

Abstract
Background
Left ventricular noncompaction cardiomyopathy (LVNC) is a genetically and phenotypically heterogeneous disease. This study aims to investigate the genetic basis and genotype‐phenotype correlations in a cohort of Chinese patients with LVNC.
Methods and Results
A total of 72 cardiomyopathy‐associated genes were comprehensively screened in 83 adults and 17 children with LVNC by targeted sequencing. Pathogenicity of the detected variants was determined according to their prevalence and American College of Medical Genetics and Genomics recommendations. Baseline and follow‐up clinical data were collected. The primary end point was a composite of death and heart transplantation. Overall, 42 pathogenic variants were identified in 38 patients (38%), with TTN,MYH7,MYBPC3, and DSP being the most commonly involved genes. At baseline, genotype‐positive adults had higher rates of atrial fibrillation and family history, and lower left ventricular ejection fraction, compared with genotype‐negative adults. During a median follow‐up of 4.2 years, more primary end points occurred in genotype‐positive adults than in genotype‐negative adults (50.0% versus 23.5%; P=0.013). Multivariable analysis demonstrated that genotype‐positive status was associated with higher risks of death and heart transplantation, independent of age, sex, and cardiac function at baseline in patients with LVNC (adjusted hazards ratio, 2.49; 95% confidence interval, 1.15–5.37; P=0.020).
Conclusions
Our study revealed a distinct genetic spectrum in Chinese patients with LVNC, with variants in TTN,MYH7,MYBPC3, and DSP being the most common. The presence of pathogenic variants is an independent risk factor for adverse outcomes and may aid in risk stratification in adult patients. Larger studies are needed to confirm these findings.
Keywords: genetics, left ventricular noncompaction, prognosis
Subject Categories: Genetics, Cardiomyopathy, Mortality/Survival
Short abstract
See Editorial by https://doi.org/10.1161/JAHA.118.010608
Clinical Perspective
What Is New?
In this large cohort study, we described a distinct genetic spectrum in Chinese patients with left ventricular noncompaction cardiomyopathy.
More than one third of the patients carried at least one pathogenic variant, with TTN, MYH7, MYBPC3, and DSP being the most commonly involved genes.
Genotype‐positive status was independently associated with increased risk of death and heart transplantation in adult patients.
What Are the Clinical Implications?
In the context of left ventricular noncompaction cardiomyopathy, genetic testing has a considerable yield in Chinese patients, as previously described in patients of European ancestry.
Genetic testing is important for better risk stratification of the individual patient and should be recommended in clinical practice.
Introduction
Left ventricular noncompaction cardiomyopathy (LVNC) is a genetically and clinically heterogeneous myocardial disorder that is present in 3% to 4% of patients with heart failure (HF).1, 2 It is characterized by a prominent trabecular meshwork and deep intertrabecular recesses communicating with the left ventricular cavity, morphologically reminiscent of early cardiac development.3, 4 The genesis of LVNC is generally thought to be caused by the arrest of myocardial compaction during the normal development of the heart.5, 6 Patients with LVNC show a wide spectrum of clinical presentations, ranging from no symptoms to thromboembolic events, end‐stage HF, or sudden cardiac death (SCD).7, 8, 9, 10
LVNC was classified as a genetic cardiomyopathy by the American Heart Association,11 and genetics plays an important role in it. Genetic defects are found in 35% to 40% of patients with isolated LVNC by molecular testing, with MYH7 as the most commonly involved gene.12, 13 Genes associated with LVNC usually include those encoding sarcomeric, cytoskeletal, or ion channel proteins, and those involved in cellular energy metabolism.4
Although LVNC has been studied for nearly a century since it was first described in 1926,14 there is still a lot to be clarified about its genetic basis and genotype‐phenotype correlations. Thus, the current study aims to investigate the molecular defects, clinical manifestations, and long‐term outcomes, as well as the relationships among them, in a cohort of Chinese patients with LVNC.
Methods
Because of privacy, the data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Study Subjects
A total of 100 unrelated patients with LVNC enrolled at Fuwai Hospital between April 2004 and May 2016 were included in the study. LVNC was diagnosed on the basis of established criteria from echocardiography and/or cardiac magnetic resonance.15, 16 The core item of these criteria is the ratio of the thickness of noncompacted/compact epicardial layer, which is >2.0 measured on echocardiography in systole or >2.3 on cardiac magnetic resonance in diastole. According to the age of onset, patients were divided into children (<18 years) and adults (≥18 years). Demographic and clinical data of all participants were collected.
The study complies with the principles of the Declaration of Helsinki and was approved by the Ethics Committee of Fuwai Hospital. Written informed consent was obtained from each participant.
Targeted Sequencing
Genomic DNA was isolated from peripheral venous blood of each participant. The coding exons and their splicing regions (adjacent 10‐bp intronic sequences) of 72 cardiomyopathy‐related genes were enriched, as determined using a custom‐designed library (Agilent Technologies, Santa Clara, CA), and were subsequently sequenced on an Illumina next‐generation sequencing platform (Illumina Inc, San Diego, CA). Sequencing reads of each individual were mapped to the human reference genome with BWA (0.7.12). After removal of polymerase chain reaction duplications with PICARD, variants were called with GATK, version 3.5, and annotated by using ANNOVAR. The mean depths of the samples were >400×, with coverage of >99.7%. Details about the genes being tested are described in Data S1.
