

Summary
Covalent DNA-protein cross-links (DPCs) impede replication fork progression and threaten genome integrity. Using Xenopus egg extracts, we previously showed that replication fork collision with DPCs causes their proteolysis, followed by translesion DNA synthesis. We show here that when DPC proteolysis is blocked, the replicative DNA helicase CMG (CDC45, MCM2–7, GINS), which travels on the leading strand template, bypasses an intact leading strand DPC. Single molecule imaging reveals that GINS does not dissociate from CMG during bypass, and CMG slows dramatically after bypass, likely due to uncoupling from the stalled leading strand. The DNA helicase RTEL1 facilitates bypass, apparently by generating singlestranded DNA beyond the DPC. The absence of RTEL1 impairs DPC proteolysis, suggesting that CMG must bypass the DPC to enable proteolysis. Our results suggest a mechanism that prevents inadvertent CMG destruction by DPC proteases, and they reveal CMG’s remarkable capacity to overcome obstacles on its translocation strand.
Graphical Abstract
A collaboration between helicases ensures that DNA damage in the form of DNA-protein crosslinks doesn’t derail replication, helping to preserve genome stability.
Introduction
DNA replication forks encounter many obstacles that challenge genome duplication. Discrete DNA lesions (e.g. pyrimidine dimers) stall the replicative DNA polymerase but not the helicase, leading to helicase-polymerase uncoupling (Byun et al., 2005; Taylor and Yeeles, 2018). Bulkier obstacles block the entire replisome, including the helicase. These include DNA interstrand cross-links (ICLs; (Fu et al., 2011)) and DNA protein cross-links (DPCs; (Duxin et al., 2014)). DPCs are formed by ultraviolet light, various chemotherapeutics (e.g. cisplatin), and by endogenous agents such as formaldehyde and topoisomerases (Ide et al., 2011; Stingele et al., 2017). Non-covalent nucleoprotein complexes (e.g. RNA polymerases and tightly bound transcription factors) also interfere with replication fork progression.
Our understanding of DPC repair is evolving rapidly. Early studies showed that DPCs smaller than ~8 kD are excised by nucleotide excision repair whereas larger DPCs require more complex pathways (Ide et al., 2011). In 2014, yeast Wss1 was identified as a DNA-dependent protease that degrades DPCs in S phase (Stingele et al., 2014). Contemporaneous experiments in Xenopus egg extracts showed that when a replication fork collides with a DPC on the leading (DPCLead) or lagging strand template (DPCLag), an unknown protease degrades the DPC to a short peptide adduct that is bypassed by translesion DNA polymerases (Figure 1A) (Duxin et al., 2014). Therefore, a protease-mediated DPC repair pathway exists that does not involve a double-strand break intermediate or recombination. Like Wss1, its vertebrate ortholog SPRTN (also named Spartan or DVC1) is a DNA-dependent protease (Lopez-Mosqueda et al., 2016; Stingele et al., 2016; Vaz et al., 2016). The protease activity of SPRTN suppresses genome instability by removing DPCs that block replication fork progression (Lessel et al., 2014; Maskey et al., 2014; Vaz et al., 2016), and SPRTN mutations cause Ruijs-Aalfs syndrome (RJALS), a human genetic disease characterized by genome instability, premature aging, and early onset liver cancer (Lessel et al., 2014). In frog egg extracts, SPRTN and the proteasome have overlapping functions in DPC proteolysis (Larsen et al., submitted; manuscript enclosed). In this setting, SPRTN activity requires that a nascent strand be extended to within a few nucleotides of the DPC, whereas proteasome activity requires DPC ubiquitylation and the presence of ssDNA near the adduct. How a DPC is selectively degraded without concomitant destruction of the neighboring, stalled replisome is unknown.
Figure 1. Disappearance of the CMG footprint at DPCLead is unaffected by proteolysis or p97 inhibition.

(A) Previous model of replication-coupled DPC repair (Duxin et al., 2014). (B) Schematic of what happens in the presence of Ub-VS (Duxin et al., 2014). (C) pDPCLead or pmeDPCLead were pre-bound with LacR to prevent one replication fork from reaching the DPC. The plasmids were replicated in mock-depleted or SPRTN-depleted egg extract containing 32P[α]-dATP and supplemented with buffer or the p97 inhibitor NMS-873 (p97i). At different times, DNA was recovered and digested with AatII and FspI, separated on a denaturing polyacrylamide gel, and visualized by autoradiography. Grey inset: Schematic of nascent leading strand products released by AatII and FspI digestion of pmeDPCLead or pDPCLead. The lower autoradiogram shows nascent leading strands generated by the rightward replication fork, and the upper autoradiogram shows both extension products. Blue bracket, CMG footprint (−30 to −37); orange bracket, products stalled at the adducted base (−1 to +1). The percentage of leading strands that approached from the −30 cluster to the −1 cluster was quantified (see methods), and the mean of n=5 experiments is graphed. Error bars represent the standard deviation. See Figure S1E for description of −1 to +12 products in lanes 7–12 and 19–24. (D) pmeDPC2xLead was replicated in SPRTN-depleted egg extracts and supplemented with buffer or p97i. At different times, plasmid-associated proteins were recovered and blotted with the indicated antibodies. Samples were also examined for DPC proteolysis (Figure S1D). A model of CMG unloading from this template is shown in Figure S1G.
Replicative helicases form hexameric rings that unwind DNA by translocating along single stranded DNA (“steric exclusion”)(O’Donnell and Li, 2018). Understanding how these essential motors overcome obstacles is an important question. The bacterial DnaB helicase, which translocates 5’ to 3’ along the lagging strand template, stalls at non-covalent nucleoprotein complexes. To overcome these obstacles, it employs two accessory helicases, UvrD and Rep, which act redundantly (Guy et al., 2009). As 3’ to 5’ helicases, Rep and UvrD assemble on the leading strand template and cooperate with DnaB in overcoming obstacles. Unlike DnaB, the eukaryotic replicative DNA helicase, CMG (a complex of CDC45, MCM2–7, and GINS), encircles and translocates 3’ to 5’ along the leading strand template (Fu et al., 2011). While isolated CMG can bypass a biotin-streptavidin (SA) complex on the lagging strand template with the assistance of MCM10, it cannot overcome a leading strand biotin-SA complex (Langston et al., 2017). Interestingly, the large T antigen DNA helicase can bypass a DPC on the translocation strand, perhaps via transient ring-opening (Yardimci et al., 2012b). Whether CMG progression past nucleoprotein complexes is assisted by accessory helicases is unknown. However, consistent with this idea, in the absence of the 5’ to 3’ helicases Rrm3 and Pif1, replisomes stall at specific locations in yeast cells (Ivessa et al., 2000). These results suggest that, like bacteria, yeast cells overcome obstacles by engaging an accessory DNA helicase that moves on the strand opposite the one hosting CMG.
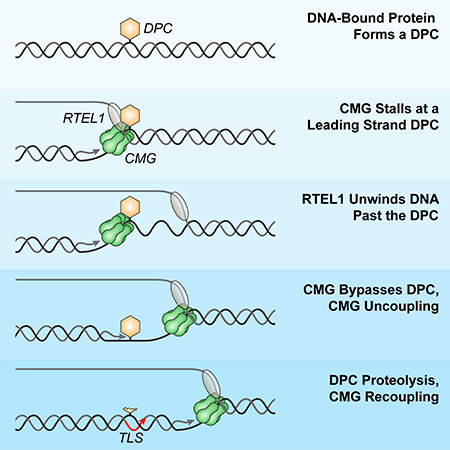
Based on experiments in Xenopus egg extracts, we previously proposed that DPC proteolysis allows the CMG helicase to move past a leading strand DPC (Figure 1A)(Duxin et al., 2014). We now show that CMG can bypass DPCLead in the absence of DPC proteolysis. Single-molecule imaging demonstrates that GINS does not dissociate during bypass and that CMG slows dramatically after bypass, likely due to temporary uncoupling from the leading strand, which stalls at the DPC. The 5’ to 3’ helicase RTEL1 facilitates CMG bypass of the DPC, and bypass is required for efficient DPC proteolysis. RTEL1 also promotes fork progression through non-covalent DNA-protein complexes. Our results require a fundamental revision of the events underlying DPC repair and reveal CMG’s remarkable capacity to overcome nucleoprotein obstacles.
Results
Leading strands rapidly approach a non-degradable DPC
To study the mechanism of DPC repair, we covalently trapped the 45 kDa DNA methyltransferase M.HpaII on its recognition site (CCGG) in a circular plasmid (Figure S1A; (Duxin et al., 2014)). When the resulting plasmid (pDPC) is replicated in Xenopus egg extracts, leading strands stall 30 nucleotides (nt) from the adducted base due to the footprint of the CMG helicase, which translocates along the leading strand template (Figure 1A; (Duxin et al., 2014)). After a pause, synthesis resumes and nascent leading strands approach to within 1 nucleotide of the DPC (“approach”), followed by translesion DNA synthesis (Figure 1A, (Duxin et al., 2014)). Because degradation of the DPC roughly coincides with approach, the results suggested that degradation of the DPC allows CMG to move beyond the adduct (Duxin et al., 2014)(Figure 1A). Surprisingly, however, when DPC proteolysis was blocked via ubiquitin depletion, the nascent leading strand still approached the adduct, albeit very inefficiently, suggesting that CMG eventually vacates its position in front of the DPC (Figure 1B; (Duxin et al., 2014)). Moreover, the nascent lagging strand was ultimately extended beyond the DPC. Therefore, the replication fork can eventually move past an intact DPC, but the fate of CMG in this process was unclear (Figure 1B).
Given the many roles of ubiquitin signaling in the DNA damage response, we wanted to block degradation of the DPC without ubiquitin depletion and examine the effect on approach. In egg extracts, the DPC is degraded redundantly by SPRTN and the proteasome (Figure S1B; Larsen et al., submitted, manuscript enclosed). To inhibit SPRTN, we immunodepleted SPRTN from extracts (Figure S1C); to inhibit the proteasome pathway, we used a DPC whose lysine residues were chemically methylated to prevent ubiquitylation (meDPC; Figure 1C, spheres with bold outlines). Under these conditions, the DPC was not degraded, and we refer to this as a “stable” DPC (Figure S1D (Larsen et al., submitted)). To ensure that a single fork encountered the stable DPC on the leading strand template (“DPCLead”), we flanked the meDPC on the right with 48 tandem copies of the Lac repressor (LacR), which blocks arrival of the leftward, converging fork (grey inset in Figure 1C; (Duxin et al., 2014)). To monitor progress of the rightward leading strand, we digested the DNA with AatII and FspI (Figure 1C; grey inset). Strikingly, leading strands approached the DPC with the same kinetics whether or not proteolysis occurred (Figure 1C; compare lanes 1–6 and 7–12, lower autoradiogram; see graph for quantification). The staggered cuts made by AatII also distinguish leading and lagging strand extension products (Figure 1C; grey inset, pink and purple arrows), which revealed that the lagging strand was readily extended past the stable DPC (Figure 1C, lanes 712, top autoradiogram). Leading strand extension past DPCLead was much slower than lagging strand extension (Figure 1C, lanes 1–6), and the former was further delayed when the DPC was stabilized (Figure 1C, lanes 7–12). Leading strand extension past the stable DPCLead was strongly inhibited by Rev1 depletion, demonstrating a requirement for TLS (Figure S1E). Collectively, these results show that when ubiquitin levels are normal, the CMG footprint disappears from the DPC and the lagging strand is extended past the DPC with the same kinetics whether or not the DPC undergoes proteolysis.
CMG bypasses a stable DPC
The disappearance of the CMG footprint at a stable DPC could be due to CMG dissociation from the DPC (in which case another helicase enables lagging strand extension past the DPC; Figure S1Fi) or CMG bypass of the DPC (Figure S1Fii). To distinguish between these possibilities, we examined approach in the presence of an inhibitor of the p97 ATPase (p97i), which extracts CMG from chromatin during replication termination and ICL repair (Fullbright et al., 2016; Maric et al., 2014; Moreno et al., 2014). If CMG has to dissociate from the stalled replisome to enable approach, p97i should delay approach. As shown in Figure 1C, p97i did not slow the kinetics of leading strand approach to or lagging strand extension past a stable DPC, suggesting that CMG bypassed the DPC. We verified that p97i prevented CMG unloading from a DPC-containing plasmid (Figure 1D), and we infer that such unloading normally occurs after DPC bypass (Figure S1G). DPC bypass did not involve new origin firing (Figure S1H), nor did it require the FANCM ATPase (Figure S1I-J) or ATR signaling (Figure S1K), suggesting bypass is mechanistically distinct from ICL traverse (Huang et al., 2013; Ling et al., 2016). Together, our data strongly suggest that CMG bypasses a bulky DPC on the translocation strand.
ssDNA downstream of a stable DPC facilitates CMG bypass
We speculated that a 5’ to 3’ accessory helicase loads onto the lagging strand template and unwinds DNA past the DPCLead to assist CMG bypass (Figure S2i). To explore this idea, we placed a second meDPC on the lagging strand template 15 nt upstream of meDPCLead (pmeDPCLag/Lead), as this should block unwinding past the DPC by a 5’ to 3’ helicase (Figure S2ii). For comparison, we placed the second DPC 15 nt downstream of the leading strand DPC (pmeDPCLead/Lag) as this should allow some DNA unwinding beyond the DPC (Figure S2iii). Consistent with our hypothesis, CMG bypass was severely inhibited by the upstream but not the downstream lagging strand DPC (Figure 2, lanes 19–24 vs. 25–30; graph for quantification). Strikingly, a 40 nt ssDNA bubble placed downstream of the DPC fully rescued bypass in the presence of the upstream DPCLag, further suggesting that DNA unwinding past the DPC is critical for bypass (Figure 2, lanes 31–36; Figure S2iv). Remarkably, bypass was now even faster than on the DNA template lacking any lagging strand obstruction (Figure 2, compare lanes 31–36 with lanes 7–12). Interestingly, two tandem meDPCs placed 15 nucleotides apart on the leading strand template also severely impaired bypass (Figure 2, lanes 13–18), suggesting that a single CMG cannot simultaneously accommodate two DPCs during bypass. Together, the data suggest that generation of ssDNA downstream of DPCLead is rate-limiting for CMG bypass, consistent with assistance by a 5’ to 3’ helicase.
Figure 2. ssDNA downstream of an intact DPC facilitates CMG bypass.

The indicated plasmids were pre-incubated with LacR, replicated in SPRTN-depleted egg extract, and analyzed as in Figure S1J. Approach was used as a proxy for CMG bypass and quantified as in Figure 1C (see methods). Figure S2 depicts the proposed events on each plasmid. The mean of n=3 experiments is graphed. Error bars represent the standard deviation.
RTEL1 promotes efficient CMG bypass of a stable DPC
Vertebrates contain six 5’ to 3’ DNA helicases (RTEL1, FANCJ, PIF1, DDX3, DDX11, and XPD). Using mass spectrometry (PP-MS), we detected all of these helicases except DDX11 on chromatin in egg extracts (Figure 3A; (Larsen et al., submitted)), and three (PIF1, RTEL1, and FANCJ) bound the plasmid in a replication-dependent fashion. Depletion of PIF1, FANCJ, or DDX3 had no significant effects on bypass of a stable DPCLead (data not shown; we did not examine XPD or DDX11). In contrast, immunodepletion of RTEL1 (Figure 3B; Figure S3A for explanation of RTEL1 isoforms in lane 1 of Figure 3B) caused leading strands to initially stall at a greater distance from the stable DPCLead (Figure 3C, −38 to −44 cluster) and greatly delayed their approach to the adduct (Figure 3C; graph for quantification). Furthermore, RTEL1 depletion delayed lagging strand advance beyond DPCLead (Figure 3D, lanes 6–10, upper autoradiogram). These results indicate that RTEL1 is required for efficient CMG bypass and that bypass allows new Okazaki fragment priming downstream of the adduct. RTEL1 depletion had no effect on the efficiency of DNA synthesis (Figure S3B). CMG bypass was rescued by wild type recombinant RTEL1 but not an ATPase deficient RTEL1 mutant (RTEL1K48R), which further inhibited bypass (Figure 3C; Figure S3C). RTEL1 depletion had only a modest effect on bypass when a single-stranded DNA bubble was placed downstream of the DPC, and it did not further inhibit bypass when meDPCLag was present upstream (Figure S3D). Our data indicate that RTEL1 enables efficient CMG bypass of intact DPCLead by generating ssDNA beyond the adduct. The bypass observed in RTEL1-depleted extracts (Figure 3C) could be due to incomplete RTEL1 depletion, the presence of partially compensating helicases, or helicase-independent bypass. The bypass defect observed when a single fork collided with DPCLead in RTEL1-depleted egg extract was rescued by IPTG, which disrupted the LacR array and allowed a second fork to converge on the DPC (Figure 3E, lanes 18–22 vs. 23–28; graph for quantification). This rescue was not due to CMG unloading as it still occurred in the presence of p97i. We conclude that DPC bypass requires ssDNA beyond the adduct, which can be created by an accessory helicase or a converging replication fork.
