

Introduction
A new paradigm for preventing atherosclerotic cardiovascular disease (ASCVD) is needed. The most recent US data show the long‐term decline in cardiovascular deaths has stopped, and has started to increase in the most at‐risk populations.1 Indeed, rising rates of obesity and diabetes mellitus in the setting of suboptimal risk factor control have resulted in a similar number of cardiovascular events occurring in those aged <65 years as ≥65 years.2 Although preventive drug therapies reduce the relative risk of cardiovascular events in primary and secondary prevention patients, the absolute risk of subsequent ASCVD events remains high.3 If nothing changes, it is projected that by 2035 nearly half the US population will have some form of cardiovascular disease and costs will double to $1.1 trillion annually.4
Systemic approaches to improving lifestyle habits and better risk factor control are clearly needed. Given the difficulty of these endeavors to date, and the persistently high burden of ASCVD when risk factor modification is started later in adulthood, we propose a new paradigm for ASCVD prevention. Based on the extensive data reviewed below, we consider that it is now time to investigate whether intensively lowering plasma apolipoprotein (apo) B lipoprotein levels in younger and early midlife adults will regress earlier stages of atherosclerosis, thereby eliminating the risk of developing clinical ASCVD events later in life. Just as an understanding of the causative agents of other diseases has allowed the eradication of a range of human scourges, this state‐of‐the‐art review will emphasize that a deep understanding of the pathogenesis of atherosclerosis can be translated into an achievable goal of eradicating ASCVD.
As a next step, we describe a proposed clinical trial to test early intervention to profoundly lower the concentration of low‐density lipoprotein (assessed by its cholesterol component, LDL‐C) and other apo B‐containing lipoprotein in individuals aged 25 to 55 years who have image‐documented preclinical atherosclerosis. Such a trial may provide the first direct evidence to support marked or even complete regression of early atherosclerosis in humans, and lay the ground work for definitive trials to support a new prevention paradigm of intensive regression therapy followed by intermittent retreatment for eradication of the clinical burden of ASCVD.
Evidence for Apo B Lipoproteins as the Root Cause of Atherosclerosis: Response‐to‐Retention
Apo B lipoproteins transport cholesterol and triglycerides in plasma. Apo B lipoproteins up to 70 nm diameter, including low‐density lipoproteins (LDL), intermediate‐density lipoproteins (IDL), smaller very‐low‐density lipoproteins (VLDL), chylomicron remnant particles, and lipoprotein (a) [Lp(a)], efficiently cross the protective endothelial layer and penetrate into the intima of the artery wall.5, 6 The probability that these particles enter and leave the arterial subendothelial space is dependent on particle size, plasma concentration, blood pressure, arterial injury, and the affinity and ability of the lipoprotein to bind proteoglycans. The equilibrium can be shifted when either lipoprotein particle concentration or intimal hyperplasia is increased (Figure 1). Normally, most LDL particles that cross the endothelium and enter the intima then return to the circulation. However, in the presence of certain cardiovascular risk factors or disturbed laminar flow, LDL particles are more likely to adhere to intimal proteoglycans and be retained in situ.7, 8 The triglyceride‐rich lipoproteins, including IDLs, chylomicron remnants, and VLDL, may have difficulty leaving the intima because of their larger size or because of entrapment by components of the arterial intima.5
Figure 1.
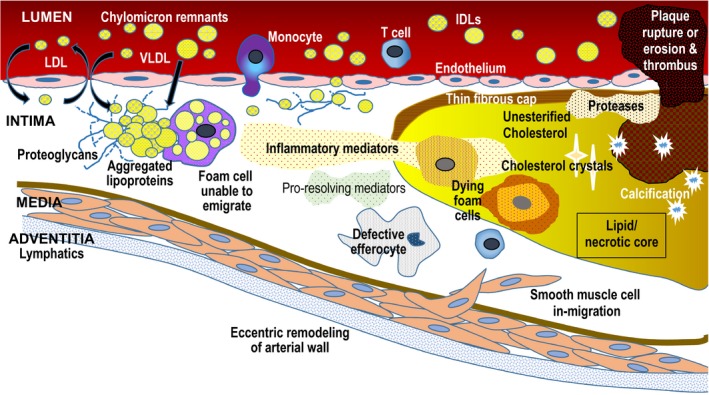
Apo B lipoprotein response‐to‐retention model of atherosclerosis initiation and progression. High plasma concentrations of apo B lipoproteins (LDL, IDL, VLDL, chylomicron remnants, Lp(a)) increase entry into intima and retention. Apo B lipoproteins bind to proteoglycans and begin aggregating, a process that accelerates once plaque begins. Retention is influenced by particle composition and diet, among other factors. Retention leads to a maladaptive cellular response leading to increased inflammation, fibrosis, and necrosis. The lipid/necrotic core forms when normal phagocytotic processes and efferocytosis are overwhelmed by continued retention and accumulation of “toxic” apo B lipoproteins. Plaque rupture or erosion can lead to formation of overlying thrombus, which can precipitate an acute clinical event. Apo indicates Apolipoprotein; IDL, intermediate lipoprotein; LDL, low‐density lipoprotein; Lp(a), Lipoprotein (a); VLDL, very‐low‐density lipoprotein.
Following retention within the arterial intima, apo B lipoproteins undergo enzymatic modifications that further accelerate accumulation and promote particle aggregation.9 Aggregation is influenced by lipoprotein particle quantity and composition, both of which are influenced by diet and adiposity.8, 10 Aggregation can also be reduced by proprotein convertase subtilisin‐like/kexin type 9 (PCSK9) inhibition.10 Aggregated apo B lipoproteins are avidly taken up by macrophages, initiating their transformation into foam cells. Apo B lipoproteins are also taken up by smooth muscle cells.
Both unesterified and esterified cholesterol from the apo B lipoproteins retained in the arterial intima account for the characteristic intracellular lipid droplets in foam cells. Lipoprotein‐derived cholesterol contributes to cholesterol‐enrichment of cell membranes, cholesterol‐rich membrane fragments, and cholesterol crystals that provoke additional maladaptive responses, such as activation of Toll‐like receptors, the NLRP3 inflammasome and interleukin‐1ß, apoptosis, and prothrombotic pathways.11, 12, 13, 14
The cholesterol and triglyceride‐rich remnant apo B lipoproteins appear to be more potent than LDL for provoking greater maladaptive immune activation for several reasons.15, 16 Lipoprotein lipase, either at the endothelial surface or within the arterial intima, degrades triglycerides into constituent fatty acids and monoacylglycerols, which generate local inflammation. Apo C‐III, an apolipoprotein present on VLDL and remnant lipoproteins that binds to lipoprotein lipase to inhibit triglyceride clearance, also increases proteoglycan binding and appears to have proinflammatory characteristics in experimental studies.17
The maladaptive inflammatory aspect of atherogenesis is downstream from the initial retention of apo B lipoprotein particles, but once present promotes cell recruitment, further plaque development, and ultimately acute ASCVD events. Aggregates of apo B lipoproteins release biologically active byproducts that are chemoattractive to macrophages, smooth muscle cells, and immunoregulatory T cells, promoting their recruitment into the developing lesion. Retained and modified apo B lipoproteins trigger the production of anti‐emigration signals by macrophages, preventing them from leaving the arterial intima, in a process reminiscent of the persistence of macrophages within tuberculous granulomata so that they do not disseminate the infection.18, 19
Persistent macrophages and other cells in atherosclerotic plaques release proatherogenic enzymes, tissue factor, and signaling molecules, inducing synthesis of more proteoglycans with increased affinity for apo B lipoproteins, other factors that enhance retention, increase fibrosis, and proteolytic enzymes.18, 19 Proteases can weaken the overlying fibrous cap, which favors plaque rupture and exposure of the procoagulant subendothelial contents, particularly tissue factor, into the blood. Moreover, the internalization of apo B lipoproteins by lesional macrophages promotes inflammatory responses and impairs resolution responses, both of which play critical roles in plaque progression. Advanced, complex atherosclerotic lesions are characterized by compensatory vascular remodeling and calcification. Neovascularization of the growing plaque and necrosis of the lipid core further contribute to plaque instability. Complicated lesions arise as advanced plaques erode or rupture with overlying thrombosis, often causing acute clinical cardiovascular events.
