

Abstract
Integrase strand transfer inhibitor (INSTI)–based regimens dominate initial human immunodeficiency virus treatment. Most INSTIs are metabolized predominantly via UDP-glucuronosyltransferases (UGTs). For drugs predominantly metabolized by UGTs, including INSTIs, in vitro data recovered from human liver microsomes (HLMs) alone often underpredict human oral clearance. While several factors may contribute, extrahepatic glucuronidation may contribute to this underprediction. Thus, we comprehensively characterized the kinetics for the glucuronidation of INSTIs (cabotegravir, dolutegravir, and raltegravir) using pooled human microsomal preparations from liver (HLMs), intestine (HIMs), and kidney (HKMs) tissues; human embryonic kidney 293 cells expressing individual UGTs; and recombinant UGTs. In vitro glucuronidation of cabotegravir (HLMs≈HKMs>>>HIMs), dolutegravir (HLMs>HIMs>>HKMs), and raltegravir (HLMs>HKMs>> HIMs) occurred in hepatic and extrahepatic tissues. The kinetic data from expression systems suggested the major enzymes in each tissue: hepatic UGT1A9 > UGT1A1 (dolutegravir and raltegravir) and UGT1A1 (cabotegravir), intestinal UGT1A3 > UGT1A8 > UGT1A1 (dolutegravir) and UGT1A8 > UGT1A1 (raltegravir), and renal UGT1A9 (dolutegravir and raltegravir). Enzymes catalyzing cabotegravir glucuronidation in the kidney and intestine could not be identified unequivocally. Using data from dolutegravir glucuronidation as a prototype, a “bottom-up” physiologically based pharmacokinetic model was developed in a stepwise approach and predicted dolutegravir oral clearance within 4.5-fold (hepatic data only), 2-fold (hepatic and intestinal data), and 32% (hepatic, intestinal, and renal data). These results suggest clinically meaningful glucuronidation of dolutegravir in tissues other than the liver. Incorporation of additional novel mechanistic and physiologic underpinnings of dolutegravir metabolism along with in silico approaches appears to be a powerful tool to accurately predict the clearance of dolutegravir from in vitro data.
Introduction
The human immunodeficiency virus (HIV) type 1 infection and the acquired immune deficiency syndrome (AIDS) are a global major public health problem. The prevalence of new HIV-1 infections and AIDS-related morbidity and mortality has considerably decreased over the past 35 years due in part to the continued development of new, highly effective HIV drugs that work by different mechanisms and the introduction of novel formulations and drug combinations (Flexner, 2019). HIV-1 infection has now evolved into a manageable disease that requires lifelong drug therapy. Thus, improving tolerability, efficacy, and cost-effectiveness of these regimens in the context of a chronic care model has become an important consideration. However, over 35 million people still live with HIV/AIDS globally (over 1 million in the United States), and over 900,000 people died of HIV-related illnesses in 2017 alone (https://www.cdc.gov/hiv/statistics/overview/index.html; https://www.who.int/gho/hiv/en/).
Due to their demonstrated clinical efficacy and excellent safety, integrase strand transfer inhibitors (INSTIs) in combination with two nucleoside/nucleotide reverse transcriptases currently dominate HIV therapy for both antiretroviral-naïve and -experienced patients (DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents, 2018). Four INSTIs (bictegravir, dolutegravir, raltegravir, and elvitegravir) have been Food and Drug Administration approved and are recommended as preferred initial regimens for most treatment-naïve HIV patients (DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents, 2018; Flexner, 2019). Cabotegravir is being developed as both an oral and long-acting injectable formulation (phase III drug development) for both the treatment and prevention of HIV infection (Flexner, 2019). Glucuronidation via uridine diphosphate-glucuronosyltransferase (UGT) enzymes (e.g., hepatic UGT1A1) is the main metabolic pathway of dolutegravir, raltegravir, and cabotegravir (Fig. 1) (Kassahun et al., 2007; Castellino et al., 2013; Bowers et al., 2016). Elvitegravir undergoes oxidation by CYP3A (Mathias et al., 2009), and both oxidation (CYP3A) and glucuronidation are involved in the metabolism of bictegravir (https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210251Orig1s000TOC.cfm).
Fig. 1.

Chemical structures of cabotegravir, dolutegravir, and raltegravir and their respective O-glucuronides (cabotegravir glucuronide, dolutegravir glucuronide, and raltegravir glucuronide). Red is the site of ether O-glucuronidation.
UGTs are typically low-affinity, high-capacity enzyme systems that have minimal consequences on drug exposure from drug perpetrators (Williams et al., 2004; Gufford et al., 2015). Those INSTIs and other drugs mainly cleared by UGTs are thought to be less susceptible to drug-drug interactions than those observed with substrates of cytochrome P450s (Williams et al., 2004; Adams et al., 2012; Trezza et al., 2015; DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents, 2018). As a result, UGT-mediated metabolism is an attractive property for new molecular entities undergoing development due to the confidence in stable metabolic elimination of the compound (Argikar et al., 2016). However, accurate in vitro to in vivo extrapolation (IVIVE) of clinical pharmacokinetics for drugs metabolized via UGT remains elusive (Argikar et al., 2016). Early-phase development of the INSTIs found that in vitro and preclinical data reported underprediction of apparent oral clearance (Laufer et al., 2009). This tendency was observed across several UGT substrates from other therapeutic classes (Boase and Miners, 2002; Soars et al., 2002; Laufer et al., 2009). This underprediction may be due to inadequate applications of the mechanistic and physiologic characteristics of the glucuronidation pathway and an inadequate understanding of the contribution of UGTs other than hepatic UGT1A1 (Izukawa et al., 2009; Court et al., 2012; Achour et al., 2017). Multiple UGTs, including UGT1A1, are expressed in multiple tissues at varying drug-metabolizing capacities, such as the liver, kidney, and intestine (Court et al., 2012; Gill et al., 2013; Margaillan et al., 2015; Drodzik et al., 2018). The possibility that UGTs in extrahepatic tissues may catalyze the metabolism of INSTIs has not been previously investigated. Further, the contribution of extrahepatic glucuronidation to overall systemic clearance of INSTIs remains unknown. The impact of factors (e.g., drug-drug interactions, organ function, and genetic variations) influence the exposure via modulation of extrahepatic UGTs remains elusive. Developing an accurate prediction framework of in vivo clearance from in vitro data of INSTIs and other UGT substrates accounting for extrahepatic metabolism is an important step to understand mechanisms influencing systemic exposure and effect of INSTIs.
The primary objective of this work was to apply an integrative approach that incorporated extrahepatic glucuronidation with our current understanding of hepatic INSTI glucuronidation to improve understanding of clearance mechanisms and predictions of in vivo pharmacokinetics from in vitro data of INSTIs. Thus, in vitro enzyme kinetic parameters were recovered to quantitatively describe the major tissue-, isoform-, and pathway-specific UGT-mediated metabolism of cabotegravir, dolutegravir, and raltegravir. Using dolutegravir as an example, the in vitro hepatic and extrahepatic glucuronidation parameters were then incorporated into a physiologically based pharmacokinetic (PBPK) model to predict clinical pharmacokinetics.
Materials and Methods
Materials and Chemicals.
