

Abstract
Background
The Sigma 1 receptor (Sigmar1) functions as an interorganelle signaling molecule and elicits cytoprotective functions. The presence of Sigmar1 in the heart was first reported on the basis of a ligand‐binding assay, and all studies to date have been limited to pharmacological approaches using less‐selective ligands for Sigmar1. However, the physiological function of cardiac Sigmar1 remains unknown. We investigated the physiological function of Sigmar1 in regulating cardiac hemodynamics using the Sigmar1 knockout mouse (Sigmar1−/−).
Methods and Results
Sigmar1−/− hearts at 3 to 4 months of age showed significantly increased contractility as assessed by left ventricular catheterization with stimulation by increasing doses of a β1‐adrenoceptor agonist. Noninvasive echocardiographic measurements were also used to measure cardiac function over time, and the data showed the development of cardiac contractile dysfunction in Sigmar1−/− hearts as the animals aged. Histochemistry demonstrated significant cardiac fibrosis, collagen deposition, and increased periostin in the Sigmar1−/− hearts compared with wild‐type hearts. Ultrastructural analysis of Sigmar1−/− cardiomyocytes revealed an irregularly shaped, highly fused mitochondrial network with abnormal cristae. Mitochondrial size was larger in Sigmar1−/− hearts, resulting in decreased numbers of mitochondria per microscopic field. In addition, Sigmar1−/− hearts showed altered expression of mitochondrial dynamics regulatory proteins. Real‐time oxygen consumption rates in isolated mitochondria showed reduced respiratory function in Sigmar1−/− hearts compared with wild‐type hearts.
Conclusions
We demonstrate a potential function of Sigmar1 in regulating normal mitochondrial organization and size in the heart. Sigmar1 loss of function led to mitochondrial dysfunction, abnormal mitochondrial architecture, and adverse cardiac remodeling, culminating in cardiac contractile dysfunction.
Keywords: bioenergetic, cardiac contractility and energetics, cardiovascular physiology, dynamics, mitochondria, Sigma 1 receptor
Subject Categories: Basic Science Research, Myocardial Biology, Physiology, Contractile function, Cell Biology/Structural Biology
Clinical Perspective
What Is New?
The Sigma 1 receptor (Sigmar1) has emerged as an interorganelle signaling protein having multiple functions, but its physiological function remained unknown.
Here we defined the physiological function of Sigmar1 in the heart.
This study shows that loss of Sigmar1 function led to mitochondrial dysfunction and abnormal mitochondrial architecture, ultimately leading to adverse cardiac remodeling.
This study reveals a potential molecular function of Sigmar1 in regulating the mitochondrial bioenergetics that are essential to maintain normal cardiac contractile function.
What Are the Clinical Implications?
Our study is the first to show the development of cardiac contractile dysfunction and cardiac fibrosis in Sigmar1−/− mice with aging.
We also found that the Sigmar1−/− hearts develop significant accumulations of irregularly shaped mitochondria and defects in mitochondrial respiratory function.
All these data demonstrate that Sigmar1 is needed to maintain normal cardiac contractility and encourage clinical research to evaluate the potential role of Sigmar1 in cardiovascular disease.
The Sigma‐1 receptor (Sigmar1) was first proposed as an opioid receptor subtype,1 but subsequent studies showed it to be a unique intracellular protein that binds diverse classes of pharmacological compounds.2 The potential physiological importance of Sigmar1 is implied by its association with many neurodegenerative diseases.3, 4, 5, 6, 7 Recessive mutations in SIGMAR1 have been identified as a causative gene for amyotrophic lateral sclerosis with or without frontotemporal lobar degeneration,4, 8 juvenile amyotrophic lateral sclerosis,5, 9 and distal hereditary motor neuropathy.10, 11, 12, 13 Subsequent studies showed that activation of Sigmar1 is protective in a variety of conditions, including Alzheimer disease, Parkinson disease, cancer, depression, amnesia, and ischemic brain injury.3, 4, 5, 6, 7 Diverse data sets suggest that Sigmar1 may be an interorganelle signaling protein having multiple functions.14, 15, 16, 17
Extensive studies have revealed multitasking functions of Sigmar1 through physical interaction with client proteins such as inositol 1,4,5‐trisphosphate receptors,18, 19 voltage‐gated calcium,20 potassium,21 and sodium22 channels, and calcium‐activated chloride channels.23 In addition, studies showed that Sigmar1 resides at the mitochondrion‐associated endoplasmic reticulum (ER) membrane playing a role in regulating the Ca2+ signaling between ER and mitochondria in Chinese hamster ovary cells.24 Within the mitochondrion‐associated ER membrane, activated Sigmar1 appears to stabilize inositol 1,4,5‐trisphosphate receptors by protecting them from proteasomal degradation24 and enhances inositol 1,4,5‐trisphosphate–dependent calcium release from ER by promoting their dissociation from the ion channel chaperone protein ankyrin B in Michigan Cancer Foundation‐7 breast tumor cell lines.19 In contrast, Sigmar1 inhibits store‐operated Ca2+ entry by attenuating STIM1 coupling to Orai1 by and reducing the Ca2+ content of the ER stores in human embryonic kidney cells.25 A recent study also showed that preincubation of macrophages with the Sigmar1 antagonist haloperidol leads to a significant inhibition of the store‐dependent Ca2+ entry induced by endoplasmic Ca2+‐ATPase inhibitors in rat peritoneal macrophages.26 Sigmar1 activation by the agonist 1,3‐di‐o‐tolylguanidine and (+)‐pentazocine decreases intracardiac ganglion neuron excitability by modulating voltage‐gated Na+ channels.27 All these studies used in vitro cell culture experiments with Sigmar1 ligands (agonist and antagonists) to define the cellular function of Sigmar1. Because all previous studies using described Sigmar1 ligands such as fluvoxamine,28 sertraline,29 (+)pentazocine,30 and haloperidol31 involve serotonin reuptake inhibitors and also have wide affinity for other receptors,32, 33 the role of Sigmar1 in the heart remains unclear despite a wealth of published literature.
The possible presence of Sigmar1 in cardiomyocytes was initially hypothesized based on experiments using radioligand‐binding assays, with the data suggesting that cardiomyocytes exhibit binding sites for Sigmar1 ligands.34, 35 Most studies addressing Sigmar1's function in cardiomyocytes have been limited to isolated heart preparations and indirect observations using nonspecific Sigmar1 ligands.35, 36, 37, 38, 39, 40 For example, Sigmar1 ligands such as (+)‐3‐3‐(3‐hydroxyphenyl)‐N‐(1‐propyl)piperidine, (+)‐pentazocine, and haloperidol were reported to alter contractility, calcium (Ca2+) influx, and rhythmic activity in cultured cardiac myocytes.35, 36, 38 Data from various studies were consistent with the hypothesis that Sigmar1 ligands can modulate cardiomyocyte contractility.
We previously reported the distribution of Sigmar1 across different organs and noted abundant expression in the heart.41, 42 We also reported that pressure overload–induced hypertrophy significantly decreased the cardiac expression of Sigmar1 and found a significant correlation between heart dysfunction and decreased Sigmar1 expression in the left ventricle (LV).14, 42, 43, 44 Treatment with the neurosteroid dehydroepiandrosterone or with pentazocine was cardioprotective in the context of pressure overload–induced cardiac dysfunction. Sigmar1 activation by treatment with fluvoxamine or SA4503 improved cardiac function and reduced cardiac hypertrophy in a transverse aortic constriction mouse model of cardiac injury.14, 42, 43, 44, 45, 46 Moreover, Sigmar1 gene expression was upregulated in the heart by strong stress stimuli such as immobilization and hypoxia.47 Despite a wealth of published literature, the role of Sigmar1 in the heart is unclear, as these compounds are serotonin reuptake inhibitors and have extensive affinity to other receptors.32, 33
Two separate lines of Sigmar1 global gene‐ablated (Sigmar1−/−) mice have been generated by gene targeting (Oprs1tm1Lmon/Oprs1tm1Lmon)48 or gene trapping (Oprs1Gt(IRESBetageo)33Lex/Oprs1Gt(IRESBetageo)33Lex).49 With both strategies, Sigmar1 homozygous knockout lines were viable, fertile, and did not display an overt phenotype compared with wild‐type littermates, but these studies were limited to cursory observations of limited sample sizes.48, 49 To date, no studies detailing the cardiac phenotype of these Sigmar1−/− mice have been reported. In the present study using Sigmar1−/− mice, we studied the functional consequences of Sigmar1 ablation in regulating cardiac function, morphology, and ultrastructure. We also observed changes in mitochondrial dynamics and bioenergetics. Our results suggest the importance of Sigmar1 in regulating mitochondrial bioenergetics essential to maintain cardiac contractile function.
Materials and Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure. Other researchers may contact the corresponding author about the data and methodological questions.
Materials
The materials we used are the following: Cell Lytic M (Sigma‐Aldrich, St. Louis, MO), Protease Inhibitor Cocktail (Roche, Basel, Switzerland), precast 7.5% to 15% Criterion Gels (BioRad, Hercules, CA), Alexa Fluor 488 wheat germ agglutinin kit (Invitrogen, Carlsbad, CA), 4′,6‐diamidino‐2‐phenylindole (Invitrogen), oligomycin (Sigma‐Aldrich), carbonyl cyanide‐p‐trifluoromethoxy‐phenylhydrazone (FCCP) (Sigma‐Aldrich), rotenone (Sigma‐Aldrich), antimycin A (Sigma‐Aldrich), Ponceau S (Acros Organics, Geel, Belgium), Calcium Green‐5N (Invitrogen), and Vectashield Hardset (H1400; Vector Labs, Burlingame, CA).
Animals
The global Sigmar1 knockout (Sigmar1−/−) mice generated by gene trapping (Oprs1Gt(IRESBetageo)33Lex/Oprs1Gt(IRESBetageo)33Lex)49 were obtained from the Mutant Mouse Resource Regional Center. Because these mice were in a mixed background (C57BL6×129S/SvEv), we backcrossed with C57BL6 for 10 generations. Both male and female mice consisting of Sigmar1−/− (n=70) and wild‐type (Wt) (n=70) littermate control mice were used. All procedures for handling animals complied with the Guide for Care and Use of Laboratory Animals and were approved by the ACUC Committee of LSU Health Sciences Center‐Shreveport. All animals were cared for according to the National Institutes of Health guidelines for the care and use of laboratory animals.