Figure 3. RTEL1 is required for efficient CMG bypass.

(A) Recovery of 5’ to 3’ helicases in the mass spectrometry dataset of Larsen et al. (submitted). Relative abundance of each protein in the specified conditions is expressed as a z-score with yellow indicating higher abundance. Where indicated, Geminin was added to block replication initiation. (B) Mock-depleted, RTEL1-depleted, and RTEL1-depleted egg extracts supplemented with wild-type RTEL1 or RTEL1-K48R were blotted with RTEL1 and ORC2 (loading control) antibodies. (C) pmeDPCLead was replicated in the indicated extracts, and supplemented with buffer, wild-type RTEL1, or RTEL1-K48R. Leading strand approach was visualized as in Figure S1J and quantified as in Figure 1C. The mean of n=3 independent experiments is graphed. Error bars represent the standard deviation. (D) pmeDPCLead or pmeDPCLag was replicated in the indicated extracts and analyzed as in Figure 1C. CMG bypass was quantified as in Figure 1C. (E) pmeDPC was replicated in the indicated extracts and supplemented with IPTG (at 5 minutes after replication initiation) and/or p97i, as indicated. Leading strand approach was visualized as in Figure S1J and quantified as in Figure 1C. The mean of n=3 independent experiments is graphed. Error bars represent the standard deviation. The slower CMG bypass observed in RTEL1-depleted extract containing IPTG relative to mock-depleted extract (light blue vs. red graph) is largely accounted for by the fact that CMG progression through the lacO array is delayed by LacR, even in the presence of IPTG, as seen from the slower appearance of resolved, linear species in Figure S3F (compare light blue vs. red graphs).
M.HpaII likely interacts intimately with both strands of the double helix (Klimasauskas et al., 1994). Therefore, covalent coupling of M.HpaII to one strand is expected to hyperstabilize the underlying DNA duplex. To test whether RTEL1 disrupts such a hyperstable duplex, we examined DPCLag, in which M.HpaII should stabilize the underlying DNA without blocking the translocation strand. As shown in Figure 3D, RTEL1 depletion delayed bypass of DPCLag, but the delay was less pronounced than at DPCLead (Figure 3D and Figure S3E). Collectively, these observations suggest that RTEL1 not only helps CMG disrupt hyperstable DNA duplex but also to overcome obstacles that are covalently attached to CMG’s translocation strand.
Efficient DPC proteolysis requires RTEL1
The kinetics of CMG bypass were identical whether or not the DPC was degraded (Figure 1C). This observation strongly implies that CMG normally bypasses the DPC before proteolysis, and it raised the question of whether bypass might be a pre-requisite for proteolysis. To address this, we replicated a plasmid containing non-methylated DPCLead in extract containing SPRTN so that both proteolysis pathways were active. The extract was mock-depleted or depleted of RTEL1. At different times, we isolated the plasmid under stringent conditions so that only covalently attached M.HpaII was recovered. After DNA digestion, immunoblotting revealed M.HpaII polyubiquitylation, followed by a decline in M.HpaII levels, which reflects replication-dependent proteolysis (Figure 4A, lanes 1–6; Larsen et al., submitted). Strikingly, M.HpaII degradation was delayed 15–20 min in extracts depleted of RTEL1 (Figure 4A). This defect was rescued by wild type RTEL1 but not the ATPase mutant. RTEL1 was required for efficient CMG bypass even when the DPC could be degraded, consistent with CMG bypass normally preceding DPC proteolysis (Figure 4B). In the absence of RTEL1, M.HpaII proteolysis was delayed but not eliminated (Figure 4A), which is explained by the substantial CMG bypass that still occurred in these conditions (Figure 4B). Together, the data indicate that RTEL1-mediated unwinding past the DPC is essential for its efficient proteolysis.
Figure 4. RTEL1 is required for efficient DPC proteolysis.

(A) pDPC2xLead was replicated in the indicated extracts and supplemented with buffer, wild-type RTEL1, or RTEL1-K48R. Plasmid was recovered under stringent conditions, the DNA digested, and the resulting samples blotted for HpaII. Signal from the entire lane was quantified, and peak signal was assigned a value of 100%. The mean of n=3 independent experiments is graphed. Error bars represent standard deviation. (B) Parallel reactions to those in (A) were supplemented with [α−32P]-dATP. Leading strand approach was visualized as in Figure S1J and quantified as in Figure 1C. The mean of n=3 independent experiments is graphed. (C) pmeDPC2XLead was replicated in the indicated extracts. Samples were processed by the pulldown procedure described in (A). Short and long (lower panel) exposures of the same blot are shown. (D) pmeDPC2xLead or pmeDPC2xLead Lead were replicated in non-depleted extract. Plasmid pull downs were performed as in (A) and presented as in (C). (E) pDPC2XLead was replicated in the indicated extracts, and plasmid pull-down was performed as in (A). RTEL1 depletion was verified in Figure S4B. (F) pDPC2XLead was replicated in the indicated egg extracts that also contained DMSO or MG262. Stringent plasmid pull-down was performed as in (A).
RTEL1-dependent DPC bypass promotes SPRTN activity
To examine whether RTEL1 is required for SPRTN activity, we examined meDPC, which is not susceptible to the proteasome but can be degraded by SPRTN (Larsen et al., submitted). The action of SPRTN was visible from the appearance of a specific M.HpaII degradation fragment that was absent in ΔSPRTN extract (Figure 4C; Larsen et al., submitted). In the absence of RTEL1, accumulation of the SPRTN-specific fragment was delayed (Figure 4C; compare lanes 1–5 and 11–15). As seen for DPC bypass, these defects were rescued by wild type but not ATPase deficient RTEL1, indicating that DNA unwinding past the DPC by RTEL1 is required for SPRTN activity. To address whether CMG bypass itself is required, we examined the effect of tandem leading strand meDPCs, which severely impair CMG bypass (Figure 2), presumably without affecting RTEL1’s ability to unwind past the adducts. As shown in Figure 4D, tandem meDPCs severely inhibited the appearance of the SPRTN-specific M.HpaII fragment compared to single meDPCs, which allow CMG bypass. Our data indicate that RTEL1-dependent DNA unwinding and CMG bypass of the DPC are both required for the SPRTN pathway.
We next addressed whether RTEL1 affects the proteasome pathway. A time course revealed that in the absence of RTEL1, the appearance of highly ubiquitylated M.HpaII species was delayed, suggesting that RTEL1 is required for efficient DPC ubiquitylation (Figure 4E). To specifically examine the effect of RTEL1 on the proteasome pathway, we replicated pDPC2xLeads in SPRTN depleted-extract with or without RTEL1 depletion and examined DPC proteolysis, using MG262 addition as a positive control for proteasome inhibition. As shown in Figure 4F, RTEL1 depletion stabilized M.HpaII to a similar extent as MG262 in ΔSPRTN extract (compare lanes 11–15 with 16–20), consistent with RTEL1 functioning in the proteasome pathway. Finally, in RTEL1-depleted extracts, chromatin-binding of SPRTN was reduced, and binding of the PSA3 proteasome subunit was delayed (Figure S4A). Together, our experiments indicate that RTEL1 is required for both proteolysis pathways.
We wanted to know whether RTEL1 regulates DPC proteolysis independently of promoting CMG bypass. We therefore employed a substrate containing a ssDNA gap across from the DPC, in which DPC proteolysis by SPRTN and the proteasome occurs in the absence of a replication fork (Figure S4C, lanes 1–6; Larsen et al., submitted). Importantly, in this replication-independent setting, RTEL1 depletion had no effect on DPC ubiquitylation and proteolysis (Figure S4C) or production of the SPRTN-specific M.HpaII degradation fragment (Figure S4D). These results suggest that RTEL1 does not directly regulate SPRTN or the proteasome but does so indirectly, by stimulating CMG bypass.
Direct observation of DPC bypass by single molecule analysis
To investigate CMG dynamics during DPC repair, we developed a single-molecule assay called KEHRMIT (Kinetics of the Eukaryotic Helicase by Real-time Molecular Imaging and Tracking) that is similar to an approach developed in yeast extracts (Duzdevich et al., 2015). DNA replication was initiated on stretched λ DNA (Figure 5Ai-iii) using GINS-depleted egg extract reconstituted with active, recombinant GINS labeled on its Psf3 subunit with Alexa Fluor 647 (Figure 5B, Figure S5A-D; rGINSAF647 labeling efficiency ≥ 90%). After ~2 minutes, when only a few origins per λ DNA had fired, we flowed in GINS-depleted extract lacking rGINSAF647 to remove background fluorescence and prevent further origin firing (Figure 5Aiv). Subsequent imaging of AF647 (Figure 5Av) revealed that each replication origin gave rise to two diffraction-limited spots of comparable intensity that traveled in opposite directions (Figure 5C, green). Spots photobleached in a single step, indicating they contain a single rGINSAF647 molecule (Figure S5E). CMGs travelled at an average velocity of ~400 nt per min (Figure 5D), consistent with fork rates on immobilized DNA templates (Loveland et al., 2012). Furthermore, rGINSAF647 migrated at the leading edge of nascent DNA tracts (Figure 5C, blue; see legend), indicating that each AF647 spot represents CMG at a replication fork. Labeled CMGs traveled on average 5.3 kilo-bases (kb) before disappearing (Figure 5E). For molecules where the DNA remained attached until the end of the experiment, the nascent DNA tract continued to grow even after loss of AF647 signal (Figure S5F). Given that there is no free pool of GINS during the imaging phase (Figure 5Aiv-v), the loss of AF647 signal reflects photobleaching and not GINS exchange. We conclude that GINS is highly processive and that KEHRMIT is a powerful means to examine CMG dynamics during replication.
Figure 5. KEHRMIT – a single molecule assay for CMG dynamics.

(A) Schematic of KEHRMIT assay. (B) Coomasie-stained SDS-PAGE gel of recombinant GINS before and after sortase labeling of Psf3 with AF647, which shifts its mobility (arrow). (C) Kymogram of a replication bubble from a KEHRMIT experiment. Green, GINSAF647 signal. Blue, Fen1mKikGR - a fluorescent protein that binds nascent DNA (Loveland et al., 2012). (D-E) Beeswarm plots of CMG speed and processivity (i.e. distance travelled) measured via KEHRMIT (dots represent n=218 individual helicase molecules). Blue line, mean; gray box, 95% Confidence Interval (CI) estimated by bootstrapping.
To examine the mechanism of DPC repair, we immobilized linear DNA containing two site-specific DPCs labeled at their C-termini with Alexa Fluor 568 (AF568) (Figure 6A). If an origin fires between the two lesions, both helicases encounter a DPCLead (Figure 6B, top); otherwise, the inward moving CMG first encounters DPCLag and then DPCLead (Figure 6B, bottom). We first conducted KEHRMIT on meDPC in SPRTN-depleted extract to inhibit DPC proteolysis and thereby maximize the possibility of observing bypass. Strikingly, we saw many instances in which CMG paused at DPCLead and then underwent bypass (Figure 6C). Interestingly, CMG slowed down dramatically after bypass, as described below. To quantify bypass events and determine their timing, we applied the following, stringent criteria: (1) both CMGAF647 and DPCAF568 signals were present before, during, and after bypass; (2) CMG travelled at least 1-pixel (~500 nt) away from the DPC after bypass; (3) CMG translocated for at least 3 time points (3 min) before and after bypass. Based on this algorithm, 42–56% of CMGs that encountered meDPCLead in SPRTN-depleted extract unambiguously bypassed the intact DPC (“BID” events) (Figure 6D, S6A), and CMG paused at meDPCLead for 15 minutes on average before undergoing bypass (Figure 6E). We observed four other classes of events (Figure 6D, see legend for a detailed description; S6B for examples). Many of these non-BID events probably involve meDPCLead bypass that could not be detected due to the slow rate of CMG progression after bypass, the stringent criteria for BID events, and the premature loss of CMG or DPC signal due to photobleaching (Figure S6C) or DNA breakage. Therefore, the actual DPCLead bypass efficiency was probably substantially higher than 50%.
Figure 6. Direct observation of DPC bypass by CMG.

(A) Stretched and immobilized DNA-DPC substrate. DNA was stained with sytox orange (top panel). The DPC was labeled on its C-terminus with AF568 (middle panel). Merge, bottom panel. (B) Cartoons depicting how the location of initiation determines whether CMGs encounter DPCLead (top) or DPCLag followed by DPCLead (bottom) (C) Kymogram of a meDPC substrate undergoing replication in SPRTN-depleted extract from an origin that fired between the DPCs. Both CMGs undergo DPC bypass. Images were acquired at 1 frame/min. Green, AF647; pink, AF568. (D) Quantification of five different classes of CMG-DPCLead encounters in n=2 independent biological repeats: (i) BID, Bypass of intact DPC, representing unambiguous bypass events; (ii) DD+, DPC disappeared first, followed by CMG departure from the pause site. When proteolysis was inhibited (meDPC, ΔSPRTN extract), DD+ events likely involve DPC bypass but do not meet the BID criteria due to DPC photobleaching; (iii) DD-, DPC disappeared first, without CMG departure from the pause site, including potential bypass events where CMG photobleached or DNA ruptured soon after the DPC signal vanished; (iv) CD, CMG disappeared first, likely due to photobleaching, obscuring any subsequent bypass events; (v) DT, CMG and DPC disappeared together, including events where the experiment ended or the DNA ruptured before bypass could be detected. Due to rounding errors, probabilities may not add up to 100%. N represents the number of molecules. (E) Beeswarm plot of the time needed to bypass meDPCLead or meDPCLag in SPRTN-depleted extract. Blue lines and gray boxes correspond to the mean and the 95% CI for the mean, respectively. N represents the number of molecules. (F) Same as (C) but showing a kymogram in which an origin fired to the right of both lesions. The leftward-moving helicase (green) first bypassed a meDPCLag in ~1 min, then reached a meDPCLead where it paused. (G) Beeswarm plot of DPC lifetime after CMG arrival at the lesion. Blue lines, gray boxes, and N as in (E). (H) Beeswarm plot of CMG speed during approach to (Appr.) and departure from (Dep.) DPC lesions versus the speed of aphidicolin-uncoupled helicases. Blue lines, gray boxes, and N as in (E). The aphidicolin condition was performed on λ DNA. (I) Kymogram of CMG-meDPCLead encounter (SPRTN-depleted extract) that resulted in DPC bypass and CMG uncoupling, followed by apparent re-coupling (white arrow).
When CMG encountered meDPCLag in SPRTN-depleted egg extract, it paused on average for only three minutes before moving past the adduct (Figure 6E-F), consistent with ensemble analysis of leading strands (Duxin et al., 2014). In 44–49% of cases, CMG unambiguously bypassed meDPCLag (Figure S6D, BID). In 47–51% of meDPCLag encounters, meDPCLag became mobile after helicase bypass and then tracked with CMG (Figure S6D, B+M; Figure S6E). Since the DNA template is immobilized via 3’-biotins, meDPCLag probably becomes mobile when the outward moving replisomes reaches the end of the template, which liberates and allows chromatin compaction of the sister chromatid containing the bypassed DPC (Figure S6F). Taking into account BID and B+M events, meDPCLag bypass efficiency exceeded 95% (Figure S6D). In conclusion, KEHRMIT shows that CMG efficiently bypasses stable DPCLead and DPCLag, and that this process does not involve GINS dissociation.
