

Abstract
Over-expression and activation of matrix metalloproteinase-9 (MMP-9) is associated with multiple diseases, and can serve as a stimulus to activate nanomaterials for sensing and controlled release. In order to achieve autonomous therapeutics with improved space-time targeting capabilities, several features need to be considered beyond the introduction of an enzyme-cleavable linker into a nanostructure. We introduce guiding principles for a customizable platform using supramolecular peptide nanostructures with three modular components to achieve tunable kinetics and morphology changes upon MMP-9 exposure. This approach enables: (1) fine-tuning of kinetics through introduction of ordered/disordered structures, (2) a 12-fold variation in hydrolysis rates achieved by electrostatic (mis) matching of particle and enzyme charge, and (3) selection of enzymatic reaction products that are either cell-killing nanofibers or that disintegrate. These guiding principles, which can be rationalized and involve exchange of just a few amino acids, enable systematic customization of enzyme-responsive peptide nanostructures for general use in performance optimization of enzyme-responsive materials.
Keywords: MMP-responsive, enzyme kinetics, electrostatic enzyme interactions, supramolecular organization, morphology change, self-assembling peptides
Graphical Abstract
Peptide amphiphiles spontaneously form spherical micelles or worm-like micelles depending on the amino acid residues in the MMP-9 cleavable segment, which also dictates the fiber formation or disassembly of post-enzymatic products. The rate of MMP-9 hydrolysis is modulated using (mis) matching electrostatic charges of the cationic (blue) and anionic (red) nanostructures and the enzyme, and further fine-tuned by varying the degree of supramolecular order to impact the enzyme’s accessibility to the substrate.
Nanomaterials impact biomedicine by taking advantage of their inherent chemical and physical properties to achieve increased circulation lifetime and selective bio-distribution in vivo, and dictate cellular uptake mechanisms,1 all of which cannot be attained by molecular drugs alone. Another beneficial feature of nanomaterials is the ability to incorporate stimuli-responsive functionalities to enhance selectivity in targeting diseased cells, and manipulate drug release profiles.2–5 In particular, an inherent biological stimulus such as (over-) expression of enzymes, can serve as a marker for diseased cells, as well as a trigger to facilitate desired changes in the nanostructure.6 Understanding the relationship between properties of nanomaterials, such as size, shape, and charge, with their bio-distribution or cellular uptake patterns have allowed researchers to develop refined systems to target specific organs and cells. For example, Discher et al. described that the increase in length of flexible filomicelles up to 8 μm increased the circulation lifetime in vivo in mice, in comparison to shorter filomicelles and spherical vesicles.7 Likewise, a thorough investigation is necessary in order to develop design rules for nanomaterials that can engage with the enzymatic stimuli with varying degrees of affinity in order to predetermine response kinetics for the desired enzyme-responsive action (i.e. disassembly, morphology switch, etc.).6
For instance, an over-expression and activation of matrix metalloproteinase-9 (MMP-9), an enzyme that is crucial to normal behavior of cells such as degradation of extracellular matrix,8 is associated with multiple diseases including cancer metastasis,9 cardiovascular diseases,10 arthritis,11 etc. Since the introduction of polymeric MMP-responsive materials by Hubbell et al.,12,13 there have been numerous strategies to exploit this highly disease-relevant enzyme for bio-medical applications.14–21 One of the challenges in designing MMP-9 responsive nanomaterials is optimizing a cleavable segment that meets the enzyme specificity,22 and is concurrently compatible with the nanoparticle system. Another, largely overlooked but important aspect of particle design is regulating the susceptibility of the nanoparticle to the enzyme stimuli through the manipulation of electrostatic properties of nanoparticles to attract or repel enzymes of opposite or same charge,23 and to control the degree of supramolecular organization of the nanoparticle to increase or limit enzyme access to the particle, and ultimately influence the observed reaction and response kinetics.24
A further design aspect is the morphology of the particle pre- and post-cleavage; there is increasing evidence that enzymatically triggered formation of nanofibers on tumor cells can cause cytotoxic effects,15,25 and Xu et al. have extensively studied this mechanism to overcome drug-resistance in cancer cells,26,27 while disintegrating particles may be beneficial for controlled drug release.19,20 The anti-cancer activity of the nanofibers depends on the kinetics of fiber formation which in turn depend on the interaction between the enzyme and the precursor, as well as the self-assembling ability of the post-enzymatic product.28 By employing these strategies, the response behavior of the enzyme-responsive nanomaterials can be optimized to achieve selective and controlled rate of drug release, and to introduce additional therapeutic functionalities.
Thus, by using rational design of peptide sequences, we present here a modular platform to customize surface charge, supramolecular organization, and enzyme specificity of peptide nanostructures. We demonstrate the significance of these properties in showing that simple, few amino acid replacements can systematically control enzyme engagement and susceptibility to the enzymatic action which dictates the response kinetics and, in addition, can dictate the action of the nanostructures (degradation or β-sheet formation) to ultimately influence cells’ fate.
RESULTS AND DISCUSSION
Rational design of peptide sequences.
For this study, we designed self-assembling peptide amphiphiles29 that form stable nanostructures under physiological conditions, and undergo morphological change or degradation upon MMP-9 hydrolysis of the peptides, building on our previous work which demonstrated that peptide micelles can encapsulate doxorubicin and transform into fibrous drug depots upon MMP-9 action.21,30 Thus, we created a modular system in which the peptide sequences contain three segments: (1) cationic or anionic hydrophilic segment to modulate enzyme engagement, (2) MMP-9 cleavable segment with ordered or disordered regions to influence enzyme kinetics and predetermine self-assembly or dis-assembly of post-cleavage product and, and (3) hydrophobic segment to drive self-assembly of precursor and of post-enzymatic self-assembling product (with potential to bind hydrophobic drugs21) (Figure 1).
Figure 1.

Sequence dependent peptide nanostructures. (A) Chemical structure and (B) cartoon of self-assembling peptide amphiphiles that respond to MMP-9 action. Positive (blue) or negative (red) charges on the nanoparticle electrostatically recruit or repel MMP-9 to influence enzyme kinetics. Self-assembling (purple) and MMP-9 cleavable segments (gray and/or orange) dictate the susceptibility of the nanostructures to MMP-9 hydrolysis by forming ordered/disordered structures, and control the fiber formation or disassembly of the post-enzymatic products. Red dash line indicates the scissile bond.
To demonstrate the ability to electrostatically recruit MMP-9 (pI = 5.7, net negative charge in physiological pH)31 to the peptide particles, we designed a hydrophilic segment in the peptide amphiphiles with cationic or anionic C termini. Each peptide was given a positive (1-3 AK/PK) or negative (1-3 AD/PD) charge using two lysine or two aspartic acid residues, thus creating 12 sequences (Table 1). In addition, these surfaces charges increase solubility of the peptide nanostructures and can be customized to potentially influence cellular uptake and bio-distribution.
Table 1.
Single letter code of 12 peptides (red dash line indicates expected cleavage site), critical aggregation concentrations of peptides determined using pyrene (Figure S9 in SI), and zeta potentials of 5 mM peptides in 2% PBS (pH 7.4) at 25°C (Figure S8 in SI).
| Peptide Sequence | Charge | CAC* | Zeta