In Vivo Cardiac Function and β‐Adrenergic Responsiveness
Invasive hemodynamic studies were performed in the intact animals as described.50, 51 The right femoral artery and vein were cannulated with polyethylene tubing (PE‐90) for measuring systemic arterial pressure. To assess myocardial performance, closed‐chest animals were studied with a high‐fidelity 1.4F Millar micromanometer‐tipped catheter placed retrograde across the aortic valve into the LV (via right carotid cutdown). Hemodynamic parameters were determined during increasing doses of dobutamine using 3‐minute constant infusions (0.1 μL/min per gram body weight), and the average value during the final 30 seconds of each infusion was used. Online heart rate, telemetry, and LV developed pressure and ±dP/dt, were archived on a Macintosh computer using Matlab software (Mathworks, Natick, MA), and the investigators were blinded to genotype.50, 51
Echocardiography
Cardiac echocardiography was performed on isoflurane‐anesthetized mice with a VisualSonics Vevo 2100 Imaging System (Toronto, ON, Canada) using a 40‐MHz transducer to assess cardiac functional parameters as described.50, 52, 53 Briefly, 2‐dimensional directed M‐mode echocardiographic images along the parasternal short axis were recorded by investigators blinded to genotype to determine LV size and systolic function. M‐mode measurements included the LV internal dimensions in diastole and systole (LVID;d and LVID;s, respectively) as well as the diastolic thickness of LV posterior wall and diastolic intraventricular septum thickness. Percentage fractional shortening was calculated as [(LVID;d−LVID;s)/LVID;d]×100. LV mass and volumes were determined from M‐mode measurements using proprietary VisualSonics (Toronto, ON, Canada) software.50, 53
Mitochondria Isolation
Mitochondria in Sigmar1−/− and Wt hearts were isolated as described previously.52 Briefly, hearts were harvested, homogenized in mannitol/sucrose‐ ethylene glycol tetraacetic acid (EGTA) buffer (225 mmol/L mannitol, 75 mmol/L sucrose, 5 mmol/L HEPES, and 1 mmol/L EGTA, pH 7.4) and subjected to differential centrifugation. Finally, mitochondria were lysed with 1× cell lytic M (Sigma‐Aldrich) containing protease and phosphatase inhibitors.52
Mitochondrial Respiration
Mitochondrial oxygen consumption rate was measured with an XF24 Extracellular Flux Analyzer (Seahorse Biosciences, North Billerica, MA) as described previously.52, 54, 55, 56 Heart mitochondria were isolated using mannitol/sucrose‐EGTA buffer (225 mmol/L mannitol, 75 mmol/L sucrose, 5 mmol/L HEPES, and 1 mmol/L EGTA, pH 7.4) by differential centrifugation as described above. Mitochondria (50 μg/well) were seeded in XF24 culture plates, and respiration was measured in mitochondrial assay buffer (220 mmol/L mannitol, 7 mmol/L sucrose, 10 mmol/L KH2PO4, 5 mmol/L MgCl2, 2 mmol/L HEPES, 1 mmol/L EGTA, 0.2% fatty acid–free bovine serum albumin, pH 7.4). Mitochondrial oxygen consumption rate (OCR) was measured and plotted at basal conditions followed by sequential addition of 1 μg/mL oligomycin (ATP‐synthase inhibitor), 4 μmol/L FCCP (a mitochondrial uncoupler), and 0.5 μmol/L rotenone (a complex I inhibitor) plus 0.5 μmol/L antimycin A (a complex III inhibitor). The OCR values were normalized to total protein content in the corresponding wells and expressed as picomoles/minute per microgram protein.52
Histological Analysis
Hearts from deeply anesthetized 6‐month‐old mice were harvested in diastole, fixed in 10% buffered formalin, and embedded in paraffin. Serial 5‐μm heart sections from each group were stained with Masson trichrome52, 53 and Sirius red.52, 57 Fibrosis and collagen deposition within sections were quantitated using ImageJ software (National Institutes of Health, Bethesda, MD) as described previously.50, 57 Fibrosis was quantified by blue‐stained areas, and nonstained myocyte areas from each section using color‐based thresholding. To quantitate collagen deposition, red‐stained areas and nonstained myocyte areas were determined using color‐based thresholding. The percentage of total fibrosis area was calculated as the blue‐stained areas divided by total surface area from each section. The percentage of collagen deposition area was calculated as the red‐stained areas divided by total surface area from each section.52
Wheat Germ Agglutinin Staining
Briefly, paraffin‐embedded heart tissue sections (5 μmol/L) were dewaxed and hydrated, and antigen was retrieved by boiling at 100°C in 10 mmol/L sodium citrate buffer (pH 6) for 30 minutes.52 The sections were then blocked with blocking solution (1% BSA, 0.1% cold water fish skin gelatin, and 1% Tween 20 in PBS) for 1 hour at room temperature followed by incubation with Alexa Fluor 488 wheat germ agglutinin (5 μg/mL, Invitrogen) and 4′,6‐diamidino‐2‐phenylindole (Invitrogen) for 1 hour at room temperature. The slides were then washed with PBS and mounted with Vectashield Hard Set (Vector Laboratories, Burlingame, CA). Stained tissue sections were observed and evaluated by an investigator blinded to genotype for wheat germ agglutinin staining on a Leica TCS SP5 spectral confocal microscope using a ×63 oil objective lens where images were acquired with Leica LAS (AF 2.6.3) software. All cardiomyocyte cross‐sectional areas were measured in each ×63 confocal field to calculate the average size of cardiomyocytes in ImageJ (v1.49) software (NIH).58
Transmission Electron Microscopy
For electron microscopic analysis, mice were anesthetized with isoflurane, and the hearts were sequentially first perfused with 1% paraformaldehyde and 2% glutaraldehyde in the cardioplegic buffer as described previously.52, 53 Hearts were perfused with 1% paraformaldehyde and 2% glutaraldehyde in 0.1 mol/L cacodylate buffer (pH 7.2), postfixed in 1% OsO4, and processed for thin sectioning. Multiple sections were counterstained with uranium and lead salts and then examined with a Hitachi 7600 (Chiyoda, Tokyo, Japan) transmission electron microscope. Images were acquired with an AMT digital camera. Multiple sections were cut from 2 to 4 mice of mixed sex, and >20 fields were observed by a blinded observer. For the electron microscopic analysis, we used 2 mice for Sigmar1−/− (male and female) and 2 mice for Wt (male and female). A blinded observer analyzed and quantitated 22 microscopic fields containing 1538 mitochondria for Sigmar1−/− hearts and 21 microscopic fields containing 2537 mitochondria for Wt hearts to measure mitochondria size, area, density, and size distribution using ImageJ software (NIH). Mitochondrial density was measured by percentage of the areas taken by mitochondria compared to those of the cardiomyocytes.
ATP Measurements
Cellular and mitochondrial ATP contents were measured using the ATP Bioluminescence Assay Kit HS II (Roche) in freshly prepared whole‐cell lysates and mitochondrial suspensions from hearts according to manufacturer's instructions.52, 59, 60 Freshly excised hearts were homogenized using a glass/Teflon homogenizer (Wheaton, Millville, NJ) to prepare whole‐cell and mitochondria lysates by methods described previously.52 ATP content was measured on a FLUOstar OPTIMA microplate reader (BMG Labtech, Ortanberg, Germany), and ATP levels in samples were normalized to protein content as measured by Bio‐Rad protein assay reagent.52, 59
Mitochondrial Electron Transport Chain Complex Activity Assays
Isolated mitochondria were used to measure mitochondrial electron transport chain complex activities spectrophotometrically, as described previously.61, 62, 63 Complex I activity was determined by measuring NADH (100 μmol/L) oxidation at 340 nm for 2 minutes in an assay mixture containing 10 μg mitochondrial protein in 50 mmol/L potassium phosphate buffer (pH 7.5), 1 mg/mL BSA, 250 μmol/L KCN, and 60 μmol/L decylubiquinone in a 1‐mL cuvette on a DS‐11 FX+ spectrophotometer (DeNovix, Wilmington, DE). In parallel, NADH oxidation was measured in the presence of complex I inhibitor 10 μmol/L rotenone in the above assay mixture for 2 minutes. Specific complex I activity was calculated as rotenone‐sensitive activity expressed as nanomoles/minute per milligram of mitochondrial protein.61, 62, 63 Complex II activity was determined by measuring the reduction of 2,6‐dichlorophenolindophenol coupled to complex II catalyzed reduction of decylubiquinone. Briefly, 5 μg mitochondrial protein in 50 mmol/L potassium phosphate buffer (pH 7.5), 1 mg/mL BSA, and 20 mmol/L succinate was incubated at 37°C for 10 minutes following measurement of 2,6‐dichlorophenolindophenol reduction in the presence of 10 μmol/L antimycin A, 10 μmol/L rotenone, 250 μmol/L KCN, 80 μmol/L 2,6‐dichlorophenolindophenol, and 60 μmol/L decylqubiquinone at 600 nm for 3 minutes.62, 63 Complex III activity was determined by measuring reduction of cytochrome C (75 μmol/L) at 550 nm for 2 minutes in reaction mixture containing 5 μg mitochondrial protein in 50 mmol/L potassium phosphate buffer (pH 7.5), 250 μmol/L KCN, 100 μmol/L EDTA, and 60 μmol/L decylubiquinol. In parallel, in the same assay mixture reduction of cytochrome C was measured in the presence of complex III inhibitor 10 μmol/L antimycin A for 2 minutes at 550 nm. Specific complex III activity was calculated as antimycin A–sensitive activity expressed as nanomoles/minute per milligram of mitochondrial protein.61, 62, 63 Extinction coefficient values for NADH (8/[mmol·cm]), 2,6‐dichlorophenolindophenol (19.1/[mmol·cm]) and reduced cytochrome C (18.5/[mmol·cm]) were used to calculate complex I, II, and III activities, respectively.62, 63
Protein Extraction and Western Blot Analyses
Total proteins were prepared from hearts harvested and lysed with Cell Lytic M (Sigma‐Aldrich) lysis buffer supplemented with Complete Protease Inhibitor Cocktail (Roche) as described previously.52, 53 The heart homogenates were centrifuged at 14 000g for 15 minutes, and protein content of the soluble lysates was measured using the modified Bradford protocol/reagent relative to a BSA standard curve (BioRad). Protein lysates were separated on SDS‐PAGE using precast 7.5% to 15% gels (BioRad) and transferred to polyvinylidene fluoride membranes (BioRad). Membranes were blocked for 1 hour in 5% nonfat dried milk and exposed to primary antibodies overnight. The following primary antibodies were used for immunoblotting: anti‐Drp1 (1:1000, 14647, Cell Signaling Technology, Danvers, MA), anti‐pDrp‐Ser637 (1:1000, 79‐951, ProSci, Poway, CA), anti‐pDrp‐Ser616 (1:1000, PA5‐64821, Invitrogen), anti‐Mfn2 (1:1000, 9482, Cell Signaling Technology), anti‐OPA1 (1:1000, 80471, Cell Signaling Technology), anti‐β‐Actin (1:1000, sc‐47778, Santa Cruz Biotechnology, Santa Cruz, CA), anti‐OXPHOS (1:1000, ab110413, Abcam, Cambridge, UK), anti‐PDH (1:1000, ab110416, Abcam), anti‐COXIV (1:1000, 4844, Cell Signaling Technology), anti‐Periostin (1:500, NBP1‐300042, Novus Biologicals, Littleton, CO). Membranes were then washed, incubated with alkaline‐phosphatase‐conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA), exposed with ECF reagent (Amersham, Little Chalfont, UK), and, finally, detected on a ChemiDoc™ Touch Imaging System (BioRad, Hercules, CA). Ponceau S protein stain of the transfer membrane was used as a loading control. Densitometry on scanned membranes was done using ImageJ software.
RNA Isolation and Quantitative Real‐Time Polymerase Chain Reaction Analysis
Total RNA was isolated with TRI reagent (Molecular Research Center) according to the manufacturer's protocol. Quantitative real‐time polymerase chain reaction was performed with a CFX‐96 instrument (Bio‐Rad) using TaqMan probes (Applied Biosystems, Foster City, CA) for cardiac fetal genes (Nppa, Nppb, and Myh6; Applied Biosystems) as described previously.53 Data were normalized to Gapdh (Applied Biosystems) content and expressed as fold increase over the Wt group.
Mitochondrial Ca2+ Uptake and Swelling Assay
Mitochondrial Ca2+ uptake was measured with Calcium Green 5N (Molecular Probes, Eugene, OR) as previously described.52 Cardiac mitochondria (100 μg) were suspended in KCl buffer (125 mmol/L KCl, 20 mmol/L HEPES, 2 mmol/L MgCl2, 2 mmol/L potassium phosphate, and 40 μmol/L EGTA, pH 7.2), 200 nmol/L Calcium Green‐5N, 7 mmol/L pyruvate, and 1 mmol/L malate. Mitochondria were challenged with additions of CaCl2, and Calcium Green‐5N fluorescence was monitored using the BioTek Synergy IV plate reader (BioTek, Winooski, VT). Ca2+ pulses of 20 nmol/mg of mitochondrial protein were added every 180 seconds using an autoinjector, and mitochondrial Ca2+ retention capacity was quantitated as described previously.52
Mitochondrial swelling assay was performed in isolated mitochondria as described previously.52 Mitochondria isolated from hearts were suspended in swelling buffer (120 mmol/L KCl, 10 mmol/L Tris pH 7.4, 5 mmol/L KH2PO4), 7 mmol/L pyruvate, 1 mmol/L malate, and 10 μmol/L EDTA. Mitochondrial swelling was induced by challenging the mitochondria with 200 μmol/L CaCl2, and the absorbance at 540 nm was monitored as described previously.52
Statistics
Data are expressed as mean±SEM. All statistical tests were done with GraphPad Prism software (La Jolla, CA). All data were tested for normality using the Kolmogorov‐Smirnov test, and data that passed the normality assumption were analyzed using a Student t test (P<0.05) for 2 groups and groups of 3 or more with 1‐way ANOVA, followed by the Tukey post hoc test. Hemodynamics data were analyzed using a mixed, 2‐factor ANOVA with repeated measures on the second factor, and post hoc comparisons were performed by Bonferroni posttests.51, 64, 65 For certain data sets with smaller sample sizes (n=3‐6), the Kruskal‐Wallis test was applied, and data were presented in graphs showing median and interquartile ranges. A value of P<0.05 was considered statistically significant.
Results
Sigmar1 Global Knockout Mouse
We obtained the global knockout mice generated by gene trapping (Oprs1Gt(IRESBetageo)33Lex/Oprs1Gt(IRESBetageo)33Lex) (Figure S1A)49 and backcrossed with C57BL6 for 10 generations because the commercially available mice are on a mixed background (C57BL6×129S/SvEv). We confirmed Sigmar1 knockdown in the heart by Western blot analysis of the heart cell lysate (Figure S1B), using Wt littermate mice as controls.
Invasive Hemodynamic Assessment of Cardiac Function in the Sigmar1−/− Mice
Cardiac function was assessed at 3 to 4 months of age by LV catheterization both under basal conditions and after stimulation with increasing doses of the β1‐adrenoceptor agonist dobutamine. At baseline, Sigmar1−/− mice showed significantly increased systolic blood pressure compared with Wt controls (Figure 1C). Although Sigmar1−/− mice showed slightly higher maximal LV pressure, mean arterial blood pressure, maximum rates of LV contraction (dP/dtmax) and relaxation (dP/dtmin), the differences were not statistically significant (Figure 1). No differences were found in basal (Figure 1A) or dobutamine‐stimulated heart rates within groups (Figure 2A), suggesting that the changes in hemodynamic parameters were not due to intrinsic heart rate differences. At all doses of dobutamine, LV pressure, systolic blood pressure, and mean arterial pressure were significantly higher in Sigmar1−/− compared with Wt, although in neither genotype did these parameters modulate in response to dobutamine dose. In both groups, dP/dtmax increased with dobutamine, with the Sigmar1−/− mice achieving significantly higher values at dobutamine doses of 4 to 32 ng/[g·min] (Figure 2E). Interestingly, diastolic indices such as dP/dtmin, LV end‐diastolic pressure, and τ were not different between Sigmar1−/− and Wt mice at baseline (Figure 1F through 1H) or with dobutamine stimulation (Figure 2F through 2H). Taken together, we found that young Sigmar1−/− mice have enhanced contractility compared with Wt, although lusiotropy appears to be unaffected by the absence of Sigmar1.
Figure 1.
