

Abstract
Background
Transforming growth factor beta (TGF‐β) is an important cytokine in mediating the cardiac fibrosis that often accompanies pathogenic cardiac remodeling. Cardiomyocyte‐specific expression of a mutant αB‐crystallin (CryABR 120G), which causes human desmin‐related cardiomyopathy, results in significant cardiac fibrosis. During onset of fibrosis, fibroblasts are activated to the so‐called myofibroblast state and TGF‐β binding mediates an essential signaling pathway underlying this process. Here, we test the hypothesis that fibroblast‐based TGF‐β signaling can result in significant cardiac fibrosis in a disease model of cardiac proteotoxicity that has an exclusive cardiomyocyte‐based etiology.
Methods and Results
Against the background of cardiomyocyte‐restricted expression of CryABR 120G, we have partially ablated TGF‐β signaling in cardiac myofibroblasts to observe whether cardiac fibrosis is reduced despite the ongoing pathogenic stimulus of CryABR 120G production. Transgenic CryABR 120G mice were crossed with mice containing a floxed allele of TGF‐β receptor 2 (Tgfbr2 f/f). The double transgenic animals were subsequently crossed to another transgenic line in which Cre expression was driven from the periostin locus (Postn) so that Tgfbr2 would be ablated with myofibroblast conversion. Structural and functional assays were then used to determine whether general fibrosis was affected and cardiac function rescued in CryABR 120G mice lacking Tgfbr2 in the myofibroblasts. Ablation of myofibroblast specific TGF‐β signaling led to decreased morbidity in a proteotoxic disease resulting from cardiomyocyte autonomous expression of CryABR 120G. Cardiac fibrosis was decreased and hypertrophy was also significantly attenuated, with a significant improvement in survival probability over time, even though the primary proteotoxic insult continued.
Conclusions
Myofibroblast‐targeted knockdown of Tgfbr2 signaling resulted in reduced fibrosis and improved cardiac function, leading to improved probability of survival.
Keywords: cell signaling, fibrosis, myofibroblast, protein aggregation, transforming growth factor‐beta
Subject Categories: Fibrosis, Pathophysiology, Heart Failure
Clinical Perspective
What Is New?
We have studied in detail cardiac fibrosis in a well‐defined mouse model of proteotoxicity, induced by cardiomyocyte‐specific expression of a mutation in αB crystallin that causes human desmin‐related cardiomyopathy.
Given that transforming growth factor beta (TGF‐β) signaling is essential for a fibrotic response, we have ablated transforming growth factor beta signaling specifically in cardiac myofibroblasts to establish how the signaling pathway functions in cardiomyocyte proteotoxic‐based cardiac disease process.
What Are the Clinical Implications?
These findings define the role of myofibroblast‐specific TGF‐β signaling and point to the efficacy of developing myofibroblast‐specific therapeutic interventions while leaving the cardiomyocyte unaffected.
Therapeutic targeting of a receptor for TGF‐β, Tgfbr2 alone in cardiac myofibroblasts as an antifibrotic treatment might provide the needed selectivity and specificity that limits side effects associated with systemic TGF‐β inhibition.
Introduction
Heart disease often occurs as a consequence of chronic or acute neurohormonal, mechanical, and genetic stress and remains a primary cause of death in developed and developing countries.1 Cardiovascular disease stemming from many primary etiologies is often accompanied by the sporadic or diffuse presence of fibrotic tissue or scar formation.2 Continued fibrotic development adversely affects cardiac function, altering the heart's anatomical structure and, as fibrosis progresses, increases chamber stiffness and decreases ventricular compliance.3 The adult mammalian heart is composed of many cell types, the most abundant being endothelial cells, cardiomyocytes, fibroblasts, and perivascular cells.4 Fibroblasts constitute around 15% of the nonmyocyte cell population, equating to nearly 11% of the total cells of the heart.5 They are a major contributor to the mature connective matrix of the myocardium, and cardiomyocyte health and function are dependent upon a supportive extracellular matrix (ECM) elaborated by the fibroblasts. Classically, the fibroblast has been rather ill‐defined as a connective tissue cell derived from the primitive mesoderm.6 In situ in the myocardium, fibroblasts are visualized as small‐sized, flattened cells that lack a basement membrane. They are tightly interspaced between, above, and below the cardiomyocytes, extending multiple cellular processes that form a dense network consistent with their main function, which, in the healthy heart, is to generate and maintain the ECM by producing the fibrillar collagens type I and II. These ECM proteins are generated by activated fibroblasts, also referred as myofibroblasts.
Transforming growth factor beta (TGF‐β) is a profibrotic cytokine. Members of the TGF‐β superfamily are ubiquitously expressed and control multiple processes that are involved in both the formation and maintenance of cardiac fibroblasts and cardiomyocytes. The importance of TGF‐β signaling in heart failure and cardiac disease is well documented,7, 8 but nothing is known about how TGF‐β signaling in the myofibroblasts impacts a chronic, genetically induced cardiomyopathy that occurs as a result of a sarcomere‐based proteotoxic process, such as desmin‐related cardiomyopathy induced by mutant αB‐crystallin (CryABR120G) expression.9, 10 Almost all related data are based on acute surgical interventions such as the induction of pressure overload or myocardial infarction.11, 12, 13 When monoclonal antibodies were used to target and reduce global TGF‐β signaling, significant antifibrotic effects in a stressed heart were observed.11, 14 Recently, a genetic model of hypertrophic cardiomyopathy was also used to confirm the general importance of TGF‐β signaling in the fibrotic processes that accompany remodeling.14
In this study, we restricted a loss of function of TGF‐β signaling to myofibroblasts to discern their role in mediating a fibrotic response to cardiac disease induced by CryABR120G expression that occurs exclusively in a different cell type, the cardiomyocyte. In recent studies, a functional periostin allele driving tamoxifen (TAM)‐inducible Cre recombinase expression whose transcriptional activity is largely restricted to activated myofibroblasts was validated and used to ablate TGF‐β receptors 1 and 2 (TGF‐βr1 and 2, respectively) in those cells.12, 15, 16 We used this locus to carry out myofibroblast‐restricted targeted ablation of TGF‐β signaling in a well‐defined genetic model of heart disease and failure that is characterized by the slow development of cardiac fibrosis over a period of months as a result of cardiomyocyte‐restricted expression of CryABR120G expression.10, 17, 18 Hearts undergo slow, but extensive, fibrosis over 7 months. We generated transgenic (Tg) mice by crossing CryAB R120G and Tgfbr2‐flox animals with mice containing a TAM‐inducible Cre recombinase (Mer‐cre‐Mer; mcm) expression cassette within the periostin genetic locus (Postnmcm). We show that ablating TGF‐β signaling in myofibroblasts can reduce cardiac fibrosis and hypertrophy in a mouse model of human cardiac proteotoxicity, improving both cardiac hemodynamics and probability of survival.
Methods
Data, analytical methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure through the facilities of Cincinnati Children's Hospital Medical Center (Cincinnati, OH). Although some materials may remain available, J.R. is not able to guarantee that he will be able to provide them at a future time.
Animals and Echocardiography
Myofibroblast‐targeted–inducible Tg mice were generated as previously described.15 The TAM‐inducible, periostin‐driven cre (Postnmcm) mouse was crossed into a targeted Tgfbr2‐loxP background so that Tgfbr2 could be ablated upon induction of Cre expression.19 Those mice were then crossbred to a FVB/N background for at least 6 generations before being crossed with the CryABR120G mice (FVB/N). The triple Tg offspring were identified by PCR. Both male and female mice were used in all experimental procedures, and they were subdivided blindly to experimental groups as per the genotypes indicated in the figures. TAM mouse chow at an effective treatment dosage of 40 mg/kg body weight (Envigo‐TD.130860; Envigo RMS, Inc, Indianapolis, IN) was used to activate the Postnmcm locus, thereby inducing Cre recombinase in activated myofibroblasts. TAM chow was fed starting from 7 to 8 weeks postbirth and continued throughout the experimental periods. Cardiac ultrasound was performed on isoflurane‐anesthetized mice at 5 months of age with a VisualSonics Vevo 2100 Imaging System (VisualSonics, Toronto, Ontario, Canada) using a 40‐MHz transducer20 by a trained sonographer blinded to genotype. Two‐dimensional directed M‐mode echocardiographic images along the parasternal long axis were recorded to determine left ventricular (LV) size and systolic function. M‐mode measurements included the LV internal dimensions in systole and diastole as well as the diastolic thickness LV posterior wall and diastolic intraventricular septum thickness. Percentage fraction shortening was calculated as [(LV internal dimensions in diastole−LV internal dimensions in systole)/LV internal dimensions in diastole]×100%. The apical 4‐chamber view was used to assess the mitral valve using both pulsed‐wave and tissue Doppler. In pulsed‐wave Doppler mode, we measured peak velocities of early (E) and late diastolic flow (A) and calculated the E/A ratio. Animals were handled in accord with the principles and procedures of the Guide for the Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee at Cincinnati Children's Hospital approved all experimental procedures.
Antibody Validation
Multiple lines of evidence were used to validate the commercially available antibodies used. For example, ablation of Tgfbr2 in cardiomyocytes decreased expression of TGF‐βr2 by western blots while abolishing cardiomyocyte staining with anti‐TGF‐βr2 antibody (ab61213 Rabbit mAb; Abcam, Cambridge, MA; Figure 1). Similarly phospho (p)‐Smad3 (small mothers against decapentaplegic homolog 3; S423/425 [C52A9] Rabbit mAb #9520; Cell Signaling Technology, Danvers, MA), p‐TAK1 (transforming growth factor beta–activated kinase 1; S412 #9339; Cell Signaling Technology), TAK1 (ab109526 Rabbit mAb; Abcam), and p‐p38 (Thr180‐Tyr182 Rabbit mAb #4092; Cell Signaling Technology) were validated by treating fibroblasts with TGF‐β (20 ng/mL; R&D Systems, Minneapolis, MN) and confirming that activation of the above proteins’ syntheses could be detected by the cognate antibodies in the appropriate sample (Figure 1).