Evidence for a Causal Role of Apo B Lipoproteins in Atherosclerosis: Epidemiology and Genetics
Atherogenesis often begins in childhood, and some individuals begin to develop advanced plaque in late adolescence or early adulthood.20, 21 The rate of plaque progression and occurrence of ASCVD events depends on the level of and duration of exposure to apo B lipoproteins, presence of other cardiovascular risk factors, and genetic predisposition.22, 23 In countries with long‐term exposure to atherogenic diets, most adults have advanced fibrocalcific plaque by age 50.21, 24 Without risk factor intervention, clinical atherothrombotic events commonly begin occurring when men are in their 6th and 7th decades, and women are in their 7th and 8th decades.25
Plasma apo B lipoprotein levels, as reflected in total cholesterol, LDL‐C, non‐high density lipoprotein (HDL)‐C, and apo B levels, are associated with greater subclinical atheroma burden and increased risk of ASCVD events in all age, sex, and race and ethnicity groups and regions.23, 26, 27, 28 Both the apo B lipoprotein level and the cholesterol content of the particles are associated with increased ASCVD.29 The increase in the relative risk of ASCVD per increment increase in total cholesterol or LDL‐C depends on the level and duration of exposure and the presence of other risk factors.21, 23, 30, 31, 32
Chronic overnutrition impairs clearance of cholesterol and triglyceride rich lipoproteins as well as causing obesity.33 Obesity predisposes to abnormal glucose metabolism and defective insulin action, which along with diet, further contributes to increased plasma levels of VLDL and cholesterol and triglyceride‐rich remnant apo B lipoproteins and maladaptive inflammation.5, 33, 34 Elevated plasma concentrations of both fasting and non‐fasting triglyceride‐rich apo B lipoproteins are associated with higher ASCVD risk.
The fundamental causal role of apo B lipoproteins is further supported by observations that individuals with genetically lower LDL‐C or apo B lipoprotein levels have lower lifetime risk of coronary heart disease, despite the presence of other risk factors.16, 21, 35 Recent data suggest atherosclerosis still develops in the absence of risk factors when LDL‐C levels are >60 mg/dL.31 It is noteworthy that contemporary aboriginal populations living a subsistence lifestyle have low plasma levels of apo B lipoproteins and high levels of inflammatory markers, including C‐reactive protein, because of chronic infection, yet have little evidence of subclinical atherosclerosis.36 Likewise, in animal models, atherosclerosis develops in the presence of tobacco smoke, immune derangements, or hypertension only when plasma levels of apo B lipoproteins are elevated.18
Evidence for Role of Apo B Lipoproteins in Plaque Regression
Atherosclerotic plaques require a continuous supply of apo B lipoproteins from plasma to progress.18 Reducing the inflow of toxic apo B lipoproteins into the arterial intima allows normal scavenger and phagocytic clearance mechanisms to clear the apo B lipoprotein overload (Figure 2).37 In advanced mouse atherosclerotic plaques, large reductions in plasma apo B lipoprotein levels induce macrophage foam cells to undergo phenotypic changes into resolution‐promoting cells. These cells have similarities to the M2 activated state, which finally allows these cells to emigrate into adventitial lymphatics and transmigrate back into the lumen.19, 38, 39, 40 Some properties of these resolution‐promoting macrophages that promote regression include effective clearance of dead lesional cells by the process of efferocytosis and the quelling of lesional inflammation. In‐migration of healthy, non‐lipid‐laden macrophages also contribute to more effective efferocytosis.41 This leads to plaques that are less necrotic and more stable against rupture and erosion. Inflammatory cells leave and smooth muscle cells migrate into the subendothelium and fibrosis resolves. The role of macrophage‐specific cholesterol removal in regression of human plaque is not yet clear, though in preclinical models, if there is no functional HDL there is little regression despite lowering apo B lipoproteins.42 Plaque stabilization occurs as the lipid‐rich core shrinks, reducing maladaptive inflammatory cell infiltration and microvascularization, and the thin fibrous cap thickens.
Figure 2.
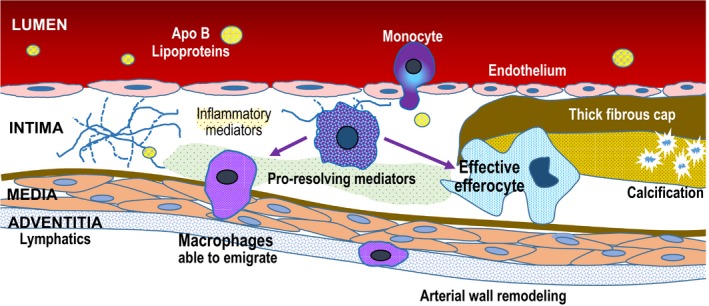
Mechanisms of regression following apo B lipoprotein reduction. Dramatic reduction in plasma concentrations of LDL‐C and other apo B lipoproteins leads to decreased subendothelial entry and retention. Decreased levels of “toxic” apo B lipoproteins allows normal phagocytic and inflammation resolving mechanisms to “heal” the plaque. Decreased foam cell formation in the intima allows macrophages to migrate into adventitial lymphatics. Increased in‐migration of monocytes that become healthy macrophages results in effective efferocytosis to remove necrotic debris. Apo indicates apolipoprotein.
In animal models, reducing LDL‐C levels to below 25 mg/dL (0.6 mmol/L) has been shown to completely regress early atheromata (Figure 3) and normalize vascular function, with return of nitric oxide sensitivity and improved nitric oxide synthase cofactor bioavailability.6, 43, 44, 45 In models of more advanced atheromata, substantial regression can occur but residual stabilized plaque remains and vascular function continues to be abnormal.6, 44, 46 Aggressive LDL‐C reductions cause almost immediate loss of foam cells through emigration, and over the longer term, resolution of necrotic regions, infiltration of smooth muscle cells, and reductions in cholesterol clefts and fibrosis.40, 47, 48
Figure 3.
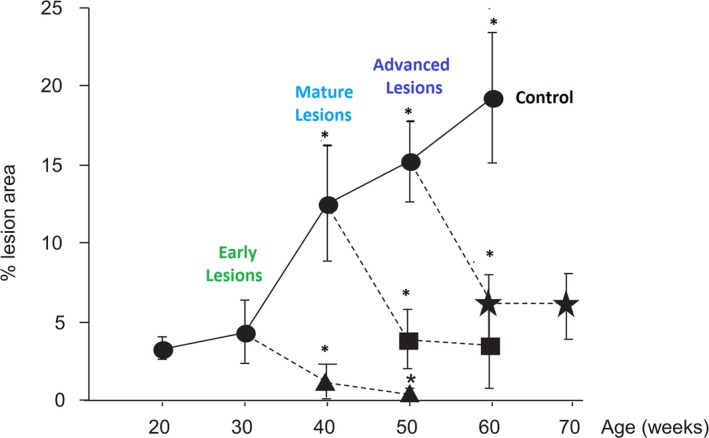
Complete regression of early plaque lesions when intensive cholesterol lowering to 11 to 55 mg/dL starts at 30 weeks, compared with substantial although not complete regression of later stages of plaque when intensive cholesterol lowering is initiated at 40 or 50 weeks. Adapted from Björkegren et al44 with permission. Copyright ©2014 PLOS Genetics. ***P<0.001.
Animal experimentation and human epidemiologic studies have shown that endothelial dysfunction and vascular stiffness develop before blood pressure levels begin to rise.49, 50, 51 These findings suggest that regression of early atherosclerosis could also prevent or delay the later development of hypertension, and thus hypertension's clinical sequelae. Indeed, several statin trials have found reductions in blood pressure and hypertension incidence in statin‐treated patients.52 In animal models, PCSK9 deficiency has been shown to reduce apo B lipoprotein levels, atherosclerosis development, and endothelial dysfunction.53 PCSK9 inhibitors have also recently been shown to improve endothelial function in proportion to the magnitude of LDL‐C lowering.54
In human clinical trials, intensive LDL‐C lowering with statins, statins combined with ezetimibe, and PCSK9 monoclonal antibodies have been shown to modestly reduce atheroma volume in proportion to the magnitude of LDL‐C decrease in individuals with clinical coronary artery disease.55, 56 Indeed, there appears to be no lower LDL‐C limit for regression of human coronary atheroma, with greater reductions in plaque volume to LDL‐C levels as low as 15 mg/dL (0.4 mmol/L) in trials of statins and PCSK9 monoclonal antibodies added to statin therapy. A systematic review of 50 regression trials found that significant plaque regression occurred in trials with an average treatment period of 20 months.57
As will be discussed below, current imaging methods may underestimate beneficial changes in plaques after treatments, particularly those that affect cellular composition (such as loss of macrophages or a change in their inflammatory state) or replacement of a necrotic core by a thicker fibrous cap. Less intensive statin therapy slows atheroma progression in primary prevention in children and adults.58, 59 Carotid artery specimens from statin‐treated individuals show favorable pathophysiologic changes including plaque stabilization similar to those observed in animal studies, and these changes are associated with decreased risk of ASCVD events.60
Greater percent regression, as assessed by intravascular ultrasound, occurs in women with a lower plaque burden, suggesting more responsiveness to aggressive LDL‐C lowering in earlier stages of plaque development.61 Indeed, recent data suggest that high‐intensity statin therapy has a greater effect on non‐calcified plaque than on more advanced, calcified lesions.62 Animal studies suggest that complete plaque resolution in humans may be possible only in early stages, rather than in later stages when there is a more extensive burden of fibrocalcific plaque.44, 63 The more rapid and extensive plaque stabilization and regression of earlier lesions appears to result from reductions in lipid core volume as well as resolution of maladaptive inflammation and early fibrotic changes.64, 65, 66
Patients with clinically manifested ASCVD in the secondary prevention regression trials continue to have a large burden of atherosclerosis despite substantial LDL‐C lowering, and remain at high risk of recurrent cardiovascular events. Up to 35% of patients experiencing a coronary event will die within a year (Figure 4).56, 67, 68 Recent studies have found the pathophysiology of acute coronary events is changing as patients are increasingly treated with statins, such that plaque erosion, rather than rupture of unstable plaque, is emerging as a major cause of recurrent coronary events.69 These findings suggest secondary prevention efforts will be insufficient for ASCVD eradication. Indeed, in recent PCSK9 monoclonal antibody trials, the continuing high rate of cardiovascular events is more likely to be explained by the persistence of a high burden of atherosclerosis than from inadequate LDL‐C lowering.67
Figure 4.
