

Abstract
Background
Angiotensin II type 1 receptor (AT 1R) autoantibody (AT1‐AA) was first identified as a causative factor in preeclampsia. Unlike physiological ligand angiotensin II (Ang II), AT1‐AA can induce vasoconstriction in a sustained manner, causing a series of adverse effects, such as vascular injury and poor placental perfusion. However, its underlying mechanisms remain unclear. Here, from the perspective of AT 1R internalization, the present study investigated the molecular mechanism of sustained vasoconstriction induced by AT 1R autoantibody.
Methods and Results
In the current study, we used the vascular‐ring technique to determine that AT1‐AA‐positive IgG, which was obtained from the sera of preeclamptic patients, induced long‐term vasoconstriction in endothelium‐intact or endothelium‐denuded rat thoracic arteries. The effect was caused by prolonged activation of AT 1R downstream signals in vascular smooth muscle cells, including Ca2+, protein kinase C, and extracellular signal‐regulated kinase 1 and 2. Then, using subcellular protein fractionation, cell surface protein biotinylation, and total internal reflection fluorescence, we found that AT1‐AA‐positive IgG resulted in significantly less AT 1R internalization than in the Ang II treatment group. Moreover, through use of fluorescent tracing and bioluminescence resonance energy transfer, we found that AT1‐AA‐positive IgG cannot induce the recruitment of β‐arrestin1/2, which mediated receptor internalization. Then, the effect of sustained AT 1R activation induced by AT1‐AA‐positive IgG was rescued by overexpression of β‐arrestin2.
Conclusions
These data suggested that limited AT 1R internalization resulting from the inhibition of β‐arrestin1/2 recruitment played an important role in sustained vasoconstriction induced by AT1‐AA‐positive IgG, which may set the stage for avoiding AT 1R overactivation in the management of preeclampsia.
Keywords: angiotensin receptor, autoantibody, internalization, preeclampsia, vasoconstriction
Subject Categories: ACE/Angiotension Receptors/Renin Angiotensin System, Preeclampsia, Cell Signalling/Signal Transduction
Clinical Perspective
What Is New?
Angiotensin II type 1 receptor autoantibody–positive IgG from preeclamptic patients can limit angiotensin II type 1 receptor internalization by attenuating the recruitment of β‐arrestin1/2.
Limited angiotensin II type 1 receptor internalization is a novel mechanism in receptor overactivation and sustained vasoconstriction induced by angiotensin II type 1 receptor autoantibody.
What Are the Clinical Implications?
Continuous vasoconstriction caused by sustained AT1R activation is an important pathological mechanism in hypertension.
The promotion of angiotensin II type 1 receptor internalization might be a new therapeutic target for angiotensin II type 1 receptor autoantibody–positive hypertension.
Preeclampsia, a serious pregnancy‐specific medical condition characterized by hypertension, proteinuria, and multiple organ failure in the third trimesters,1 affects 2% to 8% of pregnancies worldwide.2 However, its cause and pathogenesis remain unclear. Previous research indicated that sustained vasoconstriction inducing abnormal vascular tone and remodeling were the vital pathogenesis of preeclampsia.3 Sustained vasoconstriction not only affected maternal blood vessels, leading to vascular damage and high blood pressure, but also produced a poor intrauterine growth environment by increasing resistance to fetal perfusion.4, 5 However, the specific molecular mechanisms are not well understood.
The renin–angiotensin system is an important hormone system that maintains blood pressure by regulating vascular tone.6 Angiotensin II type 1 receptor (AT1R) is abundantly distributed in smooth muscle cells. Under normal conditions, AT1R can be activated by angiotensin II (Ang II) and trigger intracellular signal transduction to induce vascular constriction, such as protein kinase C (PKC) and extracellular signal‐regulated kinase 1 and 2 (ERK1/2) activation, as well as Ca2+ signal generation.7, 8 However, in some pathological conditions, AT1R was overactivated and resulted in sustained vasoconstriction. Previous studies had verified that AT1R autoantibody (AT1‐AA) was involved in the overactivation of AT1R. AT1‐AA was first discovered in preeclamptic patients,9 which demonstrated an agonist‐like effect by binding to the second extracellular loop of AT1R (AT1R‐ECII). Another study found that administration of AT1‐AA isolated from preeclamptic patients can imitate preeclampsia in mice,10 indicating the pathogenic role of AT1‐AA in hypertension. Our previous study observed that AT1‐AA can directly induce vasoconstriction in a long‐term manner compared with Ang II group,11 which may be the key mechanism underlying hypertension and vascular injury in preeclampsia. However, the underlying molecular mechanism remains unknown.
It has been reported that AT1R internalization upon Ang II stimulation is the major mechanism of preventing sustained receptor activation.12, 13 β‐arrestin1 and β‐arrestin2 (β‐arrestin1/2), which are negative adaptors of G Protein‐Coupled Receptors (GRCRs), are universally expressed in all mammalian tissues.14 Their recruitment can uncouple AT1R from G protein, thus terminating or attenuating G protein–mediated signaling (desensitization) and initiating clathrin‐mediated endocytosis (internalization) of AT1R.15, 16 Therefore, we hypothesized that AT1‐AA can limit internalization of AT1R by attenuating the recruitment of β‐arrestin1/2, resulting in sustained AT1R activation and vasoconstriction.
In this study, we compared the amount of internalized AT1R upon Ang II stimulation alone to AT1‐AA stimulation alone using 3 methods. Then, through fluorescent tracing and bioluminescence resonance energy transfer (BRET), we detected the capability of β‐arrestin1/2 recruitment upon AT1‐AA stimulation. Because the AT1R blocker has been contraindicated in treatment of preeclampsia because of its influence of severe kidney maldevelopment in the fetus,17, 18, 19 our study may help outline the mechanism of AT1‐AA–induced overactivation of AT1R and offer a new therapeutic approach to treating preeclampsia.
Materials and Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Preparation of IgG From Plasma
Based on the results of ELISA detection, AT1‐AA‐positive IgG was purified respectively from the sera of AT1‐AA‐posive preeclamptic patients (n=14) using a MAbtrap Kit (Amersham, Piscataway, NJ) (The detailed experimental methods are described in Data S1). In order to ensure homogeneity throughout the experiment, the total IgGs were mixed together to gain a sufficient number of AT1‐AA‐positive IgG. Negative IgGs from AT1‐AA–negative individuals with normal pregnancy (n=20) were also purified and mixed as controls.
Animals
Healthy Sprague Dawley rats (n=36) 12 weeks (±4 days) of age were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. Rats were housed in controlled 12 hours light/dark conditions with a constant temperature (21±3°C) with ad libitum access to water and food. Rats were excluded if their serum was found to be AT1‐AA‐positive by ELISA and were assigned to different groups at random. All experiments involving animals were performed in compliance with the National Institutes of Health Guidelines on the Use of Laboratory Animals and were approved by the Ethics Committee of Capital Medical University, Beijing, China.
Vascular‐Ring Isolation Technology
Thoracic aortic rings were isolated according to the protocols of our previous study.20 In some cases, the intimal surface of the ring was rubbed with a cotton‐covered syringe needle to remove the endothelium. We administered the different stimuli, specifically: (1) administration of AT1R‐ECII: AT1R‐ECII (10 μmol/L) and AT1‐AA‐positive IgG (1 μmol/L) were mixed at 37°C for 30 minutes and centrifuged at 15 294g for 15 minutes. Finally, the supernatant was added to the prepared vascular rings. (2) Losartan blocking: Losartan (10 μmol/L) was pre‐incubated in the vascular rings for 15 minutes, and then AT1‐AA‐positive IgG was added. We recorded changes in isometric force in each vascular ring by using LabChart version 7 software (ADInstruments, Bella Vista, New South Wales, Australia).
Immunoprecipitation
The aim of this experiment was to determine whether AT1‐AA‐positive IgG can bind to AT1R. The vascular smooth muscle cells (VSMCs) were isolated from rat thoracic aortae by the explant technique, and the cells at passages 3 to 6 were used in all experiments. The cells were lysed by regular immunoprecipitation lysis buffer (200 mmol/L Tris‐HCl pH 8.0, 137 mmol/L NaCl, 1% NP‐40, 2 mmol/L EDTA) containing protease inhibitor cocktail. Then we added AT1‐AA‐positive IgG, negative IgG, and primary anti‐AT1R (2 μg, sc‐57036; Santa Cruz Biotechnology, USA), and incubated the mixture overnight at 4°C respectively. The next day, Protein A/G agarose beads (sc‐2003; Santa Cruz Biotechnology) were added to the lysates and incubated the mixture for 2 hours at 4°C. After that, the agarose beads were collected, and they were washed 3 times with immunoprecipitation lysis buffer and boiled with 2×loading buffer for 10 minutes. Proteins were separated by SDS‐PAGE and by Western blot with use of anti‐AT1R antibody (1:1000, Abcam, UK).
Plasmid Construction
Plasmids encoding RFP‐tagged human AT1R were constructed in pcDNA3.1 by LKL Biotechnology Company (Beijing, China). YFP‐tagged human AT1R, RFP‐tagged human b‐arrestin1, b‐arrestin2, and RLuc‐tagged bovine b‐arrestin1, b‐arrestin2 were constructed in pcDNA3.1 and provided by Professor Jinpeng Sun's laboratory.
Fluorescent Labeling of AT1‐AA‐Positive IgG, Plasmid Transfection, and Observing the Colocation Between AT1R and AT1‐AA‐Positive IgG
AT1‐AA‐positive IgG was labeled green with the Lightning‐Link Rapid Atto 488 Antibody Labeling Kit (350‐0010, Innova Biosciences, UK) according to the manufacturer's instructions. Briefly, we prepared AT1‐AA‐positive IgG in PBS at a concentration of 1 mg/mL, then added 10 μL of LL‐Rapid modifier reagent to 100 μL of AT1‐AA‐positive IgG and mixed gently. We then put the mixture into the vial of LL‐Rapid mix and gently resuspended by withdrawing and redispensing the liquid twice with a pipette. After incubation for 15 minutes at room temperature in the dark, we added 10 μL of LL‐Rapid quencher reagent into the vial and mixed gently. The conjugates were ready to use after a 5‐minute incubation period.