Pooled human liver microsomes (HLMs) from 50 donors with mixed sex [average age: 47 years old (range: 5–83)], pooled human kidney microsomes (HKMs) from eight donors with mixed sex [average age: 54 years old (range: 42–70)], and pooled human intestinal microsomes (HIMs) from 15 donors with mixed sex [average age: 54 years old (range: 26–69)] were purchased from Xenotech, LLC (Lenexa, KS). UGT-expressing baculovirus-insect cell systems (Supersomes) were purchased from Corning Incorporated (Woburn, MA). Human embryonic kidney 293 (HEK293) cells overexpressing individual UGT1A enzymes were harvested, and microsomes were prepared as described previously (Sun et al., 2013). Cabotegravir and dolutegravir were purchased from MedChemExpress, LLC (Monmouth Junction, NJ). Raltegravir was obtained from the National Institutes of Health AIDS Reagent Program (Germantown, MD). Cabotegravir glucuronide, dolutegravir glucuronide, and raltegravir glucuronide were purchased from Toronto Research Chemicals Inc. (North York, Canada) and were 95% pure as determined by the supplier via thin-layer chromatography with nuclear magnetic resonance spectroscopy and mass spectrometry structural confirmation. Alamethicin, magnesium chloride, 8-14-dihydroxy efavirenz, nevirapine, saccharolactone, Tris-HCl, Tris base, and UDP-glucuronic acid (UDPGA) were purchased from Sigma-Aldrich (St. Louis, MO). Dimethylsulfoxide, acetonitrile, methanol, ethanol, and formic acid (all liquid chromatography–mass spectrometry grade) were purchased from Fisher Scientific (Hampton, NH). Dialysis membranes (12–14 kDa molecular mass cutoff) were purchased from Fisher Scientific (Hampton, NH). The 96-well microequilibrium HTD 96b dialysis device was obtained from HTDialysis, LLC (Gales Ferry, CT).
Liquid Chromatography–Tandem Mass Spectrometry Method Development.
A new liquid chromatography–tandem mass spectrometry (LC-MS/MS) method was developed for quantification of cabotegravir, dolutegravir, raltegravir, and their respective glucuronide metabolites. Chromatographic separation was accomplished using a Phenomenex Luna C18, 5-µM, 4.6 × 150-mm column (Torrance, CA) heated to 30°C with a binary gradient flow of 0.8 ml/min. The gradient elution began with 40:60 acetonitrile:water (both with 0.1% formic acid), increased to 90:10 at 5 minutes, and held for 0.5 minutes before returning to initial conditions for the remaining 2 minutes. Chromatographic separation was achieved within 7.5 minutes using a single LC method for all compounds (Supplemental Fig. 1). Samples were analyzed (3-µl injection volume) using the QTRAP 6500+ LC-MS/MS system (AB Sciex, Framingham, MA) with turboelectrospray source operated in both positive (confirmation) and negative (quantification) modes. The negative mode was used to improve glucuronide metabolite sensitivity and selectivity with confirmatory transitions in positive ion mode. 8-14-Dihydroxy efavirenz and nevirapine were used as internal standards for the negative and positive mode, respectively. Compound-specific instrument parameters were optimized for each analyte (Supplemental Table 1). The incubation-generated INSTI glucuronides were directly quantified using commercially available INSTI glucuronide authentic standards with a dynamic assay range of 0–2000 nM. The instrument response was linear with respect to increasing analyte concentration over the standard curve range used. The lower limit of quantification was 1 nM (cabotegravir glucuronide) or 2 nM (dolutegravir and raltegravir glucuronides). Data were acquired using Analyst software (version 1.6.3; AB Sciex) and quantified via MultiQuant software (version 3.0.2; AB Sciex). Assay accuracy was evaluated using MultiQuant software; standard and quality control samples were deemed acceptable if within 20% of the nominal value except for the lower limit of quantification, which was assessed with a 30% threshold.
Glucuronidation Kinetics in HLMs, HIMs, and HKMs.
Incubation conditions were optimized for each substrate and enzyme source to ensure linearity of metabolite formation with respect to time and protein concentration and to prevent greater than 20% substrate depletion (Supplemental Table 2). To determine the kinetics for the formation of the glucuronides, cabotegravir, dolutegravir, and raltegravir (concentrations spanning 0–2000 µM) were incubated in duplicate with each individual tissue microsomal preparation (HLM, HIM, and HKM protein concentrations shown in Supplemental Table 2) in Tris-HCl buffer (pH 7.4, 100 mM) containing MgCl2 (5 mM) and bovine serum albumin (BSA) (0.05%) with a total incubation volume of 150 µl. HLMs, HIMs, and HKMs were treated with alamethicin (50 µg/mg protein) on ice for 15 minutes. Saccharolactone (100 µM) was added to all HIM preparations. Mixtures were equilibrated at 37°C for 5 minutes, and reaction was initiated by the addition of 15 µl of UDPGA (2 mM final concentration) and incubated for 60 (cabotegravir), 30 (dolutegravir), and 20 (raltegravir) minutes. Reactions were terminated by removing 100 µl from the incubation and diluting into 300 µl of ice-cold acetonitrile (0.1% formic acid) containing internal standards nevirapine (0.2 µM) and 8,14-dihydroxy efavirenz (0.2 µM). Samples were vortex-mixed and centrifuged at 3000g for 20 minutes at 4°C. Supernatant (200 µl) was transferred to clean 96-well plates for analysis via LC-MS/MS.
Determination of Nonspecific Protein Binding in HLMs, HIMs, and HKMs.
The equilibrium dialysis method described by Gill et al. (2012) was used to determine the fraction unbound in the incubation, fu,inc, for all three drugs (cabotegravir, dolutegravir, raltegravir) in pooled HLMs, HIMs, and HKMs in the presence and absence of BSA. Each INSTI drug (10 µM) in buffer was added to the donor side of the membrane along with the relevant concentration of microsomal protein (Supplemental Table 2) with or without BSA (0.05%). Tris-HCl buffer (pH 7.4, 100 mM) containing MgCl2 (5 mM) and saccharolactone (100 µM; for HIMs only) was added to the acceptor side of the membrane. Experiments were performed in duplicate. The plate was left to equilibrate for 6 hours on a plate shaker (250 rpm) at 37°C. Aliquots (50 µl) were transferred from both the acceptor and donor sides of the membrane to 200 µl containing the internal standard nevirapine (0.2 µM) and methanol (0.1% formic acid). Samples were vortex-mixed and centrifuged at 3000 rpm for 20 minutes at 4°C. Supernatant (100 µl) was transferred to clean 96-well plates for analysis via LC-MS/MS as described earlier. Fraction unbound was calculated as follows:
Reaction Phenotyping Using a Recombinant UGT Enzyme Panel.
Cabotegravir, dolutegravir, or raltegravir (50 µM) was incubated in duplicate with each individual recombinant UGT (rUGT) enzyme (0.2 mg/ml rUGT 1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B15, 2B17) or vehicle/vector control as described earlier, except for the absence of alamethicin and saccharolactone. Reactions were initiated by the addition of 15 µl of UDPGA (2 mM final concentration) and terminated at 60 minutes by transferring 100 µl of each incubation into 300 µl of ice-cold acetonitrile (0.1% formic acid) containing internal standard and analyzed via LC-MS/MS as described earlier.
Isoform-Specific Glucuronidation Kinetics.
Ranging concentrations of cabotegravir, dolutegravir, or raltegravir (0–2000 µM) were incubated in duplicate with each UGT enzyme source—rUGT1A1, rUGT1A3, rUGT1A7, rUGT1A8, rUGT1A9, HEKUGT1A1, HEKUGT1A3 (raltegravir only), HEKUGT1A7, HEKUGT1A8, and HEKUGT1A9—using isoform-specific optimized conditions for protein concentration and incubation time (Supplemental Table 2). Incubation vehicle/vector and reaction initiation mirrored that described for the microsomal preparations except for the absence of alamethicin in rUGT preparations. The rUGT and HEKUGT incubations did not use saccharolactone. All rUGT incubations were completed using a single manufacturer lot.
Physiologically Based Pharmacokinetic Model Development.
