

Abstract
Deeper exploration of uncharacterized Gcn5-related N-acetyltransferases has the potential to expand our knowledge of the types of molecules that can be acylated by this important superfamily of enzymes and may offer new opportunities for biotechnological applications. While determining native or biologically relevant in vivo functions of uncharacterized proteins is ideal, their alternative or promiscuous in vitro capabilities provide insight into key active site interactions. Additionally, this knowledge can be exploited to selectively modify complex molecules and reduce byproducts when synthetic routes become challenging. During our exploration of uncharacterized Gcn5-related N-acetyltransferases from Pseudomonas aeruginosa, we identified such an example. We found that the PA3944 enzyme acetylates both polymyxin B and colistin on a single diaminobutyric acid residue closest to the macrocyclic ring of the antimicrobial peptide and determined the PA3944 crystal structure. This finding is important for several reasons. (1) To the best of our knowledge, this is the first report of enzymatic acylation of polymyxins and thus reveals a new type of substrate that this enzyme family can use. (2) The enzymatic acetylation offers a controlled method for antibiotic modification compared to classical promiscuous chemical methods. (3) The site of acetylation would reduce the overall positive charge of the molecule, which is important for reducing nephrotoxic effects and may be a salvage strategy for this important class of antibiotics. While the physiological substrate for this enzyme remains unknown, our structural and functional characterization of PA3944 offers insight into its unique noncanonical substrate specificity.
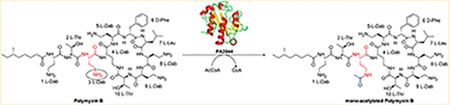
Graphical Abstract
Gcn5-related N-acetyltransferases (GNATs) make up a fascinating family of enzymes that acylate a variety of substrates ranging from small molecules to macromolecules. Members of this family are well-known for their roles in aminoglycoside antibiotic resistance, histone modification, protein acetylation, xenobiotic metabolism, and other cellular processes (reviewed in refs 1 and 2). They accomplish this by acylating a primary amine of an acceptor molecule with a donor molecule like acetyl-coenzyme A (AcCoA). While N-acylation is most common, O-acylation3,4 and S-acylation5 can also occur. The structures and functions of GNATs from all domains of life have been extensively studied; however, a significantly large number of them remain uncharacterized. On average, most organisms have approximately 20 GNATs encoded within their genomes; however, this number varies by organism and can be much lower or much greater.2 For instance, Campylobacter jejuni has four genes that encode GNATs, while Streptomyces coelicolor has 72.2 Thus, not all GNATs are conserved across organisms, which implies that the presence of certain GNATs may be tailored to the individual needs of the organism.
To gain a deeper understanding of the diversity of GNAT functions within a single bacterium, we have selected Pseudomonas aeruginosa PAO1 as a model system to further study GNATs of unknown function. The rationale for this selection is as follows. First, this bacterium is found in a variety of environments, including in water,6 in soil near the roots of plants,7 on medical devices such as catheters,8 and in the lungs of cystic fibrosis patients.9 It is a highly antibiotic-resistant bacterium that causes severe nosocomial infections, especially in immunocompromised individuals.10 Thus, knowledge gained about GNATs from this organism has important implications for agriculture and human disease as it is one of the ESKAPE11 pathogens. Second, it is genetically tractable; i.e., it can be manipulated to explore the effects of deleting or overexpressing a gene in the organism in the laboratory, and an entire curated database (Pseudomonas Genome DB, www.pseudomonas.com/12) is maintained to house information related to its study. Finally, P. aeruginosa contains 36 GNATs in its genome (Table S1), and a majority of them have been annotated as hypothetical proteins or as proteins of unknown function. Nine of them have been structurally characterized (Table S1), but the depth of functional information is even poorer.
Because of the diversity of functions of GNATs already identified to date, we expect that exploring uncharacterized GNAT proteins will expand the repertoire of substrates utilized by members of this family of proteins. Additional benefits of studying GNATs of unknown function include improving their functional annotation across diverse genomes and identifying new targeted reactions that can be utilized for enzyme-mediated chemical synthesis or biotechnological applications. In an effort to expand our knowledge of the functional space of GNATs, we previously designed a broad substrate screening assay to identify potential lead compounds to further explore the substrate specificity and catalytic activity of uncharacterized GNATs.13 While this screen has its limitations, it does provide a starting point for further GNAT functional characterization.
Here, we focused on the structural and kinetic characterization of the PA3944 enzyme. We previously screened this enzyme against our panel of potential substrates and found it could acetylate polymyxin B and E (colistin) antimicrobial peptide antibiotics as well as the dipeptide aspartame.13 Because the enzymatic activity toward polymyxins is novel, we concentrated our structural and functional characterization of PA3944 on its ability to acetylate polymyxins. These antibiotics have five potential sites for N-acetylation; therefore, we sought to further explore whether the enzyme was specific for one site or if it was promiscuous in its acetylation activity. While we do not believe the native function of this enzyme is to modify polymyxin antibiotics, we do think we can uncover clues about PA3944 substrate specificity by studying how this enzyme modifies these compounds. Because this is the first description of a polymyxin antibiotic-modifying enzyme, we also determined its crystal structure. Collectively, our study provides structural and functional information about this enzyme, which will be useful for (1) identifying other potential polymyxin-modifying GNAT enzymes and (2) engineering GNATs to selectively modify these antibiotics to salvage them in the clinic or use them for other biotechnological applications.
METHODS
Clone.
The PA3944 gene (UniProtKB entry Q9HX72) from P. aeruginosa was subcloned into the p11 vector as described previously.14 It contains an ampicillin resistance cassette, and gene expression is under control of the T7 promoter. Protein produced with this system contains an N-terminal methionine residue followed by a three-amino acid spacer (GSS), a hexahistidine tag, another four-amino acid spacer (SSGR), a tobacco etch virus (TEV) protease cleavage site (ENLYFQ/G), and a histidine spacer residue (i.e., MGSSHHHHHHSSGRENLYFQGH) prior to the first methionine of the PA3944 protein sequence.
Protein Expression and Purification for Kinetics and Mass Spectrometry Assays.
Cell Growth and Protein Expression.