Figure 1.
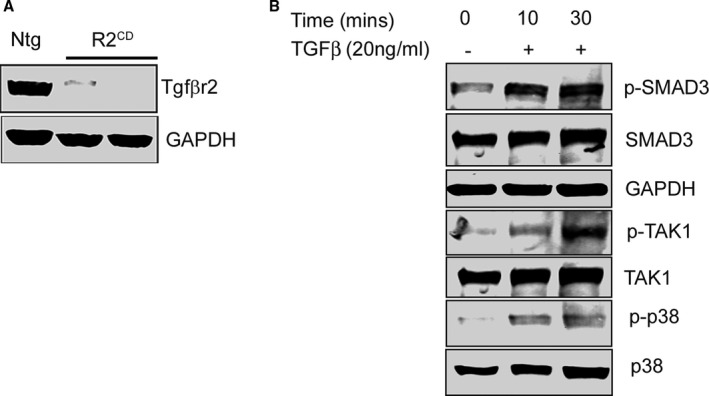
Antibody validation. A, Western blot showing transforming growth factor β receptor II (Tgfbr2) protein expression in the protein lysates from nontransgenic (Ntg) hearts and cardiomyocyte‐specific Tgfbr2‐ablated mice (R2CD). B, Validation of antibodies for phospho‐SMAD3 (p‐SMAD3, phospho‐TAK1 (p‐TAK1) and phospho‐p38 (p‐p38) after treatment of fibroblasts with TGF‐β (+). GAPDH indicates glyceraldehyde 3‐phosphate; SMAD3, small mothers against decapentaplegic homolog 3; TAK1, transforming growth factor beta–activated kinase 1; TGF‐β, transforming growth factor beta.
Histology and Immunohistochemistry
Mice were anesthetized with isoflurane, and hearts were fixed by perfusion with 10% buffered formalin. Hearts were fixed overnight at room temperature, transferred into 70% EtOH, embedded in paraffin, sectioned, and prepared for immunohistochemistry. Interstitial fibrosis was detected by staining sections with Masson's trichrome and picric acid Sirius Red staining. Images were acquired on an Olympus BX69 microscope (Olympus Corporation, Tokyo, Japan), equipped with a Nikon digital camera (Nikon, Tokyo, Japan), and NIS Elements software (Nikon Instruments Inc, Melville, NY). Immunohistochemical staining was performed on sections after heat‐activated antigen retrieval as described.21 The following primary antibodies were used: rabbit anti‐periostin (1:1000; NBP1‐30042; Novus, Novus Biologicals, Littleton, CO), rabbit anti‐phospho SMAD2/3 (1:500; D6658; Maine Medical Center Research Institute, Scarborough, ME), mouse anti‐troponin I (1:1000; Millipore, Burlington, MA), and rabbit anti‐CryAB (1:400; Assay Designs Inc, Ann Arbor, MI). They were visualized with Alexa Fluor 488– or Alexa Fluor 568–conjugated secondary antibody (Molecular Probes, Eugene, OR) directed against mouse or rabbit immunoglobulin G, and 4′,6‐diamidino‐2‐phenylindole (Invitrogen, Carlsbad, CA) was used to identify nuclei. Immunofluorescent staining for CryAB was used to quantitate the cytoplasmic area occupied by protein aggregates as described.17, 21 Ten fields per heart (n=4) were imaged using a confocal microscope system with ×60 objective for both cell‐surface and aggregate‐area measurements by a blinded observer. Cell‐surface area was measured using ImageJ analysis software (NIH, Bethesda, MD). Aggregate area was calculated by using NIS Elements AR analysis software (Nikon Instruments).
Hydroxyproline Assay
Determination of hydroxyproline content as a measure of fibrosis was performed as described.22 Briefly, tissues were hydrolysed in 6 N of HCl. Following neutralization, samples were vacuum dried and resuspended in 5 mmol/L of HCl. After the addition of chloramine T solution, sample absorbance was determined at 558 nm.
RNA Isolation and Quantitative Real‐Time PCR
Total RNA was isolated with TRI reagent (Molecular Research Center, Inc, Cincinnati, OH), according to the manufacturer's protocol, using the Precellys homogenizing kit (10409; Precellys). Quantitative real‐time PCR was performed with a CFX‐96 instrument (Bio‐Rad, Hercules, CA) using Taqman probes (Applied Biosystems, Foster City, CA) for periostin (Applied Biosystems), α‐smooth muscle actin (α‐SMA; Applied Biosystems). Cardiac fetal gene transcripts encoding β‐myosin heavy chain and natriuretic peptide precursor A and B (Applied Biosystems) were also quantitated as a measure of hypertrophic response at the gene transcript level. All data were normalized to glyceraldehyde 3‐phosphate dehydrogenase (Applied Biosystems) content and expressed as fold increase over the control group.
Cell Fractionation, SDS‐PAGE, and Immunoblotting
Soluble and insoluble protein fractions from hearts were prepared as described.23 Briefly, hearts were harvested in cold PBS (pH 7.4) containing 1% Triton‐X100, 2.5 mmol/L of EDTA, 0.5 mmol/L of PMSF, phosphatase inhibitor cocktail tablets (Roche, Indianapolis, IN), a complete protease inhibitor mixture (Roche), and then homogenized for 30 seconds. Heart extracts were centrifuged at 12 000g for 15 minutes, and supernatants were collected (soluble fraction). Pellets were dissolved in DNase (1 mg/mL in 10 mmol/L of Tris, 15 mmol/L of MgCl2) and sonicated on ice, and the protein was quantitated with a modified Bradford assay. The insoluble protein was then diluted in RIPA buffer, and 3 μg was used for subsequent electrophoresis and immunoblotting with appropriate antibodies. Protein lysates were separated on SDS‐PAGE using precast 7.5% to 15% Criterion Gels (Bio‐Rad) and transferred to PVDF membranes (Bio‐Rad). Membranes were blocked for 1 hour in 5% nonfat dried milk and exposed to primary antibodies overnight. The following primary antibodies were used for immunoblotting: anti‐TGF‐βr2 (1:500; Abcam), anti‐α‐SMA (1:1000; Sigma); anti‐periostin (1:1000; Novus); anti‐CryAB (1:5000; Assay Designs); anti‐p‐SMAD3 (1:500; Cell Signaling Technology); anti‐SMAD3 (1:1000; Abcam); anti‐p‐TAK1 (1:500; Cell Signaling Technology); anti‐TAK1 (1:1000; Abcam); anti‐p‐p38 (1:500; Cell Signaling Technology); anti‐p38 (1:1000; Cell Signaling Technology), and anti–glyceraldehyde 3‐phosphate dehydrogenase (1:10 000; Millipore). Detection was carried out using fluorescent‐conjugated secondary antibodies (LI‐COR Biosciences, Lincoln, NE) in combination with an Odyssey CLx infrared imaging system (LI‐COR Biosciences).
Isolation of Cardiomyocytes and Cardiac Fibroblasts
Cardiomyocytes and cardiac fibroblasts were isolated as described.16 Cells were collected from 12‐ to 20‐week‐old hearts by enzymatic digestion. Mice were given 100 μL of heparin (100 U/mL) by intraperitoneal injection and anesthetized with isoflurane. The heart was quickly excised and retrograde perfused using a Langendorff apparatus under constant pressure (60 mm Hg; 37°C, 4 minutes) in Ca2+‐free perfusion buffer containing 113 mmol/L of NaCl, 4.7 mmol/L of KCl, 1.2 mmol/L of MgSO4, 5.5 mmol/L of glucose, 0.6 mmol/L of KH2PO4, 0.6 mmol/L of Na2HPO4, 12 mmol/L of NaHCO3, 10 mmol/L of KHCO3, 10 mmol/L of HEPES, 10 mmol/L of 2,3‐butanedione monoxime, and 30 mmol/L of taurine. Digestion was achieved by perfusing for 3 minutes with Ca2+‐free perfusion buffer containing collagenase II (600 units/mL of collagenase II in perfusion buffer; LS004177; Worthington Biochemical Corporation, Lakewood, NJ) followed by 8 minutes of perfusion with digestion buffer containing 12.5 μmol/L of CaCl2. Subsequently, the heart was removed from the apparatus and gently teased into small pieces with fine forceps in the same enzyme solution and further dissociated mechanically using 2‐, 1.5‐, and 1‐mm‐diameter pipettes, until all large heart tissue pieces were dispersed. The digestion buffer was neutralized with buffer containing 10% FBS and 12.5 μmol/L of CaCl2, and the cell suspension was filtered through a 200‐μm mesh. Cardiomyocytes were pelleted by gravity for 20 minutes and the supernatant collected and stored on ice. Cardiomyocytes were resuspended in perfusion solution containing 5% FBS and 12.5 μmol/L of CaCl2 and subsequently allowed to settle for 20 minutes. The 2 supernatants were pooled and both cardiac fibroblast fractions were centrifuged at 500g for 7 minutes. After isolation, cells were kept on ice for further processing. Endothelial (CD 31+) and myeloid (CD 45+) cell fractions were sorted out with a Magnetic Cell Isolation and Cell Separation Kit (130‐042‐401; Miltenyi Biotec, Bergisch Gladbach, Germany), per manufacturer's instructions, with antibodies against CD31 (Miltenyi Biotec) and CD45 (Miltenyi Biotec) using the manufacturer's recommended dilutions.
Statistical Analysis
Data are expressed as mean±SEM. All statistical tests were done with GraphPad Prism software (GraphPad Software Inc, La Jolla, CA). Statistical analyses between 2 groups were carried out using the 2‐tailed Student t test. Groups of 3 or more were analyzed with 1‐way ANOVA, followed by Tukey's post hoc test. Survival probabilities were estimated with Kaplan–Meier curves, and curves were compared using the log‐rank test. A value of P<0.05 was considered to be statistically significant.