HEK293 cells were maintained in a DMEM medium supplemented with 10% fetal bovine serum. When they were grown to 70% confluence in 12‐well plastic culture dishes, we transiently transfected the cells with 0.5 μg AT1R–RFP plasmid per well by Lipofectamine 3000 transfection reagent (L300000; Thermo Fisher, USA). After 36 hours of transfection, green‐labeled AT1‐AA‐positive IgG (1 μmol/L) was added to it and incubated at 37°C for 30 minutes. After incubation, the samples were washed twice with ice‐cold PBS to remove uncombined AT1‐AA‐positive IgG. The images were obtained with a fluorescence microscope (Carl Zeiss, Oberkochen, Germany).
Intracellular Ca2+ Detection
The VSMCs were cultured in 35‐mm dishes. When confluence reached 80%, the cells were washed twice with PBS and they were incubated with a calcium indicator (Fluo‐3 AM; F1241; Thermo Fisher; 10 μmol/L in medium) for 60 minutes at 37°C. After being washed twice with PBS and added to FluoroBrite DMEM (A1896701; Thermo Fisher) containing 10% fetal bovine serum, the cells were ready for intracellular Ca2+ detection. The responses elicited by different stimulant were recorded as changes in green fluorescence intensity under a confocal microscope (UltraVIEW VoX, PerkinElmer, USA).
Subcellular‐Protein Fractionation
The VSMCs were cultured in 60‐mm dishes and they were lysed at each point in time. Subcellular proteins were fractionated by the Subcellular Protein Fractionation Kit (78840, Thermo Fisher) according to the manufacturer's instructions. The extractions were separated by SDS‐PAGE and AT1R levels were analyzed by Western blot with use of rabbit anti‐AT1R antibodies (1:1000, ab124734, Abcam, UK). Rabbit anti‐glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) and rabbit anti‐Na+/K+ ATPase from Abcam (UK) were detected as cytoplasm or membrane loading control, respectively.
Cell Surface Biotinylation Assay
The cell surface biotinylation assay was performed as previously described21, 22 with some modifications. Briefly, the VSMCs were cultured to 90% confluence in 60‐mm dishes. After washing them with ice‐cold PBS, cell surface proteins were biotinylated by incubation with 2 mg/mL of EZ‐Link sulfo‐NHS‐SS‐biotin (21331; Thermo Fisher) in PBS for 2 hours at 4°C. The cells were incubated with a medium containing AT1‐AA‐positive IgG (1 μmol/L) or Ang II (1 μmol/L) at 37°C for 30 minutes to receptor internalization. The remaining biotin on the cell surface was cleaved by incubation with cutting buffer (20 mmol/L dithiothreitol and 15 mmol/L glycine in PBS) for 2 hours at 4°C. After washing them 3 times with ice‐cold PBS, cells were harvested in a 500 μL radioimmunoprecipitation assay lysis buffer, 50 μL lysis samples were separated for input, and the rest were incubated with high‐capacity streptavidin agarose (20359; Thermo Fisher) for 1 hour at 4°C to precipitate biotinylated proteins. Finally, the collected proteins were washed with ice‐cold PBS and they were eluted from the beads by boiling in 2× loading buffer. Then, they were separated by SDS‐PAGE, and they were immunoblotted with an AT1R antibody.
Total Internal Reflection Fluorescence Microscopy
Total internal reflection fluorescence (TIRF) was used to determine the internalization of AT1R in transiently transfected cells by RFP‐fused AT1R. HEK293 cells were cultured on a 24‐mm‐diameter microscope coverglass (WHB‐6‐CS, WHB Scientific, China). After 48 hours of transfection, the cells were observed using a TIRF microscope (Olympus Corporation, Tokyo, Japan) equipped with an electron‐multiplying charge‐coupled device camera (Andor, Belfast, UK) and oil immersion objective (Olympus; magnification ×100, NA=1.49). A 101 to 103‐nm depth of field was chosen to observe AT1R fluorescence on the plasma membrane but not in cytoplasm. After stimulant administration, we monitored and pictured continuous quantifications of fluorescent receptors on the cell membrane for 2 000 s with use of MetaMorph software version 7.8.8.0 (Molecular Devices, Sunnyvale, CA).
Fluorescence Tracing
HEK293 cells were cultured on a 15‐mm‐diameter microscope coverglass and transfected with 1 μg pcDNA3.1‐AT1R‐YFP constructs and 1 μg pcDNA3.1‐β‐arrestin1‐RFP or β‐arrestin2‐RFP. After 36 hours of transfection, AT1‐AA‐positive IgG (1 μmol/L), Ang II (1 μmol/L), and negative IgG (1 μmol/L) were added to the cells and incubated at 37°C for 10 minutes. Then, the cells were washed twice with ice‐cold PBS and fixed with 4% paraformaldehyde at room temperature for 20 minutes. After that, the cells were washed with PBS 3 times. The images were obtained using a Leica Microsystems laser scanning confocal microscope (LAS AFTCS SP8).
Bioluminescence Resonance Energy Transfer
The AT1R‐YFP and β‐arrestin‐1‐RLuc or β‐arrestin‐2‐RLuc plasmids were transiently transfected into HEK293 cells. After 24‐hour transfection, the AT1R‐YFP and β‐arrestin‐1‐RLuc or AT1R‐YFP and β‐arrestin‐2‐RLuc cotransfected cells were resuspended and seeded into 96‐well plates (3603, Axygen, USA). After another 24 hours, the cells were washed with FluoroBriteTM DMEM (A1896701, Thermo Fisher, USA) and incubated in Ang II, AT1‐AA‐positive IgG, or negative IgG, respectively. BRET between RLuc and YFP was measured after the addition of the RLuc substrate coelenterazine‐h (5 μmol/L, S2011, Promega, USA). The BRET signal was calculated as the ratio of emission of YFP (527 nm) to RLuc (370–480 nm).
Statistical Analysis
The Graph‐Pad Prism 5 software package (GraphPad, Inc, San Diego, CA) was used to graph data and for statistical analysis. One‐way ANOVA followed by Bonferroni post hoc test was performed to determine statistical differences in the signal changes of PKC and ERK1/2 over the long‐term observation period in each group of different stimulants (eg, Ang II, AT1‐AA‐positive IgG, and negative IgG). One‐way repeated‐measure ANOVA was used to determine statistical differences in the signal changes of [Ca2+]i at different time points. The independent‐sample t tests were used to calculate statistical differences between the 2 groups. All P<0.05 were considered statistically significant. Data were expressed as the mean±SEM.
Results
AT1‐AA‐Positive IgG From Preeclamptic Patients Induced Isolated Aortic‐Ring Constriction in a Sustained Manner by Mainly Acting on VSMCs
To confirm the effect of AT1‐AA on vascular constriction, we performed an isolated aortic‐ring experiment. First, we isolated AT1‐AA‐positive IgG and negative IgG from preeclamptic patients and healthy individuals, respectively (Table S1 and Figure S1). Second, AT1‐AA was purified from AT1‐AA‐positive IgG by the peptide corresponding to the second extracellular loop of human AT1R (AT1R‐ECII), and linked to Sepharose 4B CNBr‐activated gel.23 Then, AT1‐AA‐positive IgG and AT1‐AA were identified and compared by SDS‐PAGE fractionation, ELISA, and the change of intracellular calcium ([Ca2+]i) (Figure S2). The results demonstrated that their biological activities were similar. Furthermore, by the doubling dilution of AT1‐AA‐positive IgG and negative IgG, we found that AT1‐AA‐positive IgG has a high capacity to combine with human AT1R‐ECII (Figure S3), suggesting that 1 μmol/L AT1‐AA‐positive IgG had the biggest combination effect while the negative IgG cannot bind to human AT1R‐ECII with the corresponding concentration. Therefore, we performed further experiments using 1 μmol/L AT1‐AA‐positive IgG and negative IgG.
As shown in Figure 1B and 1C, administration of Ang II (1 μmol/L) and AT1‐AA‐positive IgG (1 μmol/L) significantly constricted the isolated rat thoracic aortic ring. In contrast with the sharply developing and transient vasoconstriction (about 4 minutes, Figure 1H) caused by Ang II, AT1‐AA‐positive IgG induced a more slowly developing and longer‐term increase in vascular tension (which did not return to baseline until the end of our observation period, >30 minutes, Figure 1H). Negative IgG (1 μmol/L) did not induce vasoconstriction (Figure 1A), AT1R‐ECII removed the contractile effect of AT1‐AA‐positive IgG by immune adsorption, and losartan (AT1R‐specific antagonist) also blocked the effect (Figure 1E and 1F).
Figure 1.
