we identified the individual steps of the DNA cleavage reaction using single-molecule fluorescence assays, and found that the cleavage site is selected by protein-induced flexibility of DNA, not an intrinsic property of DNA itself, during the DNA bending step. Our work not only provides mechanistic insight into target recognition of type II topoisomerases, but also presents an experimental approach for elucidating the molecular mechanisms by which other enzymes that interact with DNA or RNA recognize their target sites.
Highlights.
Reaction rates for individual steps in the DNA cleavage reaction are characterized
Kinetic pathway of divalent ions in the DNA cleavage reaction is revealed
Sequence-preferential DNA bending is induced for the cleavage site selection
It is determined by protein-DNA interactions rather than by the intrinsic property
ACKNOWLEDGEMENTS
We thank all the members of our labs for their kind discussion and help, and S. Hohng (Seoul National University) for the contribution to this work at early stage. This work was supported by a grant from the National R&D Program for Cancer Control of Ministry for Health and Welfare of Korea (1520090), and National Research Foundation of Korea (NRF) Grant funded by the Korean Government (MSIP) (No. 2016R1A5A1007318 and 2018R1D1A1B07051186), the GIST Research Institute to S.L., and by NIH research grants GM033944 and GM126363 to N.O.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Bax BD, Chan PF, Eggleston DS, Fosberry A, Gentry DR, Gorrec F, Giordano I, Hann MM, Hennessy A, Hibbs M, et al. (2010). Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466, 935–940. [DOI] [PubMed] [Google Scholar]
- Berger JM, Gamblin SJ, Harrison SC, and Wang JC (1996). Structure and mechanism of DNA topoisomerase II. Nature 379, 225–232. [DOI] [PubMed] [Google Scholar]
- Boedicker JQ, Garcia HG, Johnson S, and Phillips R (2013). DNA sequence-dependent mechanics and protein-assisted bending in repressor-mediated loop formation. Phys Biol 10, 066005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg KD, Hendricks C, Burgin AB, and Osheroff N (2002). Human topoisomerase IIα possesses an intrinsic nucleic acid specificity for DNA ligation. Use of 5' covalently activated oligonucleotide substrates to study enzyme mechanism. J Biol Chem 277, 31201–31206. [DOI] [PubMed] [Google Scholar]
- Bromberg KD, and Osheroff N (2001). DNA cleavage and religation by human topoisomerase IIα at high temperature. Biochemistry 40, 8410–8418. [DOI] [PubMed] [Google Scholar]
- Bromberg KD, Velez-Cruz R, Burgin AB, and Osheroff N (2004). DNA ligation catalyzed by human topoisomerase IIα. Biochemistry 43, 13416–13423. [DOI] [PubMed] [Google Scholar]
- Burden DA, and Osheroff N (1999). In vitro evolution of preferred topoisomerase II DNA cleavage sites. J Biol Chem 274, 5227–5235. [DOI] [PubMed] [Google Scholar]
- Capranico G, and Binaschi M (1998). DNA sequence selectivity of topoisomerases and topoisomerase poisons. Biochim Biophys Acta 1400, 185–194. [DOI] [PubMed] [Google Scholar]
- Deweese JE, Burgin AB, and Osheroff N (2008a). Human topoisomerase IIα uses a two-metal-ion mechanism for DNA cleavage. Nucleic Acids Res 36, 4883–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deweese JE, Burgin AB, and Osheroff N (2008b). Using 3'-bridging phosphorothiolates to isolate the forward DNA cleavage reaction of human topoisomerase IIα. Biochemistry 47, 4129–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deweese JE, and Osheroff N (2009). The DNA cleavage reaction of topoisomerase II: wolf in sheep's clothing. Nucleic Acids Res 37, 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deweese JE, and Osheroff N (2010). The use of divalent metal ions by type II topoisomerases. Metallomics 2, 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong KC, and Berger JM (2007). Structural basis for gate-DNA recognition and bending by type IIA topoisomerases. Nature 450, 1201–1205. [DOI] [PubMed] [Google Scholar]
- Fortune JM, Dickey JS, Lavrukhin OV, Van Etten JL, Lloyd RS, and Osheroff N (2002). Site-specific DNA cleavage by Chlorella virus topoisomerase II. Biochemistry 41, 11761–11769. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M, Simon I, and Bondos S (2011). Dynamic protein-DNA recognition: beyond what can be seen. Trends Biochem Sci 36, 415–423. [DOI] [PubMed] [Google Scholar]