Results
A Fibrotic Response in the CryABR120G Model of Desmin‐Related Cardiomyopathy
Cardiomyocyte‐specific expression of CryABR120G in the mouse and human causes desmin‐related cardiomyopathy, whose functional pathology and proteotoxic effects have been well described.18, 24 Cardiac disease develops slowly over a period of months in the mouse model, with functional deficits becoming apparent in months 4 to 5 and death attributed to heart failure occurring in months 7 to 8.21, 25 We utilized well‐characterized markers to define the appearance of fibrosis in CryABR120G hearts and found that significant upregulation of the fibrotic markers, periostin and α‐SMA, occurred by 4 months in hearts. Immunostaining for periostin as well as histological analyses using Sirius Red staining detected abundant fibrosis in sections prepared from 4‐month‐old CryABR120G hearts when compared with its matched non‐Tg (Ntg) control (Figure 2A). Western blots using protein isolated from left ventricles of 4‐month‐old hearts showed significant increases in both periostin (Figure 2B and 2D) and α‐SMA (Figure 2C and 2E). Hydroxyproline content of the cardiac tissue, an indicator of collagen content, showed a 2.5‐fold increase compared with values found in Ntg hearts (Figure 2F). These findings were further supported by quantitation of Sirius Red staining (Figure 3A) and quantitative PCR analysis of α‐SMA and periostin gene expression in left ventricles of 4‐month‐old CryABR120G and Ntg controls (Figure 3B and 3C). As expected at this mid‐stage of disease, we confirmed significant cardiac hypertrophy and quantitated cardiomyocyte cross‐sectional areas using wheat germ agglutinin staining (Figure 4A and 4B). CryABR120G‐expressing cardiomyocytes at 5 months were ≈2‐fold larger than Ntg control cardiomyocytes (P<0.001), and heart weight to body weight ratios were also significantly elevated in CryABR120G animals (Figure 4C through 4E).
Figure 2.
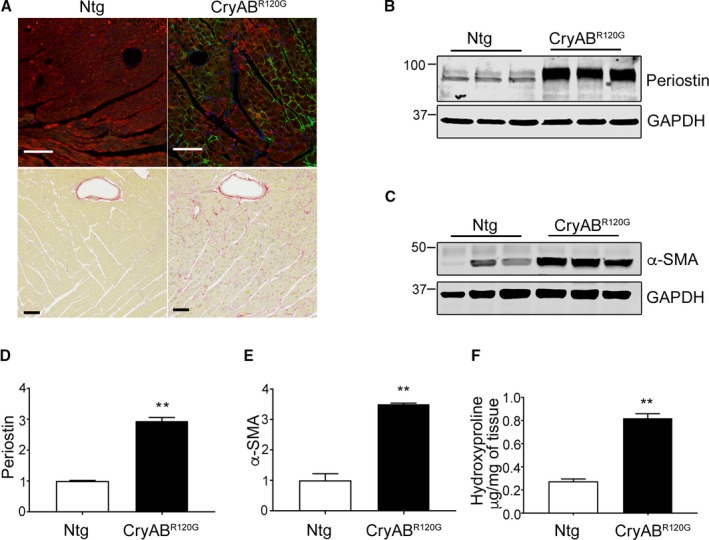
Protein markers of fibrosis in mutant αB‐crystallin (CryABR 120G) mice. A, Periostin (green) and Sirius Red–stained left ventricle sections from 4‐month‐old nontransgenic (Ntg) and CryABR 120G hearts. In periostin immunohistochemistry sections, cardiomyocytes are labeled with troponin I (red) and nuclei with DAPI (blue). The scale bar is 50 μm for the upper panels and 100 μm for the lower panels. B, Expression of the fibrosis markers, periostin and (C) α‐smooth muscle actin (α‐SMA), in left ventricles derived from 4‐month‐old Ntg and CryABR 120G hearts. D, Quantitative analysis of the periostin and (E) α‐SMA western blots: shown are the ratio values using GAPDH to normalize loading variation (n=6). F, Hydroxyproline levels derived from 4‐month‐old left ventricles (n=5). **P<0.001 vs Ntg control. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; GAPDH, glyceraldehyde 3‐phosphate.
Figure 3.
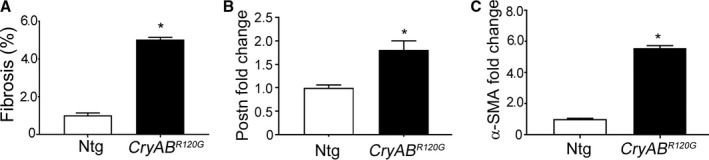
Fibrosis in mutant αB‐crystallin (CryABR 120G) mice. A, Quantification of fibrosis from the images obtained using Sirius Red–stained sections from 4‐month‐old nontransgenic (Ntg) and CryABR 120G hearts shown in Figure 1 (n=5). B and C, Quantitative PCR analysis of profibrotic transcript levels of periostin (Postn) and α‐smooth muscle actin (α‐SMA) in left ventricles derived from 16‐week‐old Ntg (n=3) and CryABR 120G hearts (n=4). *P<0.05 vs Ntg control. The Ntg control was arbitrarily set to 1.
Figure 4.
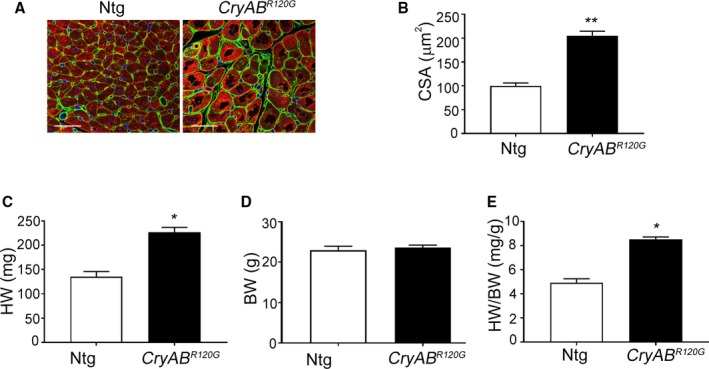
Cardiac hypertrophy in mutant αB‐crystallin (CryABR 120G) mice. A, Wheat germ agglutinin (green) staining in cardiac sections from 5‐month‐old nontransgenic (Ntg) and CryABR 120G hearts. Cardiomyocytes are stained with troponin I (red) and the nuclei with DAPI (blue). Scale bar is 20 μm. B, Assessment of cross‐sectional areas (CSA) of cardiomyocytes shown in (A) (n=5). C through E, Quantitation of heart weight (HW), body weight (BW), and heart weight to the body weight ratio (HW/BW) of the 5‐month‐old Ntg and CryABR 120G hearts (n=5). *P<0.05; **P<0.001 vs Ntg control. DAPI indicates 4′,6‐diamidino‐2‐phenylindole.
To extend these data and more fully define the fibrotic response in the cardiomyocyte‐restricted CryABR120G hearts, we performed a transcript array using 3‐month‐old mice, which is before functional deficits can be detected by cardiac echocardiography.21, 25 A number of transcripts encoded proteins that were previously annotated as playing a role in fibrosis (Figure 5A). The complete quantitative data are shown in Table 1. Specifically, a number of genes associated with TGF‐β signaling were significantly upregulated, including Tgfb1 (P=0.044), Tgfb2 (P=0.002), and Tgfb3 (P=0.025), in CryABR120G hearts. Apart from TGF‐β cytokines, profibrotic and ECM component genes, such as collagens,26 chemokines,27 connective tissue growth factor (Ctgf), and metalloproteinases,28 were also significantly upregulated.
Figure 5.
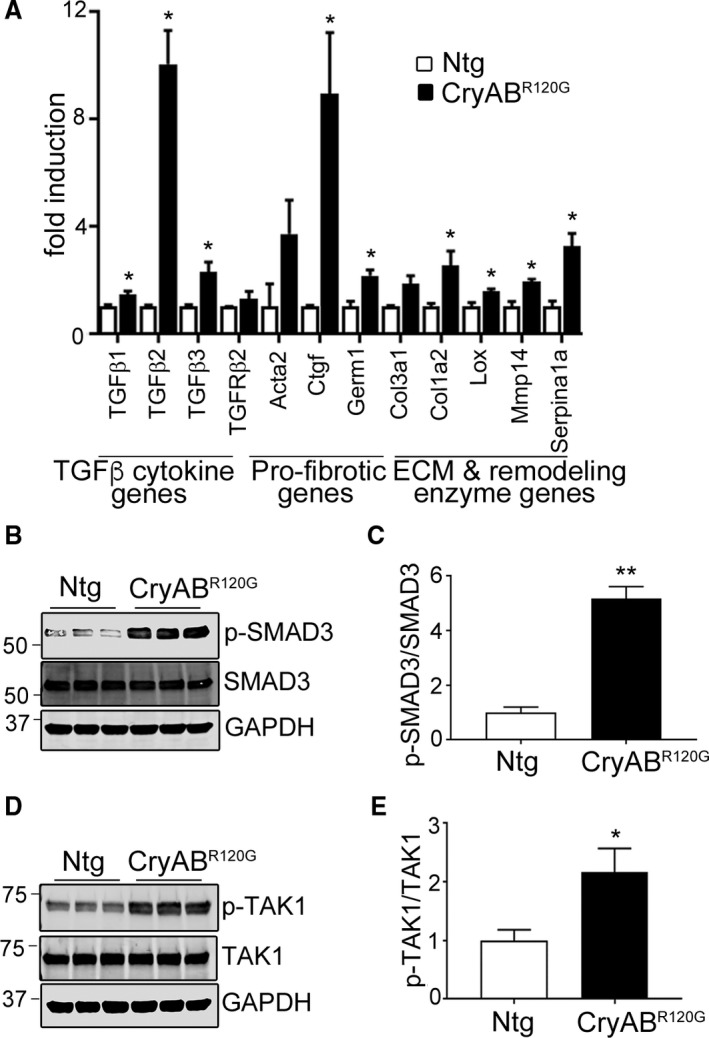
Fibrotic gene expression and TGF‐β signaling in mutant αB‐crystallin (CryABR 120G) mice. A, PCR array results of upregulated fibrogenic transcripts in ventricles from 3‐month‐old nontransgenic (Ntg) and CryABR 120G mice (n=3). Complete quantitative data are shown in Table 1. B and C, Expression of a downstream target of TGF‐β signaling, phospho‐SMAD3 (p‐SMAD3) from 3‐month‐old Ntg and CryABR 120G hearts. D and E, Expression of a downstream target of noncanonical TGF‐β signaling, p‐TAK1. The Student unpaired t test was used to compare groups (n=6). *P<0.05 vs Ntg control; **P<0.001 vs Ntg control. ECM indicates extracellular matrix; GAPDH, glyceraldehyde 3‐phosphate; SMAD3, small mothers against decapentaplegic homolog 3; TAK1, transforming growth factor beta–activated kinase 1; TGF‐β, transforming growth factor beta.