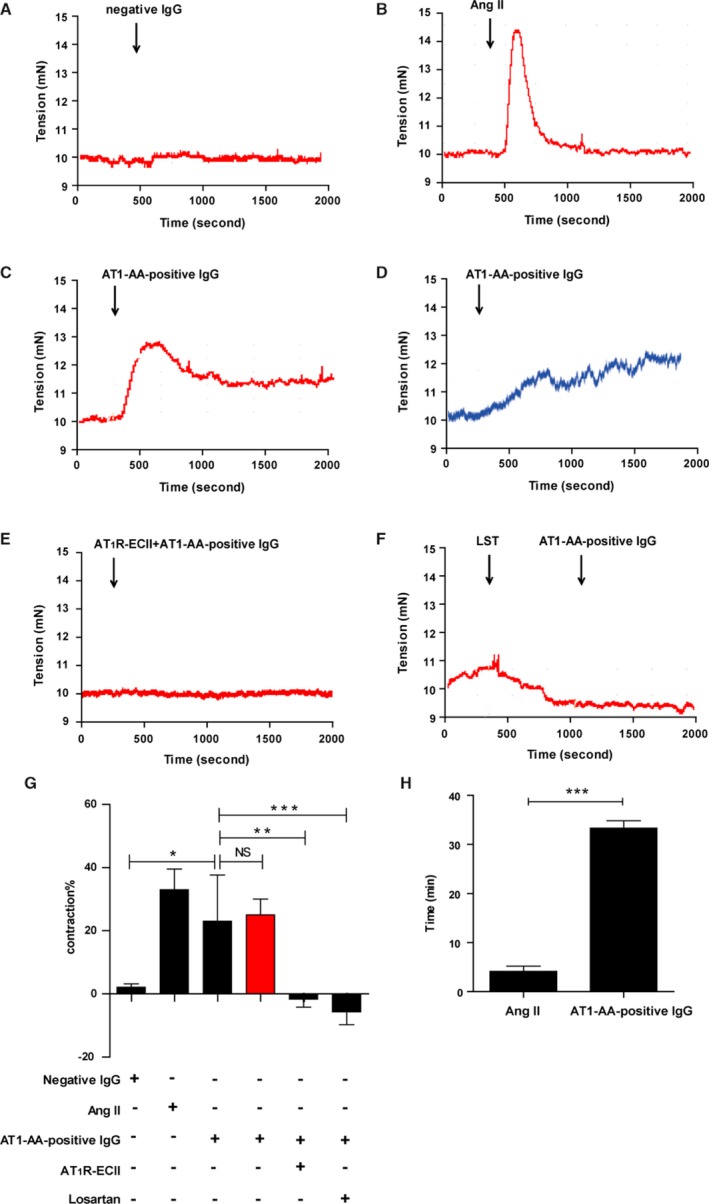
Representative data of isolated thoracic aortic‐ring constriction. Negative IgG did not induce vasoconstriction in isolated thoracic aortic rings (A). The tension of the ring increased within 2 minutes and then decreased to baseline level in less than 4 minutes under treatment with Ang II (B). With AT1‐AA‐positive IgG stimulation, the endothelium‐intact arterial ring reached maximum tension in 3 minutes and then showed a slight decrease but remained elevated until the end of the observation period (C). Endothelium‐denuded arterial rings also appeared to have sustained vasoconstriction upon AT1‐AA‐positive IgG stimulation (D). AT1R‐ECII neutralized the effect of AT1‐AA‐positive IgG (E). Pre‐incubation with 10 μmol/L losartan (LST), a specific AT1R antagonist, also blocked the effect of AT1‐AA‐positive IgG (F). Six independent results are summarized in a histogram (G). AT1‐AA‐positive IgG had similar vasoconstrictive effects by AT1R. There was no significant (NS) difference in vasoconstriction on arterial rings with either intact or damaged endothelia (red column) (date are presented as mean±SEM, *P<0.05; **P<0.01; ***P<0.001 vs AT1‐AA‐positive IgG with intact endothelia group, 1‐way ANOVA with Bonferroni post hoc test). The time courses of vasoconstrictions between Ang II and AT1‐AA‐positive IgG were profoundly different (H) (n=6, ***P<0.001 vs AT1‐AA‐positive IgG with intact endothelia group, t test). Ang II indicates angiotensin II; AT1‐AA, angiotensin II type 1 receptor autoantibody.
In addition, to demonstrate whether AT1‐AA‐positive IgG exerted sustained vasoconstriction via VSMCs rather than via the vascular endothelium, we performed an additional experiment to remove the vascular endothelium from the isolated aortic ring. It showed that acetylcholine exerted concentration‐dependent relaxation effects against norepinephrine‐induced contractions of arterial rings whose endothelia were intact, but not of arterial rings from which the endothelia had been removed (Figure S4); AT1‐AA‐positive IgG induced vasoconstriction in endothelium‐intact and endothelium‐denuded arterial rings (Figure 1C and 1D); there were no significant differences between them (Figure 1G, red column). These indicated that AT1‐AA‐positive IgG induced sustained vasoconstriction by mainly acting on VSMCs, and therefore we performed further mechanism studies on VSMCs.
AT1‐AA–Positive IgG Can Combine With AT1R in Rat VSMCs and Activate Downstream Signals in a Sustained Manner
We identified VSMCs by positive immunostaining of α‐smooth muscle actin and calponin (Figure S5). To validate whether AT1‐AA‐positive IgG directly bound to AT1R, we performed immunoprecipitation assay in VSMCs. After immunoprecipitating AT1‐AA‐positive IgG, AT1R was detected in VSMCs (Figure 2A). We next incubated RFP‐tagged AT1R‐overexpressed HEK293 cells with atto488‐conjugated AT1‐AA‐positive IgG at 37°C for 30 minutes. Fluorescence microscopy images showed that AT1‐AA and AT1R were colocalized on the surface of the cell (Figure 2B), indicating that AT1‐AA‐positive IgG can directly bind to AT1R.
Figure 2.
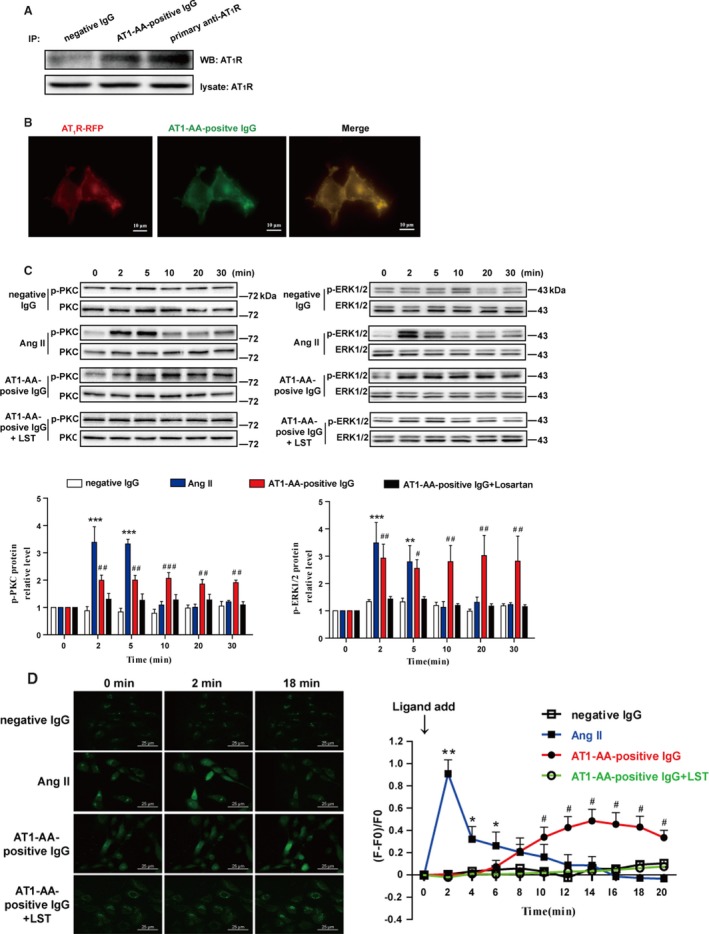
AT1‐AA‐positive IgG can directly bind to AT1R (A and B) and provoke downstream signal activation in a sustained manner (C and D). Immunoprecipitation assay showed that AT1‐AA‐positive IgG can directly bind to AT1R (A). RFP‐tagged AT1R (red)–overexpressed cells were incubated with atto488‐conjugated AT1‐AA‐positive IgG (green) at 37°C for 30 minutes. Confocal images showed that the red and green puncta were universally colocalized on the cell surface, and rarely colocalized in cytoplasm (B). Western blot data showed that phosphorylated PKC and ERK1/2 were significantly elevated at 2 and 5 minutes and decreased after 5 minutes because of Ang II stimulation. However, with AT1‐AA‐positive IgG stimulation, phosphorylated PKC and ERK1/2 were considerably elevated at 2 minutes and remained at high levels for 30 minutes. Negative IgG had no effect on PKC or ERK1/2 phosphorylation. Losartan (LST, 10 μmol/L) blocked the elevated PKC or ERK1/2 phosphorylation induced by AT1‐AA‐positive IgG. Total PKC and ERK1/2 were detected as loading controls and used to normalize phosphorylation signals via densitometry quantification. At each point in time, phosphorylated PKC and ERK1/2 were calculated and compared as percentages of the control at 0 minute. We summarized 5 independent results in a histogram (C). [Ca2+]i increased in response to stimulation with Ang II or AT1‐AA‐positive IgG. Both Ang II and AT1‐AA‐positive IgG induced an increase in [Ca2+]i, but the high level persisted longer in the AT1‐AA‐positive IgG group than in the Ang II group (˃10 minutes vs 4 minutes). The line chart summarizes the data of 6 independent experiments (D). (all conducted in rat VSMCs, data are presented as mean±SEM, *P<0.05; **P<0.01; ***P<0.001 vs 0 minute in Ang II group; # P<0.05; ## P<0.01; ### P<0.001 vs 0 minute in AT1‐AA‐positive IgG group, 1‐way ANOVA with Bonferroni post hoc test was used to test the signal changes of phosphorylated PKC and ERK1/2, and 1‐way repeated‐measures ANOVA was used to test the signal changes of [Ca2+]i). Ang II indicates angiotensin II; AT1‐AA, angiotensin II type 1 receptor autoantibody; ERK1/2, extracellular signal‐regulated kinase 1 and 2; IP, immunoprecipitation; PKC, protein kinase C; RFP, red fluorescence protein; VSMCs, vascular smooth muscle cells; WB, Western blot; F, Total fluorescence intensity; F0, fluorescence intensity baseline before the ligand was added.