To address whether CMG bypasses DPCLead when proteolysis is not impaired, we examined a non-methylated DPC in extract containing SPRTN. As expected, signatures involving early loss of DPCLead increased when proteolysis was allowed (Figure 6D; DD- and DD+). In this setting, 11–14% of CMGs exhibited unambiguous DPCLead bypass (Figure 6D; BID). We suspect that many DD+ events represent bona-fide bypass, but failed to meet the BID criteria because DPC proteolysis occurred before CMG traveled far enough beyond the DPC. In support of this interpretation, the probabilities of BID and DD+ events add up to 40–45% for degradable DPCLead – similar to the likelihood of unambiguous bypass (BID 42–56%) for stabilized DPCLead (Figure 6D). As noted above, any bypass events in the DT, DD-, and CD categories go undetected due to the premature disappearance of DPC and/or CMG signal. Thus, 11–14% is likely a gross underestimate of the true efficiency with which CMG bypassed DPCLead. In conclusion, our results are consistent with the model that CMG bypasses DPCLead prior to proteolytic processing.
Differential Kinetics of DPCLead vs. DPCLag Proteolysis
In these experiments, the AF568 fluorescence signal disappears when the C-terminus of HpaII is degraded, providing a single-molecule measurement of DPC proteolysis. In most instances, the loss of the AF568 signal was due to DNA replication and not photobleaching (Figure S6G). Importantly, DPCLag fluorescence disappeared ~7 minutes after CMG arrival, whereas DPCLead disappearance took ~25 minutes (Figure 6G). This result is consistent with our finding that proteolysis depends on bypass (Figure 4D) and that bypass takes much longer for DPCLead than DPCLag (Figure 6E). As expected, when the DPC was methylated and examined in SPRTN-depleted extract, both meDPCLead and meDPCLag were greatly stabilized (Figure 6G). In conclusion, the difference between DPCLag and DPCLead destruction is accounted for by the different kinetics with which these lesions are bypassed, consistent with bypass being required for proteolysis.
CMG slows down after DPCLead bypass
We investigated how CMG’s velocity is affected by collision with DPCLead and DPCLag. Strikingly, while CMG continued moving at 400–500 nt/min after bypassing DPCLag, it slowed to ~80 nt/min after bypassing DPCLead (Figure 6H). We speculated that CMG slowing was due to its uncoupling from leading strand synthesis, which pauses for an extended time at DPCLead but not DPCLag (Figure 1C; (Duxin et al., 2014)). To independently assess how CMG uncoupling from the leading strand impacts helicase velocity, we added aphidicolin, a potent inhibitor of replicative polymerases. In the presence of aphidicolin, CMG slowed dramatically (Figure S6H) to an average speed of only ~55 nt/min (Figure 6H) despite continued high processivity (Figure S6I). The data strongly imply that the slow rate of CMG translocation after DPCLead bypass is due to CMG uncoupling. In 5% of cases, we observed that after DPCLead bypass, CMG traveled slowly for several minutes before resuming rapid translocation, indicative of recoupling (Figure 6I). The small number of putative recoupling events is consistent with the low efficiency of TLS after extracts have been depleted. Our results suggest that after CMG bypasses DPCLead, CMG uncouples from the leading strand and slows down, minimizing ssDNA generation during TLS past the peptide adduct.
RTEL1 promotes efficient CMG progression past non-covalent nucleoprotein complexes
As shown in Figure 3D, RTEL1 is required to efficiently bypass a lagging strand DPC, probably because it helps unwind the DNA underlying the DPC. If this interpretation is correct, RTEL1 should also be required for replicative bypass of non-covalent nucleoprotein complexes that stabilizes the duplex. To test this prediction, we replicated a plasmid containing an array of 32 lacO sites bound by LacR. At different times, plasmid was recovered and cut with XmnI before native gel electrophoresis (Figure 7A). In this setting, replication forks converge on the outer edges of the LacR array, generating a discrete X-shaped intermediate whose mobility decreases as forks slowly progress through the array (Figure 7B, lanes 1–6; (Dewar et al., 2015)). When forks meet, the X-shaped species are converted into linear daughter molecules. As shown in Figure 7B, RTEL1 depletion severely delayed the accumulation of linear molecules, and this effect was partially rescued by RTEL1WT but not RTEL1K48R. To examine fork progression through the LacR array at higher resolution, DNA was nicked near the lacO sites with Nt.BspQI, and the radioactive nascent strands were separated on a urea PAGE gel, which reveals fork pausing ~30 nt upstream of each lacO site (Figure 5C, lanes 1–6; (Dewar et al., 2015)). Based on this analysis, fork progression through the array was also severely compromised in extracts lacking RTEL1, and the defect was reversed with RTEL1WT but not RTEL1K48R (Figure 7C, red boxes). Replisome progression does not require LacR proteolysis (Figure S7A). We conclude that RTEL1 is required for the eviction of non-covalent nucleoprotein complexes.
Figure 7. RTEL1 is required for CMG bypass of non-covalent nucleoprotein complexes.

(A) Top, structures generated with and without XmnI digestion before and after forks progress through the LacR array. (B) pLacO32 was pre-incubated with LacR and replicated in the indicated egg extracts containing [α−32P]-dATP. DNA was recovered, digested with the single cutter XmnI, resolved by native agarose gel electrophoresis, and visualized by autoradiography. (C) DNA samples from (B) were nicked with Nt. BspQI to release the rightward leading strand (red arrow), separated on a denaturing polyacrylamide gel, and visualized by autoradiography. lacO sites are located ~30 nt downstream of each stalling product (Dewar et al., 2015). (D) Model of replisome bypass of nucleoprotein barriers. When the replisome encounters a non-covalent nucleoprotein complex, RTEL1 and CMG cooperate to unwind the DNA underlying the protein, leading to its displacement and immediate resumption of fork progression. At a covalent DPC, RTEL1 translocates along the undamaged lagging strand template, exposing ssDNA that facilitates CMG bypass. Given the stable interaction of pol ε (grey oval) with CMG (Langston et al., 2014), we envision that it bypasses the DPC with CMG. After CMG bypass, the DPC undergoes proteolysis by SPRTN or the proteasome. Finally, the leading strand is extended past the peptide adduct using translesion synthesis polymerases.
Discussion
Our results establish a comprehensive framework to understand how the vertebrate replisome overcomes covalent and non-covalent nucleoprotein obstacles (Figure 7D).
Disruption of non-covalent nucleoprotein complexes by RTEL1
The most common nucleoprotein obstacles encountered by replication forks are non-covalent. We showed that CMG progression past a LacR array requires RTEL1, which translocates 5’ to 3’ on the lagging strand template. Together with prior work (Guy et al., 2009; Ivessa et al., 2000), a conserved mechanism emerges in which replication forks employ an accessory helicase on the strand opposite the one hosting the replicative DNA helicase. In egg extracts and cells, RTEL1 is constitutively associated with replication forks in the absence of experimentally-induced DNA damage (Figure 3A; (Vannier et al., 2013)), suggesting RTEL1 travels with the replisome. We propose that RTEL1 unwinds the DNA underlying non-covalent nucleoprotein complexes, thereby disrupting the obstacle and allowing fork progression.
CMG bypasses DPCs on both strands
Less commonly, forks encounter covalent DPCs. We previously showed that the disappearance of the CMG footprint at DPCLead correlates with the latter’s proteolysis, and that when DPCLead proteolysis is blocked by ubiquitin depletion, loss of the footprint is dramatically delayed. On this basis, we proposed that CMG progression past DPCLead requires DPC proteolysis (Duxin et al., 2014). However, we now show that when ubiquitin levels are normal, CMG can readily bypass a stable DPCLead. Thus, when DPC proteolysis is blocked by inhibiting the SPRTN and proteasome pathways, the CMG footprint disappears, even in the absence of p97 activity, implying that CMG bypasses the DPC without dissociating. In support of this interpretation, single-molecule imaging shows that the same molecule of CMG that encounters a DPCLead also travels past the adduct. Our previous observation that loss of the CMG footprint was inhibited when DPC proteolysis was blocked by a non-specific DUB inhibitor was most likely due to pleiotropic consequences of ubiquitin depletion (Duxin et al., 2014).
We envision two possible mechanisms of DPCLead bypass. In one, CMG threads the denatured DPC through its central channel (Figure S7B). Because most DPCs should be attached to DNA at an internal amino acid, this mechanism would require CMG’s central pore to accommodate ssDNA and two polypeptide chains. Notably, the time required for DPC bypass is not influenced by methylation status (Figure S6J compare meDPC vs DPC; Figure 1C for ensemble data) or when MG262 was added to block proteasome activity (Data not shown), suggesting that ubiquitylation of the DPC does not adversely affect bypass. This observation disfavors the “threading” model, which predicts that ubiquitylated DPC would be much more difficult to accommodate in CMG’s central channel, even after it is unfolded. In a second model, the MCM2–7 ring opens, allowing CMG to slide past the DPC (Figure S7C). This model is consistent with current evidence that the MCM2–7 ring opens during replication licensing and initiation (Bochman and Schwacha, 2008; Fu et al., 2011; Samel et al., 2014). Future experiments will be required to distinguish between the above models.
We recently discovered that upon collision of a replication fork with a DPC, the E3 ubiquitin ligase TRAIP promotes DPC ubiquitylation and proteolysis (Larsen et al., submitted). TRAIP-dependent DPC ubiquitylation does not require RTEL1 and therefore occurs independently of CMG bypass (Larsen et al., submitted). Surprisingly, ubiquitylation of the DPC by TRAIP appears to contribute to efficient CMG bypass. Given that DPC methylation, which prevents DPC ubiquitylation, does not affect bypass (Figure 1C), we speculate that methylation mimics the effect of ubiquitylation on bypass. The role of post-translational modifications in DPC bypass is an important area of future investigation.
The requirement for RTEL1 in CMG bypass is largely abolished by a converging fork or when a ssDNA bubble is placed downstream of the DPC, suggesting that the primary function of RTEL1 is to generate ssDNA downstream of the lesion. We favor the idea that ssDNA created beyond the DPC allows the breached CMG to re-engage with DNA beyond the DPC. Given that the non-catalytic N-terminal tier of CMG resides at the leading edge of the fork (Douglas et al., 2018; Georgescu et al., 2017), the N-terminus might re-close around ssDNA while the C-terminal ATPase domain is still engaged in bypassing the DPC. Such a mechanism would help avoid accidental dissociation of CMG during DPC bypass.
Although CMG movement past DPCLead and DPCLag both constitute “bypass” events, the underlying mechanisms are different. While CMG pauses briefly at DPCLag, it stalls for an extended period at DPCLead. In addition, the absence of RTEL1 causes greater CMG stalling at DPCLead vs. DPCLag. Given CMG’s translocation along the leading strand template (Fu et al., 2011), these data indicate that the primary role of RTEL1 at DPCLag is to assist CMG in disrupting non-covalent interactions between the DPC and the underlying DNA. In contrast, bypassing a leading strand DPC additionally requires RTEL1-dependent CMG stepping over or around the covalent DPC-DNA linkage. We previously investigated the effect of biotin-streptavidin roadblocks on CMG translocation. Whereas CMG bypass of these obstacles on the lagging strand template takes only 5–10 minutes, the same barriers on the leading strand template stall CMG for ~20–40 minutes (Fu et al., 2011), as seen for DPCs. In summary, although CMG can overcome lagging and leading strand barriers, the latter is more complex as it likely involves profound remodeling of CMG and/or the DPC.
CMG slows down after uncoupling from the leading strand
At sites of leading strand DNA damage or when DNA synthesis is inhibited, CMG uncouples from the point of synthesis, leading to ssDNA generation and ATR checkpoint activation (Byun et al., 2005; Taylor and Yeeles, 2018). However, the dynamics of the uncoupled CMG are unknown. We showed that in the presence of aphidicolin, the rate of CMG translocation slows 8-fold. Given the slow DNA unwinding by uncoupled prokaryotic DNA helicases (Graham et al., 2017; Stano et al., 2005), optimal CMG activity likely requires coupling with DNA synthesis. Importantly, after CMG bypass of DPCLead, the rate of CMG translocation also slowed dramatically, indicative of CMG uncoupling from the leading strand, which stalls at the DPC. Slow translocation of the uncoupled CMG limits the amount of ssDNA generated during replication stress and therefore reduces the likelihood of DNA breakage, accelerates recoupling of the leading strand with CMG after TLS, and may promote template switching by limiting physical separation of the sister chromatids.
CMG bypass is required for DPC proteolysis
The following evidence indicates that CMG bypass precedes and is essential for DPC proteolysis. First, the CMG footprint disappears at DPCs with the same kinetics whether or not proteolysis occurs (Figure 1C), implying that CMG bypass normally precedes proteolysis. Second, we observe bypass of degradable DPCs in single molecule experiments. Third, when we inhibit CMG bypass with RTEL1 depletion or tandem DPCs, proteolysis is compromised. Fourth, proteolysis takes longer for DPCLead than DPCLag, consistent with the slower kinetics of DPCLead bypass. Finally, DPC proteolysis by SPRTN requires that the leading strand advance to within a few nucleotides of the DPC (Larsen et al., submitted), which is only possible if CMG has moved out of the way. Altogether, the data strongly support the idea that CMG bypass is a pre-requisite for DPC proteolysis.
The relationship between CMG bypass and the proteasome is less clear. RTEL1 depletion impairs the production of long ubiquitin chains on the DPC, and it mimics MG262 addition in SPRTN-depleted extracts, implicating RTEL1 in the proteasome pathway. However, it is unclear whether RTEL1-dependent DNA unwinding at the DPC is sufficient to trigger proteasome activity, or whether CMG bypass is also required. Consistent with the former possibility, ssDNA is sufficient to trigger proteasome-mediated DPC proteolysis in the absence of a leading strand at the lesion (Larsen et al., submitted). A mechanism in which proteasome activity requires RTEL1 unwinding but not CMG bypass would allow the destruction of DPCs that cannot be bypassed (e.g. because they are too large).
The “bypass first” mechanism we describe here is well suited to enhance genome stability. First, CMG movement past DPCs before proteolysis might reduce the probability that the helicase is accidentally destroyed, which is crucial as there is no known pathway to reload MCM2–7 de novo in S phase. Second, if CMG bypass of DPCLead occurs but TLS fails, the lagging strand is still extended past the adduct (Figure 1C; upper autoradiogram). In this case, the leading strand could also be extended past the DPC via template switching (Figure S7D) or re-priming (Taylor and Yeeles, 2018).
RTEL1-dependent disruption of nucleoprotein complexes in vivo?
RTEL1 regulates telomere homeostasis, and hypomorphic mutations in the human gene cause the telomere shortening Hoyeraal-Hreidarsson syndrome, which is characterized by, bone marrow failure, immunodeficiency, and growth retardation (Vannier et al., 2014). RTEL1 resolves telomeric G-quadruplexes and dissolves T-loops, both of which facilitate telomere replication. While homozygous RTEL1 mutations are lethal, deletion of RTEL1’s PIP box leads to a general replication fork progression defect (Vannier et al., 2013). In egg extracts, the PIP box of RTEL1 is not required to bypass DPCs (data not shown), indicating that Xenopus RTEL1 can bind to replication forks via other means. The evidence is consistent with the possibility that RTEL1 promotes replication fork progression in vivo by disrupting covalent and/or non-covalent nucleoprotein complexes. However, the production of ssDNA downstream of a DPC by a converging fork or other accessory helicases appears to be sufficient to promote CMG bypass. Therefore, loss of RTEL1 alone may not detectably inhibit DPC repair in cells. Future work will be required to address whether any of the phenotypes observed in RTEL1-deficient mice or humans are attributable to defective bypass of nucleoprotein complexes.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for resources, reagents, and further information about the methods should be directed to the lead contact, Johannes Walter (johannes_walter@hms.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Xenopus laevis
Egg extracts were prepared using eggs from adult Xenopus laevis female frogs (Nasco Cat# LM00535) and sperm chromatin was prepared from the testes of adult Xenopus laevis male frogs (Nasco Cat #LM00715). All animals were healthy and not previously subjected to any other procedures or immunizations. No animal husbandry was performed for this study. Frogs were housed at the Satellite Amphibian Facility of the BCMP Department at Harvard Medical School in compliance with Institutional Animal Care and Use Committee (IACUC) regulations. All experiments involving animals were approved by the Harvard Medical Area IACUC and conform to the relevant regulatory standards.
Insect cell lines
Sf9 cells (Expression Systems Cat# 94–001S) and Tni cells (Expression Systems Cat# 94002S) were cultured at 27 °C for protein overexpression. Cells were cultured in ESF 921 insect cell culture medium (Fisher scientific Cat#96–001-01-CS).