Variants were described in accordance with the guidelines for mutation nomenclature of the Human Genome Variation Society (http://www.hgvs.org/). Variants were considered as polymorphisms and excluded if their minor allele frequency was ≥0.05% among East Asians in the Genome Aggregation Database.17 The pathogenicity of detected variants was determined in accordance with the recommendations of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology, and was classified as “pathogenic,” “likely pathogenic,” “uncertain significance,” “likely benign,” or “benign.”18 Criteria for classification are listed in Table S1 and S2. Variants classified as pathogenic or likely pathogenic were considered to be pathogenic in the current analysis. Patients with or without pathogenic variants were defined as genotype positive (G+) or genotype negative (G−), respectively. Sanger sequencing was used to validate the presence of pathogenic variants. The primers for the sequencing are listed in Table S3.
Follow‐Up and End Points
Follow‐up data were obtained by telephone interview or clinic visit. The last follow‐up was performed in April 2018. The primary end point was a composite of death and heart transplantation (HT). The secondary end points included all‐cause death, HT, and cardiovascular death, which included SCD, HF‐related death, and death from other cardiovascular causes. SCD was defined as witnessed sudden death with or without documented ventricular fibrillation, death within 1 hour of new symptoms, or nocturnal deaths with no antecedent history of worsening symptoms. HF‐related death was defined as death preceded by symptoms of HF lasting >1 hour.
Statistical Analysis
Continuous variables are expressed as median (interquartile range) and were compared by independent‐sample t test or Mann‐Whitney U test for abnormally distributed variables. Categorical variables are presented as proportions (%) and were compared using Pearson's χ2 test or Fisher's exact test. Univariable or multivariable Cox proportional hazards models were performed to calculate the hazard ratio and 95% confidence interval to estimate the effect of pathogenic variants on phenotypes. Survival curves were constructed in accordance with the Kaplan‐Meier method, and were compared using the log‐rank test. Factors included in the multivariate models for the outcomes were age, sex, and New York Heart Association functional class III/IV at baseline. Differences were considered significant if the 2‐sided P value was <0.05. All analyses were performed with SPSS, version 22.0 software (IBM, Armonk, NY).
Results
Study Population and Genetic Profile
The participants in this study consisted of 17 children (17%) and 83 adults (83%). A total of 72 (72%) of them were male, with a median (interquartile range) age of 42 (24–53) years. A total of 42 pathogenic variants were found in 38 (38.0%) of patients, of which 29 (69.0%) of variants were located in sarcomere genes (Table S4). Among these sarcomere genes, TTN was the most commonly involved gene (51.7%), followed by MYH7 (17.2%), MYBPC3 (13.8%), and ACTC1 (6.9%) (Figure 1A). Eight nonsarcomere genes were detected with pathogenic variants, among which DSP (23.1%) was the most commonly mutated gene, followed by DMD (15.4%), LAMP2 (15.4%), and SCN5A (15.4%). Notably, 25 (59.5%) of detected variants were novel, which were located in TTN, MYBPC3, DSP, NNT, DMD, and LAMP2.
Figure 1.
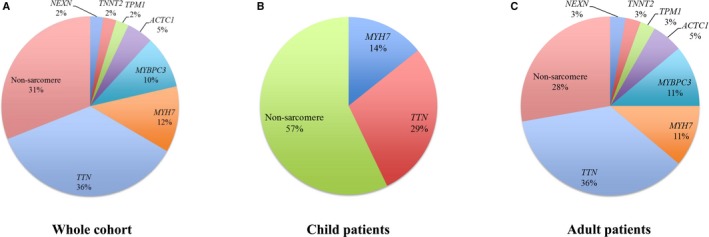
Spectrum of pathogenic variants in Chinese patients with left ventricular noncompaction cardiomyopathy. Panels show the variant spectrum in whole cohort (A), child patients (B) and adult patients (C).
There was no significant difference in the proportions of G+ status between children (35.3%, 6/17) and adults (38.6%, 32/83; P=0.801). The proportions of variants in sarcomere genes were not significantly different between children and adults (42.9% and 72.2%, respectively; P=0.190), with TTN being the predominant gene in both groups (Figure 1B and 1C). Nonsarcomere genes accounted for 57.1% and 27.8% of the variants detected in children and adults, respectively.
Genotype‐Phenotype Correlation at Baseline
Demographic and clinical characteristics of the participants are shown in Table 1. Similar characteristics were observed between G+ and G− children. For adults, atrial fibrillation and a family history of cardiomyopathy were more common; left ventricular ejection fraction was lower in G+ patients, whereas G− patients were more likely to have hypertension. Notably, 6 carriers of pathogenic variants in DSP were all found to have ventricular arrythmias (Table S5).
Table 1.