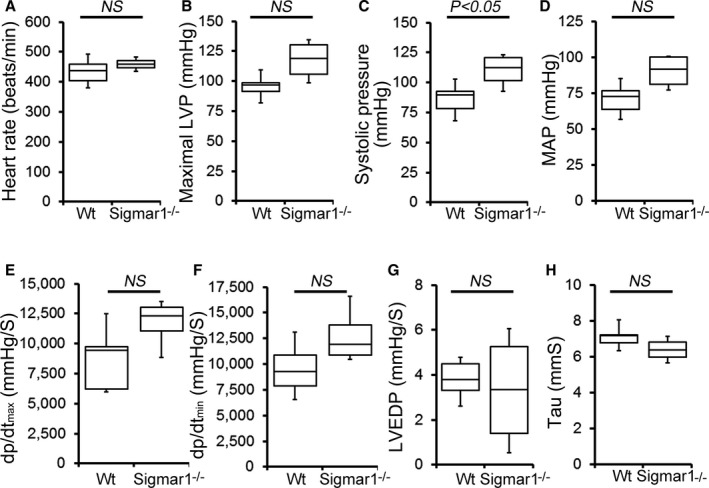
Cardiac hemodynamics in the Wt and Sigmar1−/− heart. Cardiovascular performance in anesthetized, closed‐chest mice under baseline conditions. A, Heart rate (beats/min). B, Maximal LV pressure (LVP). C, Systolic blood pressure. D, Mean arterial blood pressure (MAP). E, Maximum rate of LV contraction (dp/dtmax). F, Maximum rate of LV relaxation (dP/dtmin). G, LV end‐diastolic pressure (LVEDP). H, Relaxation time constant (τ). All measurements were carried out using 3‐ to 4‐month‐old Sigmar1−/− mice with littermate Wt mice used as controls (n=4 to 5 mice per group). Boxes represent interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by Kruskal‐Wallis test. LV indicates left ventricle; NS, not significant; Wt, wild type.
Figure 2.
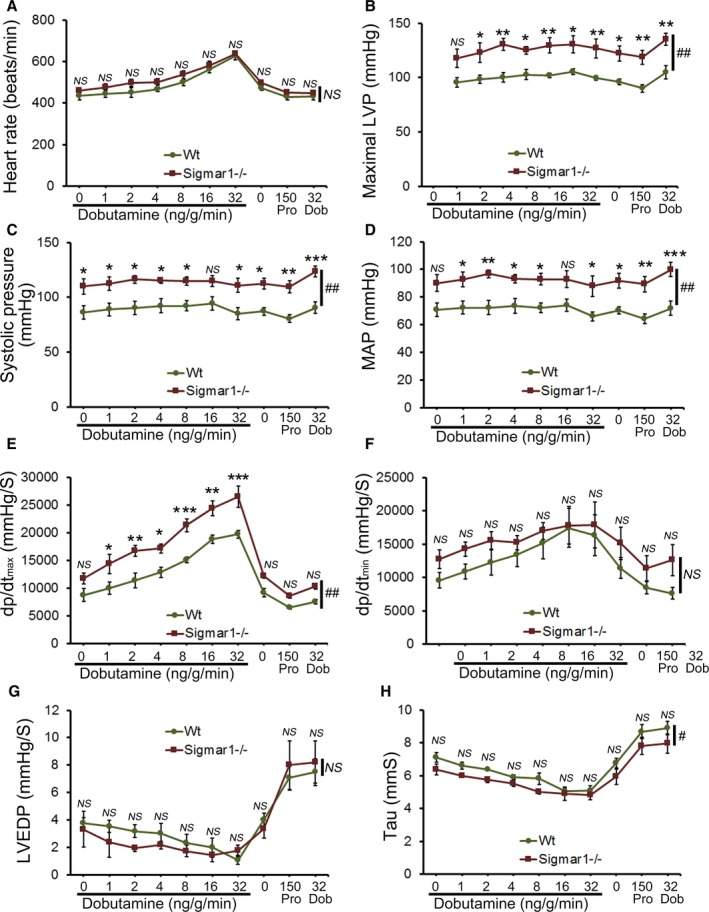
Cardiac hemodynamics in the Wt and Sigmar1−/− heart. Cardiovascular performance in anesthetized, closed‐chest mice under baseline conditions and increasing doses of the β‐adrenergic agonist dobutamine. At the conclusion of the dobutamine challenge, propranolol was delivered to produce complete β‐adrenergic blockade. A, Heart rate (beats/min). B, Maximal LV pressure (LVP). C, Systolic blood pressure. D, Mean arterial blood pressure (MAP). E, Maximum rate of LV contraction (dp/dtmax). F, Maximum rate of LV relaxation (dP/dtmin). G, LV end‐diastolic pressure (LVEDP). H, Relaxation time constant (τ). All measurements were carried out using 3 to 4 month old Sigmar1−/− mice with littermate Wt mice used as controls (n=4‐5 mice per group). Values are means±SEM. Data were analyzed using a mixed, 2‐factor ANOVA with repeated measures on the second factor, and post hoc comparisons were performed by Bonferroni posttests. *P<0.05, **P<0.01, and ***P<0.001 between Sigmar1−/− and Wt mouse for corresponding dose of dobutamine. # P<0.05 and ## P<0.01 represent significant group effect compared with Wt group. Dob indicates dobutamine; dp/dt, rate of change of pressure; LV, left ventricle; LVEDP, LV end‐diastolic volume; LVP, LV pressure; MAP, mean arterial pressure; NS, not significant; Pro, Propranolol; Wt, wild type.
Longitudinal Assessment of Sigmar1−/− Mice by Echocardiography
In vivo LV function and chamber dimensions of Sigmar1−/− and Wt mice were assessed with echocardiography in mixed‐sex cohorts at 3, 6, and 15 months of age (Figure 3). Similar to the invasive hemodynamic measurements, the youngest Sigmar1−/− were indistinguishable from Wt littermates with similar values for LV internal dimensions in diastole and systole (LVID;d and LVID;s, respectively, Figure 3A and 3B), LV fractional shortening (%FS, Figure 3C), volumes in diastole and systole (LV Vol;d and LV Vol;S, respectively, Figure 3D and 3E), and LV ejection fraction (Figure 3F). However, with aging the Sigmar1−/− mice developed progressive systolic dysfunction, evidenced by an increase in LVID;s and LV Vol;s resulting in decreased fractional shortening and ejection fraction compared with Wt littermates (Figure 3A through 3F). Interestingly, these hemodynamics chages were not accompanied by alterations in diastolic thickness of the interventricular septum and LV posterior wall with no increase in LV mass (Figure 3G through 3I) between Sigmar1−/− and Wt mice.
Figure 3.
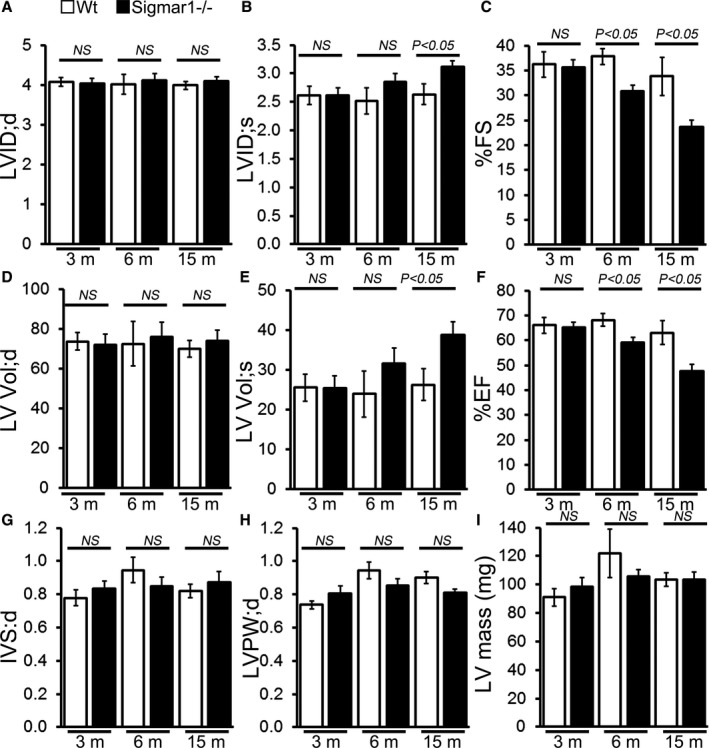
M‐mode echocardiography indices of cardiac structure and function in 3‐, 6‐, and 15‐month‐old Sigmar1−/− mice. A, LV diastolic internal dimension (LVID;d). B, LV systolic internal dimension (LVID;s). C, Percentage fractional shortening (%FS). D, LV diastolic volume (LVVol;d). E, LV systolic volume (LV Vol;s). F, Percentage ejection fraction (%EF). G, LV diastolic interventricular septum thickness (IVS;d). H, LV diastolic posterior wall thickness (LVPW;d). I, LV mass. Bars represent mean±SEM (n=5‐6 mice per group). P values were determined by Tukey's post hoc test. LV indicates left ventricle; NS, not significant; Wt, wild type.
Cardiac Fibrosis in Sigmar1−/− Mice
Because the Sigmar1−/− hearts began to show significant cardiac contractile dysfunction by 6 months of age, we evaluated cardiac morphometry by histological analysis using light microscopy with Masson trichrome and Sirius red staining (Figure 4A and 4B). Perivascular fibrosis was evident in the Sigmar1−/− hearts (Figure 4A and 4B), and expression of the fibrosis marker periostin was increased as well (Figure 4C). Heart weight–to–body weight ratios were measured as an indicator of cardiac hypertrophy and were not significantly different between Sigmar1−/− and Wt groups at 6 months of age (Figure 4D). Expression of molecular markers of cardiac hypertrophy including natriuretic peptide A, natriuretic peptide B, and α myosin heavy chain was not significantly changed in Sigmar1−/− hearts compared to Wt hearts at 6 months of age (Figure 4E through 4G). Cardiomyocyte size determined directly using wheat germ agglutinin staining of the LV demonstrated no significant difference between the 2 groups (Figure 4H). Thus, in addition to defective cardiac contractile function, the Sigmar1−/− hearts eventually undergo adverse cardiac remodeling without any changes in cardiomyocyte size.
Figure 4.
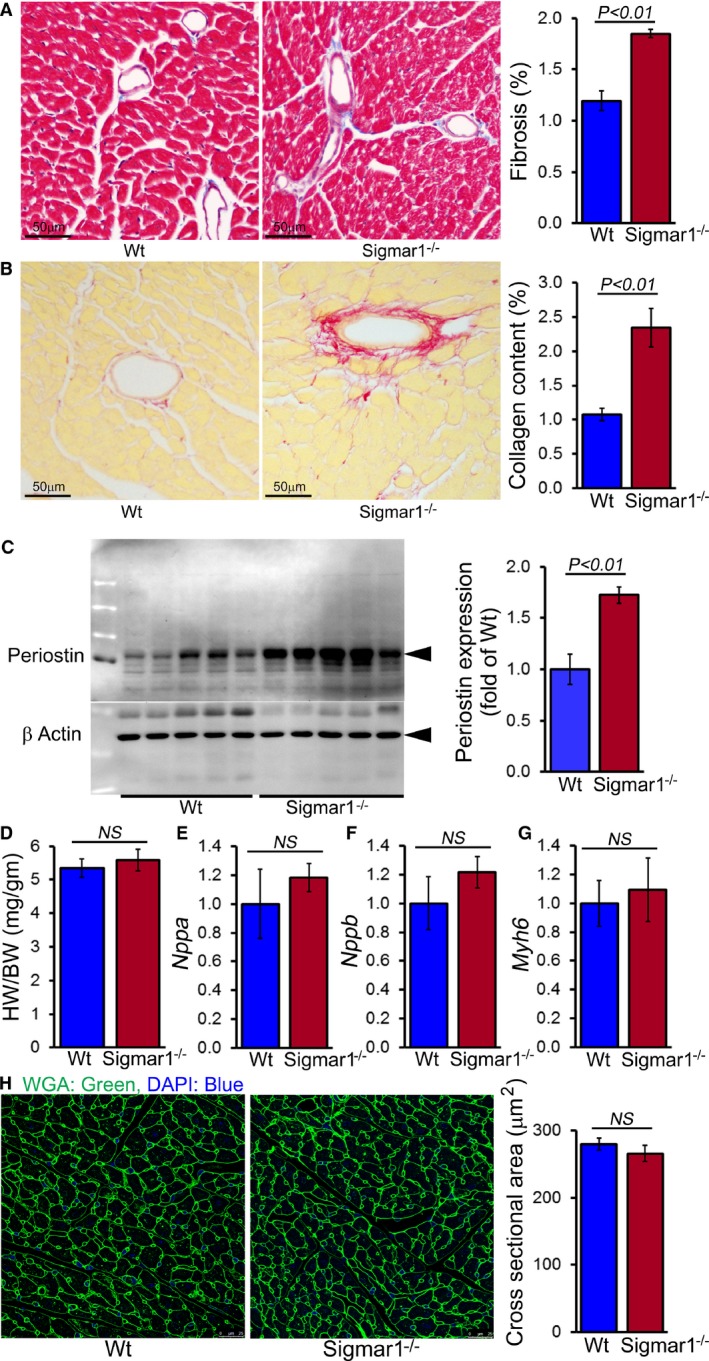
Sigmar1−/− hearts develop cardiac fibrosis at 6 months of age. A, Representative micrographs of Masson trichrome–stained LV myocardium and quantification of the cardiac fibrosis in Wt and Sigmar1−/− hearts (n=5 mice per group). Scale bars 50 μm. B, Representative micrographs of Sirius red–stained LV myocardium and quantification of the collagen deposition in Wt and Sigmar1−/− hearts (n=5 mice per group). Scale bars 50 μm. C, Representative Western blot and densitometric quantification showing significantly increased expression of periostin in Sigmar1−/− hearts (n=5 mice per group). D, Heart weight–to–body weight (HW/BW) ratio (n=7 mice per group), and mRNA expression of (E) natriuretic peptide A (Nppa), (F) natriuretic peptide B (Nppb), and (G) α‐myosin heavy chain (Myh6). Values are expressed as fold change vs Wt control (n=6 per group). H, Representative micrographs of wheat germ agglutinin (WGA, green)–stained LV myocardium and quantification of cross‐sectional area of the cardiomyocytes were carried on 10 microscoscopic fields for each heart (n=4 mice per group). Scale bars 50 μm. Bars represent mean±SEM. P values were determined by Tukey post hoc test. LV indicates left ventricle; NS, not significant; Wt, wild type.