METHODS DETAILS
Preparation of DNA constructs
To generate pDPC, we first created pJLS2 by replacing the AatII-BsmBI fragment from pJD2 (Duxin et al., 2014) with the a sequence (5’-GGGAGCTGAATGCCGCGCGAATAATGGTTTCTTAGACGT-3’) which contains a Nb.BsmI site. To generate pDPC2xLead the SacI-BssHII fragment from pJLS2 was replaced with the following sequence: 5’- CATCCACTAGCCAATTTATGCTGAGGTACCGGATTGAGTAGCTACCGGATGCTGAGGGGATCCACTAGCCAATTTATCATGG-3’. To generate pDPCLag/Lead, and pDPCLag/Lead-Bubble we first replaced the BssHII-KpnI fragment from pJLS2 with the sequence 5’-CGCGCTTAATCAGTGAGGCACCTATCTCCGGTCTGAGTCATGCGTAACTCGAGTGCTTGTAGTGGATTTAC[C5-Fluor dC]GGATTGAGTAGCTACCGGATGGTAC-3’ hybridized with either 5’- CATC[C5-Fluor dC] GGTAGCTACTCAATCCGGTAAATCCACTACAAGCACTCGAGTTACGCATGACTCAGACCG GAGATAGGTGCCTCACTGATTAAG-3’ or 5’-CATC[C5-Fluor dC]GGTAGCTACTCAATCCGGTATTAGGTGATGTTCGTGAGCTCAATGCGTACTGAGTCTGGCGAGATAGGTGCCTCACTGATTAAG −3’, respectively. The supercoiled band was purified by cesium chloride gradient ultracentrifugation. The C5-Fluor dC modified plasmids were mixed with either methylated M.HpaII or nonmethylated M.HpaII in M.HpaII reaction buffer (50 mM Tris-HCl, pH 7.5, 5 mM 2-mercaptoethanol, 10 mM EDTA) and supplemented with 100 mM S-adenosylmethionine (NEB, Ipswich, MA) for 12–18 hrs at 37 °C. pJLS2 or pJLS3 were nicked with Nt.BbvcI and ligated with an oligonucleotide containing a fluorinated cytosine (5’-TCAGCATCCGGTAGCTACTCAATC[C5-Fluor dC]GGTACC-3’) and subsequently crosslinked to M.HpaII-His6 or methylated M.HpaII-His6 to generate pDPCLead and pDPC2xLead or pmeDPCLead and pmeDPC2xLead, respectively, as previously described (Duxin et al., 2014). pJLS2 was nicked with Nb.BbvcI and ligated with an oligonucleotide containing a fluorinated cytosine (5’-TCAGCATC[C5-Fluor dC]GGTAGCTACTCAATCCGGTACC-3’) and subsequently crosslinked to methylated M.HpaII-His6 to generate pmeDPCLag, as previously described (Duxin et al., 2014). To create pmeDPCLead/Lag, pJLS2 was first nicked with Nt.BbvcI and ligated with an oligonucleotide containing a fluorinated cytosine (5’-TCAGCATC[C5-Fluor dC]GGTAGCTACTCAATCCGGTACC-3’). It was subsequently nicked with Nb.BbvcI and ligated with a second oligonucleotide containing a fluorinated cytosine (5’- TGAGGTAC[C5-Fluor dC]GGATTGAGTAGCTACCGGATGC-3’) before crosslinking to methylated M.HpaII-His6. To create pmeDPCLag/Lead pJLS2 was first nicked with Nt.BbvcI and ligated with an oligonucleotide containing a fluorinated cytosine (5’-TCAGCATCCGGTAGCTACTCAATC[C5-Fluor dC]GGTACC-3’). It was subsequently nicked with Nb.BbvcI and ligated with a second oligonucleotide containing a fluorinated cytosine (5’- TGAGGTACCGGATTGAGTAGCTAC[C5-Fluor dC]GGATGC-3’) before crosslinking to methylated M.HpaII-His6. To create pmeDPCLead/Lead or pmeDPC2xLeadLead, pJLS2 or pJLS3 was nicked with Nt.BbvcI and ligated with an oligonucleotide containing two fluorinated cytosines (5’-TCAGCATC[C5-Fluro dC]GGTAGCTACTCAATC[C5-Fluro dC]GGTACC-3’) and subsequently crosslinked to methylated M.HpaII-His6 to generate pmeDPCLead/Lead or pDPC2xLeadLead, respectively, as previously described (Duxin et al., 2014). Creation of pDPCssDNA and pmeDPCssDNA is described in (Larsen et al. submitted). Creation of pLacO32 was previously described (Dewar et al., 2015).
Xenopus egg extracts
For detailed protocols for preparation of demembranated sperm chromatin, High-Speed Supernatant (HSS), and Nucleoplasmic Extract (NPE) see (Sparks and Walter, 2018).
Preparation of demembranate sperm chromatin
Testes are harvested from 6–8 adult male Xenopus laevis frogs, and minced into tiny pieces using a clean razor blade while immersed in 1mL buffer #1 (10 mM Hepes pH 7.4, 80 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 0.2 M sucrose). The sperm are released by vortexing. The tissue is pelleted by centrifugation, and the supernatant is collected. Sperm are pelleted by centrifugation at 2600g for 15 minutes at 4 °C in a Sorvall Lynx 4000 centrifuge with swinging-bucket rotor TH13–6X50 (or equivalent). In all subsequent steps, the sperm should be kept on ice unless otherwise stated. The supernatant is discarded and the sperm pellet is resuspended in buffer #4 (10 mM Hepes pH 7.4, 80 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 2.0 M sucrose). The sperm is isolated on a sucrose step gradient in 2.5 ml thin-walled ultracentrifuge tubes (Beckman Cat# 347356): the upper sucrose layer consists of 1.7 ml Buffer #2 (10 mM Hepes pH 7.4, 80 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 2.3 M sucrose), and the bottom sucrose layer consists of 0.25 ml Buffer #3 (10 mM Hepes pH 7.4, 80 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 2.5 M sucrose). The sperm is layered on top of the sucrose cushion and spun down for 45 minutes at 93,000g at 2 °C in a tabletop ultracentrifuge (Beckman Optima Max-E or equivalent) in a swing-bucket rotor (Beckman TLS-55 or equivalent). Following centrifugation, the sperm forms a pellet at the bottom of the tube. The top half of the gradient is gently aspirated and discarded, whereas the lower half of the gradient is harvested in a clean tube and combined with the sperm pellet re-suspended in buffer #1. The sperm is diluted in buffer #1 to 12 ml and pelleted by centrifugation in a swing-bucket rotor at 3000g at 4 °C for 15 minutes. The supernatant is gently removed and the sperm is re-suspended in buffer #1 supplemented with 1mM DTT, 10 μg/ml of leupeptin (Roche Cat# 11529048001), and 10 μg/ml of aprotinin (Roche Cat# 11583794001). The sperm chromatin is de-membranated by addition of 0.4 % Triton X-100 (Sigma Cat# X100–100ML) and incubation with rotation at 4 °C for 45 minutes. Detergent is removed by pelleting the sperm through a 0.5 ml cushion of buffer #5 (10 mM Hepes pH 7.4, 80 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 0.5 M sucrose, 3.0% BSA (Fisher Cat# BP1600–100), 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM DTT). The de-membranated sperm is layered on top of the cushion and spun down for 10 minutes at 750g at room temperature in a benchtop centrifuge with swing-bucket rotor. The supernatant is discarded and the sperm is re-suspended in 0.5ml of buffer #6 (10 mM Hepes pH 7.4, 80 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 0.2 M sucrose, 3.0 % BSA, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM DTT). The sperm is pelleted for 10 minutes at 750g, and the wash procedure is repeated with buffer #6. Finally, the sperm is resuspended in 1.0ml of buffer #6 and sperm concentration is determined by adding Hoechst solution (Fisher Cat# 62249) and counting the number of sperm on a hemo-cytometer using the UV/DAPI filters on an epi-fluorescence microscope. The preparation is diluted to a final concentration of 220,000sperm/μl and 90μl aliquots are snap frozen in liquid nitrogen for subsequent use in preparing Nucleoplasmic Extract (NPE).
Preparation of High-Speed Supernatant (HSS)
Ovulation is induced in six adult female Xenopus laevis frogs according to standard protocols. Xenopus eggs are laid in water supplemented with 100 mM NaCl and harvested after 20–22 hours. Eggs are de-jellied in 1 liter of 2.2 % L cysteine pH 7.7 (Fisher Cat# ICN10144601), washed with 2 liters of 0.5 X Marc’s Modified Ringer’s (MMR) (2.5 mM Hepes pH 7.8, 50 mM NaCl, 1 mM KCl, 0.25 mM MgSO4, 1.25 mM CaCl2, 0.05 mM EDTA), then washed with 1 liter of Egg Lysis Buffer (ELB) sucrose (10 mM Hepes pH 7.7, 50 mM KCl, 2.5 mM MgCl2, 250 mM sucrose, 1 mM DTT, and 50 μg/ml cycloheximide (Sigma Cat# C7698–5G). Eggs are packed in 14-ml round-bottom Falcon tubes (Fisher Cat# 352059) by centrifugation for 1 minute at 200g in a swing-bucket benchtop centrifuge and excess buffer is aspirated. The packed eggs are supplemented with 5 μg/ml aprotinin, 5 μg/ml leupeptin, and 2.5 μg/ml cytochalasin B (Sigma Cat# C6762–10MG) and crushed by centrifugation for 20 minutes at 20,000g at 4°C in a swinging-bucket rotor in a Sorvall Lynx 4000 centrifuge (or equivalent). It is essential that the rotors be kept at room temperature until the centrifugation. After centrifugation the crushed eggs are kept on ice. The top layer of low-speed supernatant (LSS) is collected and supplemented with 50 μg/ml cycloheximide, 1 mM DTT, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 5 μg/ml cytochalasin B. The LSS is transferred to 2.5 ml thin-walled ultracentrifuge tubes (Beckman Cat# 347356) and centrifuged for 90 minutes at 260,000g at 2 °C in a tabletop ultracentrifuge with a swing-bucket rotor. After centrifugation the top lipid layer is aspirated, and the clear HSS layer is harvested, snap frozen in liquid nitrogen, and stored at −80 °C.
Preparation of Nucleoplasmic Extract (NPE)
Eggs from 20 adult female frogs are collected, washed, and de-jellied as described above for HSS, except use double the volume of L-cysteine, 0.5xMMR, and ELB-sucrose. Prepare LSS as described above, and supplement LSS with 50 μg/ml cycloheximide, 1 mM DTT, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 5 μg/ml cytochalasin B, and 3.3 μg/ml nocodazole (Sigma Cat# M1404–10MG). The extract is spun down for 15 minutes at 20,000g in a Sorvall Lynx 4000 centrifuge with swing-bucket rotor. After centrifugation the top lipid layer is aspirated and the extract is decanted into a clean tube leaving behind the black pellet. The extract is warmed up to room temperature and supplemented with ATP regeneration system: 2 mM ATP, 20 mM phosphocreatine, and 5 μg/ml phosphokinase. Nuclei are assembled by thoroughly mixing 4.5ml of extract with one 90μl aliquot of demembranated sperm chromatin and incubating at room temperature for 75–90 minutes. The nuclear assembly reaction is centrifuged for 2.5 minutes at 20,000g at 4 °C in a Sorvall Lynx 4000 in a swing-bucket rotor. The layer of nuclei formed at the top is collected and transferred to a clean tube and clarified by centrifugation at 260,000g at 2 °C for 30 minutes. Following centrifugation, the top layer of lipids is carefully aspirated and the clear NPE is harvested carefully to avoid contamination with the chromatin pellet. The NPE is snap frozen in liquid nitrogen and stored at −80 °C.
DNA replication in NPE
Plasmid DNA replication in Xenopus egg extract is carried out by supplementing high-speed supernatant (HSS) of egg cytoplasm with plasmid DNA at a final concentration of 7.5–15 ng/μL (Licensing reaction). Licensing reactions were incubated for 30 min at room temperature to assemble pre-replicative complexes (pre-RCs). To prevent licensing, Geminin was added to HSS at a final concentration of 10 μM and incubated for 10 min at room temperature prior to addition of plasmid DNA. To initiate DNA replication, 1 volume of licensing reaction was mixed with 2 volumes of nucleoplasmic extract (NPE) that had been diluted two-fold with 1xELB-sucrose (10 mM Hepes-KOH pH 7.7, 2.5 mM MgCl2, 50 mM KCl, 250 mM sucrose). For radiolabeling DNA replication products, [α−32P] dATP was added to HSS prior to the addition of plasmid DNA. For replication in the presence of LacI, 1 volume of plasmid (75 ng/μL) was incubated with an equal volume of 12 μM LacI for 30 minutes prior to transfer into HSS so that the final concentration of plasmid was 7.5 ng/μl (Duxin et al., 2014). For native agarose gel electrophoresis 0.5 μl aliquots of replication reaction were typically stopped with 5–10 volumes of replication stop buffer (8 mM EDTA, 0.13% phosphoric acid, 10% ficoll, 5% SDS, 0.2% bromophenol blue, 80 mM Tris-HCl at pH 8), treated with 1 μg/μL Proteinase K for 1 hr at 37 °C, and applied to a 0.8 agararose gel. For nascent strand analysis, 2.5 μl aliquots of replication reaction were stopped in 10 volumes of sequencing stop buffer (0.5 % SDS, 25 mM EDTA, 50 mM Tris-HCl pH 8.0) followed by addition of 1.25 μl of 190 ng/μL RNase A and incubated for 30 minutes at 37 °C. After RNase digestion, 1.25 μl of 900 ng/μL Proteinase K was added to the DNA samples and incubated overnight at room temperature. Following the Proteinase K treatment, samples were diluted to 150 μl with 10 mM Tris-HCl pH 8.0. The samples were extracted once with an equal volume of phenol/chloroform followed by one extraction with an equal volume of chloroform. The DNA was then precipitated with the addition of 0.1 volumes 3M sodium acetate pH 5.2 and 1 μl glycogen (20 mg/ml stock) and resuspended in 7.5 μl. For RTEL1 immunodepletion and rescue experiments, NPE was supplemented with ~ 200 nM recombinant wild type or mutant Xenopus RTEL1 and incubated for 15 minutes prior to replication initiation. For p97i (NMS873; Sigma Cat# SML1128) treatment, NPE was supplemented with 200 μM NMS-873 (20 mM stock) and incubated for 10 minutes prior to mixing with HSS (133.33 μM final concentration in replication mix. For MG262 (stock 20 mM; Boston Biochem.Cat# I-120) treatment, NPE was supplement with 200 μM MG262 and incubated for 15 minutes prior to mixing with HSS (133.33 μM final concentration in replication mix). A 1 mM Cdc7-i (PHA-767491; Sigma Cat# PZ0178) stock was prepared in ELB-sucrose buffer and added to replication mix at a final working concentration of 100 μM at the specified time point. For ATRi (ETP-46464; Sigma Cat# SML1321) treatment, NPE was supplemented with 100 μM ATRi and incubated for 10 minutes prior to mixing with HSS (66.67 μM final concentration in replication mix). For aphidicolin (Sigma Cat# A0781) treatment, replication mix was supplemented with 150 μM aphidicolin. For IPTG (stock 1 M; Sigma Cat# I5502) treatment, replication reactions were supplemented with 300 μM IPTG 5 minutes after replication initiation. Samples were analyzed by native 0.8% agarose gel electrophoresis. Gels were exposed to phosphorscreens and imaged on a Typhoon FLA 7000 phosphorimager (GE Healthcare). To better visualize products in Figure 3D (bottom panel), the original images were converted into a log scale for display by applying the function f(p) = log(p)*255/log (255) to each pixel (p) in the images.