Baseline Characteristics of G+ and G− Among Children and Adults
| Characteristics | Children (n=17) | Adults (n=83) | ||||
|---|---|---|---|---|---|---|
| G+ (n=6) | G− (n=11) | P Value | G+ (n=32) | G− (n=51) | P Value | |
| Age at enrollment, y | 16.0 (13.0–18.0) | 14.0 (8.0–15.0) | 0.149 | 44.0 (35.5–49.0) | 46.0 (30.0–57.0) | 0.506 |
| Age of onset, y | 15.5 (10.5–17.0) | 13.0 (8.0–15.0) | 0.180 | 39.5 (30.3–45.5) | 42.0 (28.0–52.0) | 0.660 |
| Male sex, n (%) | 6 (100.0) | 8 (72.7) | 0.515 | 22 (68.8) | 36 (70.6) | 0.859 |
| Family history of cardiomyopathy, n (%) | 1 (16.7) | 1 (9.1) | 1.000 | 9 (28.1) | 2 (3.9) | 0.002 |
| NYHA class III/IV, n (%) | 3 (50.0) | 1 (9.1) | 0.099 | 17 (53.1) | 22 (43.1) | 0.375 |
| Comorbidities, n (%) | ||||||
| Coronary artery disease | 0 (0.0) | 0 (0.0) | ··· | 1 (3.1) | 8 (15.7) | 0.143 |
| Hypertension | 0 (0.0) | 1 (9.1) | 1.000 | 0 (0.0) | 13 (25.5) | 0.001 |
| Diabetes mellitus | 0 (0.0) | 0 (0.0) | ··· | 3 (9.4) | 4 (7.8) | 1.000 |
| Hyperlipidemia | 0 (0.0) | 1 (9.1) | 1.000 | 6 (18.8) | 8 (15.7) | 0.717 |
| Other cardiomyopathies | 3 (50.0) | 3 (27.3) | 0.600 | 11 (34.4) | 10 (19.6) | 0.132 |
| Atrial fibrillation | 0 (0.0) | 0 (0.0) | ··· | 11 (34.4) | 4 (7.8) | 0.002 |
| Echocardiography | ||||||
| LVEDD, mm | 59.0 (50.0–69.8) | 47.0 (37.0–69.0) | 0.350 | 64.5 (52.3–70.0) | 61.0 (54.8–70.0) | 0.581 |
| LAD, mm | 42.0 (30.3–50.5) | 33.0 (27.0–36.0) | 0.078 | 43.5 (36.8–50.0) | 40.0 (34.0–46.5) | 0.098 |
| LVEF, % | 34.0 (20.3–61.3) | 61.0 (25.0–69.0) | 0.180 | 31.6 (25.5–44.8) | 40.0 (33.0–56.8) | 0.016 |
Data are given as median (interquartile range) unless otherwise indicated. G+ indicates genotype positive; G−, genotype negative; LAD, left atrial diameter; LVEDD, left ventricular end‐diastolic dimension; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association.
Genotype‐Phenotype Correlation for Clinical Outcomes
During a median (interquartile range) follow‐up of 4.5 (2.9–6.2) years, 32 patients (32.0%) reached the primary end point, of which 27 (27.0%) experienced death and 5 (5.0%) underwent HT. All deaths were attributable to cardiovascular causes, consisting of 20 HF‐related deaths, 6 SCDs, and 1 death attributable to another cardiovascular cause (Table 2).
Table 2.
Incidence of Primary and Secondary End Points in G+ and G− Patients
| End Point | All Patients (n=100) | Child Patients | Adult Patients | ||||
|---|---|---|---|---|---|---|---|
| G+ (n=6) | G− (n=11) | P Value | G+ (n=32) | G− (n=51) | P Value | ||
| Primary | |||||||
| Death and heart transplantation | 32 (32.0) | 2 (33.3) | 2 (18.2) | 0.584 | 16 (50.0) | 12 (23.5) | 0.013 |
| Secondary | |||||||
| All‐cause death | 27 (27.0) | 1 (16.7) | 2 (18.2) | 1.000 | 12 (37.5) | 12 (23.5) | 0.172 |
| Heart transplantation | 5 (5.0) | 1 (16.7) | 0 (0.0) | 0.353 | 4 (12.5) | 0 (0.0) | 0.020 |
| Cardiovascular death | 27 (27.0) | 1 (16.7) | 2 (18.2) | 1.000 | 12 (37.5) | 12 (23.5) | 0.172 |
| Sudden cardiac death | 6 (6.0) | 0 (0.0) | 2 (18.2) | 0.515 | 1 (3.1) | 3 (5.9) | 1.000 |
| Heart failure–related death | 20 (20.0) | 1 (16.7) | 0 (0.0) | 0.353 | 11 (34.4) | 8 (15.7) | 0.049 |
Data are given as number (percentage). G+ indicates genotype positive; G−, genotype negative.
Of the 17 children, 4 (23.5%) reached the primary end point, of whom 2 (50.0%) were G+ (Table S6). There was no significant difference in terms of the incidence of primary or secondary outcomes between G+ and G− patients (Table 2). Figure 2A displays the event‐free survival curve, demonstrating no difference in the risk of the primary end point.
Figure 2.
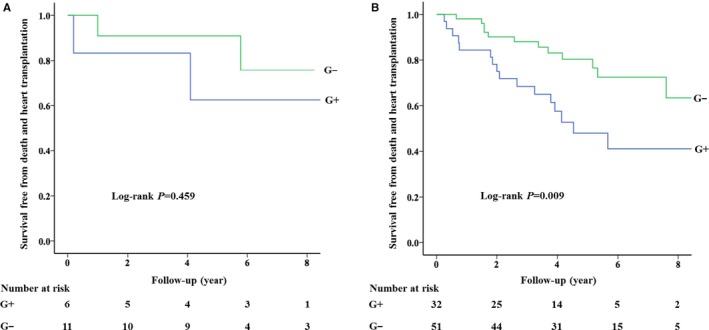
Survival curves free from death and heart transplantation in child patients (A) and adult patients (B) with left ventricular noncompaction cardiomyopathy. G+ indicates genotype positive; G−, genotype negative.