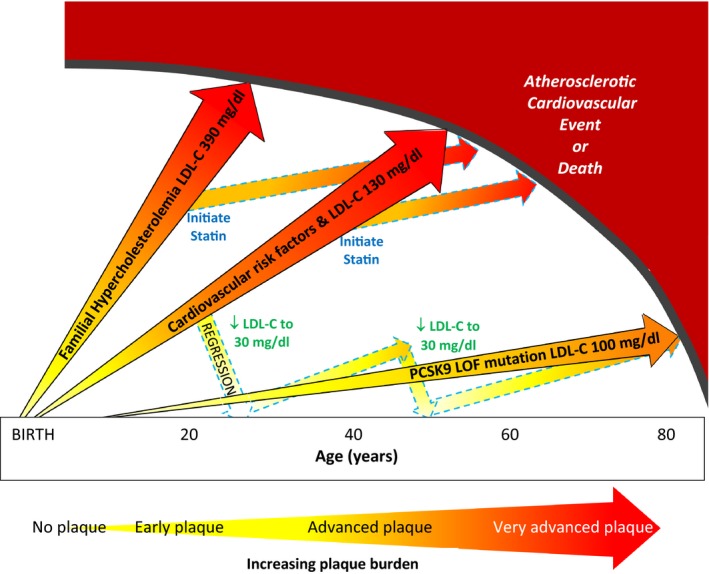
Life course trajectory of atherosclerotic progression is illustrated for individuals at very high, high, and low risk of atherosclerotic cardiovascular disease events (ASCVD). (1) individuals with heterozygous familial hypercholesterolemia who have severe LDL‐C elevation from birth have markedly accelerated atherosclerosis and premature onset of clinical ASCVD events; (2) individuals with cardiovascular risk factors from young adulthood and “average” LDL‐C levels of 130 mg/dL are more likely to experience clinical ASCVD events in early and middle age; and (3) individuals with a proprotein convertase subtilisin/kexin type 9 PCSK9 loss‐of‐function (PCSK9 LOF) mutation have lower LDL‐C levels throughout the lifespan and may be at markedly reduced risk of clinical ASCVD events. LDL‐C lowering with statins can stabilize and modestly regress plaque but does not eradicate the plaque burden and remain at increased risk of clinical ASCVD events. Intensive LDL‐C lowering to 20 to 40 mg/dL may have a greater impact on plaque regression in earlier stages of plaque. A new paradigm of “regression” treatment with intensive LDL‐C lowering earlier in the course of atherosclerosis or at younger ages could then be followed by intermittent retreatment to “maintain” a low plaque burden until late in life. LDL‐C indicates low‐density lipoprotein cholesterol (to convert to mmol/L divide by 38.65 mg/dL); PCSK9 LOF indicates proprotein convertase subtilisin/kexin type 9 loss‐of‐function mutation.
In animal models, raising functional HDL has been shown to potentiate the effect of LDL‐C reduction on plaque regression.70 In humans, however, Mendelian randomization studies have found no association between HDL‐C level and risk of cardiovascular events.71 Nor has pharmacologically increasing HDL‐C been shown to reduce cardiovascular events independent of effects on LDL‐C.72 Animal studies suggest that the functional properties of HDLs appear to be more important than the HDL‐C level per se for regressing atheroma, and some data from humans suggest that greater HDL functionality is associated with lower risk of ASCD events.73
Evidence for LDL‐C, Non‐HDL‐C, and Apo B Lowering Drug Therapy to Reduce Cardiovascular Events
It is clearly established that the LDL‐C, non‐HDL‐C, and apo B reductions induced by statins, ezetimibe and PCSK9 inhibitor therapies translate into a reduction in ASCVD events.21, 74, 75 Indeed, progressively greater magnitudes of reduction in the relative risk of cardiovascular disease correlate with progressively larger absolute reductions in LDL‐C, non‐HDL‐C, and apo B lipoprotein levels.74, 76 Non‐HDL‐C differs from LDL‐C in that non‐HDL cholesterol encompasses VLDL and remnant lipoprotein cholesterol plus LDL‐C and cholesterol in Lp(a). Importantly, in relative terms, statin therapy appears to be more effective in lower risk individuals (eg, those who are younger, since age contributes the largest component of risk in multivariate equations77), with almost twice the relative reduction in the risk of cardiovascular events in lower than in higher risk individuals.78 These data provide evidence for the concept that a greater magnitude of LDL‐C, non‐HDL‐C, or apo B lowering will have a greater relative impact on earlier, compared with later, stages of atherosclerosis, consistent with what has been found in the animal and human regression studies.79 Moreover, long‐term post‐trial follow‐up of several primary prevention statin trials provides evidence that LDL‐C lowering has a lasting impact on plaque stabilization and atherosclerotic burden and the risk of ASCVD events, or a “legacy” effect. Participants treated with statins for 3 to 5 years remains at lower cardiovascular risk over follow‐up periods of 11 to 20 years.79
Recent trials of PCSK9 inhibiting monoclonal antibodies have demonstrated that an additional 50% to 60% reduction in LDL‐C resulted in modest 15% to 20% reductions in the relative risk of cardiovascular events in statin‐treated patients with ASCVD.67 Despite reducing LDL‐C to a mean of 30 mg/dL (0.8 mmol/L) in the evolocumab group in Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk (FOURIER), the recurrent rate of major cardiovascular events was 4.5%, or 45% extrapolated to 10 years. This suggests that waiting to profoundly reduce LDL‐C, non‐HDL‐C, and apo B lipoproteins until a large burden of advanced atherosclerosis has developed has only moderate effects on the burden of atherosclerosis and does not prevent most recurrent ASCVD events. In animal models and in humans with advanced atherosclerosis, it appears that the non‐resolving inflammation in advanced plaques impairs beneficial changes that follow aggressive LDL‐C lowering, further highlighting the advantages of earlier intervention.38 Moreover, targeting maladaptive inflammation in patients with clinical ASCVD and elevated C‐reactive protein levels also appeared to have only a modest impact on recurrent cardiovascular events in the recent CANTOS (Canakinumab Anti‐inflammatory Thrombosis Outcome Study), with a 15% reduction in major cardiovascular events over 4 years.80 Taken together, these findings indicate that even potent therapies that are delayed until a large burden of advanced atherosclerosis has developed can have only moderate additional benefits on the clinical burden of ASCVD.
Advances in Non‐Invasive Imaging of Atherosclerotic Plaque
Several invasive and non‐invasive methods are available to image atherosclerotic plaque burden, composition, activity, natural history, and response to therapy. Plaque burden is consistently associated with ASCVD events using a variety of measures.47, 81 However, only some of these imaging methods are able to detect and quantify response to therapy while others have practical limitations. Coronary intravascular ultrasound is a well‐validated invasive test that has been used in numerous trials to assess drug effects on atheroma volume. In primary prevention, however, a safe, non‐invasive imaging test is required. Carotid intimal medial thickness is a well‐validated test that has been shown to detect plaque regression in children and statin‐naïve‐adults, but requires specialized technical expertise and thus not widely generalizable, and is less predictive of coronary events.82
The latest generations of coronary computed tomography angiography (CTA) scanners have high resolution for plaque and calcification and much lower radiation exposures than early generations of scanners. CTA is non‐invasive as opposed to intravascular ultrasound and optical coherence tomography, covers the entire coronary tree, and allows evaluation of the total extent of calcified and non‐calcified plaques. It is also widely available, relatively low cost, directly interpretable as to the cause of coronary heart disease events, directly demonstrates regression of visible plaque, and requires no specialized expertise to perform. Studies using CTA would be widely implementable, especially as automated reading programs under development become available.83
CTA has also emerged as the preferred choice for evaluating and characterizing composition of coronary plaques.84 In particular, measurement of low attenuation (eg, uncalcified) plaque volume by CTA is a good measure of plaque burden in younger people for several reasons. Several studies demonstrate an association between total, percent, and low attenuation coronary plaque volume assessed by CTA and the risk of future major cardiovascular events, as well as response to statins or other preventive therapy.85, 86, 87, 88 In contrast, coronary artery calcification reflects more advanced plaque, and increases (rather than decreases) with statin therapy.89, 90
CTA images have been validated against virtual histology by intravascular ultrasound and have been found to be accurate and reliable in the estimation of plaque volume.84 Semi‐automated CTA analysis of coronary plaque composition is reliable and reproducible and non‐invasive imaging of coronary CTA is a validated tool for assessment of response to drug therapy.91 The radiation dose from the latest generation of computed tomography (CT) scanners is less than a mammogram, with an exposure of 3 to 4 mSv per scan. This is less than the 6.2 mSv that the average person in the United States receives each year from natural sources like the sun, outer space, air, food, and soil, as well as from medical procedures. It is far less than the 50 mSv of radiation allowed each year for people who are exposed to radiation in their jobs.