The clone containing the PA3944 gene was transformed into Escherichia coli BL21(DE3) competent cells and stored as a glycerol stock at −80 °C until ready to use. A 5 mL starter culture of lysogeny broth (LB) medium with 100 μg/mL ampicillin was then inoculated and grown overnight at 37 °C while being shaken at 200 rpm on a benchtop shaker. The next day, 2.5 mL of starter culture was added to 250 mL of Terrific Broth (TB) with 100 μg/mL ampicillin in a 2 L glass Erlenmeyer flask and shaken at 200 rpm and 37 °C until the OD600 reached 0.6−0.8. Cells were placed on ice until they were cool, and then 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to induce protein expression at room temperature while being shaken at 150 rpm overnight. Cells were harvested by centrifugation at 2200g for 30 min at 4 °C, and the pellet was resuspended in 37.5 mL of cold lysis buffer [10 mM Tris-HCl (pH 8.3), 500 mM NaCl, 5 mM imidazole, 5% glycerol, and 5 mM β-mercaptoethanol (BME)], sonicated on ice for 5 min, and then stored at −80 °C until the protein was ready to be purified.
Protein Purification and Tag Cleavage.
The lysed cells were thawed and centrifuged at 25000g for 45 min in a Sorvall SS-34 rotor at 4 °C. The crude extract (supernatant) was loaded onto a 1 mL HisTrap FF nickel affinity column (GE Healthcare) that had been equilibrated with buffer A [10 mM Tris-HCl (pH 8.3), 500 mM NaCl, and 5 mM BME] on an ÄKTA Start FPLC instrument (GE Healthcare). Afterward, the column was washed with 5 column volumes (CV) of 5% buffer B [10 mM Tris-HCl (pH 8.3), 500 mM NaCl, 5 mM BME, and 500 mM imidazole], and protein was eluted with 100% buffer B over 5 CV. To remove imidazole, we performed a buffer exchange of the eluted protein into buffer A using a PD-10 gravity flow column (GE-Healthcare). Finally, we concentrated the protein using a 10K molecular weight cutoff (MWCO) Sartorius VivaSpin concentrator. To remove the polyhistidine tag, 5 mg of protein was incubated with TEV protease [previously purified in our laboratory using the same protocol but stored in 10 mM Tris-HCl (pH 8.3), 500 mM NaCl, 5 mM BME, 2 mM EDTA, 5 mM DTT, and 50% glycerol] in a 20:1 ratio at 4 °C overnight in cleavage buffer [50 mM Tris (pH 8.3), 1 mM DTT, 300 mM NaCl, and 5% glycerol] in a 15 mL 10K MWCO Slide-a-Lyzer Mini-dialysis device (Thermo Fisher Scientific). Cleavage buffer was exchanged once after dialysis for 2 h (we chose incubation for cleavage at 4 °C because at higher temperatures all of the protein precipitated). The next morning, the dialysate was centrifuged at 12000g for 10 min at 4 °C and the supernatant was diluted to a total volume of 3 mL with buffer A and then loaded onto the affinity column previously equilibrated with buffer A. The cleaved protein was eluted with a gradient from 0 to 30% buffer B, concentrated and exchanged into buffer without BME [10 mM Tris-HCl (pH 8.3) and 500 mM NaCl] because the BME interferes with the colorimetric enzyme kinetic assay. Protein was stored in aliquots at −80 °C. Under these storage conditions, protein was stable for at least 1 year and no change in enzymatic activity was observed.
Protein Crystallization and Size-Exclusion Chromatography.
Detailed methods for protein crystallization and size-exclusion chromatography can be found in the Supporting Information.
Data Collection and Structure Determination.
Diffraction data were collected at 100 K at the LS-CAT 21 ID-G [wavelength of 0.97856 Å, 12670 eV; Protein Data Bank (PDB) entry 6edd] and SBC-CAT 19-ID [0.97926 Å, 12661 eV; PDB entry 6edv] beamlines at the Advanced Photon Source at Argonne National Laboratory. Data were processed with HKL-2000,15 and structure determination and model building were performed with HKL-300016 coupled with MOLREP,17 BUCCANEER,18 and Fitmunk.19 Models were further refined with REFMAC520 and rebuilt with COOT.21 The quality of the models was assessed with Molprobity22 and wwPDB Validation Service.23 Molecular replacement was performed using PDB entry 3fbu (UniProt entry Q81Q99) as the search model. All structures have been deposited into the PDB as entries 6edv and 6edd (Table 1), and diffraction images have been deposited into the Integrated Resource for Reproducibility in Macromolecular Crystallography24 (proteindiffraction.org) with the following identifiers: doi.org/10.18430/m36edd and doi.org/10.18430/m36edv, respectively.
Table 1.
Data Collection and Refinement Statistics for PA3944 Structures
| 6edd | 6edv | |
|---|---|---|
| Data Collectiona | ||
| beamline | LS-CAT 21 ID-G | SBC-CAT 19 ID |
| resolution (Å) | 1.55 (1.55–1.58) | 1.35 (1.35–1.37) |
| space group | P1 | P212121 |
| wavelength (Å) | 0.97856 | 0.97926 |
| a, b, c (Å) | 36.45, 44.17, 60.12 | 36.44, 44.03, 111.64 |
| α, β, γ (deg) | 81.88, 73.32, 89.94 | 90, 90, 90 |
| completeness (%) | 96.2 (93.5) | 100.0 (99.7) |
| no. of reflections observed | 121370 | 558894 |
| no. of unique reflections | 49684 | 40378 |
| 〈I〉/〈σI〉 | 21.3 (1.9) | 46.4 (2.2) |
| CC1/2 last shell | 0.77 | 0.68 |
| redundancy | 2.4 (2.1) | 13.8 (4.6) |
| Rmerge | 0.053 (0.397) | 0.122 (0.778) |
| Wilson B factor (Å2) | 15.7 | 13.9 |
| Refinement | ||
| Rwork/Rfree | 0.166/0.194 | 0.133/0.162 |
| rmsd for bond lengths (Å) | 0.008 | 0.013 |
| rmsd for bond angles (deg) | 1.4 | 1.6 |
| mean B value (Å2) | 20 | 19 |
| no. of protein atoms | 3125 | 1513 |
| no. of water atoms | 476 | 155 |
| no. of ligand/ion atoms | 130 | 57 |
| Clashscore | 2.5 | 0.98 |
| Clashscore percentile (100) | 99 | 99 |
| rotamer outliers (%) | 0.00 | 0.00 |
| Ramachandran outliers (%) | 0.00 | 0.00 |
| residues with bad bonds/angles (%) | 0.00/0.05 | 0.00/0.00 |
| MolProbity score | 1.03 | 0.79 |
Values in parentheses are those of the highest-resolution shell.
Enzyme Kinetic Assays for the Colorimetric Method and Mass Spectrometry.
Steady-State Enzyme Kinetic Assays.