For the 83 adults, 28 (33.7%) experienced the primary end point. There was no significant difference in the incidence of the primary or secondary end points between children and adults (P=0.411, Table S7). G+ status was significantly more common in patients who reached the primary end point (57.1%, 16/28) than in those who did not (29.1%, 16/55; P=0.013; Table S8). Significantly more cases of the primary end point, HT, and HF‐related death occurred in G+ patients than in G− patients (Table 2). No difference in incidence was observed between G+ and G− patients for the other end points. Multivariable Cox regression analyses showed that G+ status and New York Heart Association class III/IV at baseline were independently associated with an increased risk of the primary end point (hazard ratio, 2.49; 95% confidence interval, 1.15–5.37; P=0.020; and hazard ratio, 2.62; 95% confidence interval, 1.13–6.08; P=0.025, respectively) (Table 3). The event‐free survival curve is displayed in Figure 2B. In addition, G+ status was associated with an increased risk of HF‐related death in univariable analyses (Table 4). After adjustment, however, this association was no longer significant. No association between G+ status and other end points was observed.
Table 3.
Multivariable Analysis of Predictors of Primary End Points
| Variable | Univariable | Multivariable | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P Value | HR | 95% CI | P Value | |
| G+ status | 2.62 | 1.23–5.54 | 0.012 | 2.49 | 1.15–5.37 | 0.020 |
| Male sex | 1.09 | 0.48–2.47 | 0.841 | 1.14 | 0.50–2.59 | 0.756 |
| Age | 1.01 | 0.99–1.03 | 0.461 | 1.01 | 0.98–1.04 | 0.629 |
| NYHA class III/IV | 2.93 | 1.29–6.68 | 0.010 | 2.62 | 1.13–6.08 | 0.025 |
CI indicates confidence interval; G+, genotype positive; HR, hazard ratio; NYHA, New York Heart Association.
Table 4.
Univariable and Multivariable Analyses of G+ Status and Secondary End Points
| Variable | Univariable | Multivariablea | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P Value | HR | 95% CI | P Value | |
| All‐cause death | 1.97 | 0.88–4.39 | 0.099 | ··· | ··· | ··· |
| Heart transplantation | NAb | NA | NA | ··· | ··· | ··· |
| Cardiovascular death | 1.97 | 0.88–4.39 | 0.099 | ··· | ··· | ··· |
| Sudden cardiac death | 0.62 | 0.06–5.92 | 0.674 | ··· | ··· | ··· |
| Heart failure–related death | 2.77 | 1.11–6.90 | 0.029 | 2.37 | 0.88–6.37 | 0.086 |
CI indicates confidence interval; G+, genotype positive; HR, hazard ratio; NA, not applicable.
Items with P<0.05 in univariable analyses were then included in the calculation for multivariable HR and 95% CI.
No heart transplantation occurred in genotype‐negative patients, so HR and 95% CI are not available.
Effect of Multiple Variants
Because only 1 of the 17 children carried >1 variant, the effect of multiple variants on phenotype was analyzed only in adults, of whom 6 were carriers of multiple variants. All of these 6 patients carried at least 1 variant in sarcomere genes. There was no significant difference in baseline characteristics or incidence of end points between carriers of multiple variants and single variant, except for a younger age at onset observed in the latter (Table 5). Univariable and multivariable Cox regression analyses showed that the risk of primary end point was significantly higher in carriers of single variant than noncarriers (hazard ratio, 2.45; 95% confidence interval, 1.08–5.54; P=0.032; Table 6). No other significant association was observed among carriers of multiple variants, single variant, and noncarriers.
Table 5.
Baseline Characteristics of Carriers of Multiple Variants and a Single Variant Among Adult Patients
| Characteristic | Adult Patients (n=83) | ||
|---|---|---|---|
| Single Variant (n=26) | Multiple Variants (n=6) | P Value | |
| Age at enrollment, y | 42.5 (35.0–46.5) | 55.0 (39.8–63.3) | 0.069 |
| Age at onset, y | 38.5 (29.5–44.0) | 52.5 (39.5–61.3) | 0.033 |
| Male sex, n (%) | 18 (69.2) | 4 (66.7) | 1.000 |
| Family history of cardiomyopathy, n (%) | 8 (30.8) | 1 (16.7) | 0.648 |
| NYHA class III/IV, n (%) | 14 (53.8) | 3 (50.0) | 1.000 |
| Comorbidities, n (%) | |||
| Coronary artery disease | 1 (3.8) | 0 (0.0) | 1.000 |
| Hypertension | 0 (0.0) | 0 (0.0) | |
| Diabetes mellitus | 2 (7.7) | 1 (16.7) | 0.476 |
| Hyperlipidemia | 4 (15.4) | 2 (33.3) | 0.310 |
| Other cardiomyopathies | 9 (34.6) | 2 (33.3) | 1.000 |
| Atrial fibrillation | 9 (34.6) | 2 (33.3) | 1.000 |
| Echocardiography | |||
| LVEDD, mm | 66.5 (56.3–72.0) | 58.5 (47.8–63.5) | 0.119 |
| LAD, mm | 43.5 (38.0–50.3) | 45.0 (35.3–50.3) | 0.981 |
| LVEF, % | 30.5 (24.8–41.8) | 41.5 (34.8–50.8) | 0.087 |
| Primary end point | |||
| Death and heart transplantation | 13 (50.0) | 3 (50.0) | 1.000 |
| Secondary end point | |||
| All‐cause death | 9 (34.6) | 3 (50.0) | 0.647 |
| Sudden cardiac death | 1 (3.8) | 0 (0.0) | 1.000 |
| Heart failure–related death | 8 (30.8) | 3 (50.0) | 0.390 |
| Cardiovascular death | 9 (34.6) | 3 (50.0) | 0.647 |
| Heart transplantation | 4 (15.4) | 0 (0.0) | 0.566 |
Data are given as median (interquartile range) unless otherwise indicated. LAD indicates left atrial diameter; LVEDD, left ventricular end‐diastolic dimension; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association.