Carotid arterial wall magnetic resonance imaging (MRI) is becoming a more commonplace method to visualize atherosclerotic plaque burden and composition (lipid‐rich necrotic core, overt calcifications, intraplaque hemorrhage and fibrous tissue visualization).92 Vessel wall MRI is reproducible, can effectively visualize all the major arterial systems, requires no ionizing radiation, has been shown to discriminate cardiovascular events, and has been used in clinical trials as a measure of drug effectiveness in plaque regression.93, 94 Additionally, MRI has proven useful in the evaluation atherosclerotic plaque microvascularization and permeability, key players in atherogenesis and rupture.
Positron emission tomography (PET) can be used to visualize maladaptive inflammation and the metabolic processes within atherosclerotic plaques.95 Radiotracers are now available to visualize distinct aspects of the atherosclerosis cascade and plaque destabilization, including macrophage‐mediated inflammatory change, hypoxia, and micro‐calcification. Of these radiotracers, 18‐F‐florodeoxyglucose (FDG) is the most common and is used to non‐invasively evaluate the metabolic activity of vascular inflammation.96 18‐F‐florodeoxyglucose uptake correlates well with macrophage content in atherosclerotic lesions and has established relationships with circulating inflammatory biomarkers. It is highly reproducible and requires only modest sample sizes to evaluate inflammatory treatment efficacy in clinical trials.81 Combined PET/MRI scanners now combine the strengths of 2 distinct imaging modalities to simultaneously offer a platform to extensively evaluate the entire atherosclerotic cascade in a single imaging session. Radiation exposure with hybrid PET/MRI scanning is lower than PET/CT and may be particularly beneficial in young patients or for serial measurements of disease progression.97
Translating the Evidence into Next Prevention Paradigm: ASCVD Eradication
The substantial body of evidence reviewed above supports apo B lipoproteins as the fundamental initiating causal factor in atherosclerosis. Moreover, early intervention to profoundly lower LDL‐C or non‐HDL‐C or apo B levels may substantially reverse, and even eradicate, earlier stages of atherosclerosis. Thus, we have proposed translating this evidence into a new paradigm for ASCVD prevention that eradicates the clinical burden of atherosclerosis by intensively lowering LDL‐C at a younger age (Figure 4).98 This new model is akin to that of cancer therapy, with an acute induction phase followed by a maintenance phase. After an intensive period of LDL‐C/non‐HDL‐C/apo B reduction to largely regress and stabilize atherosclerotic plaque, subsequent periodic retreatment every decade or so could occur as needed to suppress significant plaque re‐development and progression as the individual ages. This approach avoids the need for adherence to lifelong lipid‐lowering drug therapy for most patients and minimizes safety considerations. As discussed above, this approach seeks to approximate the life course of individuals heterozygous for PCSK9 loss‐of‐function mutations, in whom lifelong lower plasma LDL‐C levels are associated with markedly reduced risk of ASCVD events despite significant risk factor burdens.35 This approach could potentially eliminate the leading cause of death and healthcare expenditures within a generation, while addressing widening disparities in cardiovascular morbidity and mortality.
Proposed CURE ATHERO Trial
In the proposed CURE ATHERO (Curing Early Atherosclerosis) trial, we plan to translate the finding in animal models that intensive LDL‐C lowering results in extensive regression of earlier stages of plaque into a novel prevention strategy in humans. The trial will enroll younger and early midlife, obese adults with cardiovascular risk factors who have a significant burden of uncalcified atherosclerotic plaque. CURE ATHERO builds on the hypothesis that the clinical burden of atherosclerosis might be eradicated in humans in its early stages by aggressively LDL‐C lowering to a level of 20 to 40 mg/dL (0.5–1.0 mmol/L) for a relatively short intervention period of 3 years. Recent data from PCSK9 inhibitor trials suggest that this LDL‐C target is both achievable and safe over this time period.67 The primary end point of the trial, relative changes in low attenuation plaque volume measured by CTA after 3 years will be used to evaluate plaque regression.
An age‐adapted Pathologic Determinants of Atherosclerosis in Youth (PDAY) atherosclerosis likelihood risk score will be used to identify screen‐eligible women and men aged 25 to 55 years.99, 100 PDAY risk factors include age, sex, body mass index, blood pressure, non‐HDL‐C, HDL‐C, smoking, and hyperglycemia or diabetes mellitus. PDAY score ≥25 predicts a >40% chance of having a significant burden of atheroma. The PDAY risk score predicts coronary artery calcification accurately up to 25 years later.100
Eligible individuals (n=130) with measurable low attenuation plaque volume will be randomized to either: (1) intensive LDL‐C lowering to a level of 20 to 40 mg/dL (0.5–1.0 mmol/L) using lifestyle and statins±alirocumab (a PCSK9 inhibitor)±ezetimibe or (2) usual care according to the American College of Cardiology/American Heart Association's most recent cholesterol guidelines. CTAs will be performed at baseline and at 18 and 36 months.
Robust imaging and biorepositories will leverage investment in the trial for mechanistic studies and future discovery. Planned ancillary investigations include imaging studies such as PET MRI/CT to evaluate responses to intensive LDL‐C lowering on earlier stages of human plaque. Evaluations of genomic, metabolomic, lipidomic, and proteomic profiles are also planned.
CURE ATHERO will determine whether intensive LDL lowering can substantially reverse early atherosclerosis over a period of 3 years. We anticipate some participants with less advanced plaque may experience complete plaque regression based on CTA. Data from this trial will provide substantial mechanistic insights into plaque regression in younger adults and provide the basis for planning a future definitive cardiovascular regression and outcomes trials. CURE ATHERO and its ancillary studies will identify optimal treatment windows for intervention. Long‐term post‐trial follow‐up of CURE ATHERO participants will provide data on the redevelopment or progression of atherosclerotic plaque following intensive LDL‐C lowering, and insights into the need for suppressive or maintenance drug therapy or further lifestyle changes, or the most appropriate time intervals for intermittent repeat regression therapy. Importantly, CURE ATHERO will provide the foundation for future trials evaluating new therapeutic approaches to PCSK9 inhibition. CURE ATHERO should also provide insight into whether drug therapies more specifically targeting triglyceride‐rich apo B lipoproteins are also needed to reverse atherosclerosis.101
Regulatory Pathways and Guidelines
Guidance on imaging as an approvable end point has been provided by the US Food and Drug Administration and the European Medicines Agency.102, 103 General principles for regulatory approval based on imaging end points include validated atheroma measures associated with cardiovascular events, drug therapies shown to reduce cardiovascular events, and atheroma measures shown to respond to treatment in ≥2 arterial beds.81 For primary prevention, imaging measures need to be non‐invasive with low or no radiation exposure, and for large trials need to be relatively inexpensive and widely available. The most appropriate atheroma measures depend on the population and treatment studied. Examples include the finding that carotid intimal medial thickness appears to be significantly modified only in statin naïve individuals, and that coronary artery calcium increases during statin therapy.90, 104, 105 However, CT angiography, MRI, and PET/MRI or PET/CT have been shown to measure response to LDL‐C lowering therapies such as statins and PCSK9 monoclonal antibodies.81
However, population‐wide implementation of intensive early treatment to eradicate the clinical burden of ASCVD will require randomized clinical trials demonstrating marked reductions in cardiovascular events. Some guideline panels may only be persuaded by a reduction in hard events (cardiovascular death, myocardial infarction, and stroke) or total mortality. Such trials should be manageable to perform given the expectation of large reductions in relative risk in properly selected at‐risk individuals. We have estimated that a 5‐year cardiovascular outcomes trial powered at 80% for an 80% relative risk reduction could be performed in <10 000 individuals with a 0.2% annual cardiovascular event rate, a rate commonly seen in average US men aged 45 to 54 years and women aged 55 to 64 years.2, 98 CURE ATHERO can inform identification of high risk younger individuals for inclusion in a cardiovascular outcomes trial.
Conclusions
Compelling evidence supports evaluating intensive reduction of plasma apo B lipoproteins for regressing and perhaps curing early atherosclerosis. We have proposed the first human trial that will lay the groundwork for this new area of investigation into a new cardiovascular prevention paradigm aimed at eradicating the clinical burden of ASCVD. This new prevention paradigm combining an intensive induction‐phase with long‐term maintenance therapy should provide a maximum ASCVD prevention benefit with minimal inconvenience, adverse effects, and cost.