Sigmar1 Loss of Function Leads to Changes in Mitochondrial Ultrastructure
Previous studies have shown that both in vitro pharmacological inhibition of Sigmar1 and in vivo Sigmar1 knockdown resulted in a significant (≈30%) increase in motor neuron mean mitochondrial size.49 Accordingly, we examined mitochondrial ultrastructure by electron microscopy in Wt and Sigmar1−/− mouse hearts. In 6‐month‐old Wt hearts, the mitochondria are well organized and in a regular pattern along the sarcomeres. In contrast, Sigmar1−/− cardiomyocytes showed an irregularly shaped, highly fused mitochondrial network, and exhibited abnormal cristae (Figure 5A). Quantification analyses revealed that despite a lower absolute mitochondrial number (Figure 5B), the Sigmar1−/− hearts had an overall increased average mitochondrial size (Figure 5C) and mitochondrial area (Figure 5D). Consequently, the mitochondrial density in Sigmar1−/− hearts as measured by percentage mitochondrial area per total area is higher than in Wt littermates (Figure 5E). The mitochondrial size distribution frequency curve shows a significantly increased percentage of large‐size mitochondria in the Sigmar1−/− hearts compared with Wt hearts (Figure 5F). Taken together, these data demonstrate the potential impact of Sigmar1 in regulating mitochondrial organization and morphology in the heart.
Figure 5.
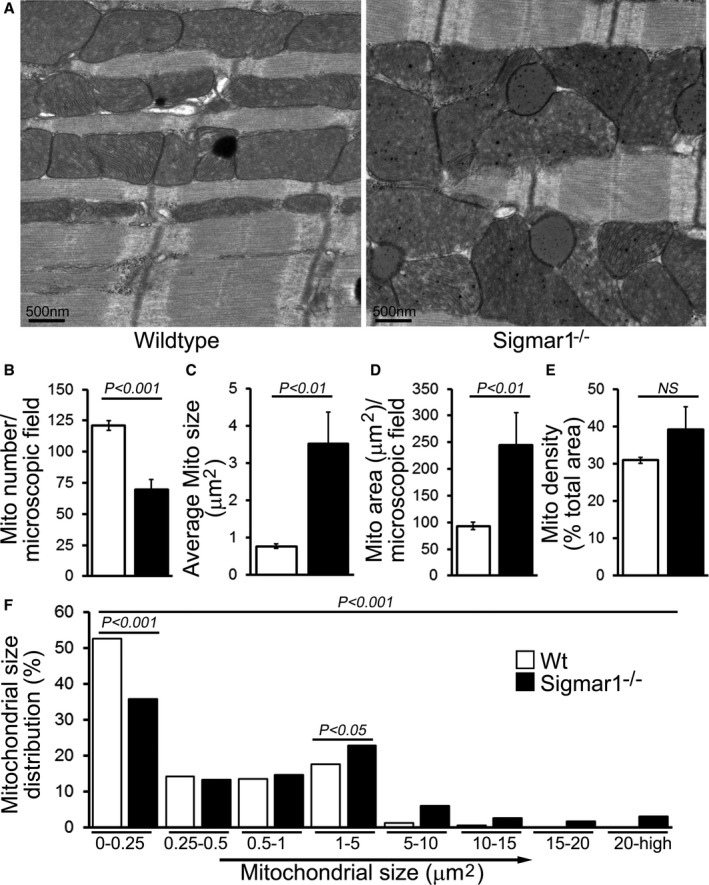
Ultrastructural analysis of Wt and Sigmar1−/− mouse hearts. A, Representative transmission electron micrographs (TEM) of hearts from Wt and Sigmar1−/− mice, respectively. Scale bars 500 nm. Representative TEM micrographs showing mitochondrial morphology quantitated as (B) mitochondria number per microscopic field, (C) average mitochondrial size (μm2), (D) mitochondrial area (μm2), (E) mitochondrial density, and (F) percentage mitochondrial size distribution in Sigmar1−/− and Wt mouse heart at 6 months of age (n=20‐23 microscopic images of ×12 400 magnification). Bars represent mean±SEM. P values were determined by Tukey post hoc test. Mito indicates mitochondria; Wt, wild type.
To further explore this concept, we examined the expression of mitochondrial fission and fusion regulatory proteins in the whole cell and mitochondrial fraction isolated from Sigmar1−/− and Wt mice at 3 and 6 months of age. Phosphorylation of GTPase dynamin‐related protein 1 (Drp1) at Ser637 in the Sigmar1−/− hearts was significantly increased in the whole cell fraction (Figure 6A and 6B). Drp1 phosphorylation at Ser616, total Drp1, and mitochondrial fusion regulatory protein OPA1 were unchanged in the whole‐cell fraction of Sigmar1−/− hearts compared to Wt hearts (Figure 6A and 6B). During mitochondrial fission, Drp1 translocates from the cytosol to prospective fission sites on the mitochondrial surface.66, 67, 68 Western blot and densitometric quantification showed decreased Drp1 in the Sigmar1−/− mitochondrial fraction compared with Wt preparations. In contrast, mitochondrial fusion regulatory protein OPA1 in the mitochondrial fraction was significantly increased in the Sigmar1−/− hearts (Figure 6C and 6D). To confirm the purity of the mitochondrial fractionation, we used GAPDH protein expression in the cytosolic fraction as a positive control and absence of GAPDH in the mitochondrial fraction as measures of purity. Western blots using the same membrane for COXIV were used to confirm the mitochondrial extracts. Ponceau S protein staining of the transfer membrane confirmed approximately equal loading and transfer across the gel.
Figure 6.
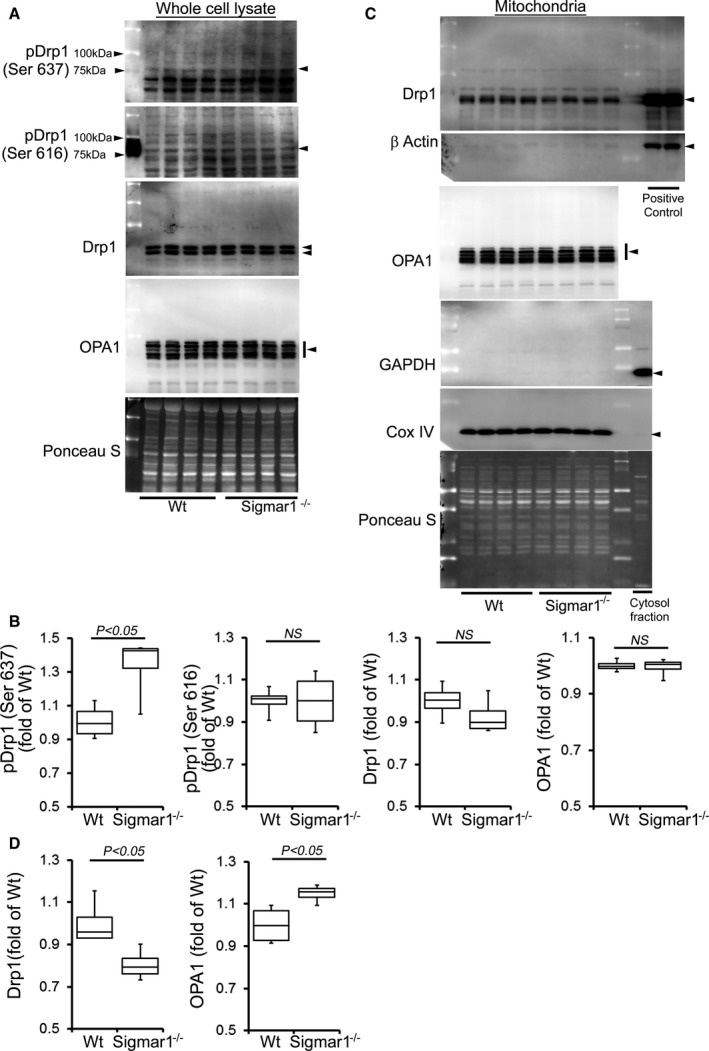
Expression of mitochondrial dynamics regulatory proteins in Sigmar1−/− mouse hearts at 3 months of age. A, Representative Western blot and (B) densitometric quantification of the expression of mitochondrial dynamic regulatory proteins in the whole‐cell fraction of Wt and Sigmar1−/− hearts: pDrp1 Ser637, pDrp1 Ser616, Drp1, and OPA1. C, Representative Western blot and (D) densitometric quantification of the expression of mitochondrial dynamic regulatory proteins in the mitochondrial fraction of Wt and Sigmar1−/− hearts: Drp1 and OPA1. Drp1 overexpressing HEK293 cells was used as a positive control for Drp1, and β‐actin was used on the same membrane. Cytosol fraction was used as a control to show purity of the mitochondrial fraction and confirmed by GAPDH. COXIV was run on the same membrane to confirm mitochondrial extracts. Ponceau S protein staining of the transfer membrane confirmed approximately equal loading across the gel (n=4 mice per group). Boxes represent interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by Kruskal‐Wallis test. NS indicates not significant; Wt, wild type.
Similarly, we also observed significantly increased Drp1 phosphorylation at Ser637 in the Sigmar1−/− hearts at 6 months of age. OPA1 expression was also significantly increased in the whole cell fraction of Sigmar1−/− hearts compared with Wt hearts (Figure 7A and 7B). Western blot and densitometric quantification showed a decreased level of Drp1 in the Sigmar1−/− mitochondrial fraction compared to Wt. In contrast, OPA1 levels in the mitochondrial fraction were significantly increased in the Sigmar1−/− hearts (Figure 7C and 7D). Therefore, the Sigmar1−/− mouse hearts showed altered expression of regulatory proteins that impact on mitochondrial dynamics and structural changes that might impact overall respiratory functions.
Figure 7.
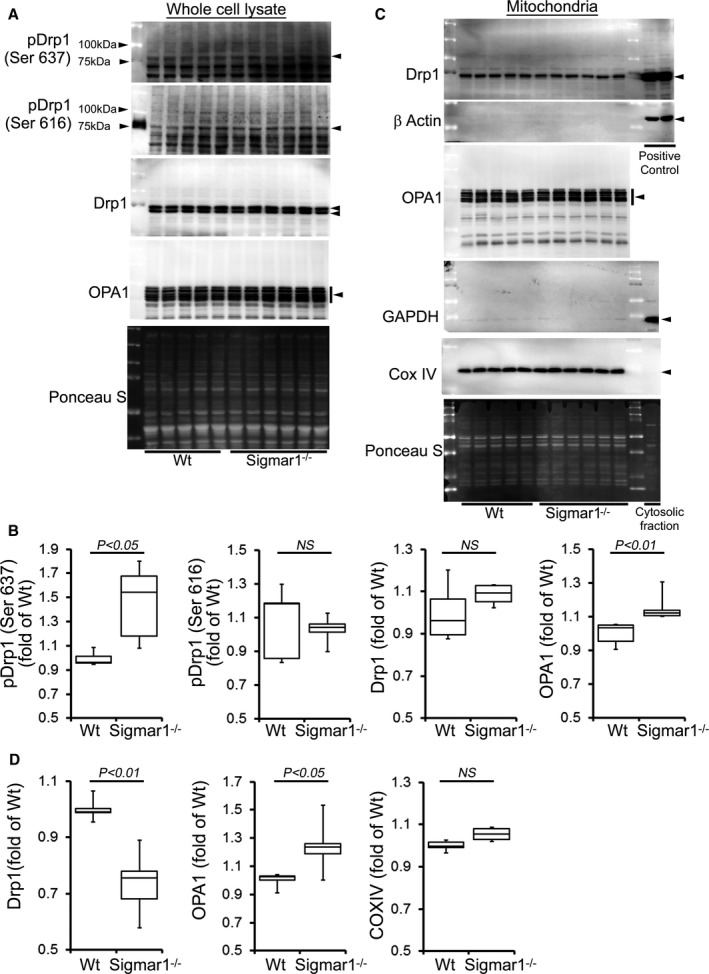
Expression of mitochondrial dynamics regulatory proteins in Sigmar1−/− mouse hearts at 6 months of age. A, Representative Western blot and (B) densitometric quantification of the expression of mitochondrial dynamic regulatory proteins in the whole‐cell fraction of Wt and Sigmar1−/− hearts: pDrp1 Ser637, pDrp1 Ser616, Drp1, and OPA1. C, Representative Western blot and (D) densitometric quantification of the expression of mitochondrial dynamic regulatory proteins in the mitochondrial fraction of Wt and Sigmar1−/− hearts: Drp1 and OPA1. Drp1‐overexpressing HEK293 cells were used as a positive control for Drp1, and β‐actin was used on the same membrane. The cytosol fraction was used as a positive control to confirm GAPDH detection. COXIV was used on the same membrane, confirming the mitochondrial extracts. Ponceau S protein staining of the transfer membrane confirmed approximately equal loading and transfer across the gel (n=5‐6 mice per group). P values were determined by Tukey post hoc test. Boxes represent interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by a Kruskal‐Wallis test. NS indicates not significant; Wt, wild type.