- Garvie CW, and Wolberger C (2001). Recognition of specific DNA sequences. Mol Cell 8, 937–946. [DOI] [PubMed] [Google Scholar]
- Hardin AH, Sarkar SK, Seol Y, Liou GF, Osheroff N, and Neuman KC (2011). Direct measurement of DNA bending by type IIA topoisomerases: implications for non-equilibrium topology simplification. Nucleic Acids Res 39, 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H, Kim H, and Myong S (2011). Protein induced fluorescence enhancement as a single molecule assay with short distance sensitivity. Proc Natl Acad Sci U S A 108, 7414–7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang W, Bae S, and Hohng S (2012). Autofocusing system based on optical astigmatism analysis of single-molecule images. Opt Express 20, 29353–29360. [DOI] [PubMed] [Google Scholar]
- Kas E, and Laemmli UK (1992). In vivo topoisomerase II cleavage of the Drosophila histone and satellite III repeats: DNA sequence and structural characteristics. EMBO J 11, 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TT, and Kim HD (2013). Measuring shape-dependent looping probability of DNA. Biophys J 104, 2068–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TT, and Kim HD (2014). Probing the elastic limit of DNA bending. Nucleic Acids Res 42, 10786–10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Dong KC, and Berger JM (2013). The role of DNA bending in type IIA topoisomerase function. Nucleic Acids Res 41, 5444–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Jung SR, Heo K, Byl JA, Deweese JE, Osheroff N, and Hohng S (2012). DNA cleavage and opening reactions of human topoisomerase IIα are regulated via Mg2+-mediated dynamic bending of gate-DNA. Proc Natl Acad Sci U S A 109, 2925–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LF, Rowe TC, Yang L, Tewey KM, and Chen GL (1983). Cleavage of DNA by mammalian DNA topoisomerase II. J Biol Chem 258, 15365–15370. [PubMed] [Google Scholar]
- Luo G, Wang M, Konigsberg WH, and Xie XS (2007). Single-molecule and ensemble fluorescence assays for a functionally important conformational change in T7 DNA polymerase. Proc Natl Acad Sci U S A 104, 12610–12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AM, Sam MD, Reich NO, and Perona JJ (1999). Structural and energetic origins of indirect readout in site-specific DNA cleavage by a restriction endonuclease. Nat Struct Biol 6, 269–277. [DOI] [PubMed] [Google Scholar]
- McClendon AK, and Osheroff N (2007). DNA topoisomerase II, genotoxicity, and cancer. Mutat Res 623, 83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miassod R, Razin SV, and Hancock R (1997). Distribution of topoisomerase II-mediated cleavage sites and relation to structural and functional landmarks in 830 kb of Drosophila DNA. Nucleic Acids Res 25, 2041–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller-Planitz F, and Herschlag D (2007). DNA topoisomerase II selects DNA cleavage sites based on reactivity rather than binding affinity. Nucleic Acids Res 35, 3764–3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munteanu MG, Vlahovicek K, Parthasarathy S, Simon I, and Pongor S (1998). Rod models of DNA: sequence-dependent anisotropic elastic modelling of local bending phenomena. Trends Biochem Sci 23, 341–347. [DOI] [PubMed] [Google Scholar]
- Nahas MK, Wilson TJ, Hohng S, Jarvie K, Lilley DM, and Ha T (2004). Observation of internal cleavage and ligation reactions of a ribozyme. Nat Struct Mol Biol 11, 1107–1113. [DOI] [PubMed] [Google Scholar]
- Nitiss JL (2009a). DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer 9, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss JL (2009b). Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 9, 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osheroff N, and Zechiedrich EL (1987). Calcium-promoted DNA cleavage by eukaryotic topoisomerase II: trapping the covalent enzyme-DNA complex in an active form. Biochemistry 26, 4303–4309. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Schevitz RW, Zhang RG, Lawson CL, Joachimiak A, Marmorstein RQ, Luisi BF, and Sigler PB (1988). Crystal structure of trp repressor/operator complex at atomic resolution. Nature 335, 321–329. [DOI] [PubMed] [Google Scholar]