Table 1.
Select Transcript Analyses
| Gene | Fold Induction | SD | P Value |
|---|---|---|---|
| Acta2 | 3.712 | 2.181 | 0.098 |
| Bcl2 | 2.176 | 2.154 | 0.438 |
| Ccl3 | 3.119 | 1.069 | 0.027 |
| Col1a2 | 1.858 | 0.519 | 0.043 |
| Col3a1 | 2.526 | 0.955 | 0.054 |
| Ctgf | 8.947 | 3.965 | 0.025 |
| Fasl | 2.438 | 0.388 | 0.022 |
| Germ1 | 2.156 | 0.377 | 0.017 |
| Hgf | 2.236 | 0.778 | 0.065 |
| Ilk | 1.079 | 0.496 | 0.806 |
| Itga3 | 1.568 | 0.153 | 0.027 |
| Itgb3 | 1.139 | 0.225 | 0.611 |
| Itgb5 | 1.515 | 0.197 | 0.015 |
| Lox | 1.554 | 0.178 | 0.036 |
| Mmp14 | 1.911 | 0.196 | 0.014 |
| Mmp3 | 2.161 | 1.172 | 0.165 |
| Myc | 4.559 | 0.739 | 0.001 |
| Serpine1 | 3.249 | 0.844 | 0.013 |
| Smad7 | 0.854 | 0.244 | 0.461 |
| Sp1 | 1.134 | 0.064 | 0.211 |
| Tgfb1 | 1.435 | 0.246 | 0.044 |
| Tgfb2 | 10.046 | 2.199 | 0.002 |
| Tgfb3 | 2.294 | 0.639 | 0.025 |
| Tgfbr2 | 1.294 | 0.271 | 0.144 |
| Tgif1 | 1.597 | 0.143 | 0.004 |
Data from a PCR array measuring transcripts associated with fibrotic response at 3 months, comparing hearts isolated from nontransgenic (Ntg) and mutant αB‐crystallin CryABR120G mice (n=3). Fold induction is expressed as mean±SD, and P values were calculated using the Student t test. The Ntg control group was arbitrarily set to 1.
The array data pointed to upregulation of TGF‐β signaling early in the fibrotic processes in CryABR120G hearts. To confirm these data and understand whether the canonical and noncanonical pathways were involved, we determined levels of representative downstream targets of TGF‐β signaling in hearts. Western blots were carried out using protein isolated from left ventricles of 3‐month‐old hearts, and we found that both the canonical SMAD‐dependent (p‐SMAD3) and noncanonical SMAD‐independent, p‐TAK1 and p‐p38 pathways were activated in CryABR120G hearts (Figures 5B through 5E and 6). At 3 months, we measured a 5‐fold increase in the ratio of p‐SMAD3 to SMAD3 (P<0.001). p‐TAK1 was similarly increased compared with TAK1, with a ≈2‐fold increase in the ratio of p‐TAK1 to TAK1 (P=0.026) and 1.58‐fold increase in the ratio of p‐p38 to p38 (P=0.012).
Figure 6.
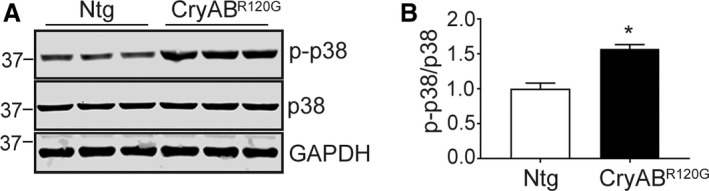
Noncanonical TGF‐β signaling in CryABR 120G hearts. A, Expression of phospho (p)‐p38 (p‐p38), p38, and the loading control GAPDH from 3‐month‐old nontransgenic (Ntg; n=5) and mutant αB‐crystallin (CryABR 120G; n=6) hearts. B, Quantification of expression of p‐p38 to p38 ratio in Ntg and CryABR 120G hearts. Values were normalized by setting the p‐p38/p38 expression ratio=1 for the Ntg hearts. The Student unpaired t test was used to compare groups. *P<0.05 vs Ntg control. GAPDH indicates glyceraldehyde 3‐phosphate.
Immunohistochemistry was used to determine the cellular distribution and location of activated p‐SMAD2/3 in CryABR120G hearts. p‐SMAD2/3 nuclear localization indicating its activation was detected in multiple cell types, including cardiac fibroblasts and myocytes (Figure 7A). Quantitation of p‐SMAD2/3–positive nuclei in individual cells of sections from Ntg and CryABR120G hearts showed a 5‐fold increase in CryABR120G cardiomyocytes (P<0.001) and a ≈4‐fold increase in noncardiomyocytes (P<0.001; Figure 7B). In order to detect activation of the signaling pathways that promote fibrosis in response to cardiac proteotoxicity, we first assessed RNA expression of profibrotic genes by quantitative PCR in isolated cardiomyocytes and noncardiomyocytes from 12‐week‐old Ntg and CryABR120G hearts. Transcripts encoding TGF‐β cytokines 2 and 3 and ECM protein RNAs, including collagen type I alpha 2 chain (Col1a2) and Ctgf, were elevated in noncardiomyocytes compared with cardiomyocytes (n=6; Table 2). Based upon these 2 results, we conclude that a model of chronic sarcomere‐based, desmin‐related cardiomyopathy elicits a characteristic fibrotic response in fibroblasts.
Figure 7.
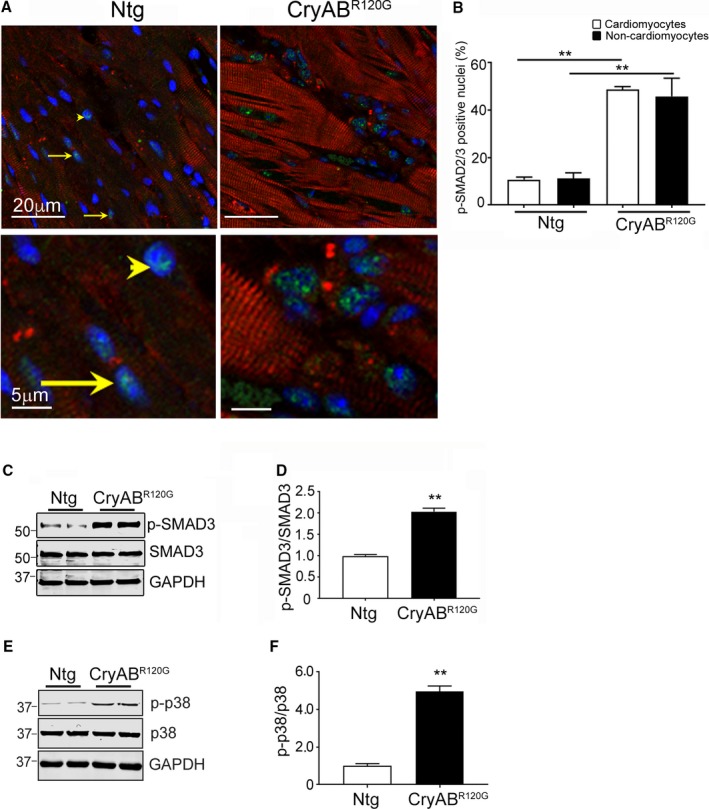
TGF‐β signaling in mutant αB‐crystallin (CryABR 120G) hearts. A, Phospho (p)‐SMAD2/3 (green) in cardiac tissue sections from 3‐month‐old nontransgenic (Ntg) and CryABR 120G hearts. Cardiomyocytes are labeled with troponin I (red) and nuclei with 4′,6‐diamidino‐2‐phenylindole, dihydrochloride (DAPI; blue). Representative p‐SMAD2/3–positive nuclei are denoted in cardiomyocytes by an arrowhead and in noncardiomyocyte nuclei by arrows. Bottom panels are enlarged areas from the top panels showing the nuclear distribution of the p‐SMAD2/3 protein. B, Activation of p‐SMAD2/3 signaling was quantified by counting p‐SMAD2/3–positive nuclei in myocyte and nonmyocyte cells (n=7 independently prepared sections for each cohort). C and D, Expression and quantification of p‐SMAD2/3 and E and F, phospho (p)‐p38 in purified fibroblasts of 3‐month‐old Ntg and CryABR 120G hearts (n=4). The Student unpaired t test was used to compare groups. **P<0.001 vs Ntg control. GAPDH indicates glyceraldehyde 3‐phosphate; SMAD3, small mothers against decapentaplegic homolog 3; TGF‐β, transforming growth factor beta.
Table 2.
Profibrotic Gene Transcripts
| Gene | Cardiomyocytes | Fibroblasts | ||||
|---|---|---|---|---|---|---|
| Fold Change | SD | P Value | Fold Change | SD | P Value | |
| Acta2 | 1.504 | 0.373 | 0.512 | 1.127 | 0.208 | 0.781 |
| Ctgf | 11.590 | 3.884 | 0.023 | 44.797 | 11.322 | 0.033 |
| Col1a2 | 12.688 | 4.771 | 0.036 | 21.698 | 5.612 | 0.041 |
| Col3a1 | 10.202 | 3.963 | 0.035 | 52.169 | 23.548 | 0.156 |
| Tgfb1 | 2.757 | 0.373 | 0.326 | 2.429 | 0.748 | 0.261 |
| Tgfb2 | 4.525 | 1.699 | 0.219 | 19.540 | 6.669 | 0.042 |
| Tgfb3 | 3.421 | 1.741 | 0.381 | 25.992 | 8.323 | 0.031 |
Data from quantitative PCR measuring profibrotic genes at 3 months using isolated cardiomyocytes and fibroblasts from nontransgenic (Ntg) and mutant αB‐crystallin CryABR120G mice (n=6). Fold induction is expressed as mean±SD.