Dolutegravir was selected as a prototype for further evaluation of the in vivo contributions of extrahepatic tissues in INSTI metabolism via PBPK modeling using Certara SimCYP (version 16.0; Certara, Inc., Princeton, NJ). Dolutegravir modeling was selected in this manuscript for a number of reasons: 1) it is the most commonly prescribed clinically, 2) it has sufficient clinical data readily available to confirm model predictions, and 3) published PBPK models are lacking. The PBPK model was parameterized initially using literature values to describe dolutegravir physicochemical properties and oxidative metabolism (Supplemental Table 3) (Castellino et al., 2013; Reese et al., 2013). The advanced dissolution, absorption, and metabolism (ADAM) model was selected for incorporation of intestinal glucuronidation and dynamic multicompartmental transit time effects. Standard SimCYP model parameters of potential enterohepatic circulation (100%) from the compound eliminated from biliary excretion were incorporated. Glucuronidation kinetics were described in a stepwise fashion via incorporation of in vitro enzyme kinetic parameters describing hepatic only; hepatic plus renal; and finally, the combination of hepatic, renal, and intestinal UGT-mediated metabolism. Simulations were conducted using the SimCYP “healthy volunteers” multiple populations [10 trials with 10 subjects (20–50 years old) in each trial] administered a single 50-mg oral dolutegravir dose under fasted conditions. Published dolutegravir clinical pharmacokinetic data with a matching dosing regimen (single fasted 50-mg oral dose in healthy volunteers) (https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204790Orig1s000TOC.cfm; Song et al., 2015) were used in the evaluation of PBPK model predictions and recovered using GetData Graph Digitizer (version 2.26.0.20; GetData Pty Ltd., Kogarah, Australia). Sensitivity analyses were conducted to quantitatively assess the impact of extrahepatic glucuronidation parameter (HIM and HKM Clint values) uncertainty on the pharmacokinetic outcomes of interest [Cmax, area under the curve (AUC), and ClPO] (Supplemental Fig. 2).
Data Analyses.
Apparent kinetic constants for glucuronidation of the test substrates were obtained via nonlinear regression by fitting Michaelis-Menten, substrate inhibition, two-site, or Hill equations to substrate concentration ([S]) versus apparent metabolite formation velocity data using Phoenix WinNonlin (version 7.0; Certara).
The Michaelis-Menten equation is as follows:
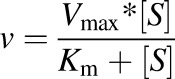 |
where v is the initial rate of reaction, Vmax is the maximum velocity, Km is the Michaelis-Menten constant (substrate concentration at 0.5 Vmax), and [S] is the substrate concentration.
Hill equation is as follows:
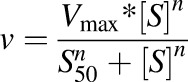 |
where S50 is the substrate concentration resulting in 50% of Vmax (analogous to Km in the previous equation), and n is the Hill coefficient.
The two-site is as follows:
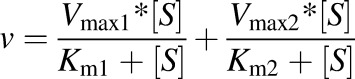 |
where Vmax1 and Vmax2 are the maximum velocities, and Km1 and Km2 are the Michaelis-Menten constants for the two sites of the enzyme.
The substrate inhibition equation is as follows:
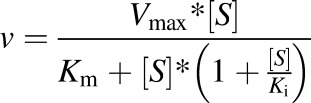 |
where Ki is the inhibition constant.
Km and S50 were corrected for nonspecific binding [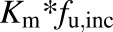 (Km,u and S50,u)]. In vitro Clint,u is calculated as (Vmax/Km,u) and Clmax,u is calculated as [Vmax × (h − 1)/Km,u + h (h − 1)1/h] (Houston and Kenworthy, 2000), where in vitro Clint,u is the unbound intrinsic clearance per microsomal protein and is calculated for substrates described by the simple Michaelis-Menten or Hill equation, respectively. Best-fit models were selected by visual inspection of the predicted versus observed data, precision of parameter estimates generated from the nonlinear regression, and Akaike information criteria. Unless noted, data are presented as the mean of duplicate incubations, with error bars showing data variability for N = 2.
(Km,u and S50,u)]. In vitro Clint,u is calculated as (Vmax/Km,u) and Clmax,u is calculated as [Vmax × (h − 1)/Km,u + h (h − 1)1/h] (Houston and Kenworthy, 2000), where in vitro Clint,u is the unbound intrinsic clearance per microsomal protein and is calculated for substrates described by the simple Michaelis-Menten or Hill equation, respectively. Best-fit models were selected by visual inspection of the predicted versus observed data, precision of parameter estimates generated from the nonlinear regression, and Akaike information criteria. Unless noted, data are presented as the mean of duplicate incubations, with error bars showing data variability for N = 2.
Scaling from In Vitro Clint to Organ Clint.
The in vitro Clint,u was used to estimate whole organ Clint as follows: in vitro Clint,u * scaling factor (MPPGL, MPPGK, or MPPI) * organ weight (liver or kidney), where MPPGL is the microsomal protein per gram of liver, MPPGK is the microsomal protein per gram of kidney, and MPPI is the microsomal protein per total intestine. The following scaling factors were used: MPPGL of 37.69 mg mics/g of liver tissue (Wood et al., 2017) (total liver weight = 1800 g) (Davies and Morris, 1993); MPPGK of 12.8 mg mics/g of renal tissue (Al-Jahdari et al., 2006) (total kidney weight = 310 g) (Davies and Morris, 1993); and MPPI of 2935.17 mg mics/total intestine (Paine et al., 1997). The microsomal scaling factors are imbedded in the SimCYP software.
Results
INSTI Glucuronidation is Tissue-Dependent.
Glucuronidation kinetic parameters were recovered with varying concentrations (0–2000 µM) of cabotegravir, dolutegravir, and raltegravir in pooled HLMs, HIMs, and HKMs to quantitatively assess tissue-specific INSTI glucuronidation (Fig. 2; Table 1). To correct the Km values derived for nonspecific protein binding in incubations (fu,inc), equilibrium dialysis experiments were performed in pooled HLMs, HIMs, and HKMs. Incubations tested in the absence of albumin found fu,inc was ≥0.85 for cabotegravir, dolutegravir, and raltegravir. Nonspecific protein binding was observed in the presence of BSA for dolutegravir and cabotegravir, whereas the fraction unbound for raltegravir was unaffected by either BSA or microsomal proteins (fu,inc was ≥0.90) (Table 1). Pooled HLMs had the greatest nonspecific protein binding for dolutegravir, and HKMs for cabotegravir. There were minimal differences in protein binding between the different organ tissues (HLMs, HIMs, HKMs) for the same substrate but noticeable changes in binding among the different INSTI substrates (Table 1). The fraction unbound (fu,inc) was then used to estimate the unbound Km (Km,u) values (Table 1), and accordingly, the in vitro Clint,u was calculated (Table 1). In the subsequent portion of the manuscript, Km is referred to as Km,u, and in vitro intrinsic clearance Clint is Clint,u.
Fig. 2.

Kinetics for the formation of glucuronides from cabotegravir (A), dolutegravir (B), and raltegravir (C) in HLMs, HIMs, and HKMs. The substrate concentration versus velocity data were fit to the Michaelis-Menten equation. Shapes (e.g. circles, triangles) represent observed data (OBS) and solid lines are predicted (PRED).
TABLE 1.
Glucuronidation kinetic parameters from pooled human microsomal preparations
Values represent the parameter estimate (S.E.) by fitting substrate concentration to the simple Michaelis-Menten equation (v = Vmax *[S]/Km + [S]) to metabolite formation velocity using Phoenix WinNonlin (version 7.0). Clint calculated as the ratio of Vmax to Km.
| Enzyme Source |
fu,inc |
Km,u |
Vmax |
Clint,u |
Clint,organ |
|---|---|---|---|---|---|
| µM | pmol/min/mg protein | µl/min/mg mics | l/h per organ | ||
| Cabotegravir | |||||
| HLMs | 0.52 | 350 (41) | 705 (35) | 2.0 | 8.1 |
| HIMs | 0.69 | NDa | 1031 (323) | NDa | NDa |
| HKMs | 0.50 | 560 (57) | 1088 (56) | 1.9 | 0.45 |
| Dolutegravir | |||||
| HLMs | 0.23 | 32 (2) | 601 (13) | 18 | 76 |
| HIMs | 0.37 | 96 (7) | 1170 (33) | 12 | 2.1 |
| HKMs | 0.29 | 47 (8) | 291 (16) | 6.2 | 1.1 |
| Raltegravir | |||||
| HLMs | 1 | 183 (23) | 1737 (63) | 9.5 | 38.7 |
| HIMs | 1 | 167 (23) | 326 (13) | 2.0 | 0.3 |
| HKMs | 0.97 | 493 (59) | 2332 (111) | 4.7 | 0.5 |
The substrate concentration versus velocity did not saturate, and the estimated Km (>6 mM) and Vmax values were unreliable. Thus, the values are not presented, and Clint,u and Clint,organ were not determined (ND).