All kinetic assays were performed in triplicate using the previously described discontinuous colorimetric method13 with minor modifications. Briefly, all reactions were performed in 50 mM Tris-HCl (pH 8.0), except for pH studies where the buffer was 50 mM Bis-Tris-propane at pH 6.5, 7.0, 8.0, 9.0, or 9.5, in a total volume of 50 μL for 10 min at 37 °C and initiated with 10 μL of enzyme. Reactions were terminated using 50 μL of 100 mM Tris-HCl (pH 8.0) and 6 M guanidine HCl to denature the protein; then 200 μL of Ellman’s reagent in buffer [0.2 mM 5,5’-dithiobis(2-nitrobenzoic acid) (DTNB), 100 mM Tris-HCl (pH 8.0), and 1 mM EDTA] was added to each sample, and then incubated at room temperature prior to measuring the absorbance at 415 nm with a fixed wavelength filter on a BioTek ELx808 microplate reader. Each molecule of CoA produced during the reaction reacts with one molecule of DTNB, which forms a 2-nitro-5-thiobenzoate (TNB2−) anion under basic conditions that absorbs at 415 nm. CoA standards (2.5 and 5 nmol) were used to determine the response factor for TNB2− at 415 nm, which was used to convert TNB2− absorbance to the number of nanomoles of CoA product produced during the enzymatic reaction. One unit of enzymatic activity is defined as 1 μmol of TNB2− produced/min. Kinetic parameters were determined by fitting data to the Michaelis−Menten equation using Origin 2017 software. Details of the concentrations of donor (AcCoA) and acceptor substrates used for each assay are described below.
The PA3944 enzyme was screened against a panel of the following compounds in Tris-HCl buffer (pH 8.0) to determine its substrate specificity and promiscuity: L-glutamate, L-aspartate, taurine, carnosine, β-alanine, aspartame, glycine, L-alanine, L-2,3-diaminopropionic acid (DAP), colistin, polymyxin B, L-2,4-diaminobutyric acid (Dab), L-ornithine, and L-lysine. Each reaction was performed in the presence of 0.5 mM AcCoA, 5 mM compound, and 0.91 μM enzyme. Substrate saturation curves were produced using 0.64 μM tagged or cleaved PA3944 enzyme and kinetic parameters were determined (Table 2). The tagged enzyme was used for substrate screening assays and pH studies, while the cleaved protein was used for substrate saturation curves. For polymyxin B tagged enzyme was used for substrate screening assays and pH studies, while the cleaved protein was used for substrate saturation curves where kinetic parameters were determined (Table 2). For polymyxin B, colistin, and Dab substrate saturation curves, the AcCoA concentration was held constant at 0.5 mM while the acceptor substrate concentration was varied from 0 to 10 mM for polymyxin B and colistin and from 0 to 20 mM for Dab. For AcCoA substrate saturation curves, the polymyxin B or colistin concentration was held constant at 5 mM while the AcCoA concentration was varied from 0 to 2 mM.
Table 2.
Comparison of Kinetic Parameters for Tagged and Cleaved PA3944 Enzyme
| PA3944 enzyme | substrate | KM (mM) | kcat(s−1) | kcat/KM(M−1 s−1) |
|---|---|---|---|---|
| tagged | polymyxin B (constant [AcCoA])a | 2.57 ± 0.23 | 0.653 | 2.54 × 102 |
| colistin (constant [AcCoA])a | 2.28 ± 0.17 | 0.672 | 2.95 × 102 | |
| DAB (constant [AcCoA])a,b | 8.60 ± 1.59 | 0.264 | 30.7 × 10 | |
| AcCoA (constant [polymyxin B])c | 0.118 ± 0.028 | 0.602 | 5.10 × 103 | |
| AcCoA (constant [colistin])c | 0.105 ± 0.017 | 0.471 | 4.49 × 103 | |
| cleaved | polymyxin B (constant [AcCoA])a | 4.00 ± 0.32 | 0.923 | 3.65 × 102 |
| colistin (constant [AcCoA])a | 2.50 ± 0.13 | 0.894 | 3.58 × 102 | |
| DAB (constant [AcCoA])a,b | 10.0 ± 1.2 | 0.391 | 3.91 × 10 | |
| AcCoA (constant [polymyxin B])c | 0.137 ± 0.020 | 0.610 | 4.45 × 103 | |
| AcCoA (constant [colistin] )c | 0.118 ± 0.021 | 0.456 | 3.86 × 103 |
The concentration of AcCoA was held constant at 0.5 mM while that of the acceptor substrate(s) was varied.
At DAB concentrations of >10 mM, substrate inhibition was observed; therefore, kinetic parameters were calculated on the basis of data collected from 0 to 10 mM DAB.
The concentration of polymyxin B or colistin was held constant at 5 mM while that of AcCoA was varied.
Enzymatic Assays for Mass Spectrometric Analysis.
Enzymatic assays were performed for 1 h at 37 °C in triplicate in a 100 μL reaction volume in the presence of 50 mM Tris-HCl (pH 8.0), 1 mM AcCoA, 1 mM polymyxin B or colistin, and 0.64 μM tagged PA3944 enzyme. Control reactions were performed in the absence of enzyme. Reactions were terminated by adding 50% methanol to the reaction mixture, and then the mixtures were frozen and stored at −80 °C. Reaction products were analyzed by mass spectrometry as described below.
Mass Spectrometry.
Reaction mixtures were extracted by solid phase extraction prior to mass spectrometry analysis. They were then combined with 450 μL of trimethylamine acetate (pH 8.0), loaded onto a Waters Corp. HLB Oasis solid phase extraction cartridge (6 mL, 200 mg), and washed with 5 mL of 100 mM triethyl acetate (pH 8.0) and 5 mL of distilled water. The peptides were eluted with 5 mL of a 70:30 acetonitrile/water solution and evaporated to near dryness in a Speedvac evaporator. The residue was dissolved in a 75% methanol/water solution for direct infusion into the mass spectrometer. Molecular weight and product ion mass spectra were acquired with a Thermo Finnigan (San Jose, CA) Advantage LCQ ion trap mass spectrometer using electrospray ionization (ESI) in the positive ion mode. The tandem mass spectral energy parameter was set to 50−60% of its maximum value for fragmentation of all the singly protonated peptides in this study. Mass spectra of all the peptide ions were acquired by direct infusion of the 3:1 methanol/water mixture. The capillary temperature was 200 °C, and the spray voltage was 4.5 kV. Molecular weight and product ion spectra were acquired from each reaction mixture and are shown in the Supporting Information.