Then, we detected the major physiological downstream signals of AT1R, including phosphorylated PKC and ERK1/2 as well as intracellular calcium ([Ca2+]i).24 Western blot data revealed that with Ang II stimulation, the phosphorylation levels of PKC and ERK1/2 greatly increased at the 2‐ and 5‐minute intervals but sharply decreased at the 10‐minute interval. Like Ang II, AT1‐AA‐positive IgG triggered increased phosphorylation of PKC and ERK1/2, but the duration of hyperphosphorylation was significantly prolonged (not recurring for 30 minutes). Incubation of negative IgG did not provoke phosphorylation. Preincubation of losartan (the AT1R‐specific antagonist) blocked the phosphorylation of PKC and ERK1/2 by AT1‐AA‐positive IgG (Figure 2C).
We observed a transient increase and rapid decrease of [Ca2+]i after Ang II administration (it returned to baseline within 6 minutes, Video S1), whereas AT1‐AA‐positive IgG caused changes of [Ca2+]i with slow rise and prolonged duration (>20 minutes, Video S2). Incubation of negative IgG with VSMCs had no effect on [Ca2+]i. Preincubation of losartan blocked the AT1‐AA–caused [Ca2+]i increase completely (Fig. 2C).
AT1‐AA‐Positive IgG Limited AT1R Internalization
To quantify AT1R internalization caused by Ang II and AT1‐AA‐positive IgG, we conducted subcellular protein fractionation using Western blot analysis. As expected, stimulation with Ang II caused a decrease of AT1R on the membrane and an increase in cytoplasm over time (Figure 3A). However, there was no statistically significant change in membrane or cytoplasm AT1R density by administration with AT1‐AA‐positive IgG (Figure 3A). These results demonstrated that AT1‐AA‐positive IgG reduced the amount of AT1R internalization compared with the Ang II treatment group.
Figure 3.
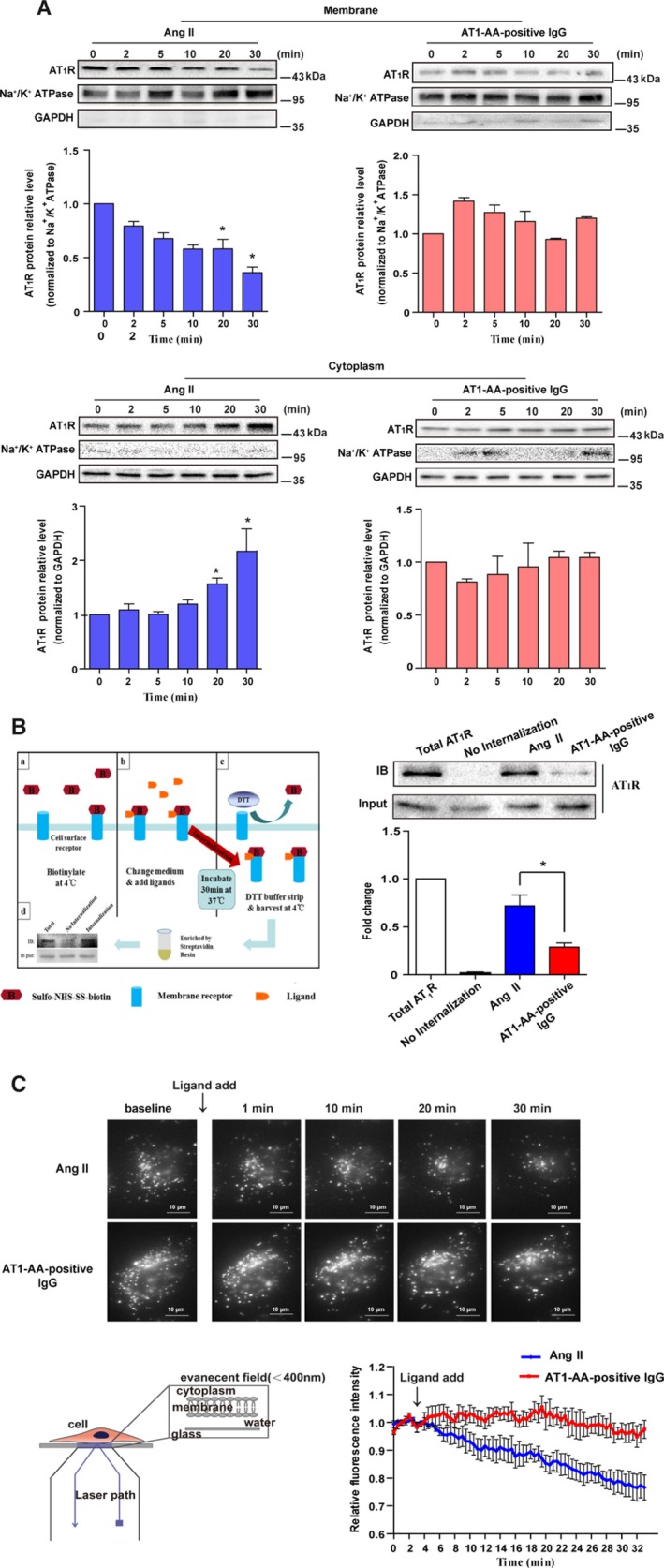
AT1‐AA limited AT1R internalization. Subcellular protein fractionation combined with Western blot analysis showed that Ang II could induce a significant decrease of AT1R in the membrane and concurrently induced a significant increase in cytoplasm (A). The change in AT1R was not detected under AT1‐AA‐positive IgG stimulation. Column statistics represented 6 independent experiments (data are presented as mean±SEM, *P<0.05 vs 0 minute, 1‐way ANOVA with Bonferroni post hoc test). Flowchart and representative immunoblotting data of AT1R intracellular trafficking detected by cell surface biotinylation are shown (B). Rat VSMCs were cultured in a 60‐mm‐diameter dish and labeled the cell surface protein with biotin. After incubation for 30 minutes at 37°C with Ang II or AT1‐AA‐positive IgG, cells were washed, lysed, and analyzed by Western blot. Data showed that AT1‐AA‐positive IgG significantly inhibited AT1R internalization compared with Ang II (t test, *P<0.05 vs Ang II, n=5). Schematic drawing, representative images, and line chart of membrane AT1R trafficking by TIRF are presented (C). Images were acquired at 1 frame/s for 33 minutes; the line chart was plotted by fluorescence intensities of AT1R–RFP in response to Ang II or AT1‐AA‐positive IgG stimulation. Although both AT1‐AA‐positive IgG and Ang II caused decreased membrane fluorescence intensity, the magnitude of the decrease was smaller after AT1‐AA‐positive IgG stimulation compared with Ang II (1% vs 25%; n=5). Ang II indicates angiotensin II; AT1R, angiotensin II type 1 receptor; AT1‐AA, angiotensin II type 1 receptor autoantibody; DTT, dithiothreitol; RFP, red fluorescence protein; TIRF, total internal reflection fluorescence; VSMCs, vascular smooth muscle cells.
To confirm our hypothesis, we designed a cell surface protein biotinylation assay based on a previous study.21 We found that the band representing AT1‐AA‐positive IgG‐induced internalization was significantly thinner than the one representing Ang II‐induced internalization (Figure 3B). It suggested that although AT1‐AA‐positive IgG directly bound to and activated cell surface AT1R as shown in Figure 2, it was distinct from Ang II in its smallest ability to induce AT1R internalization.
Finally, TIRF microscopy was used to observe AT1R internalization in real time. We specifically analyzed the fluorescence intensity of AT1R–RFP in HEK293 cells in response to Ang II or AT1‐AA‐positive IgG stimulation (Figure 3C). As the data showed, Ang II induced a significant decrease of fluorescence intensity (≈30%, Video S3), whereas AT1‐AA‐positive IgG induced such a decrease in fluorescence intensity only ≈1% (Video S4), suggesting it limited AT1R internalization. The lack of AT1‐AA‐positive IgG‐mediated internalization of AT1R might explain why AT1‐AA can induce prolonged signal activation and sustained vasoconstriction.
AT1‐AA‐Positive IgG Inhibited the Recruitment of β‐Arrestin1/2 During Activation of the Receptor
To investigate the mechanisms by which AT1‐AA‐positive IgG limited internalization of AT1R, the recruitment of β‐arrestin1/2 was assessed. By coexpressing YFP‐labeled AT1R and RFP‐labeled β‐arrestin1 or β‐arrestin2 in HEK293 cells with different stimulant at 37°C for 10 minutes, fluorescence microscopy images showed that AT1R and β‐arrestin1 and β‐arrestin2 were colocalized in the cell surface and cytoplasm (Figure 4A and 4B) under treatment with Ang II (1 μmol/L). However, for AT1‐AA‐positive IgG and negative IgG, there were rare colocalization events between AT1R and β‐arrestin1 or β‐arrestin2.
Figure 4.