Sources of Funding
Gidding is supported by the Coronary Artery Risk Development in Young Adults Study (CARDIA) (contracts HHSN268201300025C, HHSN268201300026C, HHSN268201300027C, HHSN268201300028C, HHSN268201300029C, and HHSN268200900041C from the National Heart, Lung, and Blood Institute [NHLBI], the Intramural Research Program of the National Institute on Aging [NIA], and an intra‐agency agreement between NIA and NHLBI AG0005]). Tabas is supported by grants HL075662, HL132412, HL140554, and HL127464 from the National Heart, Lung, and Blood Institute (NHLBI). Fisher is supported by NIH grants HL 084312, 131481, 129433, and DoD grant 12019098. Fayad is supported by grants P01HL131478, 1R01HL128056, 2R01HL070121, R01EB009638, R01HL119828, R01HL118440, R01HL125703, 1U01AR068043, R01HL135878, R01HL127637, R01HL135093 and R01AR068425 from the NIH and 14SFRN20780005 from the American Heart Association. Mani is supported by P01HL131478, 1R01HL128056, 2R01HL070121, 1U01AR068043, R01HL127637 from the NIH and 14SFRN20780005, 17GRNT33420119 from the American Heart Association. Rye has received support from the National Health and Medical Research Council of Australia APP1037903.
Disclosures
Robinson, MD, MPH has received research grants to Institution: Amarin, Amgen, Astra‐Zeneca, Esai, Esperion, Merck, Pfizer, Regeneron, Sanofi, Takeda (all significant) and served as a consultant for Akcea/Ionis, Amgen, Dr Reddy Laboratories, Eli Lilly, Merck, Pfizer (modest) and Regeneron and Sanofi (significant). Borén, MD, PhD has received research grant to Institution: Amgen, Astra‐Zeneca, Pfizer, Sanofi/Regeneron and NovoNordisk (all significant), and served as consultant for Eli Lilly, AstraZeneca, MSD, Amgen, NovoNordisk (modest), Zahi Fayad, PhD has received research grants to institution from Amgen, Daiichi Sankyo (all significant). Fisher, MD, PhD has served as an expert witness for Amgen (significant) and is a member of the Merck Speaker's Bureau. Gidding, MD has been a consultant for Regenxbio, not significant. Mani, PhD has received grants to institution from Daiichi Sankyo, Novartis and Aegerion (all significant) and is a consultant for Medlion Inc (modest). Nicholls, MD has received institutional grants from AstraZeneca, Amgen, Anthera, Eli Lilly, Esperion, Novartis, Cerenis, The Medicines Company, Resverlogix, InfraReDx, Roche, Sanofi‐Regeneron and LipoScience and honoraria from AstraZeneca, Eli Lilly, Anthera, Omthera, Merck, Takeda, Resverlogix, Sanofi‐Regeneron, CSL Behring, Esperion and Boehringer Ingelheim (modest). Nordestgaard, MD, DMSc has served as a consultant or has given talks sponsored by AstraZeneca, Sanofi, Regeneron, Ionis, Amgen, and Kowa (all modest). Packard, MD has received grants from Merck and Sanofi, and honoraria from Merck, Sanofi/Regeneron, Pfizer, Amgen and Daiichi‐Sankyo. Pagidipati, MD reports grants to the institution from Sanofi/Regeneron, Alexion, Amgen, Verily, Novartis. Pencina, PhD reports grants to the institution from Sanofi/Regeneron. Rye, PhD acts as a consultant for CSL Limited (modest). Tabas, MD, PhD has served as a consultant for Merck, LipimetiX Development, Inc., and Novartis (modest). Williams, MD has ownership interests in Hygieia, Inc., and Gemphire Therapeutics, Inc. (both significant) and serves on the Medical and Scientific Advisory Board for Gemphire. The remaining authors have no disclosures to report.
(J Am Heart Assoc. 2018;7:e009778 DOI: 10.1161/JAHA.118.009778.)
References
- 1. Sidney S, Quesenberry CP Jr, Jaffe MG, Sorel M, Nguyen‐Huynh MN, Kushi LH, Go AS, Rana JS. Recent trends in cardiovascular mortality in the United States and public health goals. JAMA Cardiol. 2016;1:594–599. [DOI] [PubMed] [Google Scholar]
- 2. Sniderman AD, Thanassoulis G, Williams K, Pencina M. Risk of premature cardiovascular disease vs the number of premature cardiovascular events. JAMA Cardiol. 2016;1:492–494. [DOI] [PubMed] [Google Scholar]
- 3. Robinson JG, Huijgen R, Ray K, Persons J, Kastelein JJP, Pencina MJ. Determining when to add nonstatin therapy: a quantitative approach. J Am Coll Cardiol. 2016;68:2412–2421. [DOI] [PubMed] [Google Scholar]
- 4. American Heart Association . Cardiovascular disease: A costly burden for America. Cost projections through 2035. 2017. Available at: http://www.heart.org/idc/groups/heart-public/@wcm/@adv/documents/downloadable/ucm_491543.pdf. Accessed January 10, 2018.
- 5. Nordestgaard BG. Triglyceride‐rich lipoproteins and atherosclerotic cardiovascular disease. New Insights From epidemiology, genetics, and biology. Circ Res. 2016;118:547–563. [DOI] [PubMed] [Google Scholar]
- 6. Williams KJ, Feig JE, Fisher EA. Rapid regression of atherosclerosis: insights from the clinical and experimental literature. Nat Clin Pract Cardiovasc Med. 2008;5:91–102. [DOI] [PubMed] [Google Scholar]
- 7. Williams KJ, Tabas I. Lipoprotein retention—and clues for atheroma regression. Arterioscler Thromb Vasc Biol. 2005;25:1536–1540. [DOI] [PubMed] [Google Scholar]
- 8. Borén J, Williams KJ. The central role of arterial retention of cholesterol‐rich apolipoprotein‐B‐containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. 2016;27:473–483. [DOI] [PubMed] [Google Scholar]
- 9. Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–1844. [DOI] [PubMed] [Google Scholar]
- 10. Ruuth M, Nguyen SD, Vihervaara T, Hilvo M, Laajala TD, Kondadi PK, Gisterå A, Lähteenmäki H, Kittilä T, Huusko J, Uusitupa M, Schwab U, Savolainen MJ, Sinisalo J, Lokki M‐L, Nieminen MS, Jula A, Perola M, Ylä‐Herttula S, Rudel L, Öörni A, Baumann M, Baruch A, Laaksonen R, Ketelhuth DFJ, Aittokallio T, Jauhiainen M, Käkelä R, Borén J, Williams KJ, Kovanen PT, Öörni K. Susceptibility of low‐density lipoprotein particles to aggregate depends on particle lipidome, is modifiable, and associates with future cardiovascular deaths. Eur Heart J. 2018;39:2562–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu M‐L, Reilly MP, Casasanto P, McKenzie SE, Williams KJ. Cholesterol enrichment of human monocyte/macrophages induces surface exposure of phosphatidylserine and the release of biologically‐active tissue factor‐positive microvesicles. Arterioscler Thromb Vasc Biol. 2007;27:430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun Y, Ishibashi M, Seimon T, Lee M, Sharma SM, Fitzgerald KA, Samokhin AO, Wang Y, Sayers S, Aikawa M, Jerome WG, Ostrowski MC, Bromme D, Libby P, Tabas IA, Welch CL, Tall AR. Free cholesterol accumulation in macrophage membranes activates toll‐Like receptors and p38 mitogen‐activated protein kinase and induces cathepsin K. Circ Res. 2009;104:455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rajamaki K, Lappalainen J, Oorni K, Valimaki E, Matikainen S, Kovanen PT, Eklund K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5:e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Varbo A, Benn M, Tybjærg‐Hansen A, Nordestgaard BG. Elevated remnant cholesterol causes both low‐grade inflammation and ischemic heart disease, whereas elevated low‐density lipoprotein cholesterol causes ischemic heart disease without inflammation. Circulation. 2013;128:1298–1309. [DOI] [PubMed] [Google Scholar]
- 16. Wulff AB, Nordestgaard BG, Tybjærg‐Hansen A. Loss‐of‐Function Mutations, Remnant cholesterol, low‐density lipoprotein cholesterol, and cardiovascular risk. Arterioscler Thromb Vasc Biol. 2018;38:660–668. [DOI] [PubMed] [Google Scholar]
- 17. Olkkonen VM, Sinisalo J, Jauhiainen M. New medications targeting triglyceride‐rich lipoproteins: can inhibition of ANGPTL3 or apoC‐III reduce the residual cardiovascular risk? Atherosclerosis. 2018;272:27–32. [DOI] [PubMed] [Google Scholar]
- 18. Williams K, Tabas I, Fisher E. How an artery heals. Circ Res. 2015;117:909–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moore K, Sheedy F, Fisher E. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McMahan CA, Gidding SS, Malcom GT, Tracy RE, Strong JP, McGill HC Jr; for the Pathobiological Determinants of Atherosclerosis in Youth Research Group . Pathobiological determinants of atherosclerosis in youth risk scores are associated with early and advanced atherosclerosis. Pediatrics. 2006;118:1447–1455. [DOI] [PubMed] [Google Scholar]