Micr, microsomal protein.
Cabotegravir and raltegravir appeared to be relatively low-affinity substrates for UGTs, as represented by high Km values (167–560 µM) (Table 1), whereas the Km values for dolutegravir were between 32 and 96 µM. Microsomal UGTs had the lowest affinity for cabotegravir in all three tissues compared with dolutegravir and raltegravir, suggesting slower metabolism via UGT for cabotegravir. Cabotegravir glucuronide was most efficiently (in vitro Clint) formed in HLMs and HKMs compared with HIMs. The Km values for cabotegravir glucuronidation were relatively high, and saturation was not easily achieved in the case of HIMs, suggesting that this elimination pathway is low-affinity and high-capacity, a common observation for UGT-mediated elimination routes. Thus, Km (>6 mM) and Vmax values were unreliable to estimate in vitro Clint.
Overall, the in vitro metabolism of cabotegravir in HIMs was relatively small, suggesting minimal intestinal contribution to the overall metabolic clearance. Intestinal UGTs expressed the lowest affinity (highest Km) and highest capacity (Vmax) for dolutegravir, while HLMs and HKMs showed relatively lower Km values (Table 1). Based on the in vitro Clint, dolutegravir glucuronidation was most efficient in HLMs (1.5- and 3-fold higher compared with HIMs and HKMs, respectively). The microsomal preparations suggested raltegravir glucuronidation was more similar to cabotegravir in terms of tissue involvement. Raltegravir was glucuronidated predominantly in HLMs and HKMs with lesser HIM contributions. Raltegravir glucuronidation was higher in hepatic tissue, reflected by the 4.8-fold greater in vitro Clint compared with intestinal tissue, and was 2-fold greater compared with renal tissues. Renal tissue expressed the lowest affinity (Km) but the highest UGT capacity (Vmax) for raltegravir.
The in vitro Clint was used to estimate in vivo organ Clint. In contrast to the in vitro data showing clear contributions of extrahepatic glucuronidation, the in vivo Clint data predicted from the in vitro kinetic parameters indicate a predominant role of hepatic metabolism of these drugs (Table 1).
INSTI Glucuronidation in Recombinant UGTs.
Microsomal enzyme kinetic data revealed the prominent role of extrahepatic INSTI glucuronidation and formed the basis for further evaluation of isoform-specific INSTI glucuronidation. Reaction phenotyping was conducted using recombinant UGT–expressing baculovirus-insect cell systems to qualitatively identify specific UGT enzymes responsible for formation of the respective INSTI glucuronides (Fig. 3). The reaction phenotyping showed that rUGT1A9 and rUGT1A1 catalyzed glucuronidation of cabotegravir, dolutegravir, and raltegravir at the highest rate. Other isoforms also participated to a small extent in the glucuronidation of cabotegravir (UGT1A3 ≈ UGT1A8), dolutegravir (rUGT1A3 << rUGT1A7 < rUGT1A8 and raltegravir (UGT1A7 < UGT1A8 < UGT1A3).
Fig. 3.

Formation rates of glucuronides from 50 µM cabotegravir (A), dolutegravir (B), and raltegravir (C) in a panel of rUGT isoforms (0.2 mg/ml protein). Control, vector control.
For those isoforms that showed activity (UGT1A1, UGT1A3, UGT1A7, UGT1A8, and UGT1A9), UGT isoform-specific INSTI glucuronidation was evaluated in more detail by recovering UGT enzyme-specific glucuronidation kinetic parameters. Representative substrate concentration versus velocity curves of UGT1A9 and UGT1A1 fit to Michaelis-Menten or Hill equation are shown in Fig. 4. All three INSTIs exhibited substrate inhibition kinetics in rUGT1A9 at concentrations above 1000 µM. Since these concentrations are supratherapeutic, they were removed to simplify the kinetics to fit the Hill equation. Kinetic parameters derived for all rUGTs tested are listed in Table 2. Since the determining factor influencing nonspecific binding was BSA, which was common for incubations with HLMs and the expression systems, fu,inc was not estimated for each UGT isoform. Instead, fu,inc derived from HLMs (see above) was used to account for nonspecific binding to correct the Km values derived from the expression systems. Accordingly, adjusted Km (Km,u) and Clint (Clint,u) values were calculated and are presented for each enzyme (Table 2). Because information on the specific UGT protein content was not available or provided from the supplier, the Vmax and Clint should be viewed as nominal values. To obtain insight into tissue-specific metabolism, the Km values derived from these isoforms (Table 2) were compared with those obtained from HLMs, HIMs, and HKMs (Table 1). The Km values for the formation of cabotegravir glucuronide were much higher in both HIMs and HKMs (Table 1) than any of those derived from the rUGTs (Table 2). Only the Km value of rUGT1A8 was close to that in HLMs, but given that UGT1A8 is mainly expressed in the gut wall, this relationship cannot be fully explained. The Km value for UGT1A9-mediated cabotegravir glucuronidation was more than 6-fold lower than in HLMs. Km values derived from rUGTs (rUGT1A3, 1A8, and 1A9) were close to those for dolutegravir glucuronidation in HLMs, HIMs, and HKMs. Of note, the Km value for dolutegravir glucuronidation in rUGT1A1 was much higher (216 µM). Finally, the Km values for the formation of raltegravir glucuronide in HLMs and HIMs were close to those derived from rUGT1A1 and rUGT1A9.
Fig. 4.

Kinetics for the formation of glucuronides from cabotegravir (Cabo), dolutegravir (Dolu), and raltegravir (Ralt) in UGT1A1- and UGT1A9-overexpressing baculovirus-insect cell system. The substrate concentration versus velocity data were fit to appropriate enzyme kinetic equations (see Table 2). Circles represent observed data, and solid lines are predicted.
TABLE 2.
Glucuronidation kinetic parameters from UGT overexpression in baculosomal cell system
Values represent the parameter estimate (S.E.) by fitting substrate concentration to the simple Michaelis-Menten (MM), Hill, or two-site equation, as described in the Materials and Methods, to metabolite formation velocity using Phoenix WinNonlin (version 7.0).
| Enzyme Source |
Model |
Km,u or S50,u |
Vmax |
n |
Clint,u or Clmax,u |
|---|---|---|---|---|---|
| µM | pmol/min/mg total protein | µl/min/mg protein | |||
| Cabotegravir | |||||
| rUGT1A1 | Two Site | 27 (6)a | 17 (1)a | 0.6 | |
| rUGT1A3 | MM | 46 (7) | 3 (0.1) | 0.06 | |
| rUGT1A7 | MM | 43 (5) | 10 (0.3) | 0.2 | |
| rUGT1A8 | MM | 344 (44) | 40 (2) | 0.1 | |
| rUGT1A9 | Hill | 56 (7) | 61.4 (3) | 1.26 (0) | 1.1 |
| Dolutegravir | |||||
| rUGT1A1 | MM | 216 (26) | 507 (50) | 2.3 | |
| rUGT1A3 | MM | 62 (7) | 18 (0.7) | 0.3 | |
| rUGT1A7 | MM | 9 (2) | 1 (0) | 0.1 | |
| rUGT1A8 | Hill | 44 (5) | 7 (0) | 1.9 (0) | 0.2 |
| rUGT1A9 | Hill | 39 (3) | 39 (2) | 1.9 (0) | 1.0 |
| Raltegravir | |||||
| rUGT1A1 | MM | 260 (17) | 334 (7) | 1.3 | |
| rUGT1A3 | Hill | 41 (2) | 30 (1) | 1.7 (0) | 0.55 |
| rUGT1A7 | MM | 452 (56) | 23 (1) | 0.05 | |
| rUGT1A8 | MM | 386 (57) | 39 (2) | 0.1 | |
| rUGT1A9 | Hill | 193 (12) | 459 (14) | 1.45 (0) | 1.3 |
Clint = Vmax/Km or Clmax = 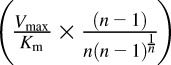 . n, Hill coefficient in the Hill equation.