RESULTS AND DISCUSSION
PA3944 Genomic Context and Protein Properties.
The PA3944 gene from P. aeruginosa PAO1 lies within a five-gene operon (PA3941−PA3945) that encodes four hypothetical proteins and one predicted acyl-CoA thioesterase II (tesB). Two of these hypothetical proteins (PA3944 and PA3945) are predicted to have a GNAT domain; however, their function is unknown, and to the best of our knowledge, they have not been previously studied. The organization of this operon is conserved in P. aeruginosa strains PAO1 (an opportunistic pathogen and widely studied laboratory strain), DK2 (isolated from Danish cystic fibrosis patients), M18 (found in the sweet melon rhizosphere and found to produce antifungal compounds), and LESB58 (a hypervirulent cystic fibrosis isolate from Liverpool). The PA3944 gene encodes a 21.9 kDa protein with a pI of 5.32 and is found in both pathogenic and nonpathogenic strains of Pseudomonas in 289 genera.25
Three-Dimensional Crystal Structure of PA3944.
We previously screened the PA3944 protein against a panel of potential substrates and found the enzyme exhibited the highest activity toward aspartame, polymyxin B, and colistin (polymyxin E).13 The rationale for including these compounds in the screening assay was that they were relatively inexpensive peptides with several potential sites for N-acylation. Because of the unusual substrate specificity that PA3944 exhibited toward polymyxin B and colistin, we sought to determine its crystal structure to learn more about how these antibiotics become acetylated. To begin, we screened this protein for crystals in the presence and absence of AcCoA/CoA and a variety of substrates identified from our previous broad substrate screening assay.13 Two structures of PA3944 with CoA were determined in different space groups: P212121 with a monomer in the asymmetric unit (PDB entry 6edv) and P1 with two monomers in the asymmetric unit (PDB entry 6edd) (Table 1).
The PA3944 protein structure consists of five α-helices that surround an antiparallel β-sheet formed by six β-strands (Figure 1), which is a typical signature of GNATs.26 The canonical V-like splay is formed between β-strands B and C, and the topology of the PA3944 structure is similar to that of the PA4794 GNAT we studied previously.27 CoA was modeled into the electron density found between helices α3 and α4 and β-strands D and F and stabilized by interactions with the following residues: Trp105, Leu107, Gly113, Arg114, Gly115, Arg118, Thr141, Asn145, Ser148, and Arg154. This CoA binding pocket is also conserved across the P. aeruginosa PA3270, PA4794, and PA2578 GNATs (Table S1).
Figure 1.

Crystal structures of PA3944 in complex with CoA. (A) Monomer assigned to an orthorhombic space group (PDB entry 6edv). α-Helices (red) are labeled from α1 to α5, β-strands (yellow) labeled from A to F, and loops colored green. CoA is shown as spheres. (B) The N-terminus (spheres) of one monomer (PDB entry 6edd, chain B, yellow) is bound to the active site of the other monomer (PDB entry 6edd, chain B, blue). CoA is shown as green sticks. The N- and C-termini are labeled as N and C, respectively, in both panels.
We performed a structural similarity search of PA3944 against the PDB using VAST28 and found the highest degree of structural similarity with two uncharacterized proteins from Bordetella pertussis (PDB entry 3juw, 24.2% sequence identity) and Bacillus halodurans (PDB entry 2qml, 10.8% sequence identity). The structure in the PDB that exhibited the highest degree of sequence identity is an uncharacterized acetyltransferase from Agrobacterium tumefaciens (PDB entry 2fsr, 32.2% sequence identity). We then used DALI29 to compare the PA3944 structure to those of all other known P. aeruginosa GNATs (Figure 2) and found the highest degree of structural similarity between PA3944 and the uncharacterized PA3270 protein (PDB entry 1yre, 22.9% sequence identity). To determine whether we could predict the possible function of PA3944 from sequence or structure, we broadened our search to include GNATs that had been previously characterized from other organisms as described in the Supporting Information. Our comparison of the amino acid sequences of the acceptor substrate binding sites of these proteins did not show any sequence similarity to PA3944; therefore, we were unable to predict a possible native function for this enzyme on the basis of previously characterized GNATs. Moreover, we observed that the topologies of the GNAT structures were highly variable and the most similar proteins adopted different conformations of some of their secondary structures (Figure 2). Thus, it was not feasible to infer function from structure for the PA3944 enzyme.
Figure 2.

Structural comparison of PA3944 with other GNATs from P. aeruginosa and other organisms with known functions. The structure of PA3944 is shown in red and black on the left of the structure being compared. The putative acceptor substrate binding region of PA3944 is colored red, and the CoA ligand is shown as black spheres. The corresponding PDB entry is shown, and gene names that correspond to the PDB entries are as follows: 1xeb (PA0115), 2fe7 (PA0478), 3owc (PA2578), 2cnm (RimI from Salmonella typhimurium), 1yre (PA3270), 2j8r (PA4866), 4l8a (PA4794), 4r87 (SpeG from Vibrio cholerae), 2eui (PA4026), 2vi7 (PA1377), 4ubr (PA4534), and 4r9m (SpeG from E. coli).
The only difference between our two PA3944 structures (P212121, PDB entry 6edv, and P1, PDB entry 6edd) was the presence of a portion of the N-terminus in the active site of the adjacent monomer of the 6edd structure (Figure 1). Here, the histidine residue that remains after tag cleavage and the first four residues of the protein (HMNAN) are observed in the electron density, and the δ−2 nitrogen of Asn2 from one chain forms a H-bond with the backbone oxygen of Ser46 on a mobile loop of the second chain. In this conformation, the ε-carbon atom of Met1 of the N-terminus bound in the active site of the opposite monomer is 5.4 Å from the sulfhydryl group of CoA. Residues Met41−Leu48 on the loop between helices α1 and α2 interact with the N-terminus and block the entrance to the active site. The N-terminus of the 6edd structure was not modeled due to a lack of electron density for the first eight residues.
Because the PA3944 protein appeared to adopt two different oligomeric states (monomer and apparent dimer) in the different crystals, we performed size-exclusion chromatography to determine its oligomeric state in solution. We found the protein eluted as a monomer regardless of pH (see Methods for more details; Figure S1). This result is consistent with PISA predictions30 that did not show any significant interactions between monomers of the P1 (6edd) structure except those connected with binding a small portion of the N-terminus. Therefore, we conclude that the PA3944 protein is a monomer in solution and the observed binding of the N-terminus in the P1 (6edd) structure is a result of crystal packing but may indicate that the protein is capable of binding a peptide or peptidelike molecule. This is further supported by the fact that the closest sequence and structural homologues with known function acetylate proteins (ribosomal protein alanine acetyltransferase from Salmonella typhimurium, UniProt entry Q8ZJW4_SALTY) or cyclic peptides [FsC-acetyl coenzyme A N(2)-transacetylase from Neosartorya fumigate, UniProt entry SIDG_ASPFU] (Figure 3).