Mitochondrial Respiratory Dysfunction in the Sigmar1−/− Hearts
To explore functional alterations directly, we isolated mitochondria from 3‐ and 6‐month‐old Sigmar1−/− and Wt mouse hearts and measured mitochondrial respiration. Real‐time OCRs in isolated mitochondria showed that basal respiration, representing the sum of all physiological mitochondrial oxygen consumption, was decreased in the Sigmar1−/− samples, indicating lower respiratory function compared with Wt hearts (Figure 8A and 8B). The injection of oligomycin, an ATP synthase inhibitor, led to a decrease in basal respiration that was reflective of oxygen consumption used to generate ATP (Figure 8C). Despite having a lower basal respiration, Sigmar1−/− mitochondria showed ATP‐linked OCR similar to Wt mitochondria. The addition of carbonyl FCCP uncouples respiration from oxidative phosphorylation and allows for the measurement of maximal OCR, which was lower in both groups of Sigmar1−/− mitochondria (Figure 8D), indicating lower overall mitochondrial activity. The extent of nonmitochondrial oxygen‐consuming processes was estimated by inhibiting the respiratory chain with rotenone and antimycin A and was not affected in Sigmar1−/− mitochondria (Figure 8E). The respiratory reserve capacity, calculated by subtracting basal OCR from FCCP‐stimulated OCR, was significantly lower in Sigmar1−/− mitochondria (Figure 8F) and ATP turnover, measured by nonmitochondrial respiration subtracted from the ATP‐linked OCR, was significantly decreased in Sigmar1−/− mitochondria (Figure 8G). The maximum respiration calculated by nonmitochondrial respiration subtracted from FCCP‐stimulated OCR was also significantly lower in Sigmar1−/− mitochondria (Figure 8H). The proton leak across the inner mitochondrial membrane, represented by nonmitochondrial respiration subtracted from the postoligomycin OCR, was significantly increased in Sigmar1−/− mitochondria (Figure 8I) at 6 months of age. We conclude that mitochondria isolated from Sigmar1−/− hearts are functionally compromised compared with normal cardiac mitochondria.
Figure 8.
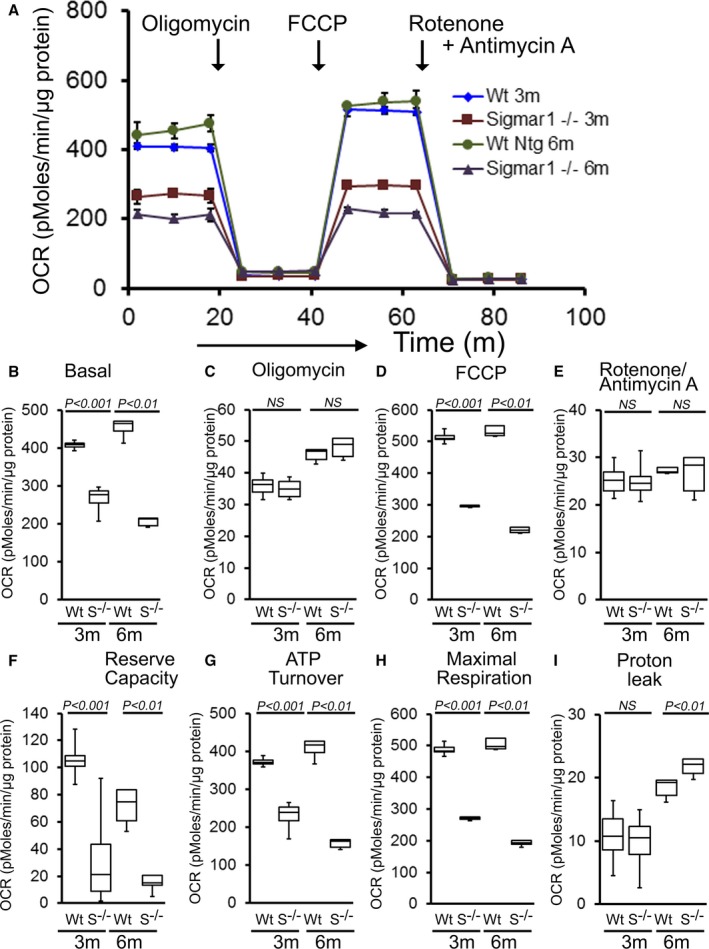
Suppression of mitochondrial respiration in 3‐ and 6‐month‐old Sigmar1−/− mouse hearts. A, Mitochondrial oxygen consumption rate (OCR) profiles in isolated mitochondria from 3‐ and 6‐month‐old Wt and Sigmar1−/− (S−/−) hearts. Arrow indicates the sequential addition of oligomycin (1 μmol/L), carbonyl cyanide‐4‐phenylhydrazone (FCCP) (4 μmol/L), and rotenone (0.5 μmol/L) plus antimycin A (0.5 μmol/L). OCR profiles are expressed as picomoles O2/[min·μg protein], and each point represents average OCR values for 3 mice. Graphs showing OCR under (B) baseline as well as with the addition of (C) oligomycin, (D) FCCP, and (E) rotenone plus antimycin A. Key parameters of mitochondrial function, including (F) reserve capacity, (G) ATP turnover, and (H) maximal respiration were significantly decreased in Sigmar1−/− mice. I, Proton leak was higher in the Sigmar1−/− mice compared with Wt mice (n=4 mice per group). Boxes represent interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by Kruskal‐Wallis test. NS indicates not significant; Wt, wild type.
Because the Sigmar1−/− hearts showed defective mitochondrial respiration, we compared oxidative phosphorylation protein levels in whole‐cell lysates using 3‐ and 6‐month‐old Sigmar1−/− and Wt mixed‐sex mouse hearts. Sigmar1−/− hearts showed significantly increased levels of Complex I expression, but Complex II, Complex III, and Complex V expression was unchanged at 3 months compared to Wt hearts (Figure 9A and 9B). Expression of the E2, E3bp, and E1α/β subunits of pyruvate dehydrogenase (PDH) complex proteins (Figure 9C and 9D) was similar in both groups of mice at 3 months. Ponceau S staining of proteins was used to visualize loading (Figure 9A and 9B). Mitochondrial Complex I activity was significantly decreased in the Sigmar1−/− hearts compared with Wt hearts at this time. Complex II and Complex III activity was not significantly different between the 2 groups (Figure 9E).
Figure 9.
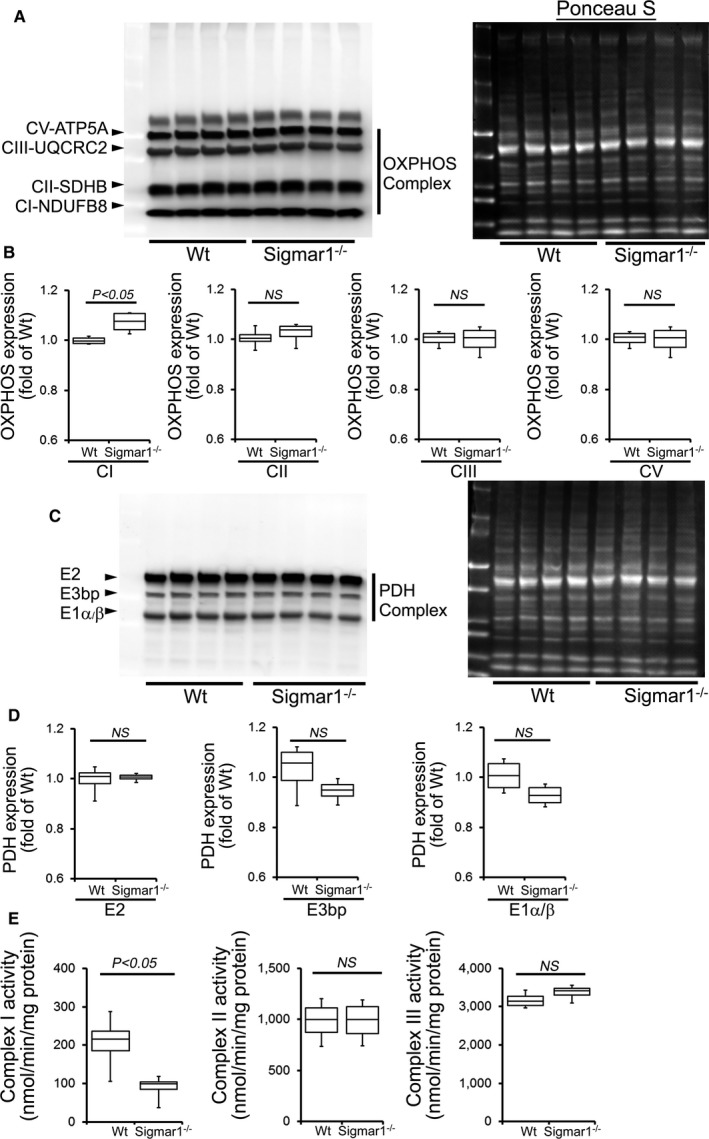
OXPHOS protein expression and complex activity in Wt and Sigmar1−/− hearts at 3 months of age. A, Representative Western blot and (B) densitometric quantification of the expression of OXPHOS protein: Complex I, Complex II, Complex III, and Complex V protein expression in 3‐month‐old Wt and Sigmar1−/− hearts. Ponceau S protein stain of the transfer membrane was used to confirm approximately equal loading (n=5‐6 mice per group). C, Representative Western blot and (D) densitometric quantification of the expression of PDH complex protein: E2, E3bp, and E1α/β protein expression in 3‐month‐old Wt and Sigmar1−/− hearts. Ponceau S protein stain of the transfer membrane was used to confirm approximately equal loading (n=5‐6 mice per group). E, Mitochondrial Complex I, II, and III activity in Wt and Sigmar1−/− hearts (n=4 mice per group). Boxes represent interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by Kruskal‐Wallis test. NS indicates not significant; OXPHOS, oxidative phosphorylation; PDH, pyruvate dehydrogenase; Wt, wild type.
Compared with Wt hearts, at 6 months of age Sigmar1−/− hearts showed significantly increased levels of Complex I, II, and III, but Complex V expression was unchanged (Figure 10A and 10B). We also found significantly increased expression of the E2, E3bp, and E1α/β subunit of PDH complex proteins (Figure 10C and 10D). Ponceau S staining of proteins was used to visualize loading. Measurement of mitochondrial and cellular ATP levels demonstrated a significantly lower ATP pool in Sigmar1−/− hearts (Figure 10E). To determine possible involvement of mitochondrial Ca2+ handling, we measured mitochondrial Ca2+ uptake and swelling in isolated mitochondria from 6‐month‐old Sigmar1−/− and Wt mouse hearts. We used Calcium Green‐5N to measure mitochondrial Ca2+ uptake.52 Ca2+ pulses of 20 nmol/mg of mitochondrial protein resulted in a rapid increase in Calcium Green‐5N fluorescence followed by a decline in the fluorescence intensity corresponding to mitochondrial Ca2+ uptake (Figure 11A). With increasing mitochondrial Ca2+ loading, extramitochondrial Ca2+ accumulates until the addition of Ca2+ leads to a sustained Ca2+ increase indicating a massive release of mitochondrial Ca2+.52 The Sigmar1−/− heart mitochondria showed similar mitochondrial Ca2+ retention capacity to Wt mitochondria (Figure 11A). Mitochondrial swelling determined by challenging isolated mitochondria with Ca2+ and measuring the decrease in light scattering also showed similar mitochondrial swelling in both groups of mice at 6 months of age (Figure 11B). These data suggest that the mitochondrial Ca2+ handling was not altered in the Sigmar1−/− hearts. Collectively, we find that the defective mitochondrial respiration in the Sigmar1−/− hearts is associated with altered expression of oxidative phosphorylation and pyruvate dehydrogenase proteins.
Figure 10.