To further delineate canonical and noncanonical TGF‐β signaling in fibroblasts/myofibroblasts, the purified fibroblast fraction (Methods) was analyzed using western blots. An ≈2‐fold increase in the ratio of p‐SMAD3/SMAD3 (P<0.001) was detected in cells isolated from CryABR120G‐expressing hearts (Figure 7C and 7D). Similarly, the p‐p38/p38 ratio was also increased by ≈4‐fold (Figure 7E and 7F), indicating at least the initial activation of TGF‐β noncanonical pathway signaling (Figure 6), consistent with elevated p‐TAK levels observed (Figure 5D and 5E).
Myofibroblast‐Specific Ablation of TGF‐βr2
Cardiomyocyte‐restricted CryABR120G mice showed increased fibrosis and induction of TGF‐β signaling and therefore constituted a suitable model for exploring the role of myofibroblast and pathologic TGF‐β signaling during the progression and course of cardiomyocyte‐driven proteotoxic cardiac disease. To test our primary hypothesis, we generated triple Tg mice in which mice carrying the α‐myosin heavy chain locus driving cardiomyocyte‐specific expression of CryABR120G were crossed into mice containing a TAM‐inducible Cre driven by the myoblast‐specific periostin promoter (Postnmcm).15 Those animals were subsequently crossed into mice containing a floxed Tgfbr2 (Figure 8A). These animals allowed us to inducibly ablate Tgfbr2 specifically in activated fibroblasts at different developmental times. To understand the efficiency of the ablation, we isolated fibroblasts (Methods) from 5‐month‐old hearts, which were fed chow with or without TAM (Figure 8B). Production of the protein in Tgfbr2‐null fibroblasts decreased by 75% (Figure 8C and 8D). We then measured p‐SMAD3 protein expression in the isolated fibroblasts using cells derived from hearts where Tgfbr2 ablation was induced 6 weeks before euthanasia. Western blot results indicated downregulation of p‐SMAD3 (Figure 8E and 8F) and p‐p38 (Figure 8G and 8H) when compared with fibroblasts isolated from uninduced (no TAM) hearts. There was no change in TGF‐βr1 mRNA expression (Figure 9A). To confirm the cell‐type specificity of ablation, we also looked at TGF‐βr1 and TGF‐βr2 transcription in cardiomyocytes from these animals and confirmed that expression levels were identical to those observed in noninduced hearts (Figure 9B and 9C).
Figure 8.
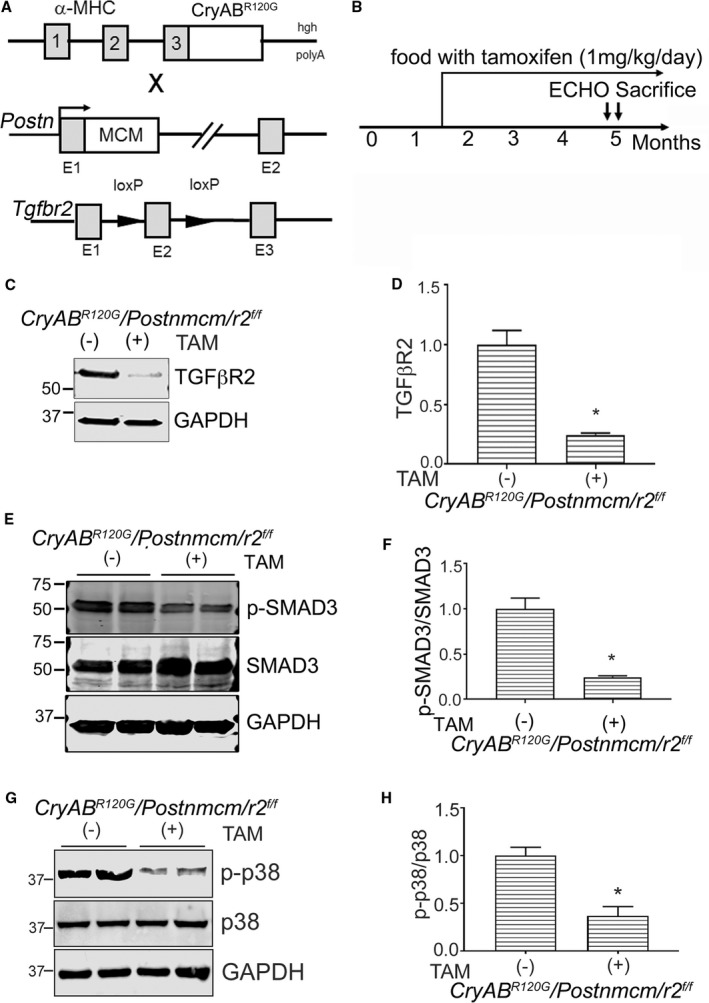
Generation of myofibroblast specific transforming growth factor β receptor II gene (Tgfbr2) (r2)–ablated mice. A, Schematic representation of the different mouse lines used. Mice with cardiomyocyte‐specific mutant α‐B‐crystallin expression (CryABR 120G) were crossed with Tgfbr2‐loxP–containing gene targeted lines and those double transgenic (Tg) lines further crossed with mice containing the periostin locus (Postn) with a tamoxifen (TAM) regulated Mer‐Cre‐Mer cassette (Postnmcm) inserted into exon (E1). B, Experimental scheme where mice were fed a TAM diet starting from 6 to 8 weeks until they were euthanitized. C and D, Western blot and quantification for Tgfβr2 protein in purified fibroblasts (n=6 separate, total hearts). E and F, Expression and quantification of SMAD3 (phospho [p]‐SMAD3/SMAD3) and (G and H) p38 signaling (p‐p38/p38) in purified fibroblasts isolated from hearts of 3‐month‐old experimental cohorts fed with TAM chow (TAM+) or normal chow (−) (n=4–6 separate, total hearts). *P<0.05 vs uninduced Cre Cry ABR 120G /Postnmcm/r2 f/f animals. GAPDH indicates glyceraldehyde 3‐phosphate; α‐MHC, α‐myosin heavy chain; SMAD3, small mothers against decapentaplegic homolog 3.
Figure 9.
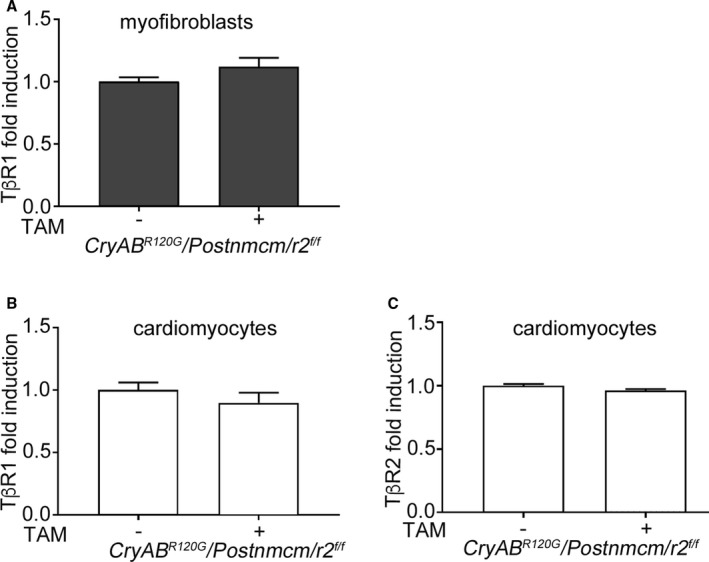
Quantitative PCR analyses of transforming growth factor β (TGF‐β) receptor 1 and 2 gene expression. A, TGF‐β receptor I (TβRI) mRNA in cardiac myofibroblasts isolated from Cre‐induced and ‐uninduced Cry ABR 120G /Postnmcm/r2 f/f mice. B and C, TβRI and TGF‐β receptor II (TβRII) mRNA in cardiomyocytes isolated from Cre‐induced (TAM+) and noninduced (−) mice, respectively (n=4). Values for the Cry ABR 120G /Postnmcm/r2 f/f control group (no tamoxifen [TAM]) was arbitrarily set to 1. The Student unpaired t test was used to compare the 2 groups.
Myofibroblast‐Specific Ablation of TGF‐βr2 Decreases Fibrosis in CryABR120G hearts
Tgfbr2 ablation was carried out early in the fibrotic process at 6 weeks (Figure 8B) and hearts analyzed 3 months later. Masson's trichome and Sirius Red staining detected abundant fibrosis in noninduced CryAB R120G /Postnmcm/r2 f/f triple Tg mice (Figure 10A), consistent with the fibrosis observed in CryABR120G Tg hearts. However, myofibroblast ablation of Tgfbr2 significantly blunted the overall cardiac fibrotic response (Figure 10A). Quantitation of fibrosis as measured by Sirius Red staining of sections derived from the left ventricles of induced triple Tg hearts showed a significant decrease (Figure 10B). After aging the experimental cohorts another 2 months, we then determined the hydroxyproline content of these ventricles and measured a decrease of >40% in the ablated samples as compared with hearts derived from noninduced triple Tg hearts (Figure 10C).
Figure 10.