. n, Hill coefficient in the Hill equation.
The low-affinity enzyme substrate concentration versus velocity did not saturate, and the estimated Km (>2 mM) and Vmax values were unreliable. Thus, only the high Km is presented and calculated for Clint,u.
INSTI Glucuronidation Kinetics in HEK Cells Expressing Individual UGTs.
Full UGT isoform-specific INSTI glucuronidation kinetics were further evaluated using selected HEK cells expressing individual UGTs (Table 3). A majority of the substrate concentration versus velocity curves fit the Michaelis-Menten equation, but some fit to atypical nonhyperbolic enzyme kinetics and were better described by the sigmoidal equation (Hill equation). The substrate concentration versus velocity curves for the glucuronidation of the INSTIs by UGT1A1 and UGT1A9 expressed in the HEK293 cell system are shown in Fig. 5, with the corresponding kinetic parameters derived for all HEKUGTs shown in Table 3. As with rUGTs, isoform-specific protein expression was not available in the subcellular fraction of the cell lines. Therefore, the presented Vmax and Clint (Table 3) are only adjusted for total microsomal protein amount and should be viewed as nominal or relative values and should not be quantitatively compared between and within cell systems (rUGTs and HEKUGTs). The Km values for cabotegravir glucuronidation in HEK cells (1A1 = 55 µM and 1A9 = 163 µM) were higher than those in rUGT1A1 and rUGT1A9 and were closer to the hepatic Km value (HLMs; 350 µM), although the values are still notably lower (by approximately 3- to 6-fold) than observed in HLMs. The Km value in intestinal tissue (millimolar range) for cabotegravir does not concur with any of the Km values derived from HEK cell UGT isoforms. The reason for this discrepancy is not clear. The Km values for dolutegravir glucuronidation in HEK cells (1A9 = 46 µM) is close to the Km value obtained from HLMs (Km = 32 µM) and HKMs (Km = 47 µM); the Km value for the other hepatic UGT examined in HEKs (1A1 = 96 µM) was 3-fold higher than in HLMs. Thus, UGT1A9 (and, to a lesser extent, 1A1) appears to be the main enzyme responsible for dolutegravir glucuronidation in the liver and kidney. Dolutegravir glucuronidation in HIMs (Km = 96 µM) appears to concur with UGT1A1 and UGT1A8 (HEK cell UGT1A1: Km = 96 µM; UGT1A8: Km = 37 µM). The Km value for raltegravir glucuronidation in UGT1A9 expressed in HEK cells (Km = 219 µM) was closer to that derived from HLMs (Km = 183 µM). The Km values derived from this enzyme (HEKUGT1A9) were about half of that observed for raltegravir glucuronidation in HKMs. This enzyme may in part explain hepatic and renal glucuronidation of raltegravir. Considering the lower Km value by UGT1A1 expressed in HEK cells and the Km in rUGT1A1 comparable to that in HLMs, it seems that both UGT1A1 and UGT1A9 are the active UGTs in raltegravir glucuronidation. UGT1A8 appears important for raltegravir glucuronidation in the gut wall (Km value in UGT1A8 expressed in HEK cells = 142 µM vs. 167 µM in HIMs).
TABLE 3.
Glucuronidation kinetic parameters from UGT overexpression in HEK cell system
Values represent the parameter estimate (S.E.) by fitting substrate concentration to the simple Michaelis-Menten (MM) or Hill equation, as described in the Materials and Methods, to metabolite formation velocity using Phoenix WinNonlin (version 7.0).
| Enzyme Source |
Model |
Km,u or S50,u |
Vmax |
n |
Clint,u or Clmax,u |
|---|---|---|---|---|---|
| µM | pmol/min/mg total protein | µl/min/mg protein | |||
| Cabotegravir | |||||
| HEKUGT1A1 | MM | 55 (9) | 7 (0) | 0.1 | |
| HEKUGT1A7 | MM | 23 (6) | 9 (0) | 0.3 | |
| HEKUGT1A8 | MM | 133 (5) | 12 (0) | 0.09 | |
| HEKUGT1A9 | MM | 163 (6) | 25 (0) | 0.2 | |
| Dolutegravir | |||||
| HEKUGT1A1 | MM | 96 (15) | 21 (1) | 0.2 | |
| HEKUGT1A7 | Hill | 2 (3) | 2 (0) | 1.4 (0) | 1.1 |
| HEKUGT1A8 | Hill | 37 (2) | 36 (1) | 1.4 (0) | 1.0 |
| HEKUGT1A9 | Hill | 46 (3) | 107 (4) | 1.7 (0) | 2.3 |
| Raltegravir | |||||
| HEKUGT1A1 | Hill | 52 (7) | 17 (1) | 1.5 (0) | 0.4 |
| HEKUGT1A3 | Hill | 22 (1) | 2 (0) | 1.7 (0) | 0.05 |
| HEKUGT1A7 | MM | 79 (8) | 6 (0) | 0.07 | |
| HEKUGT1A8 | Hill | 142 (15) | 6 (0) | 1.4 (0) | 0.1 |
| HEKUGT1A9 | Hill | 219 (29) | 24 (1.4) | 1.5 (0) | 0.06 |
Clint = Vmax/Km or Clmax = 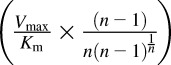 . n, Hill coefficient in the Hill equation.
. n, Hill coefficient in the Hill equation.
Fig. 5.

Kinetics for the formation of glucuronides from cabotegravir (Cabo), dolutegravir (Dolu), and raltegravir (Ralt) in UGT1A1- and UGT1A9-overexpressing human embryonic kidney cell lysates. The substrate concentration versus velocity data were fit to appropriate enzyme kinetic equation (see Table 3). Circles represent observed data, and solid lines are predicted.
Extrahepatic Glucuronidation Contributes to INSTI Metabolism.
Dolutegravir in vitro glucuronidation kinetics from microsomal data were incorporated into a physiologically based pharmacokinetic model using SimCYP to predict dolutegravir clinical pharmacokinetics. After recovering substantial dolutegravir glucuronidation in intestinal tissue, the ADAM absorption model was selected to input the intestinal glucuronidation parameters recovered in HIMs. Three PBPK models were generated to predict dolutegravir clinical pharmacokinetics: 1) in vitro hepatic glucuronidation clearance only; 2) in vitro hepatic and intestinal glucuronidation clearance; and 3) in vitro hepatic, intestinal, and renal glucuronidation clearance (Fig. 6). Empirical scaling factors were applied to the intestinal and renal glucuronidation intrinsic clearance (Clint) parameters, as these could not be input via the more mechanistic approach using the UGT isoform- and tissue-specific Km and Vmax parameters. Sensitivity analyses of these extrahepatic glucuronidation parameters revealed that model-predicted Cmax and ClPO were more sensitive to changes in intestinal Clint, while AUC was more sensitive to HKM Clint (Supplemental Fig. 1). All three models incorporated published values describing minor dolutegravir in vitro oxidative metabolism (CYP3A4) (Reese et al., 2013) and renal clearance. PBPK models underpredicted observed dolutegravir apparent oral clearance by 4.5-fold, 2-fold, or 32% by incorporating hepatic only; hepatic and intestinal; or hepatic, intestinal, and renal glucuronidation, respectively. The model incorporating glucuronidation in all three tissues predicted observed dolutegravir Cmax and AUC0-24 within 18% and 38%, respectively (Table 4).