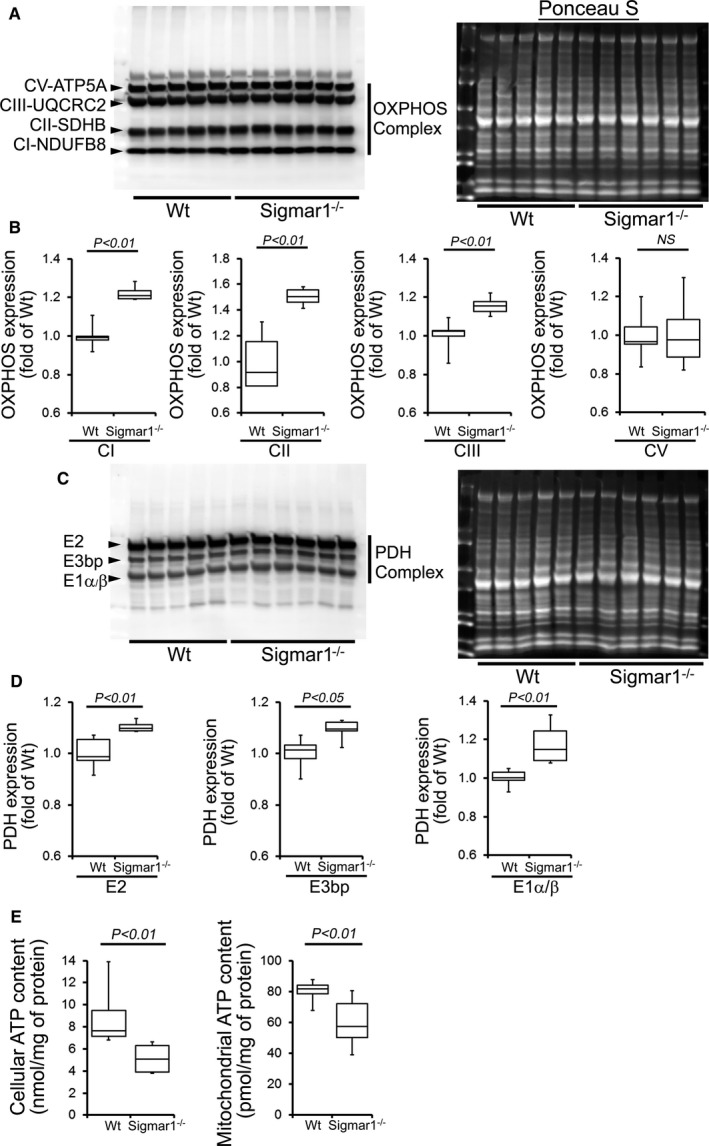
Impaired OXPHOS protein expression and ATP content in the Sigmar1−/− hearts at 6 months of age. A, Representative Western blot and (B) densitometric quantification of the expression of OXPHOS protein: Complex I, Complex II, Complex III, and Complex V protein expression in 6‐month‐old Wt and Sigmar1−/− hearts. Ponceau S protein stain of the transfer membrane was used to confirm approximately equal loading (n=5‐6 mice per group). C, Representative Western blot and (D) densitometric quantification of the expression of PDH complex protein: E2, E3bp, and E1α/β protein expression in 6‐month‐old Wt and Sigmar1−/− hearts. Ponceau S protein stain of the transfer membrane was used to confirm approximately equal loading (n=5‐6 mice per group). E, Mitochondrial and whole cellular ATP content in the Sigmar1−/− hearts (n=5‐6 mice per group). Boxes represent interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by Kruskal‐Wallis test. NS indicates not significant; OXPHOS, oxidative phosphorylation; PDH, pyruvate dehydrogenase; Wt, wild type.
Figure 11.
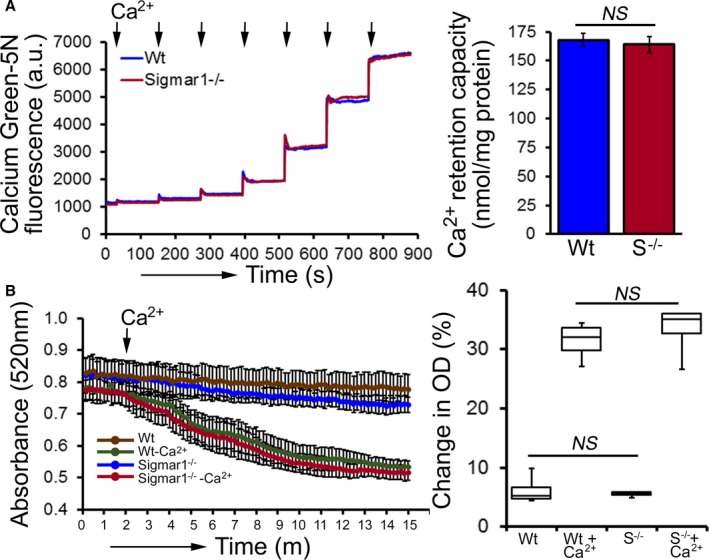
Mitochondrial Ca2+ uptake and swelling in Sigmar1−/− hearts at 6 months of age. A, Representative traces and quantification of mitochondrial Ca2+‐uptake of Sigmar1−/− and Wt hearts at 6 months of age. Fluorescence reading of Ca2+ measured with Calcium Green‐5N indicator in solution with subsequent addition of Ca2+ pulses of 20 nmol/mg of mitochondrial protein. Cumulative Ca2+ additions are shown at each arrowhead, and representative tracings are shown (n=5‐6 mice per group). Bars represent mean±SEM. P values were determined by Tukey's post‐hoc test. B, Representative images of Ca2+‐induced mitochondrial swelling isolated from the Sigmar1−/− (S−/−) and Wt hearts at 6 months of age. Mitochondrial swelling was induced by the addition of 200 μmol/L CaCl2 shown by arrow. Quantification of the mitochondrial swelling, expressed as the percentage change relative to the initial optical density (OD) in the indicated mice (n=4 mice per group). Boxes represent interquartile ranges, lines represent medians, whiskers represent ranges, and P values were determined by Kruskal‐Wallis test. a.u. indicates arbitrary units; NS, not significant; Wt, wild type.
Discussion
Given the significance of Sigmar1 in neurological diseases, in the present study we wished to define the physiological role of Sigmar1 in the heart. Our study is the first to show the development of cardiac contractile dysfunction and structural cardiac deficits in Sigmar1−/− hearts during aging. We also found that the Sigmar1−/− hearts develop significant cardiac fibrosis, accumulation of irregularly shaped mitochondria, and show defective mitochondrial respiratory function compared with to Wt hearts. All these data demonstrate a potential function of Sigmar1 in regulating regular mitochondrial organization and respiration directly or indirectly.
Earlier studies showed that exposure of isolated adult rat cardiomyocytes to a Sigmar1 ligand (eg, haloperidol) caused a marked concentration‐dependent increase in the amplitude of cell contraction.38 Our in vivo assessment of cardiac hemodynamics in young Sigmar1−/− mice showed that contractility was similar to that in Wt littermates at baseline, but stimulation with increasing doses of dobutamine resulted in increased contractility. Likewise, our echocardiographic analysis of 3‐month‐old mice did not show a significant difference in the basal cardiac function of Sigmar1−/− mice, but aging Sigmar1−/− hearts gradually developed increased systolic LV dimension and a decline in cardiac LV function. Furthermore, this dysfunction was accompanied by cardiac fibrosis in 6‐month‐old Sigmar1−/− hearts.
Balanced mitochondrial fission and fusion are indispensable to maintain normal mitochondrial network morphology and meet the metabolic needs of the cell. Sigmar1−/− hearts showed irregularly shaped highly fused large mitochondria, suggesting impairment of mitochondrial dynamics. During mitochondrial fission, the dynamin‐related GTPase Drp1 is normally recruited from the cytosol into the mitochondrial outer membrane. The mitochondrial fission‐promoting activity of Drp1 is controlled by Ser637 phosphorylation, which inhibits the Drp1 translocation to mitochondrial fission sites.69 We found that Ser637 phosphorylation was significantly increased in the Sigmar1−/− cardiac cell lysates, and Drp1 mitochondrial localization was significantly decreased in the Sigmar1−/− hearts suggesting impairment of mitochondrial fission. Conversely, mitochondrial fusion is regulated by OPA1 proteins.70 We also observed increased expression of mitochondrial OPA1, suggesting increased mitochondrial fusion in the Sigmar1−/− hearts. The bioenergetic profile of isolated mitochondria from 3‐ and 6‐month‐old Sigmar1−/− hearts is also functionally compromised compared with Wt hearts. Taken together, the Sigmar1−/− hearts showed an imbalance of mitochondrial dynamics and impairment of mitochondrial bioenergetics. Interestingly, all the reported amyotrophic lateral sclerosis–causing mutations in Sigmar1 are associated with mitochondrial dysfunction, supporting a key role of Sigmar1 in regulating mitochondrial dynamics and bioenergetics.
The hemodynamic parameters (particularly increased systolic pressure) in Sigmar1−/− mice at 3 months of age indicate that these mice are hypertensive compared with Wt mice. The mitochondrial dysfunction in Sigmar1−/− hearts at 3 months of age may be responsible for the development of high systolic pressure compared with the Wt mice. Assessment of cardiac mitochondrial function in experimental models of hypertension and heart failure showed that mitochondrial dysfunction contributes to the development of hypertension.71 Studies have also shown a positive correlation between mitochondrial structural abnormalities and alterations in mitochondrial metabolic and bioenergetic functions including decreased respiration and ATP production in the development of hypertension.71 Despite the link between damaged mitochondria and high blood pressure in the Sigmar1−/− mice, a cause‐effect relationship remains to be established. In future studies we aim to confirm the role of cardiac Sigmar1 in the development of high blood pressure using a cardiac‐specific Sigmar1 knockout mouse. Although Sigmar1−/− hearts showed markedly abnormal mitochondrial respiration at 6 months, neither the ultrastructure of the heart nor the molecular markers of hypertrophy were deranged, which is consistent with only mildly decreased LV systolic function as manifested by decreased fractional shortening as well as ejection fraction. Therefore, Sigmar1−/− young mice, despite having abnormal mitochondrial function, seem to be able to compensate for unfavorable bioenergetics. Compensation is sustained to a large degree even up to 6 months, at which time we observed a mild decrease in LV systolic function, but histology demonstrated only mild perivascular and interstitial fibrosis without activation of molecular markers of hypertrophy or increased cardiomyocyte cross‐sectional area. In future studies we aim to determine the molecular function and mechanism of Sigmar1 in the heart by a cardiomyocyte‐specific Sigmar1 knockdown in adult hearts to rule out possible compensatory changes at early ages.
Despite extensive studies, Sigmar1's subcellular localization and transmembrane topology remain obscure. Subcellular fractionation of neural tissues from mutant superoxide dismutase‐1 transgenic mice showed Sigmar1 accumulation in mitochondrial fractions.9 Immunoelectron microscopic data from the Mavlyutov group showed Sigmar1's subcellular localization to largely depend on cell and organ type.72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83 For example, Sigmar1 localized to the nuclear envelope in photoreceptor cells74 and in the nucleoplasmic reticulum as well as in the nucleus in the NSC34 cell line.82 Surprisingly, immunoelectron microscopy was unable to detect Sigmar1at the plasma membrane.72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83 Conflicting data exist regarding the localization of the Sigmar1 C‐ and N‐termini, with the Mavlyutov group showing the C‐terminus to reside inside the ER lumen and the N‐terminus in the cytosol.84 In contrast, the recently derived crystal structure proposes that the C‐terminus resides on the cytosolic side of the ER.85 These findings suggest a possible organ‐specific localization and function for Sigmar1. Further studies are required to define Sigmar1's subcellular distribution and pathophysiological role in general and with cardiac injury more specifically.
Sigmar1−/− mice were reported to develop multiple neuronal phenotypes—locomotor defects,75 significant nerve denervation,49 loss of motor neurons,49 age‐dependent motor phenotype and to show a depressive‐like behavior.48, 86 Sigmar1−/− mice also showed disruption of intracellular Ca2+ homeostasis, reduction in Ca2+ flux into the mitochondria, and lower ATP content in neuronal cells.9, 49, 75 Despite all these studies, there are no data dealing with the cardiac phenotype of Sigmar1−/− mice. The present study highlights the structure‐function relationships concerning Sigmar1's role in maintaining healthy cardiac function. Ours are the first in vivo data that bear on the structural and functional implications of the physiological functions of Sigmar1 in the heart. A significant limitation of our study is the use of global knockout mice in which we cannot rule out a cardiac impact of loss of Sigmar1 in other organs. We also do not know whether decreased mitochondrial bioenergetics underlies the morphological changes or is the result of deranged dynamics. Future studies are required to recapitulate these data using a cardiomyocyte‐specific knockout mouse. However, significant alterations in dynamics regulatory protein expression and suppression of mitochondrial respiration as early as 3 months in the Sigmar1−/− mouse hearts suggest that mitochondrial dysfunction led to cardiac dysfunction.
Sigmar1 has a significant therapeutic potential for cardiovascular disease as reflected by two Sigmar1 ligands already appear in clinical trials: cutamesine (SA4503) for ischemic stroke (phase 2)87 and SADHART‐CHF (Sertraline for Depression in Patients with Heart Failure).88, 89, 90, 91, 92, 93, 94, 95, 96 The SADHART‐CHF study demonstrated that depression remission by sertraline may improve cardiovascular outcomes in patients with chronic heart failure95, 96 but ignored sertraline's direct effect on cardiac Sigmar1 (if any). Moreover, a secondary analysis of the ENRICHD (Enhancing Recovery in Coronary Heart Disease) clinical trial showed that antidepressant treatment with the specific serotonin reuptake inhibitor sertraline (having Sigmar1 agonist activity in the nanomolar range) was associated with >40% reduction in the relative adjusted risk of death and reinfarction.14 Therefore, the present results may encourage clinical research to evaluate the potential role of Sigmar1 in cardiovascular disease.
Author Contributions
Abdullah, Alam, Aishwarya, Osinska, Peretik, and M. S. Bhuiyan performed experiments. James, Miriyala, Panchatcharam, and Lorenz analyzed data. M. A. N. Bhuiyan performed statistical analysis. Robbins and Orr contributed to reagents. Abdullah, Alam, and M. S. Bhuiyan designed experiments. Abdullah, and M. S. Bhuiyan wrote the manuscript, and all authors contributed to the preparation of the manuscript.
Sources of Funding
This work was supported by National Institutes of Health grants K99 HL122354 and R00 HL122354 to M. S. Bhuiyan; R01 HL098435 and R01 HL133497 to Orr; GM121307A to M. S. Bhuiyan, Orr, and Panchatcharam; LSUHSC‐S Malcolm Feist Cardiovascular Postdoctoral Fellowship to Abdullah; AHA Postdoctoral Fellowship to Alam; and LSUHSC‐S Malcolm Feist Predoctoral Fellowship to Aishwarya.
Disclosures
None.