Figure 3.

Pairwise sequence comparison between PA3944 and closest GNAT family proteins with known functions. Sequences are colored according to pairwise similarity from red (highly similar regions) to white (intermediately similar) to blue (highly dissimilar). Similarity was calculated as BLOSUM62 scores averaged over three residues. The CoA binding site in PA3944 is colored orange, while its acceptor substrate binding site is colored teal. The sequences are labeled with UniProt entries.
PA3944 Acetylates Polymyxin B and Colistin on a Single Diaminobutyric Acid Residue.
Polymyxins are polycationic macrocyclic antimicrobial peptides that have been isolated from strains of the bacterium Bacillus polymyxa, and they disrupt the structure of the bacterial cell membrane by interacting with its phospholipids. The most prevalent polymyxins include polymyxin B (B1 and B2) and colistin (polymyxin E1 and E2). These two families of polymyxins differ in their D-amino acid composition and in the fatty acids that are attached to the N-terminus of the peptide. Both families contain identical L-amino acid residues; however, polymyxin B also contains a D-phenylalanine residue, whereas colistin has a D-leucine residue in its place. The fatty acid composition varies for each type of polymyxin; polymyxin B1 and colistin E1 have a 6-methyloctanoyl group, whereas polymyxin B2 and colistin E2 bear a 6-methylheptanoyl residue (reviewed in ref 31).
Because polymyxins have five potential sites for N-acetylation on diaminobutryic acid (Dab) residues within their structures (Figure 4), it was unclear whether the PA3944 enzyme acetylated a single Dab or multiple Dabs on each antibiotic. Our initial hypothesis was that the enzyme might acetylate Dab5 because it is located near the D-Phe/D-Leu of polymyxin B/colistin and most closely resembles the structure of aspartame, but we could not rule out the possibility that other Dabs might be modified. To determine the site(s) of acetylation on polymyxin B and colistin, we performed the enzymatic acetylation reaction and analyzed the products of the reaction using tandem mass spectrometry. Product ion analyses of colistin and polymyxin B indicate that the predominant acetylation product of PA3944 is a singly acetylated species and occurs only once on residue 3, the primary amine of the Dab residue proximal to the macrocycle in both antibiotics.
Figure 4.

Structures of polymyxins and substrate saturation curves for PA3944. The structures of polymyxin B and colistin are shown in panels A and B, respectively. The regioselective acetylation by PA3944 occurs on the Dab residue (colored red and circled). AcCoA substrate saturation curves are shown in panel C at a constant concentration of polymyxin B (red circles) and colistin (black squares). The tagged PA3944 protein is represented as filled circles or squares, and the cleaved protein is represented by empty circles or squares. Polymyxin B and colistin substrate saturation curves are shown in panel D at a constant concentration of AcCoA. The coloring for polymyxin B, colistin, tagged protein, and cleaved protein is the same as panel C. Error bars represent the standard deviation of triplicate reactions. See Methods for more details regarding assay conditions.
Mass spectrometry analysis of the enzymatic reaction mixture containing polymyxin B yielded a singly acetylated polymyxin B1 (M + H)+ ion at m/z 1246.3 that was selected for product ion analysis. Smaller abundances of singly acetylated polymyxin ions containing sodium were observed at m/z 1267.8 and 1289.7, as well. The acetylated polymyxin B2 formed ions in much smaller abundances (Figure S2). No unmodified polymyxin B1 or B2 was observed in this mass spectrum. The product ion spectrum of the singly acetylated polymyxin B1 [m/z 1246.3 (Figure S3)] shows an ion at m/z 904.8 formed by cleavage of the side chain resulting in the loss of a neutral fragment containing the (unmodified) primary amine group that is farthest from the cyclic peptide ring. The product ion observed at m/z 762.4 is the cyclic peptide fragment formed by loss of the side chain (analogous to the ion at m/z 728.3 in the product ion spectrum of the singly acetylated colistin on Dab3 in Scheme S1). Full scan MS analysis of the control reaction mixture containing polymyxin without PA3944 enzyme showed no acetylated products.
Analysis of the enzymatic reaction mixture containing colistin showed the singly acetylated colistin E2 (M + H)+ ion at m/z 1197.8 and the singly acetylated (M + Na)+ E1 and E2 ions at m/z 1233.9 and 1219.9, respectively (Figure S4). A protonated molecular ion from unmodified colistin E2 was observed at m/z 1155.8. Full scan MS analysis of the control reaction mixture containing colistin without PA3944 enzyme showed no acetylated products. The m/z 1197.8 ion, corresponding to a singly acetylated colistin E2, was subjected to product ion analysis to determine the site of colistin acetylation (Figure S5). The cleavage of the colistin side chain produces an acylium ion at m/z 997.4 (containing the acetyl group) formed by the loss of a fragment containing the unmodified primary amine, closest to the chain terminus, analogous to the ion at m/z 904.8 observed in the product ion analysis of polymyxin. The formation of a product ion at m/z 728.3 corresponds to the unacetylated cyclic peptide fragment and demonstrates that the acetyl modification is on the primary amine closest to the cyclic peptide (Dab3) (Scheme S2).
These results strongly suggest that the enzymatic reaction of PA3944 toward both polymyxin B and colistin is specific for Dab3, the Dab residue proximal to the cyclic peptide (Figure 4), as we did not observe multiple acetylation events on either of these peptides under our described assay conditions. The colistin E2 isoform was the predominant parent species as well as the predominant acetylated species observed in the MS assays, while the relative abundances of polymyxins B1 and B2 were similar. Because both E1/B1 and E2/B2 isoforms of the antibiotics were found to be acetylated, the differences in fatty acid and D-amino acid composition do not appear to strongly affect enzymatic acetylation of these substrates. Thus, the selectivity with respect to the location of acetylation on both polymyxins was consistent. This enzymatic specificity is in contrast to the nonselective chemical methods of acetylating polymyxin antibiotics, such as acetyl chloride or acetic anhydride, which are very reactive and nonselective.
PA3944 Exhibits Similar Catalytic Efficiencies with Respect to Both Polymyxin Acceptor Substrates.