Nascent strand analysis
To nick radio-labeled nascent leading-strands, 3–4 μl of extracted and ethanol precipitated DNA (see above) at 1–2 ng μl−1 was incubated in 1x buffer 3.1 (New England BioLabs) with 0.45 units μl−1 Nb.BsmI (New England BioLabs) in a 5 μl reaction at 65 °C for 1 h. To digest radio-labeled nascent leading-strand 3–4 μl of extracted and ethanol precipitated DNA a 1–2 ng μl−1 was incubated in 1x cutsmart buffer (New England BioLabs) with 1 unit μl−1 AatII (New England BioLabs) and FspI (New England BioLabs) in a 5 μl reaction at 37 °C for 2 h. To nick rightward leading strands of pLacO32, 3–4 μl of purified DNA at 1–2 ng μl−1 was incubated in buffer 3.1 with 0.4 units μl−1 Nt.BspQI (New England BioLabs) at 37 °C for 1 h. Digestion reactions were stopped with 0.5 volumes of Sequencing Stop solution (95% formamide, 20 mM EDTA, 0.05 % bromophenol blue, 0.05% xylene cyanol FF). Nicked DNA (3.5 to 4 μl samples) was separated on 4 % (for pLacO) or 7% (pDPC) polyacrylamide sequencing gels. Gels were dried and subjected to phosphorimaging using a Typhoon FLA 7000 phosphoimager. Gels were quantified using Multi Gauge software (Fuji Photo Film Co.). For Figure 6A, 1 μl of purified DNA was used for XmnI digestion.
After leading strands stall at the −30 to −44 positions, they approach to the −1 to +1 positions relative to the DPC. Intermediates between these two clusters are also observed, but not when the DPC cannot be ubiquitylated (e.g. Figure 1C; (Duxin et al., 2014)). The question arose whether these intermediates are associated with CMGs that have bypassed or not. Importantly, our single molecule data demonstrates that CMG bypasses a DPC with similar kinetics whether or not the DPC is ubiquitylated (Figure S6J). These results indicate that these approach intermediates are generated after CMG bypass, probaly due to polymerase stalling at ubiquitylated DPCs. Therefore, to quantify the percentage of CMG that underwent bypass in Figures 1C, 2, 3C, 3E, 4B, S3D, and S3E (called “approach” in Figure 1C, where bypass had not yet been established), the radioactive signal of all leading strands located between positions +1 and −29 on the gel (reflecting CMGs that have bypassed) was divided by the radioactive signal for leading strands between positions +1 and −44 (reflecting CMGs that have stalled at the lesion or undergone bypass). In the case of pmeDPCLead/Lead (Figure 2, lanes 1318), we divided the signal between +1 of the 2nd DPC and −1 of the 1st DPC (both DPCs bypassed) by the signal between +1 of the 2nd DPC and −44 of the first DPC (bypassed and not bypassed).
Antibodies and immunodepletion
The xlRTEL1-N antibody was raised against a fragment of Xenopus laevis RTEL1 encompassing amino acids 400–654, which was tagged on its N-terminus with His6 (pJLS100). The protein fragment was overexpressed and purified from bacteria under denaturing conditions, and the antibody was raised by Pocono Rabbit Farm & Laboratory. The RTEL1 antibody was affinity purified from the serum using the RTEL1 antigen according to standard protocols. The xlRTEL1-C antibody was raised against amino acids 428–443 (Ac-HPDTSQRKKPRGDIWSC-amide) by New England Peptide. The FancM antibody was raised against (Ac-CYMFEEEMVPPENPTKRSRVS-amide) FancM peptide by Bethyl Laboratories. The following antibodies were described previously: Orc2 (Walter and Newport, 1997), CDC45 (Walter and Newport, 2000), M.HpaII (Larsen, et al., submitted), PSMA3 (Larsen, et al., submitted), SPRTN-N (Larsen, et al., submitted), Chk1-p(Ser345) (Cell Signaling Cat #2341L), and Histone H3 (Cell Signaling Cat #9715S). Mcm6 antibody was raised against a C-terminal peptide (Ac-CLVVNPNYMLED-OH) and affinity purified. The most prominent band it recognized in Western blotting of total extract was 115 kD, and it recognized the same band in protein preparations containing recombinant MCM6. Two rabbits (#34299 and #34300) were immunized with purified GINS (Pocono Rabbit Farm and Laboratory). Anti-GINS antibodies were affinity-purified from serum using GINS immobilized on AminoLink Coupling Resin (Thermo Fisher Cat# 20381). 2–4mg of purified GINS was cross-linked to 1mL of resin according to the manufacturer’s protocol. Purified antibody was dialyzed into 1x TBS buffer, concentrated to 1mg/mL final concentration, and stored at −80C.
For RTEL1 immunodepletion, 3.5 volumes of purified RTEL1 antibody (1 mg mL−1) or an equivalent amount of rabbit IgG purified from non-immunized rabbit serum (Sigma) were incubated with 1 volume of Protein A Sepharose Fast Flow (PAS) (GE Healthcare) overnight at 4°C. For FancM immunodepletion, 4 volumes of FancM (1 mg mL−1) or an equivalent amount of rabbit IgG purified from non-immunized rabbit serum (Sigma) were incubated with 1 volume of Protein A Sepharose Fast Flow (PAS) (GE Healthcare) overnight at 4°C. For SPRTN immunodepletion, 4 volumes of SPRTN serum was incubated with 1 volume of Protein A Sepharose Fast Flow (PAS) (GE Healthcare) overnight at 4°C. For mock depletion, 4 volumes of preimmune serum from matched rabbit, was used. One volume of antibody-conjugated Sepharose was then added to 5 volumes of precleared HSS or NPE and incubated for 1 hour at 4°C. The HSS or NPE was collected and incubated two more times with antibody-conjugated sepharose for a total of three rounds of depletion. The depleted HSS or NPE was collected and used immediately for DNA replication, as described above.
Protein expression and purification
M.HpaII-His6, LacI-biotin, and LacI-His6 were expressed and purified as previously described (Duxin et al., 2014). Lysine methylation of M.HpaII was carried out as described (Larsen, et al., submitted). Xenopus RTEL1 open reading frame with an N-terminal GST tag separated by a 3C cleavage site was cloned into pFastBac1 (Thermo Fisher Scientific) (pJLS101) using custom gene synthesis from Integrated DNA Technologies (IDT). The RTEL1 sequence was confirmed by Sanger sequencing. Mutants of RTEL1 were created by around-the-horn sitedirected mutagenesis, and mutations were confirmed by Sanger sequencing. The GSTRTEL1 Baculoviruses were made using the Bac-to-Bac system (Thermo Fisher Scientific) according to the manufacturer’s protocols. GST-RTEL1 and mutants were expressed in 3 L suspension cultures of Sf9 cells (Thermo Fisher Scientific) by infection with RTEL1 baculovirus for 36–48 hrs. Sf9 cells were collected via centrifugation and washed with 1XPBS and subsequently pelleted by centrifugation and flash frozen. Cell pellets were thawed and resuspended in an equal volume of 2X Lysis Buffer (100 nM HEPES pH7.5, 1 M NaO2Ac, 20 % sucrose, 0.2 % IGEPAL, 4 mM DTT, 2X Roche EDTA-free Complete protease inhibitor cocktail), 1X Lysis Buffer (50 mM HEPES pH7.5, 500 mM NaO2Ac, 10 % sucrose, 0.1 % IGEPAL, 2 mM DTT, 1X Roche EDTA-free Complete protease inhibitor cocktail) to the weight of the cell pellet. Cells were lysed by two rounds of sonication, followed by addition of ammonium sulfate (4M stock) to 200 mM final concentration and 45 μl/ml Polymin P (10 % stock) and stirred at 4 °C for 10 minutes. Lysate was cleared by ultracentrifugation at 25,000 rpm in a Beckman Ti45 rotor for 1 hour. The supernatant was subjected to ammonium sulfate precipitation using 0.2 g/ml ammonium sulfate. Proteins were pelleted by ultracentrifugation at 25,000 rpm in a Beckman Ti45 rotor for 1 hour. The supernatant was discarded and protein pellets were resuspended in 50 ml Wash Buffer A500 (25 mM HEPES pH7.5, 500 mM NaO2Ac, 10 % sucrose, 0.01 % IGEPAL, 2 mM DTT, 1X Roche EDTA-free Complete protease inhibitor cocktails). The resuspended pellet was incubated for 2 hours with 300 μl of Glutatione sepharose™ 4B (GE) at 4 °C. Following incubation, resin was first washed with 20 ml of Wash Buffer A500 and then with 10 ml of Wash Buffer A200 (25 mM HEPES pH7.5, 200 mM NaO2Ac, 10 % sucrose, 0.01 % IGEPAL, 2 mM DTT, 1X Roche EDTA-free Complete protease inhibitor cocktails). Proteins were eluted from the resin with Elution Buffer E200 (25 mM HEPES pH7.5, 200 mM NaO2Ac, 10 % sucrose, 0.005 % IGEPAL, 2 mM DTT, 20 mM L-glutathione reduced, pH adjusted to 8.0). Fractions were pooled and dialyzed against Dialysis Buffer (25 mM HEPES pH7.5, 200 mM NaO2Ac, 10 % sucrose, 0.005 % IGEPAL, 2 mM DTT) with addition of HRV 3C protease (Thermo Fisher) at 4°C for 4 hr. Aliquots of RTEL1 were flash froze n and stored at −80°C.
Plasmid pull-down
The plasmid pull-down assay was performed as described (Budzowska et al., 2015). Briefly, streptavidin-coupled magnetic beads (Invitrogen; 10 μl per pull-down) were washed three times with 50 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA pH 8, 0.02% Tween-20. Biotinylated LacI was added to the beads (4 pmol per 10 μl beads) and incubated at room temperature for 40 min. The beads were then washed four times with Pull-down Buffer (10 mM HEPES (pH 7.7), 50 mM KCl, 2.5 mM MgCl2, 250 mM sucrose, 0.25 mg/ml BSA, 0.02% Tween-20) and resuspended in 40 μl of the same buffer. The bead suspension was stored on ice until needed. At the indicated times, 4.0 μl samples of the replication reaction were withdrawn and gently mixed with LacI-coated streptavidin Dynabeads. The suspension was immediately placed on a rotating wheel and incubated for 30 min at 4 °C. The beads and associated proteins were isolated by centrifugation through a sucrose cushion (10 mM HEPES pH 7.7, 2.5 mM MgCl2, 50 mM KCl, 0.5 M sucrose, 0.02 % Tween), then washed once with Pull-down Buffer. All residual buffer was removed, and the beads were resuspended in 20 μl of 2X Laemmli sample buffer. Equal volumes of the protein samples were blotted with the indicated antibodies.
DPC pull-down
The DPC pull-down assay is a high-stringency plasmid pull-down protocol to specifically isolate DNA crosslinked HpaII from extract (Larsen et al., submitted). Briefly, Streptavidin-coupled magnetic beads (Dynabeads M-280, Invitrogen; 10 μL per pull-down) were washed twice with Wash buffer #1 (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA pH 8, 0.02 % Tween-20). Subsequently, biotinylated LacI was added to the washed beads (1 pmol per 10 μL of beads) and rotated at room temperature for 40 min. The LacI-coated beads were then washed four times with DPC pull-down buffer (20 mM Tris pH 7.5, 150 mM NaCl, 2 mM EDTA pH 8, 0.5% IPEGAL-CA630) and stored on ice. Samples (4.5 μl) from DNA replication or gap filling reactions were withdrawn and stopped in 300 μL of DPC pull-down buffer on ice. After the final time point, 10 μL of LacI-coated streptavidin Dynabeads were added to each sample and allowed to bind for 60 min at 4 °C with rotation. T he beads were subsequently washed three times with DPC pull-down buffer and then twice with Benzonase buffer (20mM Tris pH 7.5, 150mM NaCl, 2mM MgCl2, 0.02% Tween-20) before being resuspended in 12 μL Benzonase buffer containing 1 μL Benzonase (Novagen) and 1 μl streptavidin-coupled magnetic beads not coated with LacI (binds any free biotinylated LacI). Samples were incubated for 1hr at 37°C to allow for DNA digestion and DPC elution, after which the beads were pelleted and the supernatant M.HpaII eluate was mixed with 2X Laemmli sample buffer for subsequent immunoblotting analysis.
Expressing, and purifying recombinant GINS
Codon-optimized Xenopus laevis cDNAs encoding all four GINS subunits (synthesized by IDT) were cloned into a single expression plasmid (pGC128) using the MultiBac system (Trowitzsch et al., 2010). A sequence encoding a 10-aa linker, the LPETG tag for sortase labeling, and a His6 tag was added to the C-terminus of the Psf3 subunit (GGGGSGGGGS-LPETG-HHHHHH). The bacmid encoding the GINS complex was obtained by electroporating pGC128 into DH10EMBacY (or DH10Bac) electro-competent cells and purified using ZR BAC DNA miniprep kit (Zymo Research Cat# D4048).
Baculovirus encoding GINS was amplified in three stages (P1, P2, and P3) in Sf9 cells (Expression Systems Cat# 94–001S) and GINS expression levels were monitored via western blots. 500mL of Tni cell culture (Expression Systems Cat# 94–002S) at a density of 2–3 was infected with 5–10 mL P3 baculovirus (MOI>1). Cells harvested 48 hrs post-infection were pelleted at 500 × g for 15min and re-suspended in a final volume of 50mL in GINS Lysis Buffer (GLB) containing 20mM Tris-HCl pH8.0, 5% glycerol, 500mM NaCl, 20mM Imidazole, 1mM DTT, 1mM PMSF, EDTA-free cOmplete protease inhibitor cocktail (Roche Cat # 11873580001). Cells were lysed by sonication on ice and the insoluble fraction was pelleted via centrifugation for 1hr at 30,000g at 4C. The clarified lysate was incubated with 0.5mL NiNTA resin (QIAGEN Cat# 30410) for 1hr at 4C on a rotating wheel. The resin was washed 5 times with 10mL of GLB in a disposable column. The protein was eluted in 5 rounds with 500uL/each of GLB + 250 mM Imidazole; the elutions were pooled and desalted using a PD10 column (GE Healthcare Cat 17–0851-01) into 20mM Tris-HCl pH7.5, 5% glycerol, 100mM NaCl, 1mM DTT. Recombinant GINS was further purified on a MonoQ column connected to an AKTA Pure FPLC with a 100–1000mM NaCl gradient in 20mM Tris-HCl pH7.5, 5% glycerol, 1mM DTT buffer (the GINS complex eluted at ~500mM NaCl). The eluted protein was desalted using PD10 columns or dialysis into 20mM Tris-HCl pH7.5, 5% glycerol, 150mM NaCl, 1mM DTT; concentrated to ~2mg/mL, frozen in liquid nitrogen, and stored at −80C. A pellet from 0.5L of Tni cell culture yielded ~5mg of purified GINS.
Purification of HpaII for sortase labeling
A sequence encoding a 10-aa linker, the LPETG tag for sortase labeling, and a His6 tag was added to the C-terminus of HpaII (pGC220) and expressed in T7 Express cells (NEB Cat# C2566I) (Duxin et al., 2014). LPETG-tagged HpaII was affinity purified on NiNTA resin, and further purified on a MonoQ column connected to an AKTA Pure FPLC with a 100–500mM KCl gradient in 20mM HEPES pH8.2, 10% glycerol, 0.02% IGEPAL-630, 1mM DTT (HpaII eluted at ~300mM KCl). The fractions containing HpaII were pooled and dialyzed overnight into 20mM HEPES pH8.0, 100mM KCl, 10% glycerol, 0.02% IGEPAL-630, 1mM DTT; concentrated to ~2–3 mg/mL; and stored at −80C. A portion of LPETG-tagged HpaII was methylated as described above, then fluorescently labeled using sortase.
Fluorescent labeling of GINS and HpaII
Sortase-tagged proteins were conjugated to a short peptide labeled with a fluorophore (AF647 or AF568). 1mg of lyophilized GGGGYKCK peptide (synthesized by New England Peptide) was dissolved in 1mL degassed reaction buffer (20mM Hepes pH 7.5, 0.1mM EDTA, 10mM TCEP) and incubated at room temperature for 15 min. 1mg of AF647-maleimide (Thermo Fisher Cat# A20347) or 1mg of AF568-maleimide (Thermo Fisher Cat# 20341) was dissolved in 40μL degassed DMSO and added to the dissolved peptide, then incubated on a rotating wheel at room temperature for 3 hrs. The reaction was quenched with 10mM DTT. The labeled peptide was separated from unlabeled peptide and un-reacted fluorophore on a MonoQ column with a 10mM-1000mM ammonium carbonate pH 7.8–8.0 gradient (pH adjusted using 50% acetic acid). Fractions containing the labeled peptide were pooled and lyophilized, then dissolved in 50μL 20 mM HEPES pH7.5 and stored at −80C.