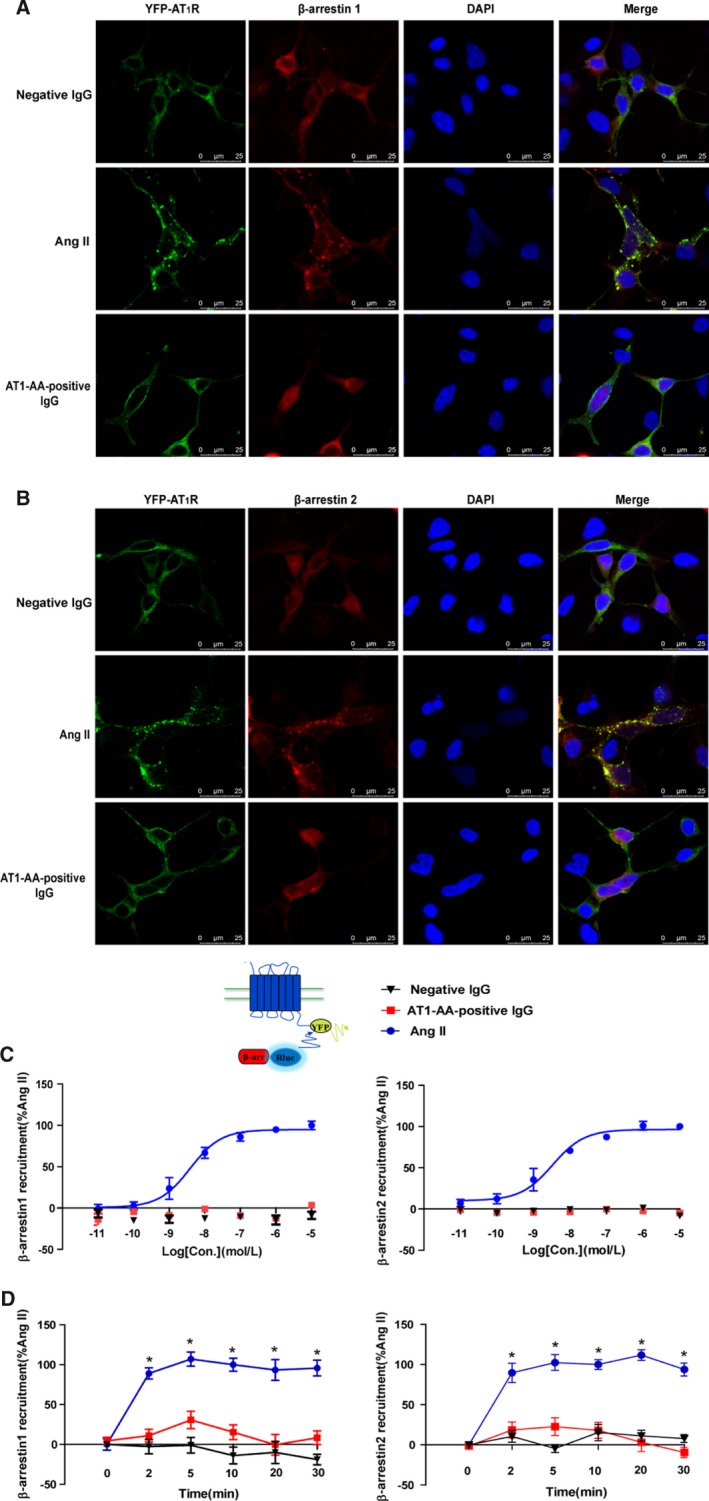
AT1‐AA‐positive IgG attenuated the recruitment of β‐arrestin1 and β‐arrestin2. Representative images of fluorescence tracking are shown. YFP‐labeled AT1R (1 μg) and RFP‐labeled β‐arrestin1 (1 μg) were co‐expressed in HEK293 cells. Treatment with Ang II (1 μmol/L) at 37°C for 10 minutes, AT1R, and β‐arrestin1 were colocalized in the cell surface and cytoplasm (middle). However, for negative IgG (upper) and AT1‐AA‐positive IgG (bottom), there was rarely colocalization between AT1R and β‐arrestin1 (A). Similarly, there was rarely colocalization between AT1R and β‐arrestin2 after treatment with negative IgG and AT1‐AA‐positive IgG (B). To confirm these findings by BRET, the relative change of BRET ratio was to represent the interaction between YFP‐labeled AT1R with RLuc‐β‐arrestin1 or β‐arrestin2. The BRET ratio with 1 μmol/L Ang II for 10 minutes was set to 100%. As concentration increased, the relative change of BRET ratio also increased upon Ang II (EC50β‐arrestin1/2=6.7±4.1/6.3±2.3 nmol/L). However, for AT1‐AA‐positive IgG, there was no significant change (C). Over time, AT1‐AA‐positive IgG (1 μmol/L) attenuated the recruitment of β‐arrestin1 and β‐arrestin2 more than Ang II did (D). (Data are presented as mean±SEM, n=4, *P<0.05 vs 0 minute in Ang II group, 1‐way ANOVA with Bonferroni post hoc test). Ang II indicates angiotensin II; AT1‐AA, angiotensin II type 1 receptor autoantibody; AT1R, angiotensin II type 1 receptor; BRET, bioluminescence resonance energy transfer; RFP, red fluorescence protein; YFP, yellow fluorescence protein.
To further confirm the AT1‐AA‐induced recruitment of β‐arrestin1/2, we recorded the relative change of BRET ratio of the interaction between YFP‐labeled AT1R with Rluc‐labeled β‐arrestin1/2. When the HEK293 cells co‐expressing YFP‐labeled AT1R and RLuc β‐arrestin1 or β‐arrestin2 were exposed to Ang II, the relative change of BRET ratio increased significantly with concentration (Figure 4C, EC50β‐arrestin1/2=6.7±4.1/6.3±2.3 nmol/L) and over time (Figure 4D, the BRET ratio with Ang II for 10 minutes set to 100%; 0 [β‐arrestin1/2: 0.1±10.9/0.0±3.7], 2 [89.1±10.4/89.7±17.9], 5 [107.1±13.2/102.4±14.6], 10 [100.0±11.4/100.0±9.1], 20 [93.5±19.5/111.7±10.0], and 30 minutes [95.8±12.2/93.9±12.1]). However, AT1‐AA‐positive IgG did not attenuate the recruitment of β‐arrestin1/2 either with the concentration (Figure 4D, not converged) or with the time (Figure 4E, 0 [β‐arrestin1/2: 4.5±7.0/−1.4±5.8], 2 [11.1±11.9/18.6±14.7], 5 [30.8±16.4/22.8±16.6], 10 [15.4±13.8/18.0±15.0], 20 [−0.5±19.4/3.3±17.2], and 30 minutes [8.4±12.8/−9.3±9.5]).
Limited AT1R Internalization Was Involved in Sustained Receptor Activation Induced by AT1‐AA‐Positive IgG
To determine whether limited AT1R internalization induced by AT1‐AA‐positive IgG was involved in sustained vasoconstriction, we investigated whether Ang II can simulate AT1‐AA‐positive IgG‐induced sustaining effect by preincubating our samples with an endocytosis inhibitor (Dynasore). Dynasore has been reported to inhibit internalization through blocking the GTPase activity of dynamin.25 As shown in Figures S6 and S7, 100 μmol/L Dynasore alone had no effect on the AT1R internalization (Figure S6) and vasoconstriction in the vascular rings (Figure S7C); but preincubation with 100 μmol/L Dynasore for 30 minutes significantly prolonged the duration of Ang II‐induced vasoconstriction and AT1R activation compared with Ang II only (Figure S7), indicating the important role of AT1R internalization in sustained AT1R activation and vasoconstriction.
Furthermore, we investigated whether the sustaining effect induced by AT1‐AA‐positive IgG can be reversed by overexpression of β‐arrestin2, which promoted AT1R internalization (Figure 5A). As shown in Figure 5, overexpression of β‐arrestin2 shortened the duration of PKC and ERK1/2 phosphorylation induced by AT1‐AA‐positive IgG, and increased [Ca2+]i, suggesting that limited AT1R internalization played a key role in sustained AT1R activation induced by AT1‐AA‐positive IgG.
Figure 5.
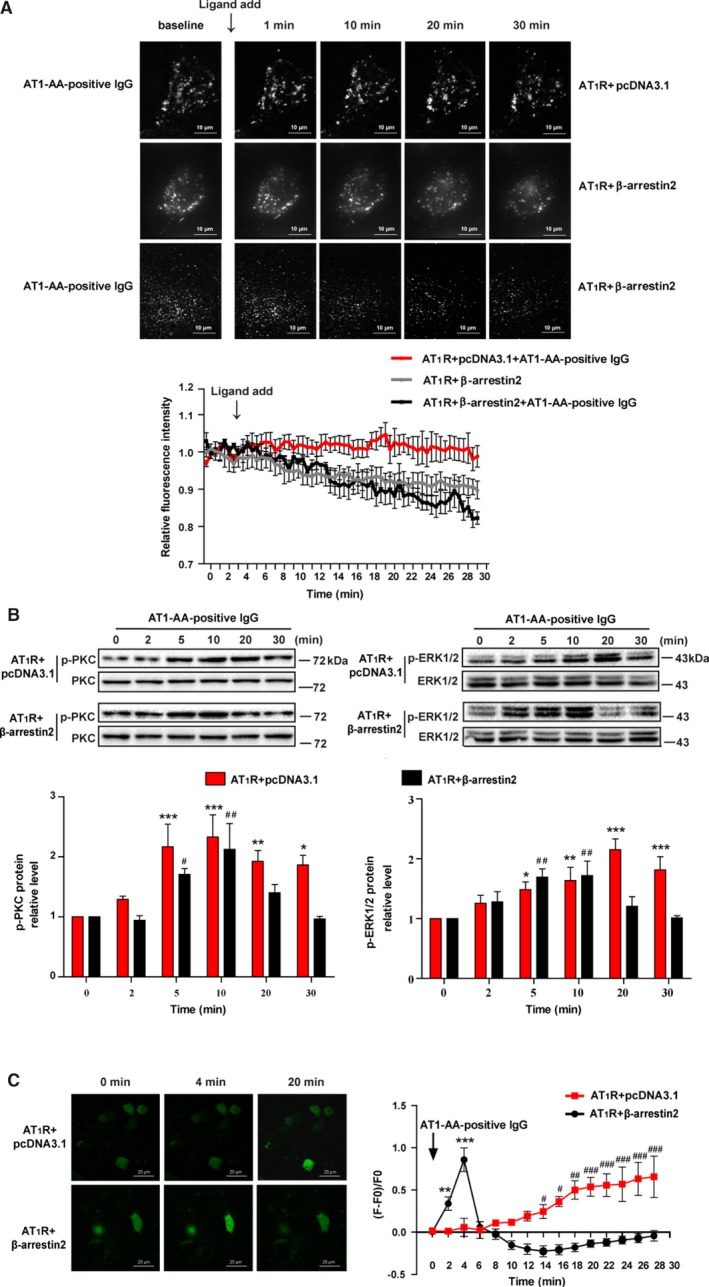
Overexpression of β‐arrestin2 abolished the AT1R sustained activation induced by AT1‐AA‐positive IgG. The representative images and line chart of AT1R internalization by TIRF after overexpression of β‐arrestin2 (1.5 μg) in HEK293 cell are shown (A). The co‐expression of RFP‐AT1R (0.5 μg) and pcDNA3.1 (1.5 μg) served as a control. After overexpression of β‐arrestin2, the AT1R internalization was increased in both the absence and presence of AT1‐AA‐positive IgG. In addition, the duration of PKC and ERK1/2 phosphorylation (B), and [Ca2+]i elevation, induced by AT1‐AA‐positive IgG was shortened (C) (data are presented as mean±SEM, n=4, *P<0.05, **P<0.01, ***P<0.001 vs 0 minute in the control group; # P<0.05, ## P<0.01, ### P<0.001 vs 0 minute in the β‐arrestin2 overexpression group, 1‐way ANOVA with Bonferroni post hoc test was used to test the signal changes of phosphorylated PKC and ERK1/2, 1‐way repeated‐measures ANOVA was used to test the signal changes of [Ca2+]i). AT1‐AA indicates angiotensin II type 1 receptor autoantibody; AT1R, angiotensin II type 1; ERK1/2, extracellular signal‐regulated kinase 1 and 2; PKC, protein kinase C; RFP, red fluorescence protein; TIRF, total internal reflection fluorescence.