Table 6.
Univariable and Multivariable Analyses of Association Between Number of Detected Variants and Clinical Outcomes
| Variable | Univariable | Multivariablea | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P Value | HR | 95% CI | P Value | |
| Multiple vs single variant | ||||||
| Death and heart transplantation | 1.27 | 0.36–4.47 | 0.710 | ··· | ··· | ··· |
| All‐cause death | 0.89 | 0.24–3.29 | 0.860 | ··· | ··· | ··· |
| Heart transplantation | NAb | NA | NA | ··· | ··· | ··· |
| Cardiovascular death | 0.89 | 0.24–3.29 | 0.860 | ··· | ··· | ··· |
| Sudden cardiac death | NAc | NA | NA | ··· | ··· | ··· |
| Heart failure–related death | 0.80 | 0.21–3.04 | 0.748 | ··· | ··· | ··· |
| Single vs no variant | ||||||
| Death and heart transplantation | 2.70 | 1.23–5.94 | 0.013 | 2.45 | 1.08–5.54 | 0.032 |
| All‐cause death | 1.87 | 0.79–4.46 | 0.156 | ··· | ··· | ··· |
| Heart transplantation | NA | NA | NA | ··· | ··· | ··· |
| Cardiovascular death | 1.87 | 0.79–4.46 | 0.156 | ··· | ··· | ··· |
| Sudden cardiac death | 0.78 | 0.08–7.55 | 0.833 | ··· | ··· | ··· |
| Heart failure–related death | 2.56 | 0.96–6.86 | 0.061 | ··· | ··· | ··· |
| Multiple vs no variant | ||||||
| Death and heart transplantation | 2.34 | 0.65–8.41 | 0.191 | ··· | ··· | ··· |
| All‐cause death | 2.34 | 0.65–8.41 | 0.191 | ··· | ··· | ··· |
| Heart transplantation | NA | NA | NA | ··· | ··· | ··· |
| Cardiovascular death | 2.34 | 0.65–8.41 | 0.191 | ··· | ··· | ··· |
| Sudden cardiac death | NA | NA | NA | ··· | ··· | ··· |
| Heart failure–related death | 3.73 | 0.96–14.45 | 0.057 | ··· | ··· | ··· |
CI indicates confidence interval; HR, hazard ratio; NA, not applicable.
Items with P<0.05 in univariable analyses were then included in the calculation of multivariable HR and 95% CI.
No heart transplantation occurred in multiple variant carriers or noncarriers, so HR and 95% CI are not available.
No sudden cardiac death occurred in multiple variant carriers, so HR and 95% CI that refer to this group are not available.
Discussion
In a large cohort of Chinese patients with LVNC, the genetic profile and correlations among genetics, clinical presentation, and long‐term outcomes were comprehensively investigated. We found that 38% of the patients carried at least one pathogenic variant, with TTN, MYH7, MYBPC3, and DSP being the most commonly involved genes. The genetic spectrum was similar between children and adults. In adults, G+ status was associated with a higher risk of death and HT, independent of age, sex, and cardiac function at baseline.
With the development of next‐generation sequencing, at least 21 genes, including sarcomeric, cytoskeletal, and ion channel genes, have been associated with LNVC.4 Genetic testing may have implications in diagnosis and family screening, so it is recommended for patients with LVNC.19 In accordance with a previously described yield of 35% to 40%,4 the overall yield of our gene panel was 38%, despite differences in patient selection and gene panel used. The yield of testing was found to be higher in children than that in adults with LVNC,12, 20 whereas it was similar between children and adults in our study. This might be explained by the rather small numbers of child patients in our study.
Our study revealed a distinct genetic spectrum in Chinese Han patients with LVNC. TTN, MYH7, MYBPC3, and DSP were the most commonly involved genes, which accounted for >60% of all detected pathogenic variants. Despite studies being performed on different ethnicities, pathogenic variants in MYH7 and MYBPC3 were frequently detected in patients with LVNC and hypertrophic cardiomyopathy,20, 21, 22 which indicated the shared genetic basis between different types of cardiomyopathy. Notably, variants in DSP were relatively common in our study. DSP encodes a critical component of desmosome structure in the myocardium and is a well‐established causal gene of arrhythmogenic cardiomyopathy.23, 24 A truncating variant in DSP has been implicated in LVNC.25 Besides, another truncating variant in DSP was found to be associated with SCD in a family.26 In our study, 3 novel truncating variants in DSP were identified in 6 patients. Unexpectedly, ventricular arrythmias were observed in all of these 6 patients, suggesting a possible genotype‐phenotype correlation. Therefore, patients with LVNC with pathogenic variants in DSP may have a high risk of arrhythmic events and should be followed up closely.