We kinetically characterized the PA3944 enzyme with respect to polymyxin B and colistin using acetyl-coenzyme A (AcCoA) as the acyl donor. Because polymyxin B and colistin structures are quite similar (Figure 4), we expected the PA3944 enzyme would exhibit comparable kinetic parameters with respect to both acceptor substrates. We found no major difference in the catalytic efficiency of the enzyme with respect to polymyxin B or colistin when comparing tagged or cleaved enzyme kinetic parameters (Table 2 and Figure 4). This indicates the differences in fatty acids at the N-terminus and D-amino acid composition within the peptide sequence of polymyxin B and colistin do not significantly alter PA3944 activity. Additionally, no significant difference in catalytic efficiency was seen when AcCoA was varied and concentrations of polymyxins were held constant regardless of whether the affinity tag was present. Thus, the presence of the affinity tag does not significantly alter PA3944 enzymatic activity.
PA3944 Is Not an Efficient Diaminobutyric Acid Acetyltransferase.
Because PA3944 acetylated Dab3 on polymyxins, we wondered whether it would acetylate free Dab amino acids more efficiently than those within the antibiotics. Dab acetyltransferases, which are GNATs and part of the ectoine biosynthetic pathway, from the halophilic bacteria Methylomicrobium alcaliphilum, Methylophaga thalassica, Methylophaga alcalica, and Halomonas elongata have been studied.32–34 We found the apparent affinity of PA3944 for Dab was 1−2 orders of magnitude lower (8.6 or 10 mM; tagged vs cleaved, Table 2) than that reported for Me. alcaliphilum, M. thalassica, and M. alcalica (0.460, 0.365, and 0.375 mM, respectively33,34), and its catalytic efficiency was 1 order of magnitude lower with respect to Dab compared to polymyxin B and colistin (Table 2). Therefore, we concluded that under our described reaction conditions the PA3944 enzyme is an inefficient Dab acetyltransferase and likely has an alternative native function.
PA3944 Expanded Substrate Screening Assays.
To further explore the substrate specificity of the PA3944 enzyme, we performed an expanded screening assay with compounds denoted in Figure 5. Similar to our previous results, the enzyme exhibited the highest activity toward aspartame, polymyxin B, and colistin, but it is not obvious why polymyxins and aspartame would be acetylated to a similar extent because their structures are quite different. The previous substrate screen13 did not include a complete series of compounds to evaluate which structural components of these molecules are important for PA3944 recognition. Therefore, we selected compounds that were commercially available to determine the structural elements necessary for acetylation of aspartame and polymyxins (Figure 5). First, we focused on molecules that represented structural components of aspartame. We found L-glutamate, L-aspartate, glycine, and alanine are very poor substrates for the PA3944 enzyme. Limited activity was observed for substrates bearing an aminoethyl group (taurine, carnosine, and diaminobutyric acid); however, β-alanine, which also contains an aminoethyl group, was a poor substrate. This indicates that there are structural elements in aspartame that are more favorable for acetylation than the simpler aminecontaining substrates we assayed (Figure 5). We are currently in the process of synthesizing aspartame analogues to learn more about the structural elements of aspartame that are recognized by PA3944.
Figure 5.

Substrate screening of PA3944. The average activity of PA3944 from triplicate reactions with respect to each substrate is shown, and more details of assay conditions can be found in Methods. Structures of each substrate are overlaid on the bar graph with the site of acetylation colored red. Only partial structures of polymyxin B and colistin are shown. Full structures of these compounds can be found in Figure 4. Error bars represent the standard deviation of triplicate reactions.
Next, we turned our attention to PA3944 activity with respect to polymyxin B and colistin. Both polymyxins contain diaminobutyric acid residues; therefore, we investigated the importance of chain length between the α-carbon and primary amine by comparing activities with respect to free amino acids glycine, L-alanine, 2,3-diaminopropionic acid (Dap), 2,4-diaminobutyric acid (Dab), L-ornithine, and L-lysine (Figure 5). We found the optimal distance between the α-carbon and the primary amine consisted of two methylenes, like that of Dab. Therefore, we determined substrate saturation curves for Dab and found that the catalytic efficiency was 1 order of magnitude lower for the single free Dab residue than when it is incorporated into the cyclic peptide of polymyxin B or colistin (Table 2). This indicates that differences in charges of substrates or additional interactions between the enzyme and polymyxin substrates enhance activity and are important for substrate specificity.
Polymyxin Acetylation Is Enzymatic, and Optimal Activity Occurs at pH 8.
GNATs use a general acid/base catalytic mechanism to acetylate their acceptor substrates. This occurs when the enzyme deprotonates a primary amine of an acceptor substrate using either an acidic active site residue or a water molecule, and then a residue like tyrosine is used to reprotonate CoA after acetyl transfer. As the pH of the buffer used for enzymatic assays increases, there is a higher likelihood that the acceptor substrate is already deprotonated and would cloud our judgment of whether the observed reaction is indeed enzymatic. Therefore, to ensure that the polymyxin acetylation reaction we observed was enzymatic, we performed a series of kinetic assays in different buffers and at different pHs. First, we measured the kinetic activity of PA3944 with respect to polymyxin B in the presence of Tris-HCl (pH 8.0) or Bis-Tris-propane (pH 8.0) buffer and compared the kinetic parameters (Figure 6A). We found the catalytic efficiencies of the enzyme in Tris-HCl (pH 8.0) and Bis-Tris-propane (pH 8.0) were 2.47 × 102 and 7.33 × 102 M−1 s−1, respectively, indicating the catalytic efficiency of the enzyme in Bis-Tris-propane buffer is nearly 3-fold higher than in Tris-HCl. Next, we performed substrate saturation curves toward polymyxin B in Bis-Tris-propane buffer at different pHs (Figure 6B). The activity of the enzyme varied with pH and did not continue to increase linearly at higher pH, which would be expected if nonenzymatic acetylation were occurring. We found the enzymatic activity toward polymyxin B at different pHs exhibited a Gaussian curve (Figure 6C) with a pH optimum at 8.0. This type of curve matches what would be expected for a general acid/base catalytic mechanism in which two residues are likely participating in the reaction. Additional plots of log Vmax as a function of pH, log Km as a function of pH, and log kcat/Km as a function of pH are presented in Figure S6. Note that substrate saturation curves at pH 6.5 and 7.0 (Figure 6B) did not reach saturation, so the apparent Vmax and Km values used to produce the curves in Figure S6 may affect the interpretation of the data. Overall, our results provide evidence that the polymyxin acetylation reaction is indeed enzymatic. Studies to identify these critical residues for enzymatic activity are currently underway in our laboratory.