Conjugation reaction: 2 nanomoles of GINS or 5 nanomoles of HpaII or meHpaII were reacted overnight at 4°C on a rotating wheel with 20× molar excess of GGGGYKCKAF647 or GGGGYKCKAF568 in the presence of 1/10× molar excess of sortase enzyme (purified as previously described (Chen et al., 2011)), supplemented with 100μL 4× sortase buffer, in a 400μL total volume. 4× Sortase Buffer consists of 80mM HEPES pH 7.5, 600mM NaCl, 40% glycerol, 4mM DTT, and 20mM CaCl2. After incubation, the reaction mixture was supplemented with NaCl to a final concentration of 500mM and imidazole to a final concentration of 10mM, and incubated with 40μL NiNTA resin for 1hr at 4C. NiNTA trapped unlabeled protein and the His6-tagged sortase whereas labeled protein and excess free peptide remained in the supernatant. Labeled protein was purified and desalted by gel filtration on a Superdex200 column in 20mM Tris-HCl pH7.5, 150mM NaCl, 10% glycerol, and 1mM DTT; then concentrated to 1mg/mL and stored at −80C for up to 1 yr.
Preparing substrate with fluorescent DPCs
The DNA substrate was cloned and assembled by tandem recombineering using the MultiBac system (Trowitzsch et al., 2010) as a 30.6 kb plasmid (pGC261) and grown in DH10β cells (NEB Cat# C3019I). The plasmid contains two sites for HpaII conjugation flanked by Nt.BbvCI sites such that a fluorinated oligo can be annealed and ligated into the backbone. 100–150μg (at ~1000 ng/uL) of pGC261 plasmid was purified using a Midi Prep kit (QIAGEN) from 250mL of bacterial culture. Supercoiled plasmid was nicked with Nt.BbvCI (NEB Cat# R0632S) and the enzyme was heat inactivated. 20-fold molar excess of modified oligo (TCAGCATCCGGTAGCTACTCAATCCGGTACC, the highlighted nucleotide is 5-fluoro-deoxycytidine, synthesized by BioSynthesis) was annealed to the nicked backbone then ligated overnight. The DNA was dialyzed in 10mM TrisHCl pH8.0, then digested with the single cutter NotI-HF (NEB Cat# R3189S) in NEB2 buffer and the enzyme was heat-inactivated. The 3’ DNA termini were biotinylated by incorporating biotin-dCTP (Thermo Fisher Cat# 19518018) and biotin-dGTP (Perkin Elmer Cat# NEL541001EA) using Klenow Fragment 3’→5’ exo- (NEB Cat# M0212S). The DNA was then purified by electrophoresis on a 0.6% agarose 1xTAE gel pre-stained with SYBR Safe (Thermo Fisher Cat# S33102), the 30.6kb linear DNA band was excised and electro-eluted in 1xTAE Buffer. The DNA was butanol-extracted several times until the volume was reduced 2–3 fold, then phenol-chloroform extracted 3 times to remove any agarose traces, followed by chloroform extraction. The DNA was ethanol-precipitated and eluted in EB (QIAGEN), and dialyzed in 10mM Tris-HCl H8.0 to remove trace contaminants. Purified DNA was conjugated to 100-fold molar excess of HpaIIAF568 or meHpaIIAF568 as described above. The reaction was run on 0.6% agarose 1xTAE gel pre-stained with SYBR Safe, the linear DNA band was excised, and the DNA was electro-eluted in 1xTAE. The eluate could be stored in the dark at 4C for a few days or at −80C for at least a few months.
Single-molecule KEHRMIT assay
Immunodepletion of endogenous GINS: For each experiment endogenous GINS was immunodepleted in 3 rounds from 50μL NPE and in 2 rounds from 70μL HSS. For each round of NPE depletion 10μL Protein A Sepharose resin (PAS, GE Healthcare) was incubated with 50μg of affinity purified anti-GINS antibody at 4C overnight. For each round of HSS depletion, 14μL PAS resin was incubated with 70μg of antibody. The resin was pelleted in a swing-bucket rotor for 30sec at 2500g and washed twice with 1xTBS, once with 1xELB sucrose, twice with 1x ELB sucrose + 0.5M NaCl, and twice with 1xELB sucrose. Immuno-depletions were performed on a rotating wheel in the cold room for 45 min/each.
Flow Cell Assembly and DNA Tethering: Flow cells were assembled using 25×75×1mm slides (VWR Cat# 48300–025), double-sided tape (Grace Bio-Labs Cat# 620001), and PEG-ylated coverslips, as previously described (Yardimci et al., 2012a). 24×60mm No. 1.5 coverslips (VWR Cat# 16004–312) were PEG-ylated using a mixture of 10% Biotin-PEG-SVA and 90% M-PEG-SVA MW5000 (Laysan Bio). Linear DNA biotinylated at both ends (λ DNA or the 30.6kb DNA substrate with dual DPC lesions) was stretched to ~90% of its contour length and immobilized onto the coverslip (Yardimci et al., 2012a). Immobilized DNA was stained with 200 nM SYTOX Green (Thermo Fisher Cat# S7020) and imaged using 488-nm laser excitation (details in the Microscopy section below). 20–50 fields of view (FOVs) near the flow cell inlet were selected and subsequently imaged during the replication experiment every 0.5–3min. The DNA stain was removed by washing the flow cell with 1xELB sucrose before any egg extracts were introduced.
Replication Reaction Mixture Assembly: After immuno-depleting HSS and NPE, the following mixtures were assembled. The ATP regeneration system (ARS) was assembled on ice by mixing 5μL 0.2M ATP (Sigma Cat# A-5394) with 10μL 1M Phosphocreatine (Sigma Cat# P-6502) and 0.5μL 5 mg/mL Creatine Phosphokinase (Sigma Cat# C-3755). ΔHSS-NPE Mix was assembled at room temperature by mixing 31μL GINS-depleted NPE with 31μL GINS-depleted HSS, and incubated at room temperature for 5min to inactivate Cdt1. The Licensing Mix was assembled by supplementing 20μL of GINS-depleted HSS with 2μL of 30-bp long dsDNA carrier (300ng/μL stock)(Loveland et al., 2012; Yardimci et al., 2012a) and 0.7μL ARS, then incubated at room temperature for 5min. The Replication Initiation Mix was assembled by adding 3μL pBlueScript carrier plasmid DNA (200ng/μL stock) (Lebofsky et al., 2011), 1μL ARS, 4μL 1xELB Sucrose, and 1μL rGINSAF647 (0.3–1.0 mg/mL stock) to 18μL of ΔHSS-NPE Mix, then incubated at room temperature for 5min. The Replication Elongation Mix was assembled by supplementing 40μL of ΔHSS-NPE Mix with 7μL pBlueScript carrier plasmid DNA (200ng/μL stock), 2μL ARS, 11μL 1xELB sucrose, and, optionally 1–2μL of Fen1mKikGR (100μM stock), and incubated at room-temperature for 5min.
DNA Replication and Imaging: After the DNA was tethered as described above, replication origins were licensed by flowing 15μL of Licensing Mix into the flow cell at 10μL/min, and incubating for 2–5min. Replication origins were fired by flowing 20μL of Replication Initiation Mix at 10μL/min, and incubating for 2–5min. Excess free rGINSAF647 was washed away by flowing 55μL of Replication Elongation Mix at 10μL/min. 2–3 minutes after starting the wash AF568 (for DPC experiments) or mKikGR (for λ DNA experiments) and AF647 were imaged every 0.5–3min for 1–3hrs. Since the PCA/PCD and glucose oxidase/catalase oxygen scavenging systems inhibited DNA replication (data not shown),oxygen scavengers were omitted and stroboscopic imaging was employed to limit photobleaching.
Microscopy: Single-molecule data was collected at the Nikon Imaging Center at Harvard Medical School on a Nikon Ti motorized inverted microscope (“Tobias”) equipped with CFI Apochromat TIRF NA1.49 oil immersion objective with 100X magnification, the Perfect Focus System for maintenance of focus over time, a Nikon TIRF illuminator, an Agilent MLC400B laser launch with 405 nm (20mW), 488 nm (50mW), 561 nm (50mW), and 647 nm (125mW) (power measured at fiber optic). Images were acquired using a ZT405/488/561/647rpc dichroic (Chroma) with an Andor DU-897 EM-CCD camera controlled by NIS Elements software.
SYTOX Green fluorescence was excited with the 488 nm laser (<0.1mW measured at the objective) for 100ms and collected with an ET525/50m emission filter (Chroma). AF568 fluorescence was excited with the 561 nm laser (0.5–1.0mW) for 100 ms and collected with an ET600/50m emission filter (Chroma). AF647 fluorescence was exited with the 647 nm laser (0.5–1.0mW) for 100 ms and collected with an ET700/75m emission filter (Chroma). Fen1mKikGR was photo-switched with the 405 nm laser (0.5–1.0 mW) for 100ms, allowed to diffuse for 400ms, then mKikGR fluorescence was excited with the 561 nm laser (0.5–1.0 mW) for 100ms and collected with a ET600/50m emission filter (Chroma) (Loveland et al., 2012).
For time-lapse experiments, images were collected every 0.5–3 min (depending on the experiment) at 20–50 different fields of view (FOVs). Multiple FOV positions were collected using a Prior Proscan II linear-encoded motorized stage. Movie frames for each FOV were aligned in ImageJ using the Image Stabilizer Plugin.
KEHRMIT data analysis
A typical KEHRMIT experiment generated a movie containing several fields of view (FOV), and each FOV initially contained ~100 double-tethered DNA molecules of the correct size (48.5kb λ DNA or 30.6kb DPC substrate extended to ~90% of their respective contour lengths). A significant portion of DNA molecules ruptured when egg extract mixtures were introduced into the flow cell. DNA molecules that were tethered too close to each other aggregated in egg extracts and were omitted from analysis. Molecules where multiple replication origins fired close to each other were also discarded. Only replication origins that gave rise to two labeled CMG molecules were included in the analysis. Active CMG molecules were detected by generating a maximum-intensity z-projection of the CMGAF647 channel where translocating CMG molecules appeared as a contiguous bright trail. Regions of interest (ROIs) corresponding to active helicases were hand-selected, automatically cropped, rotated, and individual frames were stacked to generate kymograms. The fluorescent spot image corresponding to individual CMG molecules was fitted by a 2D Gaussian and its position was determined to sub-pixel accuracy. Translocation velocity was computed by linear fitting of CMG position versus time to a straight line. Processivity was measured as the distance traveled by CMG from the beginning of the experiment until the AF647 signal disappeared due to photobleaching, DNA tether rupture, or CMG reaching the end of the DNA template. When analyzing data from aphidicolin uncoupling experiments, it was taken into account that at a low force the length of RPA-bound ssDNA is essentially the same as that of dsDNA (Lewis et al., 2017). In experiments with the 30.6kb DNA-DPC substrate, only DNA molecules containing two labeled DPCs at the expected distance were included in the analysis. Furthermore, DPCLead and DPCLag encounters were classified according to the location of the replication initiation event relative to the two lesions as illustrated in Figure 6B. CMG pauses at the DPC were identified as segments of the helicase trajectory where the CMG position was within 1px (160 nm) of the DPC. Unambiguous DPC bypass events (BID) were defined as CMG encounters with a DPCLead or DPCLag where both approach and departure could be unambiguously detected (at least 3 timepoints for each) and the corresponding velocities could be reliably measured. DD+ events were defined as CMG encounters with a DPCLead or DPCLag where the DPCAF568 signal disappeared but the CMGAF647 signal persisted, and CMG travelled at least 1px from the original site of the DPC. Conversely, DD- events were defined as CMG encounters with a DPCLead or DPCLag where the DPCAF568 signal disappeared, the CMGAF647 signal persisted, but CMG travelled less than 1px from the site of the DPC. B+M events were defined as CMG-DPCLag encounters where both CMGAF647 and DPCAF568 signals persisted and both CMG and DPC travelled more than 3px from the initial DPCLag position. Since data was acquired at 1min/frame, pause durations and DPC lifetimes are reported as integers in increments of 1min. Error-bars for mean or median values corresponding to distributions were estimated as 95% confidence intervals via bootstrapping.
KEHRMIT assay strengths and limitations
The KEHRMIT assay offers a few key advantages over fiber analysis – the most common assay used to measure the average speed of replication forks, and PhADE – a real-time assay for imaging the growth of nascent DNA tracts (PhotoActivation, Diffusion and Excitation). (i) KEHRMIT directly monitors CMG dynamics as opposed to DNA synthesis. (ii) KEHRMIT provides temporal information that reveals changes in fork speed or transient helicase pausing. (iii) KEHRMIT has a higher spatial resolution than either DNA combing or PhADE because the position of the CMG helicase on DNA can be determined with sub-pixel accuracy. However, KEHRMIT is incompatible with simultaneous imaging of SYTOX-stained DNA because at the high protein concentration present in Xenopus egg extracts cross-links SYTOX-stained DNA to the surface of PEG-functionalized glass coverslips. Instead, the DNA molecules can be stained and imaged after they are immobilized, and de-stained prior to adding egg extracts to the flow cell. Optionally, DNA can also be SYTOX-stained and imaged after replication in egg extracts and stringent washing with SDS-containing buffer (Yardimci et al., 2012a), but some double-tethered DNA molecules break during replication and subsequently cannot be visualized using SYTOX. In the labeling scheme presented here, dual-color CMGAF647 and DPCAF568 imaging is incompatible with simultaneous PhADE imaging of Fen1mKikGR bound to nascent DNA (Loveland et al., 2012) because the absorbance/emission spectra of AF568 and the photo-switched form of mKikGR overlap. Consequently, the DPCAF568 signal could not be distinguished from that of Fen1mKikGR. In addition, the use of Fen1mKikGR at very high concentration (1–3 μM) to image nascent DNA synthesis by PhADE precludes the concomitant use of any green fluorophores (such as AF488) because of overlapping absorbance/emission spectra with the green form of mKikGR.
QUANTIFICATION AND STATISTICAL ANALYSIS
All ensemble experiments were performed three or more times, except for Figure 3D which was performed twice. Replication and sequencing gels were quantified in Multi-Gauge software from Fujifilm, the values from independent experiments were averaged and plotted in Microsoft Excel with error bars representing the standard deviation (the relevant statistical details are also listed in the figure legends).
All single-molecules experiments were performed two or more times. Single molecule data was acquired using NIS Elements software from Nikon, pre-processed in ImageJ using the Image Stabilized plugin from Kang Li at Carnegie Melon University, and quantified in MATLAB software from Mathworks. All beeswarm plots represent data from individual molecules as dots (the number of molecules, N, is listed in the figure or figure legends), the mean value is depicted as a horizontal blue line, and the 95% Confidence Interval (CI) for the mean value is illustrated as a gray box. The 95% Confidence Interval for the mean was used to determine if two quantities were significantly different. The standard MATLAB implementation of bootstrapping was used to estimate the 95% CI for the mean value of measurements extracted from single-molecule experiments, as specified in the figure legends.
Supplementary Material
Figure S1. Related to Figure 1. (A) Schematic showing trapping of M.HpaII on a plasmid containing a fluorinated M.HpaII recognition site. (B) Schematic of the two replication-coupled DPC proteolysis pathways in Xenopus egg extracts (Larsen et al., submitted). (C) Mock-depleted and SPRTN-depleted egg extracts were blotted using SPRTN and Mcm6 (loading control) antibodies. (D) pmeDPC2xLead was replicated in the indicated egg extracts. At different times, plasmid was recovered under stringent conditions, the DNA digested, and the released proteins subjected to immunoblot analysis with M.HpaII antibody. (E) In Figure 1C, stabilization of the DPC led to the appearance of stalling products from +1 to +12 (lanes 7-12 and 19-24). To determine whether these products depend on TLS, pmeDPCLead was replicated in the indicated extracts supplemented with [α−32P]-dATP. Samples were processed and analyzed as in Figure 1C. This analysis showed that the +1 to +12 products were eliminated by REV1 depletion. These products were also not observed during replication of a normal DPC in SPRTN-depleted extracts containing MG262 (data not shown), suggesting they are specific to DPC methylation. (F) Alternative models to explain the disappearance of the CMG footprint at stable DPCs: CMG dissociation (i) or CMG bypass (ii). (G) Model of CMG dynamics on pmeDPC2xLead. During replication of pmeDPC2xLead, both converging forks encounter a meDPC on the leading strand template. After CMGs bypass the two-leading strand DPCs, CMGs converge, as seen during replication termination, whereupon they are ubiquitylated (probably by CRL2Lrr1 (Dewar et al., 2017)), and unloaded by p97. (H) pDPCLead was replicated in undepleted extract containing [α−32P]-dATP and supplemented with buffer or 100 μM Cdc7i (PHA767491) at zero or ten minutes after NPE extract addition, which initiates replication, and analyzed as in Figure 1C. The fact that DPC bypass was unaffected when Cdc7i was added immediately after forks reached the DPC argues that new origin firing is not required for DPC bypass. (I) A serial dilution of mock-depleted egg extract was analyzed alongside FancM-depleted egg extracts via blotting with FancM antibody. (J) pDPC2xLead was replicated in the indicated extracts supplemented with [α−32P]-dATP. At different times, DNA was recovered and nicked with Nb. BsmI, separated on a denaturing polyacrylamide gel, and visualized by autoradiography. FANCM depletion inhibited FANCD2 ubiquitylation, demonstrating that FANCM was functionally depleted (data not shown). (K) Upper panel: pDPC2xLead was replicated in the indicated extract containing [α−32P]-dATP and supplemented with either buffer or ATRi. Samples were processed and analyzed as in Figure S1J. Lower panel: same extracts without addition of [α−32P]-dATP were replicated and reaction samples were separated by SDS-PAGE and blotted with the indicated antibodies.