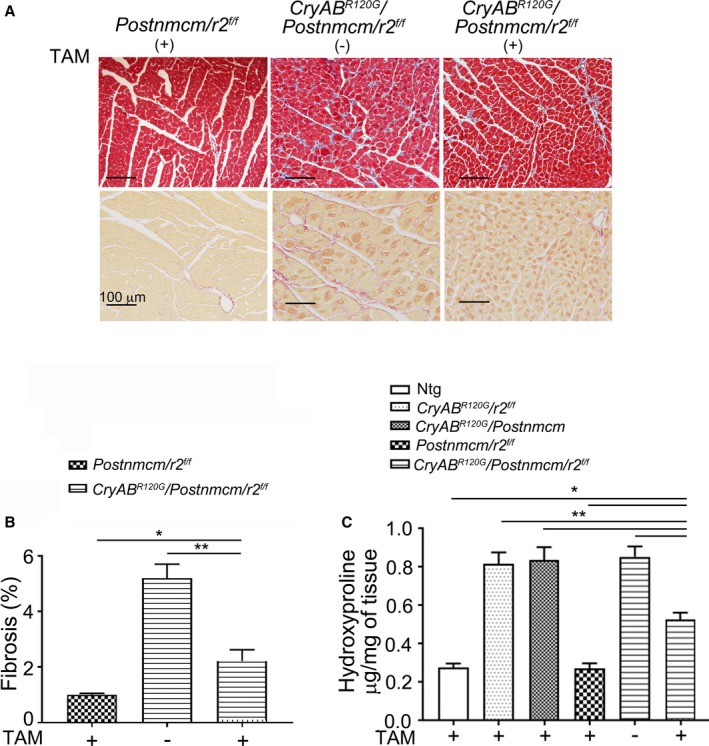
Fibrosis in myofibroblast specific transforming growth factor β receptor II gene (Tgfbr2) (r2)–ablated mutant αB‐crystallin (CryABR 120G) mice. A, Masson's trichrome and Sirius Red staining of left ventricle sections from 20‐week‐old Cry ABR 120G /Postnmcm/Tgfbr2 f/f (r2) fed with TAM chow (TAM+) or normal chow (−). B, Quantification of fibrosis from the images obtained from Sirius Red–stained sections (n=4–5). C, Hydroxyproline in hearts derived from nontransgenic (Ntg), Cry ABR 120G /Postnmcm, Cry ABR 120G /r2 f/f , Postnmcm/r2 f/f and Cry ABR 120G /Postnmcm/r2 f/f fed with TAM+ or normal chow (−) as in (A) and aged for 5 months (n=5–6), *P<0.05 vs TAM+ Cry ABR 120G /Postnmcm/r2 f/f; **P<0.001 vs TAM+ Cry ABR 120G /Postnmcm/r2 f/f.
To further define fibrotic response in these mice, we measured α‐SMA and periostin protein content (Figure 11). Western blot analyses using protein isolated from the left ventricles of 5‐month‐old hearts showed significantly decreased levels of both α‐SMA (Figure 11A and 11C) and periostin (Figures 10B and 11D) in Tgfbr2 −/− myofibroblast hearts, relative to the noninduced controls. In the absence of inducing Tgfbr2 ablation, the presence of the Postnmcm locus had no effect on the fibrotic response that was induced by CryABR120G expression.
Figure 11.
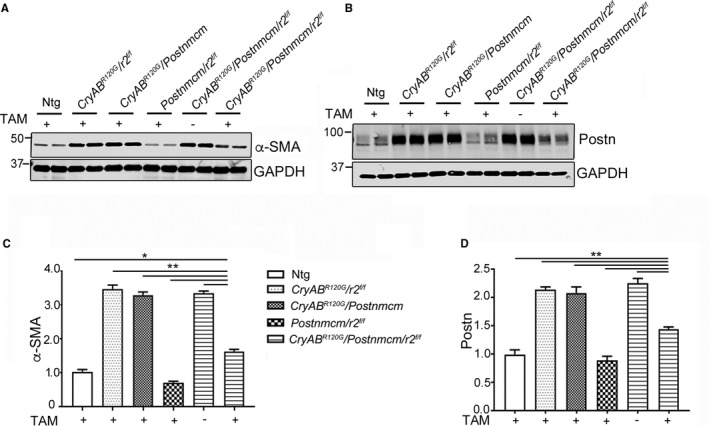
Fibrotic marker expression. A and B, Western blots of the fibrosis markers α‐smooth muscle actin (α‐SMA) and periostin (Postn), respectively, using protein isolated from 5‐month‐old left ventricles derived from the indicated experimental cohorts fed with tamoxifen (TAM)+ chow or normal chow (−) (n=4). C, Quantitative analysis of the western blots in (A) and (D) in (B), respectively, using 1‐way ANOVA followed by Tukey's post hoc test. Values were normalized using GAPDH to correct for loading variation (n=4). *P<0.05; **P<0.001 vs TAM+ Cry ABR 120G /Postnmcm/r2 f/f in pair‐wise comparisons between the indicated groups. GAPDH indicates glyceraldehyde 3‐phosphate; Ntg, nontransgenic; Postn, periostin locus.
Hypertrophy Is Decreased in the Myofibroblast‐Tgfbr2 −/− CryABR120G Hearts
As previously documented, cardiomyocyte‐specific expression of CryABR120G leads to protein aggregate formation that increases as the animals age.24 Hypertrophy is observed as early as 12 weeks in CryABR120G hearts. The myofibroblast‐restricted Tgfbr2 −/− transgene crossed into the CryABR120G line, thereby provides a unique opportunity to parse out the relative contributions of cardiomyocyte autonomous aggregate formation from the role that myofibroblast‐dependent TGF‐β signaling and subsequent fibrosis plays in the hypertrophic response. There was a striking reduction in degree of hypertrophy apparent in the intact myofibroblast‐Tgfbr2 −/− hearts compared with the noninduced controls at 5 months (Figure 12A). Whereas body weights did not differ between any of the experimental cohorts, heart weight to body weight/ratios were significantly reduced in Tgfbr2 −/− hearts (Figure 12B through 12D). Wheat germ agglutinin staining of sections derived from the left ventricles was consistent with a significant reduction in cardiomyocyte cross‐section area (Figure 12E and 12F). We analyzed expression of the hypertrophic molecular markers, β‐myosin heavy chain and natriuretic peptide precursor A and B, using RNA isolated from the ventricles and observed significant decreases in transcript levels in myofibroblast‐Tgfbr2 −/−–derived material (Figure 12G through 12I). As expected, aggregate formation in the CryABR120G model was unaffected by myoblast‐specific Tgfbr2 ablation (Figure 13A). No significant differences in ratios of aggregate area/myocyte area were observed in comparisons between the Cre‐induced or noninduced triple Tg hearts (data not shown), and proteotoxic response in terms of aggregate accumulation and insoluble and soluble CryAB accumulation were all identical between the cohorts (Figure 13B through 13E). These data point to the critical role that fibrosis itself plays in hypertrophic response when the heart is challenged by a proteotoxic stimulus.
Figure 12.
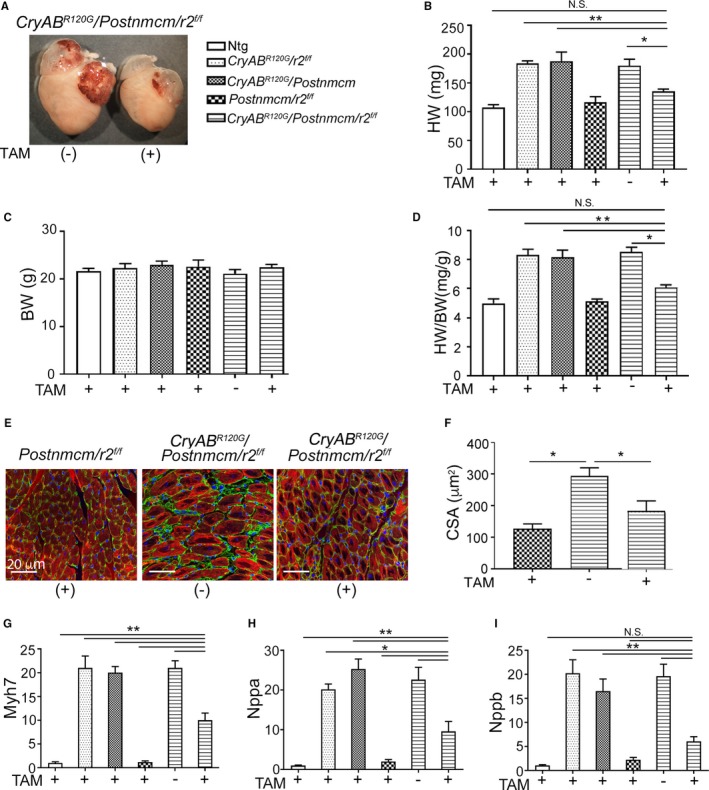
Transforming growth factor β receptor II gene (Tgfbr2) (r2) ablation in myofibroblasts prevents cardiomyocyte hypertrophy. A, Mutant αB‐crystallin gene (Cry ABR 120G) hearts with and without myofibroblast specific Tgfbr2 ablation at 5 months. B through D, Quantification of heart weights (HW), body weights (BW), and the heart weight to body weight ratios (HW/BW; n=6). E, Fluorescent wheat germ agglutinin–stained images (green) from mouse heart sections. Cardiomyocytes are labeled with troponin I (red) and nuclei by DAPI (blue). F, Assessment of cross‐section area (CSA) of cardiomyocytes from (C) in the order shown in (C; n=5). G, H and I, RNA levels of cardiac hypertrophic molecular markers β‐myosin heavy chain (Myh7), natriuretic peptide precursor A (Nppa) and B (Nppb), respectively in ventricles derived from the indicated cohorts fed with TAM+ chow or normal chow (−). (n=6). *P<0.05; **P<0.001 vs TAM+ Cry ABR 120G /Postnmcm/r2 f/f in pair‐wise comparisons between the indicated groups using 1‐way ANOVA followed by Tukey's post hoc test. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; NS, not significant; Ntg, nontransgenic.
Figure 13.