Supporting information
Figure S1. Expression of Sigmar1 in the heart.
(J Am Heart Assoc. 2018;7:e009775 DOI: 10.1161/JAHA.118.009775)
References
- 1. Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE. The effects of morphine‐ and nalorphine‐like drugs in the nondependent and morphine‐dependent chronic spinal dog. J Pharmacol Exp Ther. 1976;197:517–532. [PubMed] [Google Scholar]
- 2. Su TP. Evidence for sigma opioid receptor: binding of [3H]SKF‐10047 to etorphine‐inaccessible sites in guinea‐pig brain. J Pharmacol Exp Ther. 1982;223:284–290. [PubMed] [Google Scholar]
- 3. Prause J, Goswami A, Katona I, Roos A, Schnizler M, Bushuven E, Dreier A, Buchkremer S, Johann S, Beyer C, Deschauer M, Troost D, Weis J. Altered localization, abnormal modification and loss of function of sigma receptor‐1 in amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22:1581–1600. [DOI] [PubMed] [Google Scholar]
- 4. Luty AA, Kwok JB, Dobson‐Stone C, Loy CT, Coupland KG, Karlstrom H, Sobow T, Tchorzewska J, Maruszak A, Barcikowska M, Panegyres PK, Zekanowski C, Brooks WS, Williams KL, Blair IP, Mather KA, Sachdev PS, Halliday GM, Schofield PR. Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration‐motor neuron disease. Ann Neurol. 2010;68:639–649. [DOI] [PubMed] [Google Scholar]
- 5. Al‐Saif A, Al‐Mohanna F, Bohlega S. A mutation in sigma‐1 receptor causes juvenile amyotrophic lateral sclerosis. Ann Neurol. 2011;70:913–919. [DOI] [PubMed] [Google Scholar]
- 6. Mishina M, Ohyama M, Ishii K, Kitamura S, Kimura Y, Oda K, Kawamura K, Sasaki T, Kobayashi S, Katayama Y, Ishiwata K. Low density of sigma1 receptors in early Alzheimer's disease. Ann Nucl Med. 2008;22:151–156. [DOI] [PubMed] [Google Scholar]
- 7. Mishina M, Ishiwata K, Ishii K, Kitamura S, Kimura Y, Kawamura K, Oda K, Sasaki T, Sakayori O, Hamamoto M, Kobayashi S, Katayama Y. Function of sigma1 receptors in Parkinson's disease. Acta Neurol Scand. 2005;112:103–107. [DOI] [PubMed] [Google Scholar]
- 8. Ullah MI, Ahmad A, Raza SI, Amar A, Ali A, Bhatti A, John P, Mohyuddin A, Ahmad W, Hassan MJ. In silico analysis of SIGMAR1 variant (rs4879809) segregating in a consanguineous Pakistani family showing amyotrophic lateral sclerosis without frontotemporal lobar dementia. Neurogenetics. 2015;16:299–306. [DOI] [PubMed] [Google Scholar]
- 9. Watanabe S, Ilieva H, Tamada H, Nomura H, Komine O, Endo F, Jin S, Mancias P, Kiyama H, Yamanaka K. Mitochondria‐associated membrane collapse is a common pathomechanism in SIGMAR1‐ and SOD1‐linked ALS. EMBO Mol Med. 2016;8:1421–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gregianin E, Pallafacchina G, Zanin S, Crippa V, Rusmini P, Poletti A, Fang M, Li Z, Diano L, Petrucci A, Lispi L, Cavallaro T, Fabrizi GM, Muglia M, Boaretto F, Vettori A, Rizzuto R, Mostacciuolo ML, Vazza G. Loss‐of‐function mutations in the SIGMAR1 gene cause distal hereditary motor neuropathy by impairing ER‐mitochondria tethering and Ca2+ signalling. Hum Mol Genet. 2016;25:3741–3753. [DOI] [PubMed] [Google Scholar]
- 11. Lee JJY, van Karnebeek CDM, Drogemoller B, Shyr C, Tarailo‐Graovac M, Eydoux P, Ross CJ, Wasserman WW, Bjornson B, Wu JK. Further validation of the SIGMAR1 c.151+1G>T mutation as cause of distal hereditary motor neuropathy. Child Neurol Open. 2016;3:2329048X16669912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li X, Hu Z, Liu L, Xie Y, Zhan Y, Zi X, Wang J, Wu L, Xia K, Tang B, Zhang R. A SIGMAR1 splice‐site mutation causes distal hereditary motor neuropathy. Neurology. 2015;84:2430–2437. [DOI] [PubMed] [Google Scholar]
- 13. Horga A, Tomaselli PJ, Gonzalez MA, Laura M, Muntoni F, Manzur AY, Hanna MG, Blake JC, Houlden H, Zuchner S, Reilly MM. SIGMAR1 mutation associated with autosomal recessive Silver‐like syndrome. Neurology. 2016;87:1607–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bhuiyan MS, Fukunaga K. Targeting sigma‐1 receptor signaling by endogenous ligands for cardioprotection. Expert Opin Ther Targets. 2011;15:145–155. [DOI] [PubMed] [Google Scholar]
- 15. Chu UB, Ruoho AE. Biochemical pharmacology of the sigma‐1 receptor. Mol Pharmacol. 2016;89:142–153. [DOI] [PubMed] [Google Scholar]
- 16. Hayashi T, Fujimoto M. Detergent‐resistant microdomains determine the localization of sigma‐1 receptors to the endoplasmic reticulum‐mitochondria junction. Mol Pharmacol. 2010;77:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hayashi T, Su TP. An update on the development of drugs for neuropsychiatric disorders: focusing on the sigma 1 receptor ligand. Expert Opin Ther Targets. 2008;12:45–58. [DOI] [PubMed] [Google Scholar]
- 18. Hayashi T, Su TP. Regulating ankyrin dynamics: roles of sigma‐1 receptors. Proc Natl Acad Sci USA. 2001;98:491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu Z, Bowen WD. Role of sigma‐1 receptor C‐terminal segment in inositol 1,4,5‐trisphosphate receptor activation: constitutive enhancement of calcium signaling in MCF‐7 tumor cells. J Biol Chem. 2008;283:28198–28215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tchedre KT, Huang RQ, Dibas A, Krishnamoorthy RR, Dillon GH, Yorio T. Sigma‐1 receptor regulation of voltage‐gated calcium channels involves a direct interaction. Invest Ophthalmol Vis Sci. 2008;49:4993–5002. [DOI] [PubMed] [Google Scholar]
- 21. Aydar E, Palmer CP, Klyachko VA, Jackson MB. The sigma receptor as a ligand‐regulated auxiliary potassium channel subunit. Neuron. 2002;34:399–410. [DOI] [PubMed] [Google Scholar]
- 22. Balasuriya D, D'Sa L, Talker R, Dupuis E, Maurin F, Martin P, Borgese F, Soriani O, Edwardson JM. A direct interaction between the sigma‐1 receptor and the hERG voltage‐gated K+ channel revealed by atomic force microscopy and homogeneous time‐resolved fluorescence (HTRF(R)). J Biol Chem. 2014;289:32353–32363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang H, Cuevas J. Sigma receptors inhibit high‐voltage‐activated calcium channels in rat sympathetic and parasympathetic neurons. J Neurophysiol. 2002;87:2867–2879. [DOI] [PubMed] [Google Scholar]
- 24. Hayashi T, Su TP. Sigma‐1 receptor chaperones at the ER‐mitochondrion interface regulate Ca2+ signaling and cell survival. Cell. 2007;131:596–610. [DOI] [PubMed] [Google Scholar]
- 25. Srivats S, Balasuriya D, Pasche M, Vistal G, Edwardson JM, Taylor CW, Murrell‐Lagnado RD. Sigma1 receptors inhibit store‐operated Ca2+ entry by attenuating coupling of STIM1 to Orai1. J Cell Biol. 2016;213:65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krutetskaya ZI, Milenina LS, Naumova AA, Butov SN, Antonov VG, Nozdrachev AD. Sigma‐1 receptor antagonist haloperidol attenuates store‐dependent Ca2+ entry in macrophages. Dokl Biochem Biophys. 2018;480:162–165. [DOI] [PubMed] [Google Scholar]
- 27. Zhang H, Katnik C, Cuevas J. Sigma receptor activation inhibits voltage‐gated sodium channels in rat intracardiac ganglion neurons. Int J Physiol Pathophysiol Pharmacol. 2009;2:1–11. [PMC free article] [PubMed] [Google Scholar]
- 28. Omori IM, Watanabe N, Nakagawa A, Cipriani A, Barbui C, McGuire H, Churchill R, Furukawa TA. Fluvoxamine versus other anti‐depressive agents for depression. Cochrane Database Syst Rev. 2010;CD006114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim HS, Li H, Kim HW, Shin SE, Choi IW, Firth AL, Bang H, Bae YM, Park WS. Selective serotonin reuptake inhibitor sertraline inhibits voltage‐dependent K+ channels in rabbit coronary arterial smooth muscle cells. J Biosci. 2016;41:659–666. [DOI] [PubMed] [Google Scholar]
- 30. Hernandez LL, Appel JB. Dopaminergic involvement in the mechanism of action of pentazocine. Behav Neural Biol. 1979;26:384–400. [DOI] [PubMed] [Google Scholar]
- 31. Chertkow Y, Weinreb O, Youdim MB, Silver H. Dopamine and serotonin metabolism in response to chronic administration of fluvoxamine and haloperidol combined treatment. J Neural Transm (Vienna). 2007;114:1443–1454. [DOI] [PubMed] [Google Scholar]
- 32. Niitsu T, Iyo M, Hashimoto K. Sigma‐1 receptor agonists as therapeutic drugs for cognitive impairment in neuropsychiatric diseases. Curr Pharm Des. 2012;18:875–883. [DOI] [PubMed] [Google Scholar]
- 33. Hayashi T, Tsai SY, Mori T, Fujimoto M, Su TP. Targeting ligand‐operated chaperone sigma‐1 receptors in the treatment of neuropsychiatric disorders. Expert Opin Ther Targets. 2011;15:557–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dumont M, Lemaire S. Interaction of 1,3‐di(2‐[5‐3H]tolyl) guanidine with sigma 2 binding sites in rat heart membrane preparations. Eur J Pharmacol. 1991;209:245–248. [DOI] [PubMed] [Google Scholar]
- 35. Ela C, Barg J, Vogel Z, Hasin Y, Eilam Y. Sigma receptor ligands modulate contractility, Ca++ influx and beating rate in cultured cardiac myocytes. J Pharmacol Exp Ther. 1994;269:1300–1309. [PubMed] [Google Scholar]
- 36. Ela C, Hasin Y, Eilam Y. Apparent desensitization of a sigma receptor sub‐population in neonatal rat cardiac myocytes by pre‐treatment with sigma receptor ligands. Eur J Pharmacol. 1996;295:275–280. [DOI] [PubMed] [Google Scholar]
- 37. Fialova K, Krizanova O, Jarkovsky J, Novakova M. Apparent desensitization of the effects of sigma receptor ligand haloperidol in isolated rat and guinea pig hearts after chronic treatment. Can J Physiol Pharmacol. 2009;87:1019–1027. [DOI] [PubMed] [Google Scholar]
- 38. Novakova M, Ela C, Barg J, Vogel Z, Hasin Y, Eilam Y. Inotropic action of sigma receptor ligands in isolated cardiac myocytes from adult rats. Eur J Pharmacol. 1995;286:19–30. [DOI] [PubMed] [Google Scholar]
- 39. Novakova M, Ela C, Bowen WD, Hasin Y, Eilam Y. Highly selective sigma receptor ligands elevate inositol 1,4,5‐trisphosphate production in rat cardiac myocytes. Eur J Pharmacol. 1998;353:315–327. [DOI] [PubMed] [Google Scholar]
- 40. Novakova M, Sedlakova B, Sirova M, Fialova K, Krizanova O. Haloperidol increases expression of the inositol 1,4,5‐trisphosphate receptors in rat cardiac atria, but not in ventricles. Gen Physiol Biophys. 2010;29:381–389. [DOI] [PubMed] [Google Scholar]
- 41. Bhuiyan MS, Tagashira H, Fukunaga K. Sigma‐1 receptor stimulation with fluvoxamine activates Akt‐eNOS signaling in the thoracic aorta of ovariectomized rats with abdominal aortic banding. Eur J Pharmacol. 2011;650:621–628. [DOI] [PubMed] [Google Scholar]
- 42. Bhuiyan MS, Tagashira H, Shioda N, Fukunaga K. Targeting sigma‐1 receptor with fluvoxamine ameliorates pressure‐overload‐induced hypertrophy and dysfunctions. Expert Opin Ther Targets. 2010;14:1009–1022. [DOI] [PubMed] [Google Scholar]
- 43. Bhuiyan MS, Fukunaga K. Stimulation of sigma‐1 receptor signaling by dehydroepiandrosterone ameliorates pressure overload‐induced hypertrophy and dysfunctions in ovariectomized rats. Expert Opin Ther Targets. 2009;13:1253–1265. [DOI] [PubMed] [Google Scholar]
- 44. Tagashira H, Bhuiyan S, Shioda N, Hasegawa H, Kanai H, Fukunaga K. Sigma1‐receptor stimulation with fluvoxamine ameliorates transverse aortic constriction‐induced myocardial hypertrophy and dysfunction in mice. Am J Physiol Heart Circ Physiol. 2010;299:H1535–H1545. [DOI] [PubMed] [Google Scholar]
- 45. Tagashira H, Bhuiyan MS, Shioda N, Fukunaga K. Fluvoxamine rescues mitochondrial Ca2+ transport and ATP production through sigma(1)‐receptor in hypertrophic cardiomyocytes. Life Sci. 2014;95:89–100. [DOI] [PubMed] [Google Scholar]
- 46. Tagashira H, Shinoda Y, Shioda N, Fukunaga K. Methyl pyruvate rescues mitochondrial damage caused by SIGMAR1 mutation related to amyotrophic lateral sclerosis. Biochim Biophys Acta. 2014;1840:3320–3334. [DOI] [PubMed] [Google Scholar]
- 47. Novakova M, Bruderova V, Sulova Z, Kopacek J, Lacinova L, Kvetnansky R, Vasku A, Kaplan P, Krizanova O, Jurkovicova D. Modulation of expression of the sigma receptors in the heart of rat and mouse in normal and pathological conditions. Gen Physiol Biophys. 2007;26:110–117. [PubMed] [Google Scholar]