Discussion
Vasospasm is an important pathological feature of hypertension. In this study, our main finding was that AT1‐AA‐positive IgG isolated from the sera of preeclamptic patients reduced the amount of AT1R internalization by the inhibition of β‐arrestin1/2 recruitment compared with the Ang II treatment group, which played a key role in receptor overactivation and sustained vasoconstriction.
AT1‐AA‐positive IgG was first found in the plasma of preeclamptic patients.9 A growing number of clinical researchers have also detected high titers of AT1‐AA in several other kinds of hypertension, such as malignant hypertension,26 renal‐transplantation hypertension,27 and essential hypertension.28 Another study found that both total IgG and affinity‐purified AT1‐AAs from women with preeclampsia resulted in the appearance of key features of preeclampsia in mice, including hypertension, proteinuria, and glomerular endotheliosis (a classical renal lesion of pre‐eclampsia),10 which might be related to its effect of sustained vasoconstriction, suggesting a potential role of AT1‐AA‐positive IgG in the pathogenesis of high blood pressure. Therefore, this study aimed to investigate the molecular mechanism underlying sustained vasoconstriction induced by AT1‐AA‐positive IgG.
In the current study, the data showed that AT1‐AA‐positive IgG induced sustained vasoconstriction by mainly acting on VSMCs. Ang II can activate signals downstream of AT1R: [Ca2+]i elevation29 and PKC7 and ERK1/2 phosphorylation.24 [Ca2+]i is directly responsible for cell contraction, the activated PKC increases [Ca2+]i regulates cell proliferation, and ERK1/2 implicates the growth.24 Compared with Ang II‐induced downstream signals, we found that AT1‐AA‐positive IgG‐induced downstream signals were significantly prolonged, which might contribute to vasoconstriction and vascular remodeling and thus lead to hypertension in preeclampsia.
Under normal physiological conditions, the receptor activates downstream signals after receiving external stimuli and exerts corresponding biological effects. In order to avoid overactivation of receptors, receptor internalization is induced when exposed to the external stimuli for a long time.12 In this study, we first determined that AT1‐AA‐positive IgG did not cause a significantly greater decrease in membrane AT1R expression over time than Ang II by 3 methods, respectively: subcellular protein fractionation, cell surface protein biotinylation, and TIRF microscopy. Each of the 3 methods had its advantages and disadvantages. Subcellular protein fractionation in VSMCs is highly reliable, but there is a certain lack of accuracy; the biotin labeling method can achieve accurate quantification but does not permit real‐time observation; and while TIRF microscopy has the advantages of real‐time observation, low background signal, and quantitative calculation, the observed phenomenon does not represent 100% of the receptors in VSMCs because of the need for fluorescently fused receptors and instrumental cells. Therefore, this study combined these 3 methods to verify that AT1‐AA‐positive IgG limited the AT1R internalization. Another study supported this view partly: they observed that β‐1 adrenergic receptor autoantibodies significantly slowed receptor internalization,30 which might result in β‐1 adrenergic receptor nondesensitization.31 In addition, a large number of studies have found that AT1‐AA‐positive IgG increased the sensitivity of Ang II,32, 33 but the mechanism is not fully understood at present. Combined with our current findings in this study, we speculated that the presence of AT1‐AA limited receptor internalization and increased the potential for agonist binding to AT1R upon restimulation with Ang II, disturbing receptor desensitization.
The recruitment of β‐arrestin1/2 to AT1R plays a key role in AT1R internalization and desensitization, which can promote the separation of G proteins from receptors and thereby terminate signaling.16 In this study, using fluorescent tracing and BRET technique, unlike Ang II, we first found that AT1‐AA‐positive IgG cannot significantly induce the recruitment of β‐arrestin1/2. These data indicated that AT1R might undergo different conformational changes upon Ang II and AT1‐AA‐positive IgG. It has been reported that different conformational changes of receptors lead to distinct conformations of β‐arrestin to mediate receptor desensitization, signaling, and endocytosis.34 The cause of these different conformational changes of AT1R may be that the AT1‐AA‐binding site of AT1R is different from that of Ang II. It is reported that the AT1‐AA‐binging site is located in the extracellular loop (165–191: IHRNVFFIINTNITVCAFHYESQNSTL), and the more accurate epitope is a heptapeptide (AFHYESQ),9, 10, 35 but the Ang II binding site of AT1R was at the extracellular side between helices VI and VII.36 However, the reasons why AT1‐AA‐positive IgG inhibited β‐arrestin1/2 recruitment deserves further study.
To investigate the role of limited AT1R internalization in sustained activation and vasoconstriction, we administered an endocytosis inhibitor.25 Dynasore before Ang II treatment. As expected, preincubation with Dynasore significantly prolonged the duration of Ang II‐caused [Ca2+]i elevation and PKC and ERK1/2 phosphorylation. Moreover, it prolonged Ang II‐caused vasoconstriction. Then as reported by others, we overexpressed the β‐arrestin2 to promote the limited internalization.37 As shown in Figure 5A, overexpression of β‐arrestin2 can promote constitutive AT1R internalization in the absence of agonist. Moreover, β‐arrestin2 overexpression shortened the duration of AT1‐AA‐positive IgG‐induced [Ca2+]i elevation and PKC and ERK1/2 phosphorylation, suggesting that the sustained activation of the AT1R can be prevented by promoting receptor internalization. However, it remains possible that overexpression of β‐arrestin2 inhibited the AT1R signaling partly by blocking G‐protein recruitment.16 In the next study, we will spend more time establishing the respective contribution of the receptor internalization and G protein uncoupling to receptor desensitization, because of the complicated relationship between the 2 effects.
In summary, these findings confirmed that limited internalization of AT1R induced by AT1‐AA‐positive IgG contributed to persistent receptor activation and vascular constriction, which might be involved in the occurrence of hypertension. In addition, promotion of AT1R internalization partially inhibited AT1‐AA‐positive IgG‐mediated pathological response. It has been reported that the promotion of constitutive AT1R internalization by modified AT1R‐associated protein expression can inhibit Ang II‐mediated pathological response in mouse distal convoluted cells and plays a key role in the modulation of blood pressure.38 For this reason, we surmised that promotion of AT1R internalization might be a target of therapeutic approach for hypertension.
Limitations and Perspectives
Our study here demonstrated that AT1‐AA‐positive IgG directly combined with AT1R, and then limited AT1R internalization by attenuating the recruitment of β‐arrestin1/2, which might explain the molecular mechanism of AT1R overactivation and sustained vasoconstriction. These data strongly suggested that AT1‐AA was a risk factor worthy of attention in the pathological process of cardiovascular disease induced by AT1R overactivation and sustained vasoconstriction. However, we did not investigate the effect of AT1‐AA‐positive IgG from each patient individually, because of the insufficient number of antibodies. In the next step, we will pay more attention to the heterogeneity of AT1‐AA. Additionally, there is no valid treatment for preeclampsia patients except termination of pregnancy because AT1R blocker can cause severe kidney maldevelopment in the fetus.17, 18 From our observations, AT1‐AA was biased towards activating the Gq‐mediated signaling rather than β‐arrestin‐mediated internalization, resulting in sustained AT1R activation and vasoconstriction. AT1‐AA‐positive IgG‐induced pathological response can be rescued by promoting internalization, suggesting that it might be possible to provide a new perspective for treatment of hypertension from AT1R internalization.
Sources of Funding
This work was supported by grants from the Major Research Plan of the National Natural Science Foundation of China (grant no. 91539205) and the National Natural Science Foundation of China (NSFC, grant no. 81770393) to Liu, NSFC (grant no. 31771267) to Zhang, and NSFC (grant no. 81471478) to Yang.
Disclosures
None.
Supporting information
Data S1. Supplemental Methods
Table S1. Clinical Profiles of Patients and Healthy Controls (Normal‐Pregnancy Group)
Figure S1. AT1‐AA levels were significantly greater in preeclamptic patients. A, The P/N value of pregnant women was detected by modified ELISA, the dotted line represents the value of 2.1 (***P<0.001 vs normal pregnancy). B, The positive rate of AT1‐AA both in normal pregnancy and preeclampsia; χ2 test was used to compare preeclampsia with normal pregnancy group (***P<0.001 vs normal pregnancy).