Two novel truncating variants in LAMP2 were identified in 2 patients in our study. Deficiency in LAMP2 has been shown to cause an X‐linked lysosomal condition called Danon disease.27 The role of LAMP2 in LVNC was only described in a case report, in which a truncating LAMP2 variant was described as having been detected in a patient with LVNC and Danon disease.28 Herein, we provide additional evidence of the association between LAMP2 deficiency and LVNC.
It is well recognized that 15% to 30% of patients with LVNC could experience premature death, and ≈10% could develop severe HF that may eventually lead to HT.3, 29 In our study, 27% of the patients died and 5% underwent HT, which also revealed the considerable morbidity and mortality in the process of LVNC. Despite the relatively poor prognosis, the effect of a positive genetic test result on long‐term outcomes remains unclear in adult patients. Probst et al found that pathogenic variants were not associated with cardiac function, HF, or arrythmia events.30 Similarly, a recent study by van Waning et al showed no difference in the risk of major adverse cardiac events between adults with LVNC with and without pathogenic variants.20 In contrast, our study found that G+ status was independently associated with higher risks of death and HT in adult patients with LVNC. More than 70 sarcomere and nonsarcomere genes were screened in our study, whereas the study by Probst et al focused mainly on sarcomere genes.30 The end point in the study by van Waning et al differed from ours, with thromboembolism and arrythmia events additionally being included.20 Besides, only 6% of participants died and 2% received HT in their study, which were much less than those in our study. Such differences in screening genes and end points might at least partially explain the difference in results between these studies and ours. Our discovery is of great importance, because prognostic implications can be speculated from genetic testing. Such information can aid in risk stratification, which can then assist with therapeutic decision making and improve prognosis.
The specific mechanism underlying the adverse outcomes of G+ patients is still unclear. A plausible one is the remarkably abnormal structure and/or function of cardiomyocytes caused by the variants. For example, a defect in sarcomere genes can lead to the development of diminished force generation and myocardial fibrosis.13, 31 These changes may eventually result in contractile dysfunction and, therefore, may be associated with higher rates of HF events. In support, of all the adult patients with variants in sarcomere genes in our study, more than half had left ventricular ejection fraction <40% at baseline and one third died of HF during the follow‐up. Defects in genes encoding ion channels, such as KCNE1 and SCN5A, have been related with disturbed currents in cardiomyocytes, which may be associated with higher risk of arrhythmic events, including SCD.32, 33
Multiple pathogenic variants have been associated with more severe manifestations and worse outcomes in patients with LVNC, compared with a single variant.31, 34 In our study, the risk of death and HT in carriers of multiple variants was higher but not significantly than that in carriers of a single variant or noncarriers. Because there were only 6 adult patients carrying >1 pathogenic variant, our failure to confirm a dosage effect of pathogenic variants might have been attributable to the lack of a sufficient number of participants for the results to reach a significant level.
There were several limitations in this study. First, this was an observational study, which might have been associated with an intrinsic bias. Second, all participants were from a single tertiary center. This might have caused a lack of representativeness and limited the generalizability of the findings. Third, the sample size was relatively small, so certain conclusions could not be drawn in some subgroups, such as child patients. Studies in larger populations are needed to confirm our discoveries.
To our knowledge, this is one of the largest LVNC cohorts with detailed genetic information and long‐term follow‐up data in a Chinese Han population. We proved that, in adult patients with LVNC, G+ status was an independent risk factor for death and HT, highlighting the importance of genetic testing for better risk stratification of the individual patient.
Sources of Funding
This study was supported by the National Natural Science Foundation of China (81470460).
Disclosures
Pu reports grants from the National Natural Science Foundation of China. The remaining authors have no disclosures to report.
Supporting information
Data S1. Gene panel.
Table S1. Criteria for Classifying Pathogenic Variants According to ACMG Guideline
Table S2. Rules for Combining Criteria for Pathogenic and Likely Pathogenic Variants
Table S3. Primers Used for Sanger Sequencing Confirmation
Table S4. Pathogenic and Likely Pathogenic Variants Detected in the Cohort
Table S5. Clinical Characteristics of the Six Carriers of DSP Variant
Table S6. Characteristics of Child Patients Who Reached the Primary Endpoint
Table S7. Incidence of Primary and Secondary Endpoints in Child and Adult Patients
Table S8. Characteristics of Adult Patients Who Reached the Primary Endpoint
Acknowledgments
We would like to thank all of the staff members for data collection, data entry, and monitoring for their contribution to the current study.
(J Am Heart Assoc. 2018;7:e009910 DOI: 10.1161/JAHA.118.009910.)