Fig. 6.

Application of PBPK model to predict pharmacokinetics of a single 50-mg oral dose of dolutegravir from in vitro data. Predicted mean concentration versus time profiles (solid lines) with 95th and 5th percentiles (dashed lines) incorporating in vitro glucuronidation kinetics in a stepwise approach from HLMs (A); HLMs and HIMs (B); and HLMs, HIMs, and HKMs (C), overlaid with observed clinical data (squares) are shown.
TABLE 4.
Dolutegravir clinical pharmacokinetic outcomes recovered from observed data and PBPK model predictions from 1) HLMs only; 2) HLMs and HIMs; and 3) HLMs, HIMs, and HKMs [geometric mean and coefficient of variation (CV%) incorporating in vitro glucuronidation kinetics data from 1) HLMs only; 2) HLMs and HIMs; and 3) HLMs, HIMs, and HKMs]
|
Cmax |
AUC0-∞ |
ClPO |
t1/2 |
fm (%) |
fg |
fh |
fa |
||
|---|---|---|---|---|---|---|---|---|---|
| Liver |
Kidney |
||||||||
| µg/ml | µg·h/ml | l/h | h | % | % | % | |||
| Observed | 2.2 (43) | 43.7 (45) | 1.1 (45) | 14.4 (19) | N/A | ||||
| HLMs only | 5.8 (19) | 199.6 (55) | 0.3 (50) | 21.1 (40) | 99.2 | 0.8 | 98 | 99 | 100 |
| HLMs and HIMs | 2.7 (24) | 89.4 (55) | 0.56 (50) | 21.1 (40) | 99.2 | 0.8 | 46 | 99 | 100 |
| HLMs, HIMs, HKMs | 2.6 (24) | 60.4 (43) | 0.8 (44) | 15.3 (37) | 79.7 | 20.3 | 46 | 99 | 100 |
Discussion
This work is the first comprehensive in vitro characterization of cabotegravir, dolutegravir, and raltegravir glucuronidation in microsomes derived from liver and extrahepatic tissues, HEK293 cells expressing individual UGTs, and recombinant UGTs. We confirmed involvement of previously reported UGT isoforms and identified additional enzymes catalyzing the glucuronidation of these INSTIs. Our study is the first to report that these three INSTIs undergo substantial extrahepatic glucuronidation. Using dolutegravir in vitro hepatic and extrahepatic metabolism, we showed that a “bottom-up” PBPK model incorporating extrahepatic glucuronidation accurately predicted dolutegravir clinical pharmacokinetics from in vitro data. These results suggest the occurrence of clinically meaningful glucuronidation in tissues other than the liver—specifically, intestine and kidney. Incorporation of additional novel mechanistic and physiologic underpinnings of dolutegravir metabolism along with in silico approaches appear to be powerful tools to accurately predict the clearance of dolutegravir from in vitro data.
Our data showed that all INSTIs tested undergo efficient glucuronidation in HLMs. The INSTI hepatic UGT enzyme kinetic parameters reported in this work were similar to those values published in the literature (Kassahun et al., 2007; Reese et al., 2013; Trezza et al., 2015). Underprediction of oral clearance for UGT substrates using IVIVE approaches is quite common, and several factors may contribute to this problem (Boase and Miners, 2002; Soars et al., 2002). It is well established that UGTs are differentially expressed in hepatic and extrahepatic tissues (Guillemette et al., 2014; Margaillan et al., 2015; Drodzik et al., 2018). Therefore, it is possible that this tissue-specific expression contributes to INSTI glucuronidation. Indeed, our data are the first to demonstrate that these drugs undergo efficient extrahepatic glucuronidation in microsomes derived from human kidney and intestinal tissues. Based on the in vitro Clint displayed in Table 1: 1) HLMs and HKMs equally contribute toward cabotegravir glucuronidation, with minor involvement of HIMs; 2) HLMs and then HIMs contributed to dolutegravir glucuronidation, with minor participation from HKMs; and 3) the metabolism of raltegravir was approximately 2- and 5-fold higher in HLMs than in HKMs and HIMs, respectively. On the basis of in vivo Clint, we noted that HLMs alone provide a larger contribution to INSTI glucuronidation with minor contribution from extrahepatic tissue in contrast to the data derived in vitro. While understanding the total activity per organ is desirable, the uncertainty of the absolute UGT amount in the gut and kidney makes this comparison less accurate. In addition, the in vitro data only show the ability of the enzyme systems in those tissues to metabolize the drugs. The in vitro data suggest that the gut and kidney are clearly capable of metabolizing the drugs. However, when considering factors such as the overall size of the organ, blood flow, drug uptakes, and other physiologic parameters, it is clear that the liver still plays a major role. There is no doubt that the liver is the main contributor, but without including the gut and kidney, it is not possible to predict the pharmacokinetic behavior of these compounds accurately, as shown in our dolutegravir PBPK model.
We tested the hypothesis that extrahepatic metabolism contributes to INSTI glucuronidation in vivo. Using dolutegravir glucuronidation data derived from HLMs, HIMs, and HKMs, we developed a PBPK model to show that incorporation of extrahepatic glucuronidation substantially improves the accuracy of PBPK model predictions (Fig. 6). Although the absolute bioavailability of dolutegravir remains unknown, the high gut and liver involvement in dolutegravir glucuronidation is suggestive of significant first-pass metabolism, while kidney glucuronidation also contributes to systemic clearance (Supplemental Fig. 2). This PBPK model will be tested and validated in the future for IVIVE for other INSTIs and substrates of UGTs.
The comprehensive characterization of the INSTIs glucuronidation in recombinant UGTs (Table 2) and in HEK293 cells expressing individual UGTs (Table 3) provides important insights into isoform-specific metabolism of these drugs, as summarized in Table 5. We are aware that additional inhibition analysis could have added valuable information in addition to the Km determinations. However, there are no selective and specific inhibitors of UGT that allow unequivocal identification of individual isoforms. Our data from expressed UGTs generally agree well with published literature identifying the enzymes active in the respective INSTI glucuronidation; however, qualitative differences are noted. We found that UGT1A9 and UGT1A1 are major contributors of cabotegravir, dolutegravir, and raltegravir metabolism, with contribution from other isoforms (UGT1A3, UGT1A7, and UGT1A8). Poor predictions were observed for cabotegravir metabolism with regard to the tissue-specific relationship to specific UGT isoforms due to large differences in Km values. The supraphysiological Km values in hepatic and extrahepatic tissues along with two-site enzyme kinetics (Fig. 4) suggest multiple isoforms may be responsible for cabotegravir’s glucuronidation in the different tissues. For dolutegravir, UGT1A9 Km values were similar to those found in hepatic and renal tissue. Similarly, UGT1A1 and UGT1A9 Km values were similar to hepatic and intestinal tissue with regard to raltegravir glucuronidation. This suggests UGT1A9 may be the major isoform responsible for hepatic glucuronidation of dolutegravir and raltegravir. Considering that UGT1A9 is predominantly expressed in the kidney, this enzyme may also be the major isoform for renal glucuronidation of dolutegravir and raltegravir. UGT1A9 has been reported as undetectable in gastrointestinal tissue (Strassburg et al., 2000; Komura and Iwaki, 2011) and, thus, is unlikely to contribute to the intestinal glucuronidation of raltegravir (and dolutegravir) despite similar Km values in HIMs and UGT1A9 (Strassburg et al., 2000; Komura and Iwaki, 2011). UGT1A1 appears to participate in hepatic and intestinal glucuronidation of raltegravir. Based on the Km values in HEK cell expression system data, UGT1A8, an enzyme mainly expressed in the intestine (Strassburg et al., 2000; Komura and Iwaki, 2011), appears to be important in the intestinal glucuronidation of dolutegravir and raltegravir. It is important to note that the kinetic data obtained from baculovirus-insect Supersomes (rUGT) and HEK293 UGT-overexpressed cell lysates should be interpreted carefully because they were normalized to total protein instead of specific UGT protein amount (Tables 2 and 3). The Km values between cell systems for the same isoform broadly agree with each other, with slight differences. It appears that HEK-expressed UGT1A8 and UGT1A9 correlate better with HIMs and HLMs for dolutegravir and raltegravir glucuronidation. This additional layer of understanding may facilitate quantitative assessment of genetic variation and nongenetic perturbations to UGT metabolism caused by changes in the underlying system and may allow for better prediction of clinical impact.