- 48. Langa F, Codony X, Tovar V, Lavado A, Gimenez E, Cozar P, Cantero M, Dordal A, Hernandez E, Perez R, Monroy X, Zamanillo D, Guitart X, Montoliu L. Generation and phenotypic analysis of sigma receptor type I (sigma 1) knockout mice. Eur J Neurosci. 2003;18:2188–2196. [DOI] [PubMed] [Google Scholar]
- 49. Bernard‐Marissal N, Medard JJ, Azzedine H, Chrast R. Dysfunction in endoplasmic reticulum‐mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain. 2015;138:875–890. [DOI] [PubMed] [Google Scholar]
- 50. Bhuiyan MS, McLendon P, James J, Osinska H, Gulick J, Bhandary B, Lorenz JN, Robbins J. In vivo definition of cardiac myosin‐binding protein C's critical interactions with myosin. Pflugers Arch. 2016;468:1685–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lorenz JN, Robbins J. Measurement of intraventricular pressure and cardiac performance in the intact closed‐chest anesthetized mouse. Am J Physiol. 1997;272:H1137–H1146. [DOI] [PubMed] [Google Scholar]
- 52. Alam S, Abdullah CS, Aishwarya R, Miriyala S, Panchatcharam M, Peretik JM, Orr AW, James J, Robbins J, Bhuiyan MS. Aberrant mitochondrial fission is maladaptive in desmin mutation‐induced cardiac proteotoxicity. J Am Heart Assoc. 2018;7:e009289 DOI: 10.1161/JAHA.118.009289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez‐Caballero S, Osinska H, Cheng EH, Robbins J, Kinnally KW, Molkentin JD. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife. 2013;2:e00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 2015;12:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chandra M, Escalante‐Alcalde D, Bhuiyan MS, Orr AW, Kevil C, Morris AJ, Nam H, Dominic P, McCarthy KJ, Miriyala S, Panchatcharam M. Cardiac‐specific inactivation of LPP3 in mice leads to myocardial dysfunction and heart failure. Redox Biol. 2018;14:261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Karakikes I, Chaanine AH, Kang S, Mukete BN, Jeong D, Zhang S, Hajjar RJ, Lebeche D. Therapeutic cardiac‐targeted delivery of miR‐1 reverses pressure overload‐induced cardiac hypertrophy and attenuates pathological remodeling. J Am Heart Assoc. 2013;2:e000078 DOI: 10.1161/JAHA.113.000078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ma X, Takeda K, Singh A, Yu ZX, Zerfas P, Blount A, Liu C, Towbin JA, Schneider MD, Adelstein RS, Wei Q. Conditional ablation of nonmuscle myosin II‐B delineates heart defects in adult mice. Circ Res. 2009;105:1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. [DOI] [PubMed] [Google Scholar]
- 60. Yaniv Y, Spurgeon HA, Ziman BD, Lyashkov AE, Lakatta EG. Mechanisms that match ATP supply to demand in cardiac pacemaker cells during high ATP demand. Am J Physiol Heart Circ Physiol. 2013;304:H1428–H1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Goguadze N, Zhuravliova E, Morin D, Mikeladze D, Maurice T. Sigma‐1 receptor agonists induce oxidative stress in mitochondria and enhance complex I activity in physiological condition but protect against pathological oxidative stress. Neurotox Res. 2017. Available at: https://link.springer.com/article/10.1007/s12640-017-9838-2. Accessed September 29, 2018. [DOI] [PubMed] [Google Scholar]
- 62. Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc. 2012;7:1235–1246. [DOI] [PubMed] [Google Scholar]
- 63. Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. [DOI] [PubMed] [Google Scholar]
- 64. Sanbe A, Fewell JG, Gulick J, Osinska H, Lorenz J, Hall DG, Murray LA, Kimball TR, Witt SA, Robbins J. Abnormal cardiac structure and function in mice expressing nonphosphorylatable cardiac regulatory myosin light chain 2. J Biol Chem. 1999;274:21085–21094. [DOI] [PubMed] [Google Scholar]
- 65. Periasamy M, Reed TD, Liu LH, Ji Y, Loukianov E, Paul RJ, Nieman ML, Riddle T, Duffy JJ, Doetschman T, Lorenz JN, Shull GE. Impaired cardiac performance in heterozygous mice with a null mutation in the sarco(endo)plasmic reticulum Ca2+‐ATPase isoform 2 (SERCA2) gene. J Biol Chem. 1999;274:2556–2562. [DOI] [PubMed] [Google Scholar]
- 66. Karbowski M, Neutzner A, Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol. 2007;178:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin‐related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kanamaru Y, Sekine S, Ichijo H, Takeda K. The phosphorylation‐dependent regulation of mitochondrial proteins in stress responses. J Signal Transduct. 2012;2012:931215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105–117. [DOI] [PubMed] [Google Scholar]
- 71. Eirin A, Lerman A, Lerman LO. Mitochondrial injury and dysfunction in hypertension‐induced cardiac damage. Eur Heart J. 2014;35:3258–3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mavlyutov TA, Baker EM, Losenegger TM, Kim JR, Torres B, Epstein ML, Ruoho AE. The sigma‐1 receptor‐A therapeutic target for the treatment of ALS? Adv Exp Med Biol. 2017;964:255–265. [DOI] [PubMed] [Google Scholar]
- 73. Mavlyutov TA, Duellman T, Kim HT, Epstein ML, Leese C, Davletov BA, Yang J. Sigma‐1 receptor expression in the dorsal root ganglion: reexamination using a highly specific antibody. Neuroscience. 2016;331:148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mavlyutov TA, Epstein M, Guo LW. Subcellular localization of the sigma‐1 receptor in retinal neurons—an electron microscopy study. Sci Rep. 2015;5:10689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mavlyutov TA, Epstein ML, Andersen KA, Ziskind‐Conhaim L, Ruoho AE. The sigma‐1 receptor is enriched in postsynaptic sites of C‐terminals in mouse motoneurons. An anatomical and behavioral study. Neuroscience. 2010;167:247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mavlyutov TA, Epstein ML, Liu P, Verbny YI, Ziskind‐Conhaim L, Ruoho AE. Development of the sigma‐1 receptor in C‐terminals of motoneurons and colocalization with the N, N’‐dimethyltryptamine forming enzyme, indole‐N‐methyl transferase. Neuroscience. 2012;206:60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mavlyutov TA, Epstein ML, Verbny YI, Huerta MS, Zaitoun I, Ziskind‐Conhaim L, Ruoho AE. Lack of sigma‐1 receptor exacerbates ALS progression in mice. Neuroscience. 2013;240:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mavlyutov TA, Guo LW. Peeking into sigma‐1 receptor functions through the retina. Adv Exp Med Biol. 2017;964:285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mavlyutov TA, Guo LW, Epstein ML, Ruoho AE. Role of the sigma‐1 receptor in amyotrophic lateral sclerosis (ALS). J Pharmacol Sci. 2015;127:10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mavlyutov TA, Nickells RW, Guo LW. Accelerated retinal ganglion cell death in mice deficient in the sigma‐1 receptor. Mol Vis. 2011;17:1034–1043. [PMC free article] [PubMed] [Google Scholar]
- 81. Mavlyutov TA, Ruoho AE. Ligand‐dependent localization and intracellular stability of sigma‐1 receptors in CHO‐K1 cells. J Mol Signal. 2007;2:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mavlyutov TA, Yang H, Epstein ML, Ruoho AE, Yang J, Guo LW. APEX2‐enhanced electron microscopy distinguishes sigma‐1 receptor localization in the nucleoplasmic reticulum. Oncotarget. 2017;8:51317–51330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yang H, Fu Y, Liu X, Shahi PK, Mavlyutov TA, Li J, Yao A, Guo SZ, Pattnaik BR, Guo LW. Role of the sigma‐1 receptor chaperone in rod and cone photoreceptor degenerations in a mouse model of retinitis pigmentosa. Mol Neurodegener. 2017;12:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mavylutov T, Chen X, Guo L, Yang J. APEX2‐ tagging of sigma 1‐receptor indicates subcellular protein topology with cytosolic N‐terminus and ER luminal C‐terminus. Protein Cell. 2018;9:733–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schmidt HR, Zheng S, Gurpinar E, Koehl A, Manglik A, Kruse AC. Crystal structure of the human sigma1 receptor. Nature. 2016;532:527–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sabino V, Cottone P, Parylak SL, Steardo L, Zorrilla EP. Sigma‐1 receptor knockout mice display a depressive‐like phenotype. Behav Brain Res. 2009;198:472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Urfer R, Moebius HJ, Skoloudik D, Santamarina E, Sato W, Mita S, Muir KW; Cutamesine Stroke Recovery Study Group . Phase II trial of the sigma‐1 receptor agonist cutamesine (SA4503) for recovery enhancement after acute ischemic stroke. Stroke. 2014;45:3304–3310. [DOI] [PubMed] [Google Scholar]
- 88. Glassman AH, Bigger JT, Gaffney M, Shapiro PA, Swenson JR. Onset of major depression associated with acute coronary syndromes: relationship of onset, major depressive disorder history, and episode severity to sertraline benefit. Arch Gen Psychiatry. 2006;63:283–288. [DOI] [PubMed] [Google Scholar]
- 89. Jiang W, Krishnan R, Kuchibhatla M, Cuffe MS, Martsberger C, Arias RM, O'Connor CM; SADHART‐CHF Investigators . Characteristics of depression remission and its relation with cardiovascular outcome among patients with chronic heart failure (from the SADHART‐CHF Study). Am J Cardiol. 2011;107:545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jiang W, O'Connor C, Silva SG, Kuchibhatla M, Cuffe MS, Callwood DD, Zakhary B, Henke E, Arias RM, Krishnan R; SADHART‐CHF Investigators . Safety and efficacy of sertraline for depression in patients with CHF (SADHART‐CHF): a randomized, double‐blind, placebo‐controlled trial of sertraline for major depression with congestive heart failure. Am Heart J. 2008;156:437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. O'Connor CM, Jiang W, Kuchibhatla M, Silva SG, Cuffe MS, Callwood DD, Zakhary B, Stough WG, Arias RM, Rivelli SK, Krishnan R; SADHART‐CHF Investigators . Safety and efficacy of sertraline for depression in patients with heart failure: results of the SADHART‐CHF (Sertraline Against Depression and Heart Disease in Chronic Heart Failure) trial. J Am Coll Cardiol. 2010;56:692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Serebruany VL, Glassman AH, Malinin AI, Nemeroff CB, Musselman DL, van Zyl LT, Finkel MS, Krishnan KR, Gaffney M, Harrison W, Califf RM, O'Connor CM; Sertraline AntiDepressant Heart Attack Randomized Trial Study Group . Platelet/endothelial biomarkers in depressed patients treated with the selective serotonin reuptake inhibitor sertraline after acute coronary events: the Sertraline AntiDepressant Heart Attack Randomized Trial (SADHART) Platelet Substudy. Circulation. 2003;108:939–944. [DOI] [PubMed] [Google Scholar]
- 93. Serebruany VL, Suckow RF, Cooper TB, O'Connor CM, Malinin AI, Krishnan KR, van Zyl LT, Lekht V, Glassman AH; Sertraline Antidepressant Heart Attack Randomized Trial . Relationship between release of platelet/endothelial biomarkers and plasma levels of sertraline and N‐desmethylsertraline in acute coronary syndrome patients receiving SSRI treatment for depression. Am J Psychiatry. 2005;162:1165–1170. [DOI] [PubMed] [Google Scholar]
- 94. Swenson JR, O'Connor CM, Barton D, Van Zyl LT, Swedberg K, Forman LM, Gaffney M, Glassman AH; Sertraline Antidepressant Heart Attack Randomized Trial Group . Influence of depression and effect of treatment with sertraline on quality of life after hospitalization for acute coronary syndrome. Am J Cardiol. 2003;92:1271–1276. [DOI] [PubMed] [Google Scholar]
- 95. Xiong GL, Fiuzat M, Kuchibhatla M, Krishnan R, O'Connor CM, Jiang W; SADHART‐CHF Investigators . Health status and depression remission in patients with chronic heart failure: patient‐reported outcomes from the SADHART‐CHF trial. Circ Heart Fail. 2012;5:688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Xiong GL, Prybol K, Boyle SH, Hall R, Streilein RD, Steffens DC, Krishnan R, Rogers JG, O'Connor CM, Jiang W; SADHART‐CHF Investigators . Inflammation markers and major depressive disorder in patients with chronic heart failure: results from the Sertraline Against Depression and Heart Disease in Chronic Heart Failure study. Psychosom Med. 2015;77:808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Expression of Sigmar1 in the heart.