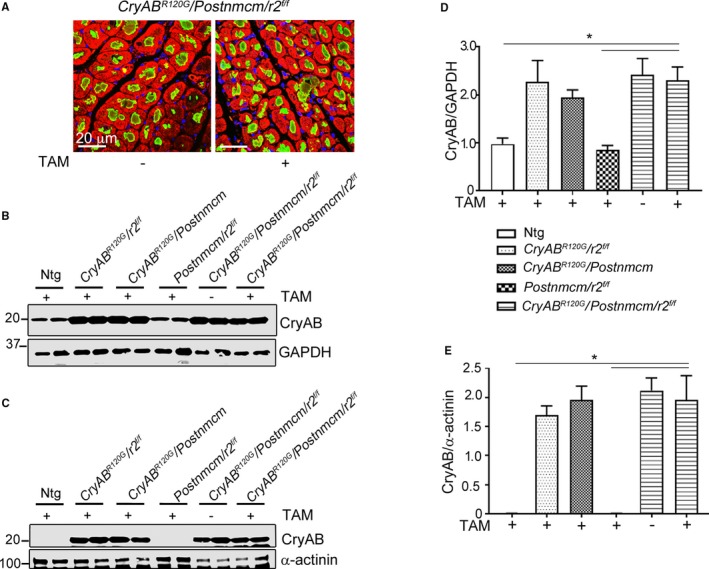
Ablation of transforming growth factor β receptor II gene Tgfbr2 (r2) in myofibroblasts does not alter misfolded protein accumulation in mutant αB‐crystallin (CryABR 120G) hearts. A, Immunofluorescent staining of CryAB (green) with α‐actinin (red) shows no difference in aggregate accumulations in cardiomyocytes derived from Cry ABR 120G /Postnmcm/r2 f/f hearts with (+) or without (−) r2 ablation in the myofibroblasts (n=4). B and C, Western blot showing CryAB protein levels present in soluble and insoluble (aggregate‐containing) fractions. D and E, Quantitation of CryAB protein expression in soluble and insoluble fractions derived from hearts of the different experimental cohorts as indicated. CryAB was not detected in fractions derived from animals not expressing the CryABR 120G. Comparisons were made using 1‐way ANOVA followed by Tukey's post hoc test (n=4). *P<0.05 vs Cre induced Cry ABR 120G /Postnmcm/r2 f/f. GAPDH indicates glyceraldehyde 3‐phosphate; Ntg, nontransgenic.
Cardiac Function and Survival in Myofibroblast‐Tgfbr2 −/− CryABR120G Hearts
Cardiac parameters were measured using standard echocardiographic procedures with 5‐month‐old animals.20, 29 As expected, LV mass in Cre noninduced double or triple Tg mice was significantly increased compared with controls, which included Ntg as well as double Tg mice lacking the CryABR120G transgene. LV mass was improved in Cre‐induced triple TG mice, with values approaching those observed in Ntg animals (Figure 14A). Myofibroblast‐specific TGF‐βr2 ablated CryAB mice showed significantly higher LV systolic function relative to Cre uninduced triple Tg mice, with preserved fractional shortening (Figure 14B). Myofibroblast ablation of TGF‐βr2 resulted in improved measures of diastolic function as well, reflected in improvement in left atrial dilation and the ventricular filling velocity (MV E/A) ratio in Cre‐induced triple Tg mice (Figure 14C and 14D). Survival probabilities in the induced, ablated animals were also significantly improved (Figure 14E). However, myofibroblast‐TGF‐βr2 −/− animals did not have a normal life span, which is not surprising considering the cell‐autonomous effects of CryABR120G expression. That is, the primary proteotoxic stimulus, expression of CryABR120G and intracardiomyocyte aggregate accumulation, was unaffected (Figure 13). However, the data do show the relative contribution of cardiac fibrosis to survival in this animal model, and, in the absence of an intact fibrotic response, survival is significantly prolonged.
Figure 14.
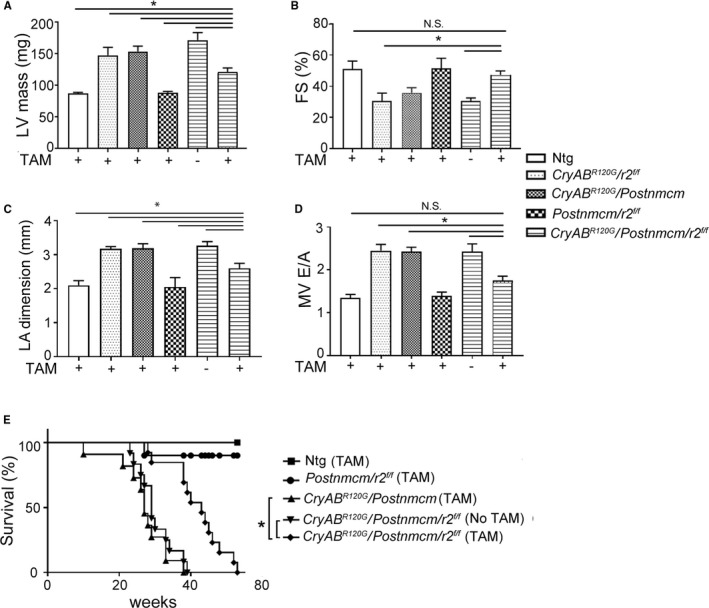
Ablation of transforming growth factor β receptor II gene Tgfbr2 (r2) in myofibroblasts preserves cardiac function and improves survival in mutant αB‐crystallin (CryABR 120G) mice. A, Left ventricular (LV) mass. B, Fractional shortening (FS) and C, Left atrial (LA) dimension and D, Ventricular filling velocity (MV E/A; n=6–9). All measurements were performed at 5 months. *P<0.05, vs tamoxifen (TAM)+ Cry ABR 120G /Postnmcm/r2 f/f in pair‐wise comparisons between the indicated groups using 1‐way ANOVA followed by Tukey's post hoc test. NS indicates not significant. E, Kaplan–Meier survival curves of the CryABR 120G mice with and without myofibroblast Tgfbr2 ablation. n=10 to 12. *P<0.05, vs comparisons between the curves using the log‐rank test. Ntg indicates nontransgenic.
Discussion
The data show that myofibroblast‐based TGF‐β signaling plays a crucial role for the progression of fibrosis that occurs as a result of a proteotoxic stimulus over a period of weeks or even months, mimicking a chronic insult that results in human cardiac disease and heart failure. Strikingly, even though the proteotoxic insult was only cardiomyocyte based, myofibroblast‐specific ablation of Tgfbr2 effectively decreased fibrosis over a period of months, resulting in decreased cardiomyocyte hypertrophy, conserved cardiac function, and increased survival probability over time.
TGF‐β signaling has been extensively studied in cardiac disease, although a majority of studies used acute, surgically induced myocardial infarction,13, 30 pressure overload–induced hypertrophy,11, 12 general inflammatory stimuli, or, very recently, a genetic model of protein sarcomeric mutation‐induced hypertrophic cardiomyopathy.14 We chose to ablate Tgfbr2 to test the hypothesis that both canonical and noncanonical signaling might be affected given that both are upregulated in myofibroblasts isolated from CryABR120G hearts. Fibroblast‐specific ablation of TGF‐β downstream signaling (SMAD3 or p38) decreases pathogenic fibrosis in either a pressure overload model or during ischemic injury, respectively.12, 30 Similarly, ablatingTgfbr1/2 in myofibroblasts preserved cardiac function in the pressure overload model.12 We were interested in determining whether these data could be extended to a chronic, long‐term genetically induced disease process. CryABR120G, when expressed specifically in cardiomyocytes, leads to a generalized deficit in protein conformation that triggers multiple, cardiomyocyte‐based pathological processes, resulting in cardiomyocyte dropout and extensive fibrosis.18, 31, 32 In this study, we were interested in understanding whether manipulating myofibroblast activation in this cardiomyocyte‐based proteotoxic disease setting could impact on disease pathogenesis. Here, we tested the hypothesis that the resultant fibrosis is predominantly fibroblast based in proteotoxic cardiac disease.
TGF‐β signaling is remarkably diverse, affecting development, morphogenesis, cell survival, growth, and proliferation.33 Given that the peptide can act in either a paracrine or autocrine manner, communication among cells can be critical to determining the net response to its activation or suppression.34 Teekakirikul et al showed that only an ≈3% reduction in interstitial fibrosis preserved cardiac function and hypertrophy in losartan‐treated mice with hypertrophic cardiomyopathy.14 Similarly, a modest decrease in interstitial fibrosis (from ≈8% to ≈5%) also reduced hypertrophy and preserved cardiac function during transverse aortic constriction–induced heart failure in mice hearts.12 In absolute terms, the area of staining decreased from a value of ≈5.5% to 2%, representing a decrease of >50% in fibrotic deposition as measured by Sirius Red staining (Figure 10).
Seidman et al have shown that sarcomeric‐based mutations can signal fibrotic processes in the noncardiomyocyte population, with TGF‐β signaling playing a necessary role.14 Two mouse models of hypertrophic cardiomyopathy caused by 2 different mutations in the α‐myosin heavy chain gene pointed to the importance of nonmyocyte TGF‐β signaling in fibrotic response.14 Genetic ablation of the periostin gene in mice reduced, but did not extinguish, nonmyocyte proliferation and fibrosis. In contrast, administration of TGF‐β‐neutralizing antibodies abrogated nonmyocyte proliferation and fibrosis. Those data showed nonmyocyte activation of TGF‐β signaling as a pivotal mechanism for increased fibrosis in hypertrophic cardiomyopathy and a potentially important factor contributing to diastolic dysfunction and heart failure. Our data extend and build upon these observations, identifying the myofibroblast as a critical target cell for ablating the fibrotic response that occurs in response to accumulation of cardiac protein aggregates, with subsequent benefits in terms of improved parameters detectable by echocardiography. Although relatively insensitive in some respects compared with magnetic resonance imaging analyses and invasive catheterization techniques, ultrasound was able to detect significant improvements in LV systolic and diastolic function. The ultimate outcome of these changes was increased probability of survival in the context of a generalized, proteotoxic response.
Our studies do not directly differentiate if resident or newly recruited fibroblasts become activated and Cre production is initiated under the prolonged experimental conditions. However, we continued feeding TAM‐supplemented chow to the experimental cohort, and, so, presumably both the resident fibroblasts as well as any additional cells recruited to the fibroblast lineage and subsequently differentiated to myofibroblasts activated periostin synthesis and Cre production. The data obtained using purified fibroblast populations did confirm the efficacy of Tgfbr2 ablation in that cell population.