Figure 6.

PA3944 activity with respect to polymyxin B in different buffers and at different pHs. (A) Comparison of polymyxin B substrate saturation curves at a constant concentration of AcCoA (0.5 mM) in Tris-HCl and Bis-Tris propane buffers at pH 8.0. Data were fit using nonlinear regression as described in Methods. (B) Polymyxin B substrate saturation curves as a function of pH in Bis-Tris propane buffers from pH 6.5 to 9.5. Data were fit using nonlinear regression as described in Methods. (C) PA3944 activity with respect to 2 mM polymyxin B and 0.5 mM AcCoA in Bis-Tris propane buffers at pH 6.5, 7.0, 8.0, 9.0, and 9.5. Data were fit to a Gaussian curve. See Methods for further details.
Polymyxins Are Heterogeneous and May Affect Reproducibility.
Because commercial polymyxins are heterogeneous mixtures of fermentation products isolated from bacteria and can adhere to polystyrene, it is important to note that different preparations of solutions of these molecules may vary in composition from batch to batch (reviewed in ref 35). While this potentially presents challenges for interpreting and reproducing kinetic parameters from different laboratories, in our hands two different PA3944 enzyme preparations and polymyxin B preparations were used to collect the enzyme kinetic assay data depicted in Figures 4–6. We observed differences in kcat and Km from the two preparations; however, the catalytic efficiencies of the enzymes were nearly identical. Thus, catalytic efficiencies may be the more useful parameter for comparing PA3944 enzymatic activity in the future from lab to lab.
The Non-Native Function of PA3944 Could Be Exploited for Biotechnology Applications.
Because of the rise in antibiotic resistance toward most antibiotics, polymyxins are being used as a last resort to treat multi-drug-resistant or extreme drug-resistant bacterial infections (for reviews, see refs 31 and 36). While we have shown that PA3944 can acetylate polymyxin antibiotics in vitro, we do not think this is its physiological function. The rationale for our conclusion is primarily based on the fact that the apparent affinity of PA3944 for both polymyxins is in the millimolar range (Table 2) and reported break points for polymyxin B or colistin for resistance of P. aeruginosa to these antibiotics is in the micromolar range [≥4 μg/mL (~3 μM) for colistin and ≥8 μg/mL (∼6 μM) for polymyxin B35]. While it is unlikely that the native function of PA3944 is to modify polymyxins, its ability to singly acetylate Dab3 on the fatty acyl tail of these important antibiotics could be exploited. For instance, in vitro chemical mechanisms of acylation of polymyxins are promiscuous, and having a controlled acylation method for modifying these antibiotics enables specific chemical transformations of these important molecules. Additionally, future directed evolution of the PA3944 enzyme may allow for selective acylation of other sites of these types of antibiotics, which could significantly improve synthetic strategies of these molecules. Furthermore, if the PA3944 enzyme is able to recognize other antimicrobial peptides in a similar manner, it could also be used to selectively modify structurally similar octapeptin antimicrobial peptides. While the native function of PA3944 remains unknown and warrants further investigation, the regioselectivity of this enzyme may be exploited for future biotechnological applications (e.g., designing reporters) and/or drug development.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the following people for their helpful discussions and/or technical assistance: George Gassner, Laura Conrad-Miller, Teaster Baird Jr., Marc O. Anderson, Jorge C. Escalante-Semerena, Robert Hancock, Reza Falsafi, Jackson Baumgartner, David T. Tran, Brian Amsler, and Andrew S. Kuhn. Additionally, M.L.K. will be forever grateful to Gerry Wright for sage advice and guidance and critical reading of the manuscript. This research used resources of the Advanced PhotonSource, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02–06CH11357. The results shown in this report are derived from work performed at SBC-CAT and LS-CAT sectors. SBC-CAT is operated by UChicago Argonne, LLC, for the DOE Office of Biological and Environmental Research under Contract DEAC02–06CH11357. LS-CAT Sector 21 is supported by the Michigan Economic Development Corp. and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Funding
Funding for this work was provided by the National Science Foundation (Grant CHE-1708863 to M.L.K. and Grant CHE-1708927 to D.P.B.) and a Facilitating Research and Creative Work at San Francisco State University Collaborative Grant (to M.L.K.). This research was also funded by federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH), Department of Health and Human Services, under Contracts HHSN272201200026C and HHSN272201700060C and by NIH Grant GM117325 (to W.M.).
Abbreviations
- GNAT
Gcn5-related N-acetyltransferase
- DTNB
5,5’-dithiobis(2-nitrobenzoic acid)
- TB
Terrific Broth
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- BME
β-mercaptoethanol
- AcCoA
acetyl-coenzyme A
- Dab
L-2,4-diaminobutyric acid
- Dap
L-2,3-diaminopropionic acid
- TNB2−
2-nitro-5-thiobenzoate
- TFA
trifluoroacetic acid
- MeOH
methanol; rmsd, root-mean-square deviation
Footnotes
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.8b00946.