References
- 1. Patrianakos AP, Parthenakis FI, Nyktari EG, Vardas PE. Noncompaction myocardium imaging with multiple echocardiographic modalities. Echocardiography. 2008;25:898–900. [DOI] [PubMed] [Google Scholar]
- 2. Kovacevic‐Preradovic T, Jenni R, Oechslin EN, Noll G, Seifert B, Attenhofer Jost CH. Isolated left ventricular noncompaction as a cause for heart failure and heart transplantation: a single center experience. Cardiology. 2009;112:158–164. [DOI] [PubMed] [Google Scholar]
- 3. Oechslin EN, Attenhofer Jost CH, Rojas JR, Kaufmann PA, Jenni R. Long‐term follow‐up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol. 2000;36:493–500. [DOI] [PubMed] [Google Scholar]
- 4. Towbin JA, Lorts A, Jefferies JL. Left ventricular non‐compaction cardiomyopathy. Lancet. 2015;386:813–825. [DOI] [PubMed] [Google Scholar]
- 5. Risebro CA, Riley PR. Formation of the ventricles. ScientificWorldJournal. 2006;6:1862–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sedmera D, McQuinn T. Embryogenesis of the heart muscle. Heart Fail Clin. 2008;4:235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pignatelli RH, McMahon CJ, Dreyer WJ, Denfield SW, Price J, Belmont JW, Craigen WJ, Wu J, El Said H, Bezold LI, Clunie S, Fernbach S, Bowles NE, Towbin JA. Clinical characterization of left ventricular noncompaction in children: a relatively common form of cardiomyopathy. Circulation. 2003;108:2672–2678. [DOI] [PubMed] [Google Scholar]
- 8. Engberding R, Yelbuz TM, Breithardt G. Isolated noncompaction of the left ventricular myocardium: a review of the literature two decades after the initial case description. Clin Res Cardiol. 2007;96:481–488. [DOI] [PubMed] [Google Scholar]
- 9. Towbin JA. Left ventricular noncompaction: a new form of heart failure. Heart Fail Clin. 2010;6:453–469, viii. [DOI] [PubMed] [Google Scholar]
- 10. Ichida F, Hamamichi Y, Miyawaki T, Ono Y, Kamiya T, Akagi T, Hamada H, Hirose O, Isobe T, Yamada K, Kurotobi S, Mito H, Miyake T, Murakami Y, Nishi T, Shinohara M, Seguchi M, Tashiro S, Tomimatsu H. Clinical features of isolated noncompaction of the ventricular myocardium: long‐term clinical course, hemodynamic properties, and genetic background. J Am Coll Cardiol. 1999;34:233–240. [DOI] [PubMed] [Google Scholar]
- 11. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB; American Heart Association, Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; Council on Epidemiology and Prevention . Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. [DOI] [PubMed] [Google Scholar]
- 12. Hoedemaekers YM, Caliskan K, Michels M, Frohn‐Mulder I, van der Smagt JJ, Phefferkorn JE, Wessels MW, ten Cate FJ, Sijbrands EJ, Dooijes D, Majoor‐Krakauer DF. The importance of genetic counseling, DNA diagnostics, and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circ Cardiovasc Genet. 2010;3:232–239. [DOI] [PubMed] [Google Scholar]
- 13. Klaassen S, Probst S, Oechslin E, Gerull B, Krings G, Schuler P, Greutmann M, Hurlimann D, Yegitbasi M, Pons L, Gramlich M, Drenckhahn JD, Heuser A, Berger F, Jenni R, Thierfelder L. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation. 2008;117:2893–2901. [DOI] [PubMed] [Google Scholar]
- 14. Grant R. An unusual anomaly of the coronary vessels in the malformed heart of a child. Heart. 1926;13:273–283. [Google Scholar]
- 15. Jenni R, Oechslin E, Schneider J, Attenhofer Jost C, Kaufmann PA. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non‐compaction: a step towards classification as a distinct cardiomyopathy. Heart. 2001;86:666–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petersen SE, Selvanayagam JB, Wiesmann F, Robson MD, Francis JM, Anderson RH, Watkins H, Neubauer S. Left ventricular non‐compaction: insights from cardiovascular magnetic resonance imaging. J Am Coll Cardiol. 2005;46:101–105. [DOI] [PubMed] [Google Scholar]
- 17. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell‐Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce‐Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano‐Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie‐Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG; Exome Aggregation Consortium . Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, Hershberger RE, Judge DP, Le Marec H, McKenna WJ, Schulze‐Bahr E, Semsarian C, Towbin JA, Watkins H, Wilde A, Wolpert C, Zipes DP. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8:1308–1339. [DOI] [PubMed] [Google Scholar]
- 20. van Waning JI, Caliskan K, Hoedemaekers YM, van Spaendonck‐Zwarts KY, Baas AF, Boekholdt SM, van Melle JP, Teske AJ, Asselbergs FW, Backx A, du Marchie Sarvaas GJ, Dalinghaus M, Breur J, Linschoten MPM, Verlooij LA, Kardys I, Dooijes D, Lekanne Deprez RH, IJpma AS, van den Berg MP, Hofstra RMW, van Slegtenhorst MA, Jongbloed JDH, Majoor‐Krakauer D. Genetics, clinical features, and long‐term outcome of noncompaction cardiomyopathy. J Am Coll Cardiol. 2018;71:711–722. [DOI] [PubMed] [Google Scholar]