- 21. Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, Hegele RA, Krauss RM, Raal FJ, Schunkert H, Watts GF, Borén J, Fazio S, Horton JD, Masana L, Nicholls SJ, Nordestgaard BG, van de Sluis B, Taskinen M‐R, Tokgözoğlu L, Landmesser U, Laufs U, Wiklund O, Stock JK, Chapman MJ, Catapano AL. Low‐density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38:2459–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salfati E, Nandkeolyar S, Fortmann SP, Sidney S, Hlatky MA, Quertermous T, Go AS, Iribarren C, Herrington DM, Goldstein BA, Assimes TL. Susceptibility loci for clinical coronary artery disease and subclinical coronary atherosclerosis throughout the life‐course. Circ Cardiovasc Genet. 2015;8:803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Navar‐Boggan AM, Peterson ED, D'Agostino RB, Neely B, Sniderman AD, Pencina MJ. Hyperlipidemia in early adulthood increases long‐term risk of coronary heart disease. Circulation. 2015;131:451–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nemetz PN, Smith CY, Bailey KR, Roger VL, Edwards WD, Leibson CL. Trends in coronary atherosclerosis: a tale of two population subgroups. Am J Med. 2015;129:307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Insull W Jr. The pathology of atherosclerosis: plaque development and plaque responses to medical treatment. Am J Med. 2009;122:S3–S14. [DOI] [PubMed] [Google Scholar]
- 26. The Emerging Risk Factors Collaboration . Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pencina M, D'Agostino R, Zdrojewski T, Williams K, Thanassoulis G, Furberg C, Peterson E, Vasan R, Sniderman A. Apolipoprotein B improves risk assessment of future coronary heart disease in the Framingham Heart Study beyond LDL‐C and non‐HDL‐C. Eur J Prev Cardiol. 2015;22:1321–1327. [DOI] [PubMed] [Google Scholar]
- 28. Yusuf S, Rangarajan S, Teo K, Islam S, Li W, Liu L, Bo J, Lou Q, Lu F, Liu T, Yu L, Zhang S, Mony P, Swaminathan S, Mohan V, Gupta R, Kumar R, Vijayakumar K, Lear S, Anand S, Wielgosz A, Diaz R, Avezum A, Lopez‐Jaramillo P, Lanas F, Yusoff K, Ismail N, Iqbal R, Rahman O, Rosengren A, Yusufali A, Kelishadi R, Kruger A, Puoane T, Szuba A, Chifamba J, Oguz A, McQueen M, McKee M, Dagenais G. Cardiovascular risk and events in 17 low‐, middle‐, and high‐income countries. N Engl J Med. 2014;371:818–827. [DOI] [PubMed] [Google Scholar]
- 29. Sniderman AD, Williams K, Contois JH, Monroe HM, McQueen MJ, de Graaf J, Furberg CD. A meta‐analysis of low‐density lipoprotein cholesterol, non‐high‐density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circ Cardiovasc Qual Outcomes. 2011;4:337–345. [DOI] [PubMed] [Google Scholar]
- 30. Prosepctive Studies Collaboration . Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta‐analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet. 2007;370:1829–1839. [DOI] [PubMed] [Google Scholar]
- 31. Fernández‐Friera L, Fuster V, López‐Melgar B, Oliva B, García‐Ruiz JM, Mendiguren J, Bueno H, Pocock S, Ibáñez B, Fernández‐Ortiz A, Sanz J. Normal LDL‐cholesterol levels are associated with subclinical atherosclerosis in the absence of risk factors. J Am Coll Cardiol. 2017;70:2979–2991. [DOI] [PubMed] [Google Scholar]
- 32. Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Borén J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AFH, Stroes E, Taskinen M‐R, Tybjærg‐Hansen A; European Atherosclerosis Society Consensus Panel . Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus Statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Taskinen M‐R, Borén J. New insights into the pathophysiology of dyslipidemia in type 2 diabetes. Atherosclerosis. 2015;239:483–495. [DOI] [PubMed] [Google Scholar]
- 34. Williams K, Wu X. Imbalanced insulin action in chronic over nutrition: clinical harm, molecular mechanisms, and a way forward. Atherosclerosis. 2016;247:225–282. [DOI] [PubMed] [Google Scholar]
- 35. Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–1272. [DOI] [PubMed] [Google Scholar]
- 36. Kaplan H, Thompson RC, Trumble BC, Wann LS, Allam AH, Beheim B, Frohlich B, Sutherland ML, Sutherland JD, Stieglitz J, Rodriguez DE, Michalik DE, Rowan CJ, Lombardi GP, Bedi R, Garcia AR, Min JK, Narula J, Finch CE, Gurven M, Thomas GS. Coronary atherosclerosis in indigenous South American Tsimane: a cross‐sectional cohort study. Lancet. 2017;389:1730–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tabas I. 2016 Russell Ross memorial lecture in vascular biology: molecular‐cellular mechanisms in the progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2017;37:183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, Gusarova V, Gromada J, Weinstock A, Moore KJ, Loke P, Fisher EA. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J Clin Invest. 2017;127:2904–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vuorio T, Nurmi H, Moulton K, Kurkipuro J, Robciuc MR, Öhman M, Heinonen SE, Samaranayake H, Heikura T, Alitalo K, Ylä‐Herttuala S. Lymphatic vessel insufficiency in hypercholesterolemic mice alters lipoprotein levels and promotes atherogenesis. Arterioscler Thromb Vasc Biol. 2014;34:1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Llodrá J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte‐derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. PNAS. 2004;101:11779–11784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kojima Y, Weissman IL, Leeper NJ. The role of efferocytosis in atherosclerosis. Circulation. 2017;135:476–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Feig JE, Rong JX, Shamir R, Sanson M, Vengrenyuk Y, Liu J, Rayner K, Moore K, Garabedian M, Fisher EA. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte‐derived cells. PNAS. 2011;108:7166–7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miller JD, Chu Y, Castaneda LE, Serrano KM, Brooks RM, Heistad DD. Vascular function during prolonged progression and regression of atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2013;33:459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Björkegren JLM, Hägg S, Talukdar HA, Foroughi Asl H, Jain RK, Cedergren C, Shang M‐M, Rossignoli A, Takolander R, Melander O, Hamsten A, Michoel T, Skogsberg J. Plasma cholesterol‐induced lesion networks activated before regression of early, mature, and advanced atherosclerosis. PLoS Genet. 2014;10:e1004201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Reis ED, Li J, Fayad ZA, Rong JX, Hansoty D, Aguinaldo J‐G, Fallon JT, Fisher EA. Dramatic remodeling of advanced atherosclerotic plaques of the apolipoprotein E‐deficient mouse in a novel transplantation model. J Vasc Surg. 2001;34:541–2A. [DOI] [PubMed] [Google Scholar]
- 46. Jung C, Christiansen S, Kaul M, Koziolek E, Reimer R, Heeren J, Adam G, Heine M, Ittrich H. Quantitative and qualitative estimation of atherosclerotic plaque burden in vivo at 7T MRI using Gadospin F in comparison to en face preparation evaluated in ApoE KO mice. PLoS One. 2017;12:e0180407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kataoka Y, Andrews J, Puri R, Psaltis PJ, Nicholls SJ. Plaque burden, microstructures and compositions underachieving very low LDL‐C levels. Curr Opin Endocrinol Diabetes Obes. 2017;24:122–132. [DOI] [PubMed] [Google Scholar]
- 48. Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, Randolph GJ, Fisher EA. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE‐deficient mice. PNAS. 2006;103:3781–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hansen L, Taylor WR. Is increased arterial stiffness a cause or consequence of atherosclerosis? Atherosclerosis. 2016;249:226–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen W, Li S, Fernandez C, Sun D, Lai C‐C, Zhang T, Bazzano L, Urbina EM, Deng H‐W. Temporal relationship between elevated blood pressure and arterial stiffening among middle‐aged Black and white adults: the Bogalusa Heart Study. Am J Epidemiol. 2016;183:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gutiérrez E, Flammer AJ, Lerman LO, Elízaga J, Lerman A, Fernández‐Avilés F. Endothelial dysfunction over the course of coronary artery disease. Eur Heart J. 2013;34:3175–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Otsuka T, Mizuno K, Shinozaki T, Kachi Y, Nakamura H. Preventive effect of pravastatin on the development of hypertension in patients with hypercholesterolemia: a post‐hoc analysis of the MEGA study. J Clin Lipidol. 2017;11:988–1006. [DOI] [PubMed] [Google Scholar]