TABLE 5.
Summary of the tissue- and isoform-specific UGTs responsible for cabotegravir, dolutegravir, and raltegravir metabolism based on relations of Km values derived from expression systems
| HLMs | HIMs | HKMs | |
|---|---|---|---|
| Cabotegravir | UGT1A1a | ND | ND |
| Dolutegravir | UGT1A9 > UGT1A1a | UGT1A3 > UGT1A8 > UGT1A1a | UGT1A9 |
| Raltegravir | UGT1A9 > UGT1A1 | UGT1A8 > UGT1A1 | UGT1A9 |
ND, not determined.
Greater than 2-fold difference in isoform UGT Km value from that observed for the tissue Km value.
The clinical relevance of pharmacogenomic variation contributing to the observed variability of integrase inhibitor disposition is limited for UGT1A1 and nearly nonexistent for UGT1A9 and the other UGTs. The UGT1A1 gene is highly polymorphic, with common genetic variants that reduce hepatic UGT1A1 activity (*6, *28, *36, and *37 are the most studied) (Wenning et al., 2009; Adams et al., 2012; Yagura et al., 2015). Chen et al. (2014) reported that dolutegravir clearance was significantly reduced and its exposure increased in carriers of low- and reduced-activity polymorphisms compared with subjects with normal activity. A similar statement is presented in the Food and Drug Administration–approved dolutegravir package insert (Food and Drug Administration, Office of Clinical Pharmacology Review, 2015). A recent study showed that UGT1A1*6 and *28 alleles individually and in combination are significantly associated with higher dolutegravir plasma trough concentrations and neuropsychiatric events (Yagura et al., 2017). A similar response was seen with raltegravir (Wenning et al., 2009; Yagura et al., 2015; Lee et al., 2016; Belkhir et al., 2018). There is still much to learn about the clinical implications of genetic variability in the UGT system and the importance in patient response or safety. First, no data are available regarding pharmacogenetics of cabotegravir, and the impact of genetic variations in other UGTs on INSTI exposure remains unstudied. Second, the tissue-specific impact of pharmacogenomic variability in extrahepatic UGT expression and implications for drug metabolism remain relatively unexplored.
In summary, significant glucuronidation occurs in various tissues throughout the body, with the liver, kidney, and intestine being particularly important for orally administered drugs. The degree of contribution from each tissue is UGT substrate- and isoform-specific and must be taken into account to improve in vitro prediction of in vivo behavior. Incorporation of in vitro hepatic, intestinal, and renal glucuronidation in a PBPK model achieved predicted dolutegravir clearance within 32%, Cmax within 17%, and AUC0-24 within 38% of observed data. Dynamic modeling and simulation approaches, along with clinical assessment, are needed to produce a useful tool to predict perturbations to the underlying system and individualize patient care.
Abbreviations
- AUC
area under the curve
- BSA
bovine serum albumin
- HEK293
human embryonic kidney 293
- HIM
human intestinal microsome
- HIV
human immunodeficiency virus
- HKM
human kidney microsome
- HLM
human liver microsome
- INSTI
integrase strand transfer inhibitor
- IVIVE
in vitro–in vivo extrapolation
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- MPPGK
microsomal protein per gram of kidney
- MPPGL
microsomal protein per gram of liver
- MPPI
microsomal protein per total intestine
- PBPK
physiologically based pharmacokinetic
- rUGT
recombinant UGT
- UDPGA
UDP-glucuronic acid
- UGT
UDP-glucuronosyltransferase
Authorship Contributions
Participated in research design: Liu, Lu, Desta, Gufford.
Conducted experiments: Liu, Lu, Watson, Gufford.
Contributed new reagents or analytic tools: Gufford, Lu, Watson, Lazarus.
Performed data analysis: Liu, Gufford, Desta.
Wrote or contributed to the writing of the manuscript: Liu, Lu, Watson, Lazarus, Desta, Gufford.
Footnotes
This work was supported in part by the Indiana Center for AIDS Research and grants from the National Institutes of Health National Institute of General Medical Sciences [Grants R01s GM078501 and GM121707 (Z.D.)]. S.N.L. and B.T.G. are supported by the National Institute of General Medical Sciences [Grant T32 GM008425]. B.T.G. is also supported by the Indiana Clinical and Translational Sciences Institute Young Investigator Award [Grant UL1 TR001108].
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Achour B, Dantonio A, Niosi M, Novak JJ, Fallon JK, Barber J, Smith PC, Rostami-Hodjegan A, Goosen TC. (2017) Quantitative characterization of major hepatic UDP-glucuronosyltransferase enzymes in human liver microsomes: comparison of two proteomic methods and correlation with catalytic activity. Drug Metab Dispos 45:1102–1112. [DOI] [PubMed] [Google Scholar]
- Adams JL, Greener BN, Kashuba ADM. (2012) Pharmacology of HIV integrase inhibitors. Curr Opin HIV AIDS 7:390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Jahdari WS, Yamamoto K, Hiraoka H, Nakamura K, Goto F, Horiuchi R. (2006) Prediction of total propofol clearance based on enzyme activities in microsomes from human kidney and liver. Eur J Clin Pharmacol 62:527–533. [DOI] [PubMed] [Google Scholar]
- Argikar UA, Potter PM, Hutzler JM, Marathe PH. (2016) Challenges and opportunities with non-CYP enzymes aldehyde oxidase, carboxylesterase, and UDP-glucuronosyltransferase: focus on reaction phenotyping and prediction of human clearance. AAPS J 18:1391–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkhir L, Seguin-Devaux C, Elens L, Pauly C, Gengler N, Schneider S, Ruelle J, Haufroid V, Vandercam B. (2018) Impact of UGT1A1 polymorphisms on Raltegravir and its glucuronide plasma concentrations in a cohort of HIV-1 infected patients. Sci Rep 8:7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boase S, Miners JO. (2002) In vitro-in vivo correlations for drugs eliminated by glucuronidation: investigations with the model substrate zidovudine. Br J Clin Pharmacol 54:493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers GD, Culp A, Reese MJ, Tabolt G, Moss L, Piscitelli S, Huynh P, Wagner D, Ford SL, Gould EP, et al. (2016) Disposition and metabolism of cabotegravir: a comparison of biotransformation and excretion between different species and routes of administration in humans. Xenobiotica 46:147–162. [DOI] [PubMed] [Google Scholar]
- Castellino S, Moss L, Wagner D, Borland J, Song I, Chen S, Lou Y, Min SS, Goljer I, Culp A, et al. (2013) Metabolism, excretion, and mass balance of the HIV-1 integrase inhibitor dolutegravir in humans. Antimicrob Agents Chemother 57:3536–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, St Jean P, Borland J, Song I, Yeo AJ, Piscitelli S, Rubio JP. (2014) Evaluation of the effect of UGT1A1 polymorphisms on dolutegravir pharmacokinetics. Pharmacogenomics 15:9–16. [DOI] [PubMed] [Google Scholar]
- Court MH, Zhang X, Ding X, Yee KK, Hesse LM, Finel M. (2012) Quantitative distribution of mRNAs encoding the 19 human UDP-glucuronosyltransferase enzymes in 26 adult and 3 fetal tissues. Xenobiotica 42:266–277. [DOI] [PubMed] [Google Scholar]