Although cardiac fibrosis was decreased by almost half by Tgfbr2 ablation, we cannot ascribe the entire beneficial effects observed in morbidity and mortality to fibrosis alone.
Considering the pleiotropic actions of TGF‐β signaling, there are potentially many pathways that could be affected and, in turn, affect the disease processes. Both canonical and noncanonical TGF‐β downstream signaling were profoundly decreased in the myofibroblasts of Cre‐induced CryAB R120G /Postn/r2 hearts. Apart from this, transcriptional analyses showed decreased expression of the cardiac hypertrophy biomarkers, natriuretic peptide precursor A, natriuretic peptide precursor B, and myosin heavy chain 7. Both decreased TGF‐β signaling and its impact on multiple pathways resulting in decreased hypertrophic gene transcription, among others, would contribute to decreased morbidity and improved survival.
Given that protein conformation‐based disease is becoming a widely recognized contributing pathology to cardiac disease,17, 35 a comprehensive and mechanistic study of the fibrosis that accompanies and impacts on the developing heart disease is warranted. Cardiomyocyte expression of CryABR120G is a primary proteotoxic stimulus resulting in the disease, desmin‐related cardiomyopathy,18, 24 and thus provides an opportunity for a comprehensive and mechanistic study of the fibrosis that accompanies and impacts the developing heart disease. After confirming activation of TGF‐β signaling as a result of CryABR120G expression, we assessed the pathway's involvement, but in a myofibroblast‐autonomous manner. We observed no detrimental effects of continuous TAM feeding in the absence of the inducible transgene in control experimental cohorts. We are thus able to ascribe the effects specifically to expression of CryABR120G and myofibroblast Tgfbr2 gene ablation driven by activation of the periostin promoter, subsequent Cre expression, and Tgfbr2 excision. We found that if TGF‐β signaling is ablated in this cell population, cardiac fibrosis is significantly blunted, with a concomitant preservation of cardiac function and architecture.
Ablation of TGF‐β signaling in the cardiac myofibroblast had no effect on the cardiomyocyte‐autonomous pathogenic process of aggregate formation (Figure 13A), which eventually led to death by mechanisms potentially related to heart failure. The data do show, however, the relative roles that fibrosis and hypertrophy play in contributing to the overall probability of survival in this model given that, in the absence of those processes but with the primary disease etiology unaffected, the survival time was almost doubled in the face of chronic CryABR120G production. These new data that bear on the cellular basis for cardiac fibrosis point to the potential of therapies targeting myofibroblast activity in cardiac disease. Such a therapeutic, translational approach might be effective even when the primary disease stimulus, in this case, proteotoxicity and protein aggregation attributed to CryABR120G expression, is restricted to the cardiomyocyte.
Sources of Funding
Funding sources include NIH‐P01HL69779, NIH‐P01HL059408, NIH‐R01HL05924, NIH‐R011062927 and a Trans‐Atlantic Network of Excellence grant from Le Fondation Leducq (Grant # 11 CVD) as well American Heart Association Postdoctoral Fellowship grants to Bhandary (16POST30710005), Meng (17POST33670035) and Valiente Alandi (16POST30180015).
Disclosures
None.
(J Am Heart Assoc. 2018;7:e010013 DOI: 10.1161/JAHA.118.010013)
References
- 1. Mortality GBD, Causes of Death C . Global, regional, and national life expectancy, all‐cause mortality, and cause‐specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1459–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gourdie RG, Dimmeler S, Kohl P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat Rev Drug Discov. 2016;15:620–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC. Myofibroblast‐mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol. 2013;10:15–26. [DOI] [PubMed] [Google Scholar]
- 4. Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, Sjostrom SL, Szewczykowska M, Jackowska T, Dos Remedios C, Malm T, Andra M, Jashari R, Nyengaard JR, Possnert G, Jovinge S, Druid H, Frisen J. Dynamics of cell generation and turnover in the human heart. Cell. 2015;161:1566–1575. [DOI] [PubMed] [Google Scholar]
- 5. Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D'Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD. Revisiting cardiac cellular composition. Circ Res. 2016;118:400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Robert L. [The fibroblast, definition of its phenotype by its “programme” of biosynthesis of the extracellular matrix]. Pathol Biol (Paris). 1992;40:851–858. [PubMed] [Google Scholar]
- 7. Bujak M, Frangogiannis NG. The role of TGF‐beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)‐beta signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gupta MK, Gulick J, Liu R, Wang X, Molkentin JD, Robbins J. Sumo E2 enzyme UBC9 is required for efficient protein quality control in cardiomyocytes. Circ Res. 2014;115:721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maloyan A, Osinska H, Lammerding J, Lee RT, Cingolani OH, Kass DA, Lorenz JN, Robbins J. Biochemical and mechanical dysfunction in a mouse model of desmin‐related myopathy. Circ Res. 2009;104:1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, Zhang D, Nakamura T, Takimoto E, Kass DA. Pivotal role of cardiomyocyte TGF‐beta signaling in the murine pathological response to sustained pressure overload. J Clin Invest. 2011;121:2301–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, Karch J, Molkentin JD. Fibroblast‐specific TGF‐beta‐Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127:3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A, Conway SJ, Graff JM, Frangogiannis NG. Opposing actions of fibroblast and cardiomyocyte Smad3 signaling in the infarcted myocardium. Circulation. 2018;137:707–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, Kim JB, Schmitt JP, Molkentin JD, Norris RA, Tager AM, Hoffman SR, Markwald RR, Seidman CE, Seidman JG. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non‐myocyte proliferation and requires TGF‐beta. J Clin Invest. 2010;120:3520–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, Lin SCJ, Aronow BJ, Tallquist MD, Molkentin JD. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Travers JG, Kamal FA, Valiente‐Alandi I, Nieman ML, Sargent MA, Lorenz JN, Molkentin JD, Blaxall BC. Pharmacological and activated fibroblast targeting of Gbetagamma‐GRK2 after myocardial ischemia attenuates heart failure progression. J Am Coll Cardiol. 2017;70:958–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, Hewett T, Robbins J. Expression of R120G‐alphaB‐crystallin causes aberrant desmin and alphaB‐crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. [DOI] [PubMed] [Google Scholar]
- 19. Ishtiaq Ahmed AS, Bose GC, Huang L, Azhar M. Generation of mice carrying a knockout‐first and conditional‐ready allele of transforming growth factor beta2 gene. Genesis. 2014;52:817–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bhuiyan MS, McLendon P, James J, Osinska H, Gulick J, Bhandary B, Lorenz JN, Robbins J. In vivo definition of cardiac myosin‐binding protein C's critical interactions with myosin. Pflugers Arch. 2016;468:1685–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gupta MK, McLendon PM, Gulick J, James J, Khalili K, Robbins J. UBC9‐mediated sumoylation favorably impacts cardiac function in compromised hearts. Circ Res. 2016;118:1894–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Accornero F, van Berlo JH, Correll RN, Elrod JW, Sargent MA, York A, Rabinowitz JE, Leask A, Molkentin JD. Genetic analysis of connective tissue growth factor as an effector of transforming growth factor beta signaling and cardiac remodeling. Mol Cell Biol. 2015;35:2154–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu N, Bitan G, Schrader T, Klärner F‐G, Osinska H, Robbins J. Inhibition of mutant alphaB crystallin‐induced protein aggregation by a molecular tweezer. J Am Heart Assoc. 2017;6:e006182 DOI: 10.1161/JAHA.117.006182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sanbe A, Osinska H, Villa C, Gulick J, Klevitsky R, Glabe CG, Kayed R, Robbins J. Reversal of amyloid‐induced heart disease in desmin‐related cardiomyopathy. Proc Natl Acad Sci USA. 2005;102:13592–13597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ranek MJ, Terpstra EJ, Li J, Kass DA, Wang X. Protein kinase G positively regulates proteasome‐mediated degradation of misfolded proteins. Circulation. 2013;128:365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ponticos M, Harvey C, Ikeda T, Abraham D, Bou‐Gharios G. JunB mediates enhancer/promoter activity of COL1A2 following TGF‐beta induction. Nucleic Acids Res. 2009;37:5378–5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frangogiannis NG. Chemokines in the ischemic myocardium: from inflammation to fibrosis. Inflamm Res. 2004;53:585–595. [DOI] [PubMed] [Google Scholar]
- 28. Keeling J, Herrera GA. Matrix metalloproteinases and mesangial remodeling in light chain‐related glomerular damage. Kidney Int. 2005;68:1590–1603. [DOI] [PubMed] [Google Scholar]
- 29. Meng Q, Bhandary B, Osinska H, James J, Xu N, Shay‐Winkler K, Gulick J, Willis MS, Lander C, Robbins J. MMI‐0100 inhibits cardiac fibrosis in a mouse model overexpressing cardiac myosin binding protein C. J Am Heart Assoc. 2017;6:e006590 DOI: 10.1161/JAHA.117.006590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, Gunaje J, Otsu K, Davis J. Fibroblast‐specific genetic manipulation of p38 mitogen‐activated protein kinase in vivo reveals its central regulatory role in fibrosis. Circulation. 2017;136:549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maloyan A, Robbins J. Autophagy in desmin‐related cardiomyopathy: thoughts at the halfway point. Autophagy. 2010;6:665–666. [DOI] [PubMed] [Google Scholar]
- 32. Sanbe A, Osinska H, Saffitz JE, Glabe CG, Kayed R, Maloyan A, Robbins J. Desmin‐related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc Natl Acad Sci USA. 2004;101:10132–10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Frangogiannis NG. Targeting the transforming growth factor (TGF)‐beta cascade in the remodeling heart: benefits and perils. J Mol Cell Cardiol. 2014;76:169–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71:549–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Terrell D, Robbins J. Protein conformation‐based disease: getting to the heart of the matter. Annu Rev Physiol. 2010;72:15–17. [DOI] [PubMed] [Google Scholar]