Supplemental methods for protein expression and purification for crystallization, protein crystallization, size-exclusion chromatography, and sequence and structural comparison of homologues; supplemental results, including mass spectrometry of acetylated polymyxin B and colistin; and Table S1, Figures S1– S6, and Schemes S1 and S2 (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Favrot L, Blanchard JS, and Vergnolle O (2016) Bacterial GCN5-Related N-Acetyltransferases: From Resistance to Regulation. Biochemistry 55, 989–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hentchel KL, and Escalante-Semerena JC (2015) Acylation of Biomolecules in Prokaryotes: a Widespread Strategy for the Control of Biological Function and Metabolic Stress. Microbiol. Mol. Biol. Rev. 79, 321–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hegde SS, Javid-Majd F, and Blanchard JS (2001) Overexpression and mechanistic analysis of chromosomally encoded aminoglycoside 2’-N-acetyltransferase (AAC(2’)-Ic) from Mycobacterium tuberculosis. J. Biol. Chem 276, 45876–45881. [DOI] [PubMed] [Google Scholar]
- (4).Daigle DM, Hughes DW, and Wright GD (1999) Prodigious substrate specificity of AAC(6’)-APH(2″), an amino-glycoside antibiotic resistance determinant in enterococci and staphylococci. Chem. Biol 6, 99–110. [DOI] [PubMed] [Google Scholar]
- (5).Gu L, Geders TW, Wang B, Gerwick WH, Hakansson K, Smith JL, and Sherman DH (2007) GNAT-like strategy for polyketide chain initiation. Science 318, 970–974. [DOI] [PubMed] [Google Scholar]
- (6).Caskey S, Stirling J, Moore JE, and Rendall JC (2018) Occurrence of Pseudomonas aeruginosa in waters: implications for patients with cystic fibrosis (CF). Lett. Appl. Microbiol 66, 537–541. [DOI] [PubMed] [Google Scholar]
- (7).Green SK, Schroth MN, Cho JJ, Kominos SK, and Vitanza-jack VB (1974) Agricultural plants and soil as a reservoir for Pseudomonas aeruginosa. Appl. Microbiol. 28, 987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mittal R, Aggarwal S, Sharma S, Chhibber S, and Harjai K (2009) Urinary tract infections caused by Pseudomonas aeruginosa: a minireview. J. Infect. Public Health 2, 101–111. [DOI] [PubMed] [Google Scholar]
- (9).Bhagirath AY, Li Y, Somayajula D, Dadashi M, Badr S, and Duan K (2016) Cystic fibrosis lung environment and Pseudomonas aeruginosa infection. BMC Pulm. Med. 16, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Obritsch MD, Fish DN, MacLaren R, and Jung R (2005) Nosocomial infections due to multidrug-resistant Pseudomonas aeruginosa: epidemiology and treatment options. Pharmacotherapy 25, 1353–1364. [DOI] [PubMed] [Google Scholar]
- (11).Rice LB (2008) Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis 197, 1079–81. [DOI] [PubMed] [Google Scholar]
- (12).Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, and Brinkman FS (2016) Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 44, D646–D653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kuhn ML, Majorek KA, Minor W, and Anderson WF (2013) Broad-substrate screen as a tool to identify substrates for bacterial Gcn5-related N-acetyltransferases with unknown substrate specificity. Protein Sci. 22, 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhang R, Skarina T, Katz J, Beasley S, Khachatryan A, Vyas S, Arrowsmith C, Clarke S, Edwards A, Joachimiak A, and Savchenko A (2001) Structure of Thermotoga maritima stationary phase survival protein SurE: a novel acid phosphatase. Structure 9, 1095–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Otwinowski Z, and Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–26. [DOI] [PubMed] [Google Scholar]
- (16).Minor W, Cymborowski M, Otwinowski Z, and Chruszcz M (2006) HKL-3000: the integration of data reduction and structure solution−from diffraction images to an initial model in minutes. Acta Crystallogr., Sect. D: Biol. Crystallogr 62, 859–866. [DOI] [PubMed] [Google Scholar]
- (17).Vagin A, and Teplyakov A (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr 30, 1022–1025. [Google Scholar]
- (18).Cowtan K (2008) Fitting molecular fragments into electron density. Acta Crystallogr., Sect. D: Biol. Crystallogr 64, 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Porebski PJ, Cymborowski M, Pasenkiewicz-Gierula M, and Minor W (2016) Fitmunk: improving protein structures by accurate, automatic modeling of side-chain conformations. Acta Crystallogr., Sect. D: Biol. Crystallogr 72, 266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, and Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Emsley P, and Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (22).Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB 3rd, Snoeyink J, Richardson JS, and Richardson DC (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Gore S, Sanz García E, Hendrickx PM, Gutmanas A, Westbrook JD, Yang H, Feng Z, Baskaran K, Berrisford JM, Hudson BP, et al. (2017) Validation of structures in the Protein Data Bank. Structure 25, 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Grabowski M, Langner KM, Cymborowski M, Porebski PJ, Sroka P, Zheng H, Cooper DR, Zimmerman MD, Elsliger M, Burley SK, and Minor W (2016) A public database of macromolecular diffraction experiments. Acta Crystallogr., Sect. D: Biol. Crystallogr 72, 1181–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, and Brinkman FS (2016) Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 44, D646–D653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Vetting MW, de Carvalho S.,L. p., Yu M, Hegde SS, Magnet S, Roderick SL, and Blanchard JS (2005) Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys 433, 212–226. [DOI] [PubMed] [Google Scholar]
- (27).Majorek KA, Kuhn ML, Chruszcz M, Anderson WF, and Minor W (2013) Structural, Functional, and Inhibition Studies of a Gcn5-related N-Acetyltransferase (GNAT) Superfamily Protein PA4794. J. Biol. Chem 288, 30223–30235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Madej T, Lanczycki CJ, Zhang D, Thiessen PA, Geer RC, Marchler-Bauer A, and Bryant SH (2014) MMDB and VAST: tracking structural similarities between macromolecular complexes. Nucleic Acids Res. 42, D297–D303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Holm L, and Laakso LM (2016) Dali server update. Nucleic Acids Res. 44, W351–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Krissinel E, and Henrick K (2007) Inference of macro-molecular assemblies from crystalline state. J. Mol. Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- (31).Velkov T, Thompson PE, Nation RL, and Li J (2010) Structure− activity relationships of polymyxin antibiotics. J. Med. Chem 53, 1898–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ono H, Sawada K, Khunajakr N, Tao T, Yamamoto M, Hiramoto M, Shinmyo A, Takano M, and Murooka Y (1999) Characterization of biosynthetic enzymes for ectoine as a compatible solute in a moderately halophilic eubacterium, Halomonas elongata. J. Bacteriol 181, 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Mustakhimov II, Rozova ON, Reshetnikov AS, Khmelenina VN, Murrell JC, and Trotsenko YA (2008) Characterization of the recombinant diaminobutyric acid acetyltransferase from Methylophaga thalassica and Methylophaga alcalica. FEMS Microbiol. Lett 283, 91–96. [DOI] [PubMed] [Google Scholar]
- (34).Reshetnikov A, Mustakhimov I, Khmelenina V, and Trotsenko YA (2005) Cloning, purification, and characterization of diaminobutyrate acetyltransferase from the halotolerant methanotroph Methylomicrobium alcaliphilum 20Z. Biochemistry (Moscow) 70, 878–883. [DOI] [PubMed] [Google Scholar]
- (35).Bakthavatchalam YD, Pragasam AK, Biswas I, and Veeraraghavan B (2018) Polymyxin susceptibility testing, interpretative breakpoints and resistance mechanisms: An update. J. Glob. Antimicrob. Resist 12, 124–136. [DOI] [PubMed] [Google Scholar]
- (36).Velkov T, Roberts KD, Thompson PE, and Li J (2016) Polymyxins: a new hope in combating Gram-negative superbugs? Future Med. Chem 8, 1017–1025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.