- 53. Sun H, Krauss RM, Chang JT, Teng B‐B. PCSK9 deficiency reduces atherosclerosis, apolipoprotein B secretion, and endothelial dysfunction. J Lipid Res. 2018;59:207–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Maulucci G, Cipriani F, Russo D, Casavecchia G, Di Staso C, Di Martino L, Ruggiero A, Di Biase M, Brunetti ND. Improved endothelial function after short term therapy with evolocumab. J Clin Lipidol. 2018;12:669–673. [DOI] [PubMed] [Google Scholar]
- 55. Puri R, Nissen SE, Shao M, Uno K, Kataoka Y, Kapadia SR, Tuzcu EM, Nicholls SJ. Impact of baseline lipoprotein and C‐reactive protein levels on coronary atheroma regression following high‐intensity statin therapy. Am J Cardiol. 2014;114:1465–1472. [DOI] [PubMed] [Google Scholar]
- 56. Nicholls SJ, Puri R, Anderson T, Ballantyne CM, Cho L, Kastelein JJ, Koenig W, Somaratne R, Kassahun H, Yang J, Wasserman SM, Scott R, Ungi I, Podolec J, Ophuis AO, Cornel JH, Borgman M, Brennan DM, Nissen SE. Effect of evolocumab on progression of coronary disease in statin‐treated patients: the GLAGOV randomized clinical trial. JAMA. 2016;316:2373–2384. [DOI] [PubMed] [Google Scholar]
- 57. Noyes AM, Thompson PD. A systematic review of the time course of atherosclerotic plaque regression. Atherosclerosis. 2014;234:75–84. [DOI] [PubMed] [Google Scholar]
- 58. Wiegman A, Hutten BA, de Groot E, Rodenburg J, Bakker HD, Buller HR, Sijbrands EJG, Kastelein JJP. Efficacy and safety of statin therapy in children with familial hypercholesterolemia: a randomized controlled trial. JAMA. 2004;292:331–337. [DOI] [PubMed] [Google Scholar]
- 59. Crouse JR III, Raichlen JS, Riley WA, Evans GW, Palmer MK, O'Leary DH, Grobbee DE, Bots ML; for the METEOR Study Group . Effect of rosuvastatin on progression of carotid intima‐media thickness in low‐risk individuals with subclinical atherosclerosis: the METEOR trial. JAMA. 2007;297:1344–1353. [DOI] [PubMed] [Google Scholar]
- 60. Sun J, Zhao X‐Q, Balu N, Neradilek MB, Isquith DA, Yamada K, Cantón G, Crouse JR, Anderson TJ, Huston J, O'Brien K, Hippe DS, Polissar NL, Yuan C, Hatsukami TS. Carotid plaque lipid content and fibrous cap status predict systemic CV outcomes. The MRI Substudy in AIM‐HIGH. JACC Cardiovasc Imaging. 2017;10:241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stegman B, Shao M, Nicholls SJ, Elshazly M, Cho L, King P, Kapadia S, Tuzcu M, Nissen SE, Puri R. Coronary atheroma progression rates in men and women following high‐intensity statin therapy: a pooled analysis of REVERSAL, ASTEROID and SATURN. Atherosclerosis. 2016;254:78–84. [DOI] [PubMed] [Google Scholar]
- 62. Zeb I, Li D, Nasir K, Malpeso J, Batool A, Flores F, Dailing C, Karlsberg RP, Budoff M. Effect of statin treatment on coronary plaque progression—a serial coronary CT angiography study. Atherosclerosis. 2013;231:198–204. [DOI] [PubMed] [Google Scholar]
- 63. Nozue T, Yamamoto S, Tohyama S, Fukui K, Umezawa S, Onishi Y, Kunishima T, Sato A, Nozato T, Miyake S, Takeyama Y, Morino Y, Yamauchi T, Muramatsu T, Hirano T, Hibi K, Terashima M, Michishita I. Impacts of age on coronary atherosclerosis and vascular response to statin therapy. Heart Vessels. 2014;29:456–463. [DOI] [PubMed] [Google Scholar]
- 64. Kini AS, Baber U, Kovacic JC, Limaye A, Ali ZA, Sweeny J, Maehara A, Mehran R, Dangas G, Mintz GS, Fuster V, Narula J, Sharma SK, Moreno PR. Changes in plaque lipid content after short‐term intensive versus standard statin therapy: the YELLOW trial (reduction in yellow plaque by aggressive lipid‐lowering therapy). J Am Coll Cardiol. 2013;62:21–29. [DOI] [PubMed] [Google Scholar]
- 65. Nozue T, Yamamoto S, Tohyama S, Fukui K, Umezawa S, Onishi Y, Kunishima T, Sato A, Nozato T, Miyake S, Takeyama Y, Morino Y, Yamauchi T, Muramatsu T, Hibi K, Terashima M, Michishita I. Comparison of arterial remodeling and changes in plaque composition between patients with progression versus regression of coronary atherosclerosis during statin therapy (from the TRUTH Study). Am J Cardiol. 2012;109:1247–1253. [DOI] [PubMed] [Google Scholar]
- 66. Tawakol A, Fayad ZA, Mogg R, Alon A, Klimas MT, Dansky H, Subramanian SS, Abdelbaky A, Rudd JHF, Farkouh ME, Nunes IO, Beals CR, Shankar SS. Intensification of statin therapy results in a rapid reduction in atherosclerotic inflammation: results of a multi‐center FDG‐PET/CT feasibility study. J Am Coll Cardiol. 2013;62:909–917. [DOI] [PubMed] [Google Scholar]
- 67. Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722. [DOI] [PubMed] [Google Scholar]
- 68. Benjamin EJ, Virani SS, Callaway CW, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O'Flaherty M, Palaniappan LP, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P. Heart disease and stroke statistics—2018 update: a report from the American Heart Association. Circulation. 2018;137:e67–e492. [DOI] [PubMed] [Google Scholar]
- 69. Quillard T, Franck G, Mawson T, Folco E, Libby P. Mechanisms of erosion of atherosclerotic plaques. Curr Opin Lipidol. 2017;28:434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hewing B, Parathath S, Barrett T, Chung WKK, Astudillo YM, Hamada T, Ramkhelawon B, Tallant TC, Yusufishaq MSS, DiDonato JA, Huang Y, Buffa J, Berisha SZ, Smith JD, Hazen SL, Fisher EA. Effects of native and myeloperoxidase‐modified apolipoprotein A‐I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2014;34:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Benn M, Nordestgaard BG. From genome‐wide association studies to Mendelian randomization: novel opportunities for understanding cardiovascular disease causality, pathogenesis, prevention, and treatment. Cardiovasc Res. 2018;114:1192–1208. [DOI] [PubMed] [Google Scholar]
- 72. Keene D, Price C, Shun‐Shin MJ, Francis DP. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta‐analysis of randomised controlled trials including 117 411 patients. BMJ. 2014;349:g4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Klancic T, Woodward L, Hofmann SM, Fisher EA. High density lipoprotein and metabolic disease: potential benefits of restoring its functional properties. Mol Metab. 2016;5:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Silverman MG, Ference BA, Im K, Wiviott SD, Giugliano RP, Grundy SM, Braunwald E, Sabatine MS. Association between lowering LDL‐C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta‐analysis. JAMA. 2016;316:1289–1297. [DOI] [PubMed] [Google Scholar]
- 75. Thanassoulis G, Williams K, Ye K, Brook R, Couture P, Lawler P, de Graaf J, Furberg C, Sniderman A. Relations of change in plasma levels of LDL‐C, non‐HDL‐C, and apo B with risk reduction from statin therapy: a meta‐analysis of randomized trials. J Am Heart Assoc. 2014;3:e000759 DOI: 10.1161/JAHA.113.000759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Boekholdt S, Arsenault B, Mora S, Pedersen T, LaRosa J, Nestel P, Simes R, Durrington P, Hitman W, Welch K, DeMicco D, Zwinderman A, Clearfield M, Downs J, Tonkin A, Colhoun H, Gotto A, Ridker P, Kastelein J. Association of LDL cholesterol, non–HDL cholesterol, and apolipoprotein B levels with risk of cardiovascular events among patients treated with statins. JAMA. 2012;307:1302–1309. [DOI] [PubMed] [Google Scholar]