- 21. Miller EM, Hinton RB, Czosek R, Lorts A, Parrott A, Shikany AR, Ittenbach RF, Ware SM. Genetic testing in pediatric left ventricular noncompaction. Circ Cardiovasc Genet. 2017;10:e001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. 2017;121:749–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fressart V, Duthoit G, Donal E, Probst V, Deharo JC, Chevalier P, Klug D, Dubourg O, Delacretaz E, Cosnay P, Scanu P, Extramiana F, Keller D, Hidden‐Lucet F, Simon F, Bessirard V, Roux‐Buisson N, Hebert JL, Azarine A, Casset‐Senon D, Rouzet F, Lecarpentier Y, Fontaine G, Coirault C, Frank R, Hainque B, Charron P. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace. 2010;12:861–868. [DOI] [PubMed] [Google Scholar]
- 24. Lopez‐Ayala JM, Gomez‐Milanes I, Sanchez Munoz JJ, Ruiz‐Espejo F, Ortiz M, Gonzalez‐Carrillo J, Lopez‐Cuenca D, Oliva‐Sandoval MJ, Monserrat L, Valdes M, Gimeno JR. Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: characterizing a phenotype. Europace. 2014;16:1838–1846. [DOI] [PubMed] [Google Scholar]
- 25. Williams T, Machann W, Kuhler L, Hamm H, Muller‐Hocker J, Zimmer M, Ertl G, Ritter O, Beer M, Schonberger J. Novel desmoplakin mutation: juvenile biventricular cardiomyopathy with left ventricular non‐compaction and acantholytic palmoplantar keratoderma. Clin Res Cardiol. 2011;100:1087–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bhonsale A, Groeneweg JA, James CA, Dooijes D, Tichnell C, Jongbloed JD, Murray B, te Riele AS, van den Berg MP, Bikker H, Atsma DE, de Groot NM, Houweling AC, van der Heijden JF, Russell SD, Doevendans PA, van Veen TA, Tandri H, Wilde AA, Judge DP, van Tintelen JP, Calkins H, Hauer RN. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy‐associated mutation carriers. Eur Heart J. 2015;36:847–855. [DOI] [PubMed] [Google Scholar]
- 27. Cheng Z, Fang Q. Danon disease: focusing on heart. J Hum Genet. 2012;57:407–410. [DOI] [PubMed] [Google Scholar]
- 28. Van Der Starre P, Deuse T, Pritts C, Brun C, Vogel H, Oyer P. Late profound muscle weakness following heart transplantation due to Danon disease. Muscle Nerve. 2013;47:135–137. [DOI] [PubMed] [Google Scholar]
- 29. Bhatia NL, Tajik AJ, Wilansky S, Steidley DE, Mookadam F. Isolated noncompaction of the left ventricular myocardium in adults: a systematic overview. J Card Fail. 2011;17:771–778. [DOI] [PubMed] [Google Scholar]
- 30. Probst S, Oechslin E, Schuler P, Greutmann M, Boye P, Knirsch W, Berger F, Thierfelder L, Jenni R, Klaassen S. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ Cardiovasc Genet. 2011;4:367–374. [DOI] [PubMed] [Google Scholar]
- 31. Miszalski‐Jamka K, Jefferies JL, Mazur W, Glowacki J, Hu J, Lazar M, Gibbs RA, Liczko J, Klys J, Venner E, Muzny DM, Rycaj J, Bialkowski J, Kluczewska E, Kalarus Z, Jhangiani S, Al‐Khalidi H, Kukulski T, Lupski JR, Craigen WJ, Bainbridge MN. Novel genetic triggers and genotype‐phenotype correlations in patients with left ventricular noncompaction. Circ Cardiovasc Genet. 2017;10:e001763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, Yamamoto S, Ozawa T, Ding WG, Toyoda F, Kawamura M, Akao M, Matsuura H, Kimura T, Kita T, Horie M. D85N, a KCNE1 polymorphism, is a disease‐causing gene variant in long QT syndrome. J Am Coll Cardiol. 2009;54:812–819. [DOI] [PubMed] [Google Scholar]
- 33. Zaklyazminskaya E, Dzemeshkevich S. The role of mutations in the SCN5A gene in cardiomyopathies. Biochim Biophys Acta. 2016;1863:1799–1805. [DOI] [PubMed] [Google Scholar]
- 34. Wang C, Hata Y, Hirono K, Takasaki A, Ozawa SW, Nakaoka H, Saito K, Miyao N, Okabe M, Ibuki K, Nishida N, Origasa H, Yu X, Bowles NE, Ichida F; for LVNC Study Collaborators . A wide and specific spectrum of genetic variants and genotype‐phenotype correlations revealed by next‐generation sequencing in patients with left ventricular noncompaction. J Am Heart Assoc. 2017;6:e006210 DOI: 10.1161/JAHA.117.006210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Gene panel.
Table S1. Criteria for Classifying Pathogenic Variants According to ACMG Guideline
Table S2. Rules for Combining Criteria for Pathogenic and Likely Pathogenic Variants
Table S3. Primers Used for Sanger Sequencing Confirmation
Table S4. Pathogenic and Likely Pathogenic Variants Detected in the Cohort
Table S5. Clinical Characteristics of the Six Carriers of DSP Variant
Table S6. Characteristics of Child Patients Who Reached the Primary Endpoint
Table S7. Incidence of Primary and Secondary Endpoints in Child and Adult Patients
Table S8. Characteristics of Adult Patients Who Reached the Primary Endpoint