- Pendleton M, Lindsey RH, Felix CA, Grimwade D, and Osheroff N (2014). Topoisomerase II and leukemia. Ann N Y Acad Sci 1310, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, Sun Y, Huang SN, and Nitiss JL (2016). Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol 17, 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasnik I, McKinney SA, and Ha T (2006). Nonblinking and long-lasting single-molecule fluorescence imaging. Nat Methods 3, 891–893. [DOI] [PubMed] [Google Scholar]
- Rohs R, Jin X, West SM, Joshi R, Honig B, and Mann RS (2010). Origins of specificity in protein-DNA recognition. Annu Rev Biochem 79, 233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Hohng S, and Ha T (2008). A practical guide to single-molecule FRET. Nat Methods 5, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn ME, Connolly BK, Gurunathan K, and Levitus M (2007). Fluorescence properties and photophysics of the sulfoindocyanine Cy3 linked covalently to DNA. J Phys Chem B 111, 11064–11074. [DOI] [PubMed] [Google Scholar]
- Sander M, and Hsieh T (1983). Double strand DNA cleavage by type II DNA topoisomerase from Drosophila melanogaster. J Biol Chem 258, 8421–8428. [PubMed] [Google Scholar]
- Sander M, and Hsieh TS (1985). Drosophila topoisomerase II double-strand DNA cleavage: analysis of DNA sequence homology at the cleavage site. Nucleic Acids Res 13, 1057–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt BH, Burgin AB, Deweese JE, Osheroff N, and Berger JM (2010). A novel and unified two-metal mechanism for DNA cleavage by type II and IA topoisomerases. Nature 465, 641–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokina M, Koh HR, Patel SS, and Ha T (2009). Fluorescent lifetime trajectories of a single fluorophore reveal reaction intermediates during transcription initiation. J Am Chem Soc 131, 9630–9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzner JR, Chung IK, and Muller MT (1990). Eukaryotic topoisomerase II preferentially cleaves alternating purine-pyrimidine repeats. Nucleic Acids Res 18, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzner JR, and Muller MT (1988). A consensus sequence for cleavage by vertebrate DNA topoisomerase II. Nucleic Acids Res 16, 5533–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafabakhsh R, and Ha T (2012). Extreme bendability of DNA less than 100 base pairs long revealed by single-molecule cyclization. Science 337, 1097–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velez-Cruz R, Riggins JN, Daniels JS, Cai H, Guengerich FP, Marnett LJ, and Osheroff N (2005). Exocyclic DNA lesions stimulate DNA cleavage mediated by human topoisomerase IIα in vitro and in cultured cells. Biochemistry 44, 3972–3981. [DOI] [PubMed] [Google Scholar]
- Vos SM, Tretter EM, Schmidt BH, and Berger JM (2011). All tangled up: how cells direct, manage and exploit topoisomerase function. Nat Rev Mol Cell Biol 12, 827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC (1985). DNA topoisomerases. Annu Rev Biochem 54, 665–697. [DOI] [PubMed] [Google Scholar]
- Wang JC (2002). Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 3, 430–440. [DOI] [PubMed] [Google Scholar]
- Waters JT, and Kim HD (2013). Equilibrium of s surface-pinned semiflexible polymer. Macromolecules 46, 6659–6666. [Google Scholar]
- Wendorff TJ, Schmidt BH, Heslop P, Austin CA, and Berger JM (2012). The structure of DNA-bound human topoisomerase II alpha: conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J Mol Biol 424, 109–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worland ST, and Wang JC (1989). Inducible overexpression, purification, and active site mapping of DNA topoisomerase II from the yeast Saccharomyces cerevisiae. J Biol Chem 264, 4412–4416. [PubMed] [Google Scholar]
- Wu CC, Li TK, Farh L, Lin LY, Lin TS, Yu YJ, Yen TJ, Chiang CW, and Chan NL (2011). Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 333, 459–462. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
IDL and Matlab scripts were used for analyzing fluorescence images from EM-CCD. Custom scripts are available upon request to the corresponding author Sanghwa Lee (sanglee@gist.ac.kr).