- Davies B, Morris T. (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10:1093–1095. [DOI] [PubMed] [Google Scholar]
- DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents (2018) Guidelines for the use of antiretroviral agents in adults and adolescents living with HIV (Updated: 2018). US Department of Health and Human Services, Washington, DC. [Google Scholar]
- Drodzik M, Busch D, Lapczuk J, Muller J, Ostrowski M, Kurzawski M, Oswald S. (2018) Protein abundance of clinically relevant drug-metabolizing enzymes in the human liver and intestine: a comparative analysis in paired tissue specimens. Clin Pharmacol Ther 104:515–524. [DOI] [PubMed] [Google Scholar]
- Flexner C. (2019) Modern human immunodeficiency virus therapy: progress and prospects. Clin Pharmacol Ther 105:61–70. [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration, Office of Clinical Pharmacology Review (2015) 204790 Dolutegravir, Tivicay. Food and Drug Administration, Silver Spring, MD. [Google Scholar]
- Gill KL, Gertz M, Houston JB, Galetin A. (2013) Application of a physiologically based pharmacokinetic model to assess propofol hepatic and renal glucuronidation in isolation: utility of in vitro and in vivo data. Drug Metab Dispos 41:744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill KL, Houston JB, Galetin A. (2012) Characterization of in vitro glucuronidation clearance of a range of drugs in human kidney microsomes: comparison with liver and intestinal glucuronidation and impact of albumin. Drug Metab Dispos 40:825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gufford BT, Barr JT, González-Pérez V, Layton ME, White JR, Jr, Oberlies NH, Paine MF. (2015) Quantitative prediction and clinical evaluation of an unexplored herb-drug interaction mechanism in healthy volunteers. CPT Pharmacometrics Syst Pharmacol 4:701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemette C, Lévesque É, Rouleau M. (2014) Pharmacogenomics of human uridine diphospho-glucuronosyltransferases and clinical implications. Clin Pharmacol Ther 96:324–339. [DOI] [PubMed] [Google Scholar]
- Houston JB, Kenworthy KE. (2000) In vitro-in vivo scaling of CYP kinetic data not consistent with the classical Michaelis-Menten model. Drug Metab Dispos 28:246–254. [PubMed] [Google Scholar]
- Izukawa T, Nakajima M, Fujiwara R, Yamanaka H, Fukami T, Takamiya M, Aoki Y, Ikushiro S, Sakaki T, Yokoi T. (2009) Quantitative analysis of UDP-glucuronosyltransferase (UGT) 1A and UGT2B expression levels in human livers. Drug Metab Dispos 37:1759–1768. [DOI] [PubMed] [Google Scholar]
- Kassahun K, McIntosh I, Cui D, Hreniuk D, Merschman S, Lasseter K, Azrolan N, Iwamoto M, Wagner JA, Wenning LA. (2007) Metabolism and disposition in humans of raltegravir (MK-0518), an anti-AIDS drug targeting the human immunodeficiency virus 1 integrase enzyme. Drug Metab Dispos 35:1657–1663. [DOI] [PubMed] [Google Scholar]
- Komura H, Iwaki M. (2011) In vitro and in vivo small intestinal metabolism of CYP3A and UGT substrates in preclinical animals species and humans: species differences. Drug Metab Rev 43:476–498. [DOI] [PubMed] [Google Scholar]
- Laufer R, Paz OG, Di Marco A, Bonelli F, Monteagudo E, Summa V, Rowley M. (2009) Quantitative prediction of human clearance guiding the development of Raltegravir (MK-0518, isentress) and related HIV integrase inhibitors. Drug Metab Dispos 37:873–883. [DOI] [PubMed] [Google Scholar]
- Lee LS, Seng KY, Wang LZ, Yong WP, Hee KH, Soh TI, Wong A, Cheong PF, Soong R, Sapari NS, et al. (2016) Phenotyping of UGT1A1 activity using raltegravir predicts pharmacokinetics and toxicity of irinotecan in FOLFIRI. PLoS One 11:e0147681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margaillan G, Rouleau M, Fallon JK, Caron P, Villeneuve L, Turcotte V, Smith PC, Joy MS, Guillemette C. (2015) Quantitative profiling of human renal UDP-glucuronosyltransferases and glucuronidation activity: a comparison of normal and tumoral kidney tissues. Drug Metab Dispos 43:611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias AA, West S, Hui J, Kearney BP. (2009) Dose-response of ritonavir on hepatic CYP3A activity and elvitegravir oral exposure. Clin Pharmacol Ther 85:64–70. [DOI] [PubMed] [Google Scholar]
- Paine MF, Khalighi M, Fisher JM, Shen DD, Kunze KL, Marsh CL, Perkins JD, Thummel KE. (1997) Characterization of interintestinal and intraintestinal variations in human CYP3A-dependent metabolism. J Pharmacol Exp Ther 283:1552–1562. [PubMed] [Google Scholar]
- Reese MJ, Savina PM, Generaux GT, Tracey H, Humphreys JE, Kanaoka E, Webster LO, Harmon KA, Clarke JD, Polli JW. (2013) In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a HIV integrase inhibitor. Drug Metab Dispos 41:353–361. [DOI] [PubMed] [Google Scholar]
- Soars MG, Burchell B, Riley RJ. (2002) In vitro analysis of human drug glucuronidation and prediction of in vivo metabolic clearance. J Pharmacol Exp Ther 301:382–390. [DOI] [PubMed] [Google Scholar]
- Song I, Borland J, Arya N, Wynne B, Piscitelli S. (2015) Pharmacokinetics of dolutegravir when administered with mineral supplements in healthy adult subjects. J Clin Pharmacol 55:490–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassburg CP, Kneip S, Topp J, Obermayer-Straub P, Barut A, Tukey RH, Manns MP. (2000) Polymorphic gene regulation and interindividual variation of UDP-glucuronosyltransferase activity in human small intestine. J Biol Chem 275:36164–36171. [DOI] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trezza C, Ford SL, Spreen W, Pan R, Piscitelli S. (2015) Formulation and pharmacology of long-acting cabotegravir. Curr Opin HIV AIDS 10:239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenning LA, Petry AS, Kost JT, Jin B, Breidinger SA, DeLepeleire I, Carlini EJ, Young S, Rushmore T, Wagner F, et al. (2009) Pharmacokinetics of raltegravir in individuals with UGT1A1 polymorphisms. Clin Pharmacol Ther 85:623–627. [DOI] [PubMed] [Google Scholar]
- Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, Peterkin V, Koup JR, Ball SE. (2004) Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos 32:1201–1208. [DOI] [PubMed] [Google Scholar]
- Wood FL, Houston JB, Hallifax D. (2017) Clearance prediction methodology needs fundamental improvement: trends common to rat and human hepatocytes/microsomes and implications for experimental methodology. Drug Metab Dispos 45:1178–1188. [DOI] [PubMed] [Google Scholar]
- Yagura H, Watanabe D, Ashida M, Kushida H, Hirota K, Ikuma M, Ogawa Y, Yajima K, Kasai D, Nishida Y, et al. (2015) Correlation between UGT1A1 polymorphisms and raltegravir plasma trough concentrations in Japanese HIV-1-infected patients. J Infect Chemother 21:713–717. [DOI] [PubMed] [Google Scholar]
- Yagura H, Watanabe D, Kushida H, Tomishima K, Togami H, Hirano A, Takahashi M, Hirota K, Ikuma M, Kasai D, et al. (2017) Impact of UGT1A1 gene polymorphisms on plasma dolutegravir trough concentrations and neuropsychiatric adverse events in Japanese individuals infected with HIV-1. BMC Infect Dis 17:622. [DOI] [PMC free article] [PubMed] [Google Scholar]