{kind=link}
Figure S2. Related to Figure 2. Proposed action of a 5’ to 3’ helicase (pink) on four different substrates (i-iv). Since there are only 4 bp between the DPC attachment site and the bubble in pmeDPCLag/Lead-Bubble, arrival of CMG should melt the duplex surrounding the DPC (iv).
{kind=link}
Figure S3. Related to Figure 3. (A) Egg extracts contain multiple RTEL1 isoforms, as seen in lane 1 and Figure 3B, lane 1. To distinguish them, we depleted egg extract with an antibody raised against an N-terminal fragment of RTEL1 (α-RTEL1-N, lane 2; used in all experiments except lane 3 of this panel) or an antibody raised against a C-terminal peptide (α-RTEL1-C, lane 3), and blotted with α-RTEL1-N or ORC2 (loading control) antibodies. Unlike α-RTEL1-N, α-RTEL1-C depleted only the largest RTEL1 isoform, consistent with the presence of multiple isoforms (as seen in mice (Ding et al., 2004)), only the largest of which has the C-terminal extension against which the antibody was raised. Depletion of extracts with α-RTEL1-C antibody (lane 3) had no effect on CMG bypass (data not shown), demonstrating that the shorter isoforms were sufficient to perform this function. (B) Incorporation of [α−32P]-dATP during pmeDPCLead replication in mock-depleted or RTEL1-depleted extract was quantified and graphed. The mean of n=5 experiments is graphed. Error bars represent the standard deviation. (C) Coomassie blue-stained SDS-PAGE of purified RTEL1 wild-type and RTEL1-K48R. The RTEL1 and co-purifying GST-tag bands are indicated. (D) pmeDPCLead, pmeDPCLag/Lead, and pmeDPCLag/Lead-Bubble were replicated in the presence of [α−32P]-dATP in the indicated extracts and analyzed as in Figure S1J. The mean of n=3 experiments is graphed. Error bars represent the standard deviation. (E) Quantification of CMG bypass in Figure 3D. (F) DNA samples from Figure 3E were digested with the single-cutter AflIII, separated on a non-denaturing agarose gel, visualized by autoradiography, and linear products were quantified. The mean of n=3 experiments is graphed. Error bars represent the standard deviation.
{kind=link}
Figure S4. Related to Figure 4. (A) pDPC2XLead was replicated in the indicated extracts. At different times, plasmid-associated proteins were recovered under non-stringent conditions and blotted with the indicated antibodies. (B) Extracts used in Figure 4E were supplemented with [α−32P]-dATP and used to replicate pDPC2xLead. DNA samples were separated on a native agarose gel and subjected to autoradiography. The gel shows that in the absence of RTEL1, replication products accumulated as slow mobility intermediates instead of undergoing dissolution into monomeric open circular (OC) and supercoiled (SC) plasmids, reflecting a defect in CMG bypass and lack of DNA unwinding of the 165 bp that separates the DPCs (see cartoon). (C) pDPCssDNA was incubated directly in the indicated extracts without first licensing in HSS, which prevents replication initiation due to the high concentration of Geminin in NPE (Arias and Walter, 2005). Chromatin was isolated by the stringent pull-down procedure described in Figure 4A and analyzed for M.HpaII levels. RTEL1 depletion had no effect on the rate of M.HpaII destruction. (D) Same as in (C) except that M.HpaII conjugated to the gapped plasmid was methylated to inhibit the proteasome pathway. Short and long (larger excerpt) exposures of the same blot are shown. The result shows that in the setting of ssDNA, the SPRTN-specific product was generated at the same rate whether or not RTEL1 was present
{kind=link}
Figure S5. Related to Figure 5. (A-B) pmeDPC2xLead was replicated in the indicated extracts containing [32P]-dATP and supplemented with buffer or rGINSAF647. At the indicated times, DNA was recovered, resolved on a native agarose gel (A) or a sequencing gel (B), and visualized by autoradiography. Endogenous GINS depletion abolished and recombinant GINSAF647 fully rescued DNA replication (A). rGINSAF647 also supported loss of the CMG footprint (B). (C) The efficiency of DPC bypass by CMG from panel B and two additional experiments (n=3 total experiments) was quantified as described in Figure 2B and graphed. (D) A serial dilution of mock-depleted and GINS-depleted NPE was supplemented with buffer, 300 nM (1×) or 600 nM (2×) AF647-labeled rGINS, or 300 nM (1×) unlabeled rGINS, and analyzed by western blotting with antibodies against the Psf1 subunit of GINS. (E) Integrated fluorescence intensity traces for individual CMG molecules traveling along λ DNA illustrated that AF647 photobleached in a single step. (F) Kymogram of a replication bubble (as in Figure 5C) illustrating that the replication fork continues to travel (blue) after AF647 (green) photobleaching (arrow).
{kind=link}
Figure S6. Related to Figure 6. (A) Example of automated analysis of a DPCLead bypass event. (Left) Kymogram of the CMG-DPCLead encounter. (Middle) CMG and DPC positions were determined by fitting the centroid of the corresponding fluorescent signal. (Right) Plot of CMG-DPC distance. Approach (green), pause (red), and departure (blue) phases were detected by an automated algorithm (See methods). (B) Examples of BID, DD+, DD-, CD, and DT events. (C) Photobleaching kinetics for DPCAF568 and GINSAF647 (N=200 molecules were analyzed in each case). T1/2 denotes the half-life extracted from the exponential decay fit (solid lines) to the raw data (dots). The experiment was conducted in egg extract with DPCAF568 and GINSAF647 non-specifically bound to the glass coverslip using the same laser power and frame rate as experiments described in Figure 6. These data suggest that AF647 photobleaching is a major contributor to CD events. Although AF568 photobleaching contributes to DD+/− events for stabilized DPCs, it plays only a minor role in DD+/− events for degradable DPCs. (D) Quantification of three different classes of CMG-DPCLag encounters from n=2 independent experiments: (i) Bypass of intact DPC (BID), representing unambiguous bypass events; (ii) CMG bypassed the DPC position followed by DPC mobilization due to sister chromatid separation (B+M). (iii) DPC disappeared first, followed by CMG departure from the pause site (DD+). These are probably bypass events where the DPC was degraded before CMG departure. N represents the number of molecules. (E) Two examples of B+M events at meDPCLag in SPRTN-depleted extract. (F) Model of how B+M events result from sister chromatid separation when the outward-moving CMG reaches the end of the DNA substrate. (G) Histograms of meDPC lifetime in mock-depleted extract in the absence of replication (N=199 molecules) (+geminin control to block origin firing) versus meDPCLead in SPRTN-depleted extracts (N=151 molecules) and nonmethylated DPCLead in mock-depleted extract (N=183 molecules), both in the presence of replication. In all cases, DPC lifetime was measured from the beginning of the experiment. The peak at 90-100 min corresponds to DPCAF568 molecules that survived the entire 100-min experiment. Blue lines and gray boxes correspond to the mean and the 95% CI. Given that the DPC lifetime in reactions with active proteolysis (bottom panel) is significantly shorter than the DPC lifetime when proteolysis has been inhibited (middle panel) or when replication has been inhibited (top panel), we conclude that the loss of DPC signal in replication reactions where both SPRTN and the proteasome are active is largely due to replication-coupled proteolysis and not AF568 photobleaching. (H) Replication origins firing in extracts supplemented with vehicle (left) or 150 μM aphidicolin (right). CMG and nascent DNA signals are shown in green and blue respectively (same as Figure 5C). The absence of blue signal in the +Aph. experiment confirms the inhibition of DNA synthesis. (I) Beeswarm plot of CMG processivity in the presence of aphidicolin. Blue lines and gray boxes correspond to the mean and the 95% CI. N represents the number of molecules. (J) Beeswarm plot of the time needed to bypass methylated DPCLead in SPRTN-depleted extract versus nonmethylated DPCLead in undepleted extract. Blue lines and gray boxes as in (I). N represents the number of molecules.
{kind=link}
Figure S7. Related to Figure 6. (A) pLacO32 was pre-incubated with LacR and replicated in the indicated extracts containing [α−32P]-dATP and supplemented with DMSO or MG262. At the indicated times, DNA was recovered, digested with the single cutter XmnI, resolved by native agarose gel electrophoresis, and visualized by autoradiography. Accumulation of the linear species was quantified, the mean of n=3 experiments is graphed. Error bars represent the standard deviation. Immunodepletion of SPRTN and addition of proteasome inhibitor had no effect on the conversion of X-shaped intermediates to fully replicated daughter molecules, indicating that LacR proteolysis was not required for replication fork progression through the array. (B) Bypass model in which CMG denatures the DPC and threads the resulting polypeptide chains through the central channel of the MCM2-7. (C) Bypass model in which the MCM2-7 ring transiently opens, possibly due to dissociation of GINS and/or CDC45 (not shown), allowing the helicase to translocate or be towed (not shown) past the DPC. (D) Model for template switching at a DPC. If CMG bypasses the DPC but TLS fails, the leading strand anneals to the nascent lagging strand. After extension of the leading strand (blue arrow), it undergoes dissolution and re-anneals to the leading strand template, thereby bypassing the adduct.
{kind=link}
Highlights.
Replicative helicase CMG bypasses DNA-protein crosslinks on its translocation strand
RTEL1, an accessory helicase, facilitates bypass by unwinding DNA beyond the DPC
CMG slows after bypass, likely due to uncoupling from DNA synthesis
Bypass triggers DPC proteolysis and prevents accidental degradation of CMG
Acknowledgements
We thank Niels Mailand, Peter Burgers, Joe Loparo, and the Walter laboratory for feedback on the manuscript, Seungwoo Chang for sortase, Ravi Amunugama for characterizing the FANCM antibody, and Livio Dukaj for Fen1mKikGR. Single-molecule data was collected at the Nikon Imaging Center (NIC) at Harvard Medical School with technical assistance by Jennifer Waters and Talley Lambert. J.L.S. and G.C. were supported by postdoctoral fellowships from the Damon Runyon Cancer Research Foundation and the Jane Coffin Childs Memorial Fund, respectively. J.C.W. is supported by NIH grant GM80676. J.C.W. is an investigator of the Howard Hughes Medical Institute. J.P.D. received funding from the European Research Council (grant agreement No 715975) and the Novo Nordisk Foundation (Grant agreement NNF14CC0001).