Figure S2. The AT1‐AA and AT1‐AA‐positive IgG were compared. A, The combining capacity with AT1R‐ECII was detected by ELISA, the numerals 1, 2, 3, 4, and 5 represent AT1‐AA‐positive IgG, AT1‐AA, negative control, positive control, and blank control, respectively. B, The purity was analyzed using SDS‐PAGE the characters M, 1, and 2 represent marker, AT1‐AA‐positive IgG, and AT1‐AA, respectively. C, The biological activity was compared by intracellular Ca2+ detection (n=3).
Figure S3. AT1‐AA‐positive IgG had high capacity to combine with human AT1R‐ECII. AT1‐AA‐positive and negative IgG were diluted with a serial concentration and detected the amount of binding with AT1R‐ECII by ELISA. The OD value (405 nm) was positively correlated with the degree of binding. By fitting the curve and selecting a 95% CI, 2 dashed lines divided the image into 4 intervals. Horizontal dashed line: confidence limit of background scatter. Vertical dashed line: specificity cut‐off.
Figure S4. Intact and damaged endothelia of vascular rings were identified. A, Acetylcholine exerted concentration‐dependent relaxation effects against norepinephrine‐induced contractions of arterial rings with intact endothelia. B, SNP exerted relaxation effects against NE‐induced contractions of vascular rings with their endothelia removed, whereas Ach did not show concentration‐dependent relaxation effects. Ach indicates acetylcholine; NE, norepinephrine; SNP, sodium nitroprusside.
Figure S5. VSMCs were identified. A, VSMCs successfully developed from the aortic tissue of rats. B, The cultured cells were immunofluorescence stained for markers of VSMCs (α‐SMA and calponin). α‐SMA indicates α‐smooth muscle actin; VSMCs, vascular smooth muscle cells.
Figure S6. The endocytic inhibitor Dynasore abolished Ang II‐induced AT1R internalization. Representative images and line chart of membrane AT1R trafficking by TIRF are shown. The images were acquired at 1 frame/s for 33 minutes. The ligand was added at the 3‐minute mark. Data show that administration of Ang II did not induce AT1R internalization after 30 minutes of pre‐incubation with Dynasore. AT1R indicates angiotensin II type 1; TIRF, total internal reflection fluorescence.
Figure S7. Ang II induced persistent AT1R activation and sustained vasoconstriction after endocytosis inhibition. We pre‐incubated an endocytosis inhibitor, Dynasore, with a vascular ring or VSMCs before administering Ang II. A, With Dynasore pre‐incubation, Ang II prolonged the duration of PKC and ERK1/2 hyperphosphorylation (n=6; *P<0.05 vs 0 minute). B, With Dynasore pre‐incubation, Ang II prolonged [Ca2+]i elevation (n=5; * P<0.05;** P<0.01 vs baseline; # P<0.05 vs Ang II group,). C, With Dynasore pre‐incubation, Ang II induced prolonged vasoconstriction (n=6, ** P<0.01 vs Ang II group); ns indicates not significant. Ang II indicates angiotensin II; AT1R, angiotensin II type 1 receptor; ERK1/2, extracellular signal‐regulated kinase 1 and 2; PKC, protein kinase C; VSMCs, vascular smooth muscle cells.
Video S1. The [Ca2+]i increased in response to stimulation with Ang II (1 μmol/L).
Video S2. The [Ca2+]i increased in response to stimulation with AT1‐AA (1 μmol/L).
Video S3. The internalization of AT1R in transiently transfected cells by RFP‐fused AT1R upon Ang II (1 μmol/L) stimulation. The red puncta represent the RFP‐fused AT1R on the membrane.
Video S4. The internalization of AT1R in transiently transfected cells by RFP‐fused AT1R upon AT1‐AA (1 μmol/L) stimulation. Best viewed with Windows Media Player.
Acknowledgments
We would like to thank Taiyuan Central Hospital, Shanxi, China, for helping with clinical‐sample collection. We also thank Professor Jinpeng Sun (Shandong University School) and Professor Haihong Ye (Capital Medical University) for providing us with the plasmid and making suggestions on this study. We would like to thank Wang (Shanxi Medical University) and Zimulinda Mukurarinda U. Victorien (Capital Medical University), for providing language help.
(J Am Heart Assoc. 2019;8:e011179 DOI: 10.1161/JAHA.118.011179.)
Contributor Information
Suli Zhang, Email: sueney716@126.com.
Huirong Liu, Email: liuhr2000@ccmu.edu.cn.
References
- 1. Lambert G, Brichant JF, Hartstein G, Bonhomme V, Dewandre PY. Preeclampsia: an update. Acta Anaesthesiol Belg. 2014;65:137–149. [PubMed] [Google Scholar]
- 2. WHO . WHO Recommendations for Prevention and Treatment of Pre‐Eclampsia and Eclampsia. Geneva: World Health Organization; 2011. [PubMed] [Google Scholar]
- 3. Huppertz B. Placental origins of preeclampsia: challenging the current hypothesis. Hypertension. 2008;51:970–975. [DOI] [PubMed] [Google Scholar]
- 4. Hypertension in pregnancy . Report of the American College of obstetricians and gynecologists’ task force on hypertension in pregnancy. Obstet Gynecol. 2013;122:1122–1131. [DOI] [PubMed] [Google Scholar]
- 5. Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre‐eclampsia. Lancet. 2010;376:631–644. [DOI] [PubMed] [Google Scholar]
- 6. Te RL, van Esch JH, Roks AJ, van den Meiracker AH, Danser AH. Hypertension: renin‐angiotensin‐aldosterone system alterations. Circ Res. 2015;116:960–975. [DOI] [PubMed] [Google Scholar]
- 7. Goodfriend TL, Elliott ME, Catt KJ. Angiotensin receptors and their antagonists. N Engl J Med. 1996;334:1649–1654. [DOI] [PubMed] [Google Scholar]
- 8. Spat A, Enyedi P, Hajnoczky G, Hunyady L. Generation and role of calcium signal in adrenal glomerulosa cells. Exp Physiol. 1991;76:859–885. [DOI] [PubMed] [Google Scholar]
- 9. Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103:945–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, Popek EJ, Hicks MJ, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibodies induce pre‐eclampsia in pregnant mice. Nat Med. 2008;14:855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang S, Zheng R, Yang L, Zhang X, Zuo L, Yang X, Bai K, Song L, Tian J, Yang J, Liu H. Angiotensin type 1 receptor autoantibody from preeclamptic patients induces human fetoplacental vasoconstriction. J Cell Physiol. 2013;228:142–148. [DOI] [PubMed] [Google Scholar]
- 12. Torres‐Tirado D, Ramiro‐Diaz J, Knabb MT, Rubio R. Molecular weight of different angiotensin II polymers directly determines: density of endothelial membrane AT1 receptors and coronary vasoconstriction. Vascul Pharmacol. 2013;58:346–355. [DOI] [PubMed] [Google Scholar]
- 13. Hein L, Meinel L, Pratt RE, Dzau VJ, Kobilka BK. Intracellular trafficking of angiotensin II and its AT1 and AT2 receptors: evidence for selective sorting of receptor and ligand. Mol Endocrinol. 1997;11:1266–1277. [DOI] [PubMed] [Google Scholar]
- 14. Jean‐Charles PY, Kaur S, Shenoy SK. G protein‐coupled receptor signaling through beta‐arrestin‐dependent mechanisms. J Cardiovasc Pharmacol. 2017;70:142–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goodman OJ, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. Beta‐arrestin acts as a clathrin adaptor in endocytosis of the beta2‐adrenergic receptor. Nature. 1996;383:447–450. [DOI] [PubMed] [Google Scholar]
- 16. Thomas WG, Qian H. Arresting angiotensin type 1 receptors. Trends Endocrinol Metab. 2003;14:130–136. [DOI] [PubMed] [Google Scholar]
- 17. Vendemmia M, Garcia‐Meric P, Rizzotti A, Boubred F, Lacroze V, Liprandi A, Simeoni U. Fetal and neonatal consequences of antenatal exposure to type 1 angiotensin II receptor‐antagonists. J Matern Fetal Neonatal Med. 2005;18:137–140. [DOI] [PubMed] [Google Scholar]
- 18. Daikha‐Dahmane F, Levy‐Beff E, Jugie M, Lenclen R. Foetal kidney maldevelopment in maternal use of angiotensin II type I receptor antagonists. Pediatr Nephrol. 2006;21:729–732. [DOI] [PubMed] [Google Scholar]
- 19. Ueki N, Takeda S, Koya D, Kanasaki K. The relevance of the Renin‐Angiotensin system in the development of drugs to combat preeclampsia. Int J Endocrinol. 2015;2015:572713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang X, Wang F, Chang H, Zhang S, Yang L, Wang X, Cheng X, Zhang M, Ma XL, Liu H. Autoantibody against AT1 receptor from preeclamptic patients induces vasoconstriction through angiotensin receptor activation. J Hypertens. 2008;26:1629–1635. [DOI] [PubMed] [Google Scholar]
- 21. Kuruvilla R, Zweifel LS, Glebova NO, Lonze BE, Valdez G, Ye H, Ginty DD. A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of TrkA trafficking and retrograde signaling. Cell. 2004;118:243–255. [DOI] [PubMed] [Google Scholar]
- 22. Levy‐Toledano R, Caro LH, Hindman N, Taylor SI. Streptavidin blotting: a sensitive technique to study cell surface proteins; application to investigate autophosphorylation and endocytosis of biotin‐labeled insulin receptors. Endocrinology. 1993;133:1803–1808. [DOI] [PubMed] [Google Scholar]