- 77. Goff DC Jr, Lloyd‐Jones DM, Bennett G, Coady S, D'Agostino RB Sr, Gibbons R, Greenland P, Lackland DT, Levy D, O'Donnell CJ, Robinson JG, Schwartz JS, Shero ST, Smith SC Jr, Sorlie P, Stone NJ, Wilson PWF. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63:2935–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Collaborators Cholesterol Treatment Trialists . The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta‐analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Packard CJ, Weintraub WS, Laufs U. New metrics needed to visualize the long‐term impact of early LDL‐C lowering on the cardiovascular disease trajectory. Vascul Pharmacol. 2015;71:37–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida‐Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 81. Doris MK, Dweck MR, Fayad ZA. The future of imaging in cardiovascular disease intervention trials: 2017 and beyond. Curr Opin Lipidol. 2016;27:605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang Y, Guallar E, Qiao Y, Wasserman BA. Is Carotid intima‐media thickness as predictive as other noninvasive techniques for the detection of coronary artery disease? Arterioscler Thromb Vasc Biol. 2014;34:1341–1345. [DOI] [PubMed] [Google Scholar]
- 83. Ahmadi A, Narula J. Primary and secondary prevention, or subclinical and clinical atherosclerosis. JACC Cardiovasc Imaging. 2017;10:447–450. [DOI] [PubMed] [Google Scholar]
- 84. Leipsic J, Abbar S, Achenbach S, Cury R, Earls JP, Mancini GJ, Nieman K, Pontone G, Raff GL. SCCT guidelines for the interpretation and reporting of coronary CT angiography: a report of the Society of Cardiovascular Computed Tomography Guidelines Committee. J Cardiovasc Comput Tomogr. 2014;8:342–358. [DOI] [PubMed] [Google Scholar]
- 85. Nadjiri J, Hausleiter J, Jähnichen C, Will A, Hendrich E, Martinoff S, Hadamitzky M. Incremental prognostic value of quantitative plaque assessment in coronary CT angiography during 5 years of follow up. J Cardiovasc Computed Tomogr. 2016;10:97–104. [DOI] [PubMed] [Google Scholar]
- 86. Williams MC, Hunter A, Shah ASV, Assi V, Lewis S, Smith J, Berry C, Boon NA, Clark E, Flather M, Forbes J, McLean S, Roditi G, van Beek EJR, Timmis AD, Newby DE. Use of coronary computed tomographic angiography to guide management of patients with coronary disease. J Am Coll Cardiol. 2016;67:1759–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chow BJW, Small G, Yam Y, Chen L, McPherson R, Achenbach S, Al‐Mallah M, Berman DS, Budoff MJ, Cademartiri F, Callister TQ, Chang H‐J, Cheng VY, Chinnaiyan K, Cury R, Delago A, Dunning A, Feuchtner G, Hadamitzky M, Hausleiter J, Karlsberg RP, Kaufmann PA, Kim Y‐J, Leipsic J, LaBounty T, Lin F, Maffei E, Raff GL, Shaw LJ, Villines TC, Min JK. Prognostic and therapeutic implications of statin and aspirin therapy in individuals with nonobstructive coronary artery disease: results from the CONFIRM (Coronary CT Angiography Evaluation For Clinical Outcomes: an International Multicenter Registry) Registry. Arterioscler Thromb Vasc Biol. 2015;35:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hulten E, Villines TC, Cheezum MK, Berman DS, Dunning A, Achenbach S, Al‐Mallah M, Budoff MJ, Cademartiri F, Callister TQ, Chang H‐J, Cheng VY, Chinnaiyan K, Chow BJW, Cury RC, Delago A, Feuchtner G, Hadamitzky M, Hausleiter J, Kaufmann PA, Karlsberg RP, Kim Y‐J, Leipsic J, Lin FY, Maffei E, Plank F, Raff GL, Labounty TM, Shaw LJ, Min JK. Usefulness of coronary computed tomography angiography to predict mortality and myocardial infarction among Caucasian, African and East Asian Ethnicities (from the CONFIRM [Coronary CT Angiography Evaluation for Clinical Outcomes: an International Multicenter] Registry). Am J Cardiol. 2013;111:479–485. [DOI] [PubMed] [Google Scholar]
- 89. Nakahara T, Dweck MR, Narula N, Pisapia D, Narula J, Strauss HW. Coronary artery calcification: from mechanism to molecular imaging. JACC Cardiovasc Imaging. 2017;10:582–593. [DOI] [PubMed] [Google Scholar]
- 90. Auscher S, Heinsen L, Nieman K, Vinther KH, Løgstrup B, Møller JE, Broersen A, Kitslaar P, Lambrechtsen J, Egstrup K. Effects of intensive lipid‐lowering therapy on coronary plaques composition in patients with acute myocardial infarction: assessment with serial coronary CT angiography. Atherosclerosis. 2015;241:579–587. [DOI] [PubMed] [Google Scholar]
- 91. Li Z, Hou Z, Yin W, Liu K, Gao YT, Xu H, Yu F, Ma Z, Yu W, Yang L, Lu B. Effects of statin therapy on progression of mild noncalcified coronary plaque assessed by serial coronary computed tomography angiography: a multicenter prospective study. Am Heart J. 2016;180:29–38. [DOI] [PubMed] [Google Scholar]
- 92. Raggi P, Baldassarre D, Day SM, de Groot E, Fayad Z. Non‐invasive imaging of atherosclerosis regression with magnetic resonance to guide drug development. Atherosclerosis. 2016;251:476–482. [DOI] [PubMed] [Google Scholar]
- 93. Mani V, Muntner P, Gidding SS, Aguiar SH, El Aidi H, Weinshelbaum KB, Taniguchi H, van der Geest R, Reiber JHC, Bansilal S, Farkouh M, Fuster V, Postley JE, Woodward M, Fayad ZA. Cardiovascular magnetic resonance parameters of atherosclerotic plaque burden improve discrimination of prior major adverse cardiovascular events. J Cardiovasc Magn Reson. 2009;11:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Fayad ZA, Mani V, Woodward M, Kallend D, Abt M, Burgess T, Fuster V, Ballantyne CM, Stein EA, Tardif J‐C, Rudd JHF, Farkouh ME, Tawakol A. Safety and efficacy of dalcetrapib on atherosclerotic disease using novel non‐invasive multimodality imaging (dal‐PLAQUE): a randomised clinical trial. Lancet. 2012;378:1547–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Evans N, Tarkin J, Chowdhury M, Warburton E, Rudd J. PET imaging of atherosclerotic disease: advancing plaque assessment from anatomy to pathophysiology. Curr Atheroscler Rep. 2016;18:30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rudd J, Fayad Z. Imaging atherosclerotic plaque inflammation. Nat Clin Pract Cardiovasc Med. 2008;5:S11–S17. [DOI] [PubMed] [Google Scholar]
- 97. Robson PM, Dey D, Newby DE, Berman D, Li D, Fayad ZA, Dweck MR. MR/PET imaging of the cardiovascular system. JACC Cardiovasc Imaging. 2017;10:1165–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Robinson JG, Gidding SS. Curing atherosclerosis should be the next major cardiovascular prevention goal. J Am Coll Cardiol. 2014;63:2779–2785. [DOI] [PubMed] [Google Scholar]
- 99. McMahan CA, Gidding SS, Fayad ZA, Zieske AW, Malcom GT, Tracy RE, Strong JP, McGill HC Jr; for the Pathobiological Determinants of Atherosclerosis in Youth Research Group . Risk scores predict atherosclerotic lesions in young people. Arch Intern Med. 2005;165:883–890. [DOI] [PubMed] [Google Scholar]
- 100. Gidding SS, Rana JS, Prendergast C, McGill H, Carr JJ, Liu K, Colangelo LA, Loria CM, Lima J, Terry JG, Reis JP, McMahan CA. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) risk score in young adults predicts coronary artery and abdominal aorta calcium in middle age: the CARDIA Study. Circulation. 2016;133:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Madsen CM, Varbo A, Nordestgaard BG. Unmet need for primary prevention in individuals with hypertriglyceridaemia not eligible for statin therapy according to European Society of Cardiology/European Atherosclerosis Society guidelines: a contemporary population‐based study. Eur Heart J. 2018;39:610–619. [DOI] [PubMed] [Google Scholar]
- 102. European Medicines Agency . Guideline on clinical investigation of medicinal products in the treatment of lipid disorders. 2016. EMA/CHMP/748108/2013, Rev. 3; Committee for Medicinal Products for Human Use (CHMP). Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/07/WC500209944.pdf. Accessed January 10, 2018.
- 103. Food and Drug Administration . Clinical Trial Imaging Endpoint Process Standards Guidance for Industry. Draft guidance March 2015 Clinical/Medical Revision 1. 2015. Available at: https://www.fda.gov/downloads/drugs/guidances/ucm268555.pdf. Accessed January 10, 2018.
- 104. Kastelein JJP, Akdim F, Stroes ESG, Zwinderman AH, Bots ML, Stalenhoef AFH, Visseren FLJ, Sijbrands EJG, Trip MD, Stein EA, Gaudet D, Duivenvoorden R, Veltri EP, Marais AD, de Groot E; the ENHANCE Investigators . Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008;358:1431–1443. [DOI] [PubMed] [Google Scholar]
- 105. Smilde TJ, van Wissen S, Awollersheim H, Trip MD, Kastelein JJP, Stalenhoef AFH. Effect of aggressive versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolemia (ASAP): a prospective, randomised, double‐blind trial. Lancet. 2001;357:577–581. [DOI] [PubMed] [Google Scholar]