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arias EE, and Walter JC (2005). Replication-dependent destruction of Cdt1 limits DNA replication to a single round per cell cycle in Xenopus egg extracts. Genes Dev 19, 114–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochman ML, and Schwacha A (2008). The Mcm2–7 complex has in vitro helicase activity. Mol Cell 31, 287–293. [DOI] [PubMed] [Google Scholar]
- Byun TS, Pacek M, Yee MC, Walter JC, and Cimprich KA (2005). Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev 19, 1040–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I, Dorr BM, and Liu DR (2011). A general strategy for the evolution of bond-forming enzymes using yeast display. Proc Natl Acad Sci U S A 108, 11399–11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar JM, Budzowska M, and Walter JC (2015). The mechanism of DNA replication termination in vertebrates. Nature 525, 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar JM, Low E, Mann M, Raschle M, and Walter JC (2017). CRL2(Lrr1) promotes unloading of the vertebrate replisome from chromatin during replication termination. Genes Dev 31, 275–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Schertzer M, Wu X, Gertsenstein M, Selig S, Kammori M, Pourvali R, Poon S, Vulto I, Chavez E, et al. (2004). Regulation of murine telomere length by Rtel: an essential gene encoding a helicase-like protein. Cell 117, 873–886. [DOI] [PubMed] [Google Scholar]
- Douglas ME, Ali FA, Costa A, and Diffley JFX (2018). The mechanism of eukaryotic CMG helicase activation. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxin JP, Dewar JM, Yardimci H, and Walter JC (2014). Repair of a DNA-protein crosslink by replication-coupled proteolysis. Cell 159, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duzdevich D, Warner MD, Ticau S, Ivica NA, Bell SP, and Greene EC (2015). The dynamics of eukaryotic replication initiation: origin specificity, licensing, and firing at the single-molecule level. Mol Cell 58, 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YV, Yardimci H, Long DT, Guainazzi A, Bermudez VP, Hurwitz J, van Oijen A, Scharer OD, and Walter JC (2011). Selective Bypass of a Lagging Strand Roadblock by the Eukaryotic Replicative DNA Helicase. Cell 146, 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullbright G, Rycenga HB, Gruber JD, and Long DT (2016). p97 Promotes a Conserved Mechanism of Helicase Unloading during DNA Cross-Link Repair. Mol Cell Biol 36, 2983–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu R, Yuan Z, Bai L, de Luna Almeida Santos R, Sun J, Zhang D, Yurieva O, Li H, and O’Donnell ME (2017). Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation. Proc Natl Acad Sci U S A 114, E697–E706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JE, Marians KJ, and Kowalczykowski SC (2017). Independent and Stochastic Action of DNA Polymerases in the Replisome. Cell 169, 1201–1213 e1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CP, Atkinson J, Gupta MK, Mahdi AA, Gwynn EJ, Rudolph CJ, Moon PB, van Knippenberg IC, Cadman CJ, Dillingham MS, et al. (2009). Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol Cell 36, 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, and Seidman MM (2013). The DNA Translocase FANCM/MHF Promotes Replication Traverse of DNA Interstrand Crosslinks. Mol Cell 52, 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide H, Shoulkamy MI, Nakano T, Miyamoto-Matsubara M, and Salem AM (2011). Repair and biochemical effects of DNA-protein crosslinks. Mutat Res 711, 113–122. [DOI] [PubMed] [Google Scholar]
- Ivessa AS, Zhou JQ, and Zakian VA (2000). The Saccharomyces Pif1p DNA helicase and the highly related Rrm3p have opposite effects on replication fork progression in ribosomal DNA. Cell 100, 479–489. [DOI] [PubMed] [Google Scholar]
- Klimasauskas S, Kumar S, Roberts RJ, and Cheng X (1994). HhaI methyltransferase flips its target base out of the DNA helix. Cell 76, 357–369. [DOI] [PubMed] [Google Scholar]
- Langston LD, Mayle R, Schauer GD, Yurieva O, Zhang D, Yao NY, Georgescu RE, and O’Donnell ME (2017). Mcm10 promotes rapid isomerization of CMG-DNA for replisome bypass of lagging strand DNA blocks. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston LD, Zhang D, Yurieva O, Georgescu RE, Finkelstein J, Yao NY, Indiani C, and O’Donnell ME (2014). CMG helicase and DNA polymerase epsilon form a functional 15-subunit holoenzyme for eukaryotic leading-strand DNA replication. Proc Natl Acad Sci U S A 111, 15390–15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebofsky R, van Oijen AM, and Walter JC (2011). DNA is a co-factor for its own replication in Xenopus egg extracts. Nucleic Acids Res 39, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessel D, Vaz B, Halder S, Lockhart PJ, Marinovic-Terzic I, Lopez-Mosqueda J, Philipp M, Sim JC, Smith KR, Oehler J, et al. (2014). Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat Genet 46, 1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JS, Spenkelink LM, Schauer GD, Hill FR, Georgescu RE, O’Donnell ME, and van Oijen AM (2017). Single-molecule visualization of Saccharomyces cerevisiae leading-strand synthesis reveals dynamic interaction between MTC and the replisome. Proc Natl Acad Sci U S A 114, 10630–10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling C, Huang J, Yan Z, Li Y, Ohzeki M, Ishiai M, Xu D, Takata M, Seidman M, and Wang W (2016). Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell Discov 2, 16047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Mosqueda J, Maddi K, Prgomet S, Kalayil S, Marinovic-Terzic I, Terzic J, and Dikic I (2016). SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveland AB, Habuchi S, Walter JC, and van Oijen AM (2012). A general approach to break the concentration barrier in single-molecule imaging. Nature methods 9, 987–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maric M, Maculins T, De Piccoli G, and Labib K (2014). Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 346, 1253596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskey RS, Kim MS, Baker DJ, Childs B, Malureanu LA, Jeganathan KB, Machida Y, van Deursen JM, and Machida YJ (2014). Spartan deficiency causes genomic instability and progeroid phenotypes. Nat Commun 5, 5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno SP, Bailey R, Campion N, Herron S, and Gambus A (2014). Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science 346, 477–481. [DOI] [PubMed] [Google Scholar]
- O’Donnell ME, and Li H (2018). The ring-shaped hexameric helicases that function at DNA replication forks. Nat Struct Mol Biol 25, 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samel SA, Fernandez-Cid A, Sun J, Riera A, Tognetti S, Herrera MC, Li H, and Speck C (2014). A unique DNA entry gate serves for regulated loading of the eukaryotic replicative helicase MCM2–7 onto DNA. Genes & development 28, 1653–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks J, and Walter JC (2018). Extracts for Analysis of DNA Replication in a Nucleus-Free System. Cold Spring Harb Protoc. [DOI] [PubMed] [Google Scholar]
- Stano NM, Jeong YJ, Donmez I, Tummalapalli P, Levin MK, and Patel SS (2005). DNA synthesis provides the driving force to accelerate DNA unwinding by a helicase. Nature 435, 370–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, Bellelli R, Alte F, Hewitt G, Sarek G, Maslen SL, Tsutakawa SE, Borg A, Kjaer S, Tainer JA, et al. (2016). Mechanism and Regulation of DNA-Protein Crosslink Repair by the DNADependent Metalloprotease SPRTN. Mol Cell 64, 688–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, Bellelli R, and Boulton SJ (2017). Mechanisms of DNA-protein crosslink repair. Nat Rev Mol Cell Biol 18, 563–573. [DOI] [PubMed] [Google Scholar]
- Stingele J, Schwarz MS, Bloemeke N, Wolf PG, and Jentsch S (2014). A DNA-dependent protease involved in DNA-protein crosslink repair. Cell 158, 327–338. [DOI] [PubMed] [Google Scholar]
- Taylor MRG, and Yeeles JTP (2018). The Initial Response of a Eukaryotic Replisome to DNA Damage. Mol Cell 70, 1067–1080 e1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowitzsch S, Bieniossek C, Nie Y, Garzoni F, and Berger I (2010). New baculovirus expression tools for recombinant protein complex production. J Struct Biol 172, 45–54. [DOI] [PubMed] [Google Scholar]
- Vannier JB, Sandhu S, Petalcorin MI, Wu X, Nabi Z, Ding H, and Boulton SJ (2013). RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science 342, 239–242. [DOI] [PubMed] [Google Scholar]
- Vannier JB, Sarek G, and Boulton SJ (2014). RTEL1: functions of a disease-associated helicase. Trends Cell Biol 24, 416–425. [DOI] [PubMed] [Google Scholar]
- Vaz B, Popovic M, Newman JA, Fielden J, Aitkenhead H, Halder S, Singh AN, Vendrell I, Fischer R, Torrecilla I, et al. (2016). Metalloprotease SPRTN/DVC1 Orchestrates ReplicationCoupled DNA-Protein Crosslink Repair. Mol Cell 64, 704–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, and Newport J (2000). Initiation of eukaryotic DNA replication: origin unwinding and sequential chromatin association of Cdc45, RPA, and DNA polymerase alpha. Mol Cell 5, 617–627. [DOI] [PubMed] [Google Scholar]
- Walter J, and Newport JW (1997). Regulation of replicon size in Xenopus egg extracts. Science 275, 993–995. [DOI] [PubMed] [Google Scholar]
- Yardimci H, Loveland AB, van Oijen AM, and Walter JC (2012a). Single-molecule analysis of DNA replication in Xenopus egg extracts. Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardimci H, Wang X, Loveland AB, Tappin I, Rudner DZ, Hurwitz J, van Oijen AM, and Walter JC (2012b). Bypass of a protein barrier by a replicative DNA helicase. Nature 492, 205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Related to Figure 1. (A) Schematic showing trapping of M.HpaII on a plasmid containing a fluorinated M.HpaII recognition site. (B) Schematic of the two replication-coupled DPC proteolysis pathways in Xenopus egg extracts (Larsen et al., submitted). (C) Mock-depleted and SPRTN-depleted egg extracts were blotted using SPRTN and Mcm6 (loading control) antibodies. (D) pmeDPC2xLead was replicated in the indicated egg extracts. At different times, plasmid was recovered under stringent conditions, the DNA digested, and the released proteins subjected to immunoblot analysis with M.HpaII antibody. (E) In Figure 1C, stabilization of the DPC led to the appearance of stalling products from +1 to +12 (lanes 7-12 and 19-24). To determine whether these products depend on TLS, pmeDPCLead was replicated in the indicated extracts supplemented with [α−32P]-dATP. Samples were processed and analyzed as in Figure 1C. This analysis showed that the +1 to +12 products were eliminated by REV1 depletion. These products were also not observed during replication of a normal DPC in SPRTN-depleted extracts containing MG262 (data not shown), suggesting they are specific to DPC methylation. (F) Alternative models to explain the disappearance of the CMG footprint at stable DPCs: CMG dissociation (i) or CMG bypass (ii). (G) Model of CMG dynamics on pmeDPC2xLead. During replication of pmeDPC2xLead, both converging forks encounter a meDPC on the leading strand template. After CMGs bypass the two-leading strand DPCs, CMGs converge, as seen during replication termination, whereupon they are ubiquitylated (probably by CRL2Lrr1 (Dewar et al., 2017)), and unloaded by p97. (H) pDPCLead was replicated in undepleted extract containing [α−32P]-dATP and supplemented with buffer or 100 μM Cdc7i (PHA767491) at zero or ten minutes after NPE extract addition, which initiates replication, and analyzed as in Figure 1C. The fact that DPC bypass was unaffected when Cdc7i was added immediately after forks reached the DPC argues that new origin firing is not required for DPC bypass. (I) A serial dilution of mock-depleted egg extract was analyzed alongside FancM-depleted egg extracts via blotting with FancM antibody. (J) pDPC2xLead was replicated in the indicated extracts supplemented with [α−32P]-dATP. At different times, DNA was recovered and nicked with Nb. BsmI, separated on a denaturing polyacrylamide gel, and visualized by autoradiography. FANCM depletion inhibited FANCD2 ubiquitylation, demonstrating that FANCM was functionally depleted (data not shown). (K) Upper panel: pDPC2xLead was replicated in the indicated extract containing [α−32P]-dATP and supplemented with either buffer or ATRi. Samples were processed and analyzed as in Figure S1J. Lower panel: same extracts without addition of [α−32P]-dATP were replicated and reaction samples were separated by SDS-PAGE and blotted with the indicated antibodies.
Figure S2. Related to Figure 2. Proposed action of a 5’ to 3’ helicase (pink) on four different substrates (i-iv). Since there are only 4 bp between the DPC attachment site and the bubble in pmeDPCLag/Lead-Bubble, arrival of CMG should melt the duplex surrounding the DPC (iv).
Figure S3. Related to Figure 3. (A) Egg extracts contain multiple RTEL1 isoforms, as seen in lane 1 and Figure 3B, lane 1. To distinguish them, we depleted egg extract with an antibody raised against an N-terminal fragment of RTEL1 (α-RTEL1-N, lane 2; used in all experiments except lane 3 of this panel) or an antibody raised against a C-terminal peptide (α-RTEL1-C, lane 3), and blotted with α-RTEL1-N or ORC2 (loading control) antibodies. Unlike α-RTEL1-N, α-RTEL1-C depleted only the largest RTEL1 isoform, consistent with the presence of multiple isoforms (as seen in mice (Ding et al., 2004)), only the largest of which has the C-terminal extension against which the antibody was raised. Depletion of extracts with α-RTEL1-C antibody (lane 3) had no effect on CMG bypass (data not shown), demonstrating that the shorter isoforms were sufficient to perform this function. (B) Incorporation of [α−32P]-dATP during pmeDPCLead replication in mock-depleted or RTEL1-depleted extract was quantified and graphed. The mean of n=5 experiments is graphed. Error bars represent the standard deviation. (C) Coomassie blue-stained SDS-PAGE of purified RTEL1 wild-type and RTEL1-K48R. The RTEL1 and co-purifying GST-tag bands are indicated. (D) pmeDPCLead, pmeDPCLag/Lead, and pmeDPCLag/Lead-Bubble were replicated in the presence of [α−32P]-dATP in the indicated extracts and analyzed as in Figure S1J. The mean of n=3 experiments is graphed. Error bars represent the standard deviation. (E) Quantification of CMG bypass in Figure 3D. (F) DNA samples from Figure 3E were digested with the single-cutter AflIII, separated on a non-denaturing agarose gel, visualized by autoradiography, and linear products were quantified. The mean of n=3 experiments is graphed. Error bars represent the standard deviation.
Figure S4. Related to Figure 4. (A) pDPC2XLead was replicated in the indicated extracts. At different times, plasmid-associated proteins were recovered under non-stringent conditions and blotted with the indicated antibodies. (B) Extracts used in Figure 4E were supplemented with [α−32P]-dATP and used to replicate pDPC2xLead. DNA samples were separated on a native agarose gel and subjected to autoradiography. The gel shows that in the absence of RTEL1, replication products accumulated as slow mobility intermediates instead of undergoing dissolution into monomeric open circular (OC) and supercoiled (SC) plasmids, reflecting a defect in CMG bypass and lack of DNA unwinding of the 165 bp that separates the DPCs (see cartoon). (C) pDPCssDNA was incubated directly in the indicated extracts without first licensing in HSS, which prevents replication initiation due to the high concentration of Geminin in NPE (Arias and Walter, 2005). Chromatin was isolated by the stringent pull-down procedure described in Figure 4A and analyzed for M.HpaII levels. RTEL1 depletion had no effect on the rate of M.HpaII destruction. (D) Same as in (C) except that M.HpaII conjugated to the gapped plasmid was methylated to inhibit the proteasome pathway. Short and long (larger excerpt) exposures of the same blot are shown. The result shows that in the setting of ssDNA, the SPRTN-specific product was generated at the same rate whether or not RTEL1 was present
Figure S5. Related to Figure 5. (A-B) pmeDPC2xLead was replicated in the indicated extracts containing [32P]-dATP and supplemented with buffer or rGINSAF647. At the indicated times, DNA was recovered, resolved on a native agarose gel (A) or a sequencing gel (B), and visualized by autoradiography. Endogenous GINS depletion abolished and recombinant GINSAF647 fully rescued DNA replication (A). rGINSAF647 also supported loss of the CMG footprint (B). (C) The efficiency of DPC bypass by CMG from panel B and two additional experiments (n=3 total experiments) was quantified as described in Figure 2B and graphed. (D) A serial dilution of mock-depleted and GINS-depleted NPE was supplemented with buffer, 300 nM (1×) or 600 nM (2×) AF647-labeled rGINS, or 300 nM (1×) unlabeled rGINS, and analyzed by western blotting with antibodies against the Psf1 subunit of GINS. (E) Integrated fluorescence intensity traces for individual CMG molecules traveling along λ DNA illustrated that AF647 photobleached in a single step. (F) Kymogram of a replication bubble (as in Figure 5C) illustrating that the replication fork continues to travel (blue) after AF647 (green) photobleaching (arrow).
Figure S6. Related to Figure 6. (A) Example of automated analysis of a DPCLead bypass event. (Left) Kymogram of the CMG-DPCLead encounter. (Middle) CMG and DPC positions were determined by fitting the centroid of the corresponding fluorescent signal. (Right) Plot of CMG-DPC distance. Approach (green), pause (red), and departure (blue) phases were detected by an automated algorithm (See methods). (B) Examples of BID, DD+, DD-, CD, and DT events. (C) Photobleaching kinetics for DPCAF568 and GINSAF647 (N=200 molecules were analyzed in each case). T1/2 denotes the half-life extracted from the exponential decay fit (solid lines) to the raw data (dots). The experiment was conducted in egg extract with DPCAF568 and GINSAF647 non-specifically bound to the glass coverslip using the same laser power and frame rate as experiments described in Figure 6. These data suggest that AF647 photobleaching is a major contributor to CD events. Although AF568 photobleaching contributes to DD+/− events for stabilized DPCs, it plays only a minor role in DD+/− events for degradable DPCs. (D) Quantification of three different classes of CMG-DPCLag encounters from n=2 independent experiments: (i) Bypass of intact DPC (BID), representing unambiguous bypass events; (ii) CMG bypassed the DPC position followed by DPC mobilization due to sister chromatid separation (B+M). (iii) DPC disappeared first, followed by CMG departure from the pause site (DD+). These are probably bypass events where the DPC was degraded before CMG departure. N represents the number of molecules. (E) Two examples of B+M events at meDPCLag in SPRTN-depleted extract. (F) Model of how B+M events result from sister chromatid separation when the outward-moving CMG reaches the end of the DNA substrate. (G) Histograms of meDPC lifetime in mock-depleted extract in the absence of replication (N=199 molecules) (+geminin control to block origin firing) versus meDPCLead in SPRTN-depleted extracts (N=151 molecules) and nonmethylated DPCLead in mock-depleted extract (N=183 molecules), both in the presence of replication. In all cases, DPC lifetime was measured from the beginning of the experiment. The peak at 90-100 min corresponds to DPCAF568 molecules that survived the entire 100-min experiment. Blue lines and gray boxes correspond to the mean and the 95% CI. Given that the DPC lifetime in reactions with active proteolysis (bottom panel) is significantly shorter than the DPC lifetime when proteolysis has been inhibited (middle panel) or when replication has been inhibited (top panel), we conclude that the loss of DPC signal in replication reactions where both SPRTN and the proteasome are active is largely due to replication-coupled proteolysis and not AF568 photobleaching. (H) Replication origins firing in extracts supplemented with vehicle (left) or 150 μM aphidicolin (right). CMG and nascent DNA signals are shown in green and blue respectively (same as Figure 5C). The absence of blue signal in the +Aph. experiment confirms the inhibition of DNA synthesis. (I) Beeswarm plot of CMG processivity in the presence of aphidicolin. Blue lines and gray boxes correspond to the mean and the 95% CI. N represents the number of molecules. (J) Beeswarm plot of the time needed to bypass methylated DPCLead in SPRTN-depleted extract versus nonmethylated DPCLead in undepleted extract. Blue lines and gray boxes as in (I). N represents the number of molecules.
Figure S7. Related to Figure 6. (A) pLacO32 was pre-incubated with LacR and replicated in the indicated extracts containing [α−32P]-dATP and supplemented with DMSO or MG262. At the indicated times, DNA was recovered, digested with the single cutter XmnI, resolved by native agarose gel electrophoresis, and visualized by autoradiography. Accumulation of the linear species was quantified, the mean of n=3 experiments is graphed. Error bars represent the standard deviation. Immunodepletion of SPRTN and addition of proteasome inhibitor had no effect on the conversion of X-shaped intermediates to fully replicated daughter molecules, indicating that LacR proteolysis was not required for replication fork progression through the array. (B) Bypass model in which CMG denatures the DPC and threads the resulting polypeptide chains through the central channel of the MCM2-7. (C) Bypass model in which the MCM2-7 ring transiently opens, possibly due to dissociation of GINS and/or CDC45 (not shown), allowing the helicase to translocate or be towed (not shown) past the DPC. (D) Model for template switching at a DPC. If CMG bypasses the DPC but TLS fails, the leading strand anneals to the nascent lagging strand. After extension of the leading strand (blue arrow), it undergoes dissolution and re-anneals to the leading strand template, thereby bypassing the adduct.