- 23. Fu ML, Leung PS, Wallukat G, Bergstrom G, Fu H, Schulze W, Herlitz H. Agonist‐like activity of antibodies to angiotensin II receptor subtype 1 (AT1) from rats immunized with AT1 receptor peptide. Blood Press. 1999;8:317–324. [DOI] [PubMed] [Google Scholar]
- 24. Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S. Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin Sci (Lond). 2007;112:417–428. [DOI] [PubMed] [Google Scholar]
- 25. Dutta D, Donaldson JG. Search for inhibitors of endocytosis: intended specificity and unintended consequences. Cell Logist. 2012;2:203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fu ML, Herlitz H, Schulze W, Wallukat G, Micke P, Eftekhari P, Sjogren KG, Hjalmarson A, Muller‐Esterl W, Hoebeke J. Autoantibodies against the angiotensin receptor (AT1) in patients with hypertension. J Hypertens. 2000;18:945–953. [DOI] [PubMed] [Google Scholar]
- 27. Dragun D, Muller DN, Brasen JH, Fritsche L, Nieminen‐Kelha M, Dechend R, Kintscher U, Rudolph B, Hoebeke J, Eckert D, Mazak I, Plehm R, Schonemann C, Unger T, Budde K, Neumayer HH, Luft FC, Wallukat G. Angiotensin II type 1‐receptor activating antibodies in renal‐allograft rejection. N Engl J Med. 2005;352:558–569. [DOI] [PubMed] [Google Scholar]
- 28. Zhu F, Sun Y, Wang M, Ma S, Chen X, Cao A, Chen F, Qiu Y, Liao Y. Correlation between HLA‐DRB1, HLA‐DQB1 polymorphism and autoantibodies against angiotensin AT(1) receptors in Chinese patients with essential hypertension. Clin Cardiol. 2011;34:302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li L, Pang XB, Chen BN, Gao L, Wang L, Wang SB, Wang SB, Liu DP, Du GH. Pinocembrin inhibits angiotensin II‐induced vasoconstriction via suppression of the increase of [Ca2+]i and ERK1/2 activation through blocking AT(1)R in the rat aorta. Biochem Biophys Res Commun. 2013;435:69–75. [DOI] [PubMed] [Google Scholar]
- 30. Bornholz B, Weidtkamp‐Peters S, Schmitmeier S, Seidel CA, Herda LR, Felix SB, Lemoine H, Hescheler J, Nguemo F, Schafer C, Christensen MO, Mielke C, Boege F. Impact of human autoantibodies on beta1‐adrenergic receptor conformation, activity, and internalization. Cardiovasc Res. 2013;97:472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Magnusson Y, Wallukat G, Waagstein F, Hjalmarson A, Hoebeke J. Autoimmunity in idiopathic dilated cardiomyopathy. Characterization of antibodies against the beta 1‐adrenoceptor with positive chronotropic effect. Circulation. 1994; 89:2760–2767. [DOI] [PubMed] [Google Scholar]
- 32. Wenzel K, Rajakumar A, Haase H, Geusens N, Hubner N, Schulz H, Brewer J, Roberts L, Hubel CA, Herse F, Hering L, Qadri F, Lindschau C, Wallukat G, Pijnenborg R, Heidecke H, Riemekasten G, Luft FC, Muller DN, Lamarca B, Dechend R. Angiotensin II type 1 receptor antibodies and increased angiotensin II sensitivity in pregnant rats. Hypertension. 2011;58:77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cunningham MJ, Williams JM, Amaral L, Usry N, Wallukat G, Dechend R, LaMarca B. Agonistic autoantibodies to the angiotensin II type 1 receptor enhance angiotensin II‐induced renal vascular sensitivity and reduce renal function during pregnancy. Hypertension. 2016;68:1308–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cahill TR, Thomsen AR, Tarrasch JT, Plouffe B, Nguyen AH, Yang F, Huang LY, Kahsai AW, Bassoni DL, Gavino BJ, Lamerdin JE, Triest S, Shukla AK, Berger B, Little JT, Antar A, Blanc A, Qu CX, Chen X, Kawakami K, Inoue A, Aoki J, Steyaert J, Sun JP, Bouvier M, Skiniotis G, Lefkowitz RJ. Distinct conformations of GPCR‐beta‐arrestin complexes mediate desensitization, signaling, and endocytosis. Proc Natl Acad Sci USA. 2017;114:2562–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tian M, Sheng L, Huang P, Li J, Zhang CH, Yang J, Liao YH, Li LD. Agonistic autoantibodies against the angiotensin AT1 receptor increase in unstable angina patients after stent implantation. Coron Artery Dis. 2014;25:691–697. [DOI] [PubMed] [Google Scholar]
- 36. Balakumar P, Jagadeesh G. Structural determinants for binding, activation, and functional selectivity of the angiotensin AT1 receptor. J Mol Endocrinol. 2014;53:R71–R92. [DOI] [PubMed] [Google Scholar]
- 37. Zhang X, Wang F, Chen X, Li J, Xiang B, Zhang YQ, Li BM, Ma L. Beta‐arrestin1 and beta‐arrestin2 are differentially required for phosphorylation‐dependent and ‐independent internalization of delta‐opioid receptors. J Neurochem. 2005;95:169–178. [DOI] [PubMed] [Google Scholar]
- 38. Tamura K, Wakui H, Azushima K, Uneda K, Haku S, Kobayashi R, Ohki K, Haruhara K, Kinguchi S, Matsuda M, Yamashita A, Umemura S. Angiotensin II type 1 receptor binding molecule ATRAP as a possible modulator of renal sodium handling and blood pressure in pathophysiology. Curr Med Chem. 2015;22:3210–3216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Methods
Table S1. Clinical Profiles of Patients and Healthy Controls (Normal‐Pregnancy Group)
Figure S1. AT1‐AA levels were significantly greater in preeclamptic patients. A, The P/N value of pregnant women was detected by modified ELISA, the dotted line represents the value of 2.1 (***P<0.001 vs normal pregnancy). B, The positive rate of AT1‐AA both in normal pregnancy and preeclampsia; χ2 test was used to compare preeclampsia with normal pregnancy group (***P<0.001 vs normal pregnancy).
Figure S2. The AT1‐AA and AT1‐AA‐positive IgG were compared. A, The combining capacity with AT1R‐ECII was detected by ELISA, the numerals 1, 2, 3, 4, and 5 represent AT1‐AA‐positive IgG, AT1‐AA, negative control, positive control, and blank control, respectively. B, The purity was analyzed using SDS‐PAGE the characters M, 1, and 2 represent marker, AT1‐AA‐positive IgG, and AT1‐AA, respectively. C, The biological activity was compared by intracellular Ca2+ detection (n=3).
Figure S3. AT1‐AA‐positive IgG had high capacity to combine with human AT1R‐ECII. AT1‐AA‐positive and negative IgG were diluted with a serial concentration and detected the amount of binding with AT1R‐ECII by ELISA. The OD value (405 nm) was positively correlated with the degree of binding. By fitting the curve and selecting a 95% CI, 2 dashed lines divided the image into 4 intervals. Horizontal dashed line: confidence limit of background scatter. Vertical dashed line: specificity cut‐off.
Figure S4. Intact and damaged endothelia of vascular rings were identified. A, Acetylcholine exerted concentration‐dependent relaxation effects against norepinephrine‐induced contractions of arterial rings with intact endothelia. B, SNP exerted relaxation effects against NE‐induced contractions of vascular rings with their endothelia removed, whereas Ach did not show concentration‐dependent relaxation effects. Ach indicates acetylcholine; NE, norepinephrine; SNP, sodium nitroprusside.
Figure S5. VSMCs were identified. A, VSMCs successfully developed from the aortic tissue of rats. B, The cultured cells were immunofluorescence stained for markers of VSMCs (α‐SMA and calponin). α‐SMA indicates α‐smooth muscle actin; VSMCs, vascular smooth muscle cells.
Figure S6. The endocytic inhibitor Dynasore abolished Ang II‐induced AT1R internalization. Representative images and line chart of membrane AT1R trafficking by TIRF are shown. The images were acquired at 1 frame/s for 33 minutes. The ligand was added at the 3‐minute mark. Data show that administration of Ang II did not induce AT1R internalization after 30 minutes of pre‐incubation with Dynasore. AT1R indicates angiotensin II type 1; TIRF, total internal reflection fluorescence.
Figure S7. Ang II induced persistent AT1R activation and sustained vasoconstriction after endocytosis inhibition. We pre‐incubated an endocytosis inhibitor, Dynasore, with a vascular ring or VSMCs before administering Ang II. A, With Dynasore pre‐incubation, Ang II prolonged the duration of PKC and ERK1/2 hyperphosphorylation (n=6; *P<0.05 vs 0 minute). B, With Dynasore pre‐incubation, Ang II prolonged [Ca2+]i elevation (n=5; * P<0.05;** P<0.01 vs baseline; # P<0.05 vs Ang II group,). C, With Dynasore pre‐incubation, Ang II induced prolonged vasoconstriction (n=6, ** P<0.01 vs Ang II group); ns indicates not significant. Ang II indicates angiotensin II; AT1R, angiotensin II type 1 receptor; ERK1/2, extracellular signal‐regulated kinase 1 and 2; PKC, protein kinase C; VSMCs, vascular smooth muscle cells.
Video S1. The [Ca2+]i increased in response to stimulation with Ang II (1 μmol/L).
Video S2. The [Ca2+]i increased in response to stimulation with AT1‐AA (1 μmol/L).
Video S3. The internalization of AT1R in transiently transfected cells by RFP‐fused AT1R upon Ang II (1 μmol/L) stimulation. The red puncta represent the RFP‐fused AT1R on the membrane.
Video S4. The internalization of AT1R in transiently transfected cells by RFP‐fused AT1R upon AT1‐AA (1 μmol/L) stimulation. Best viewed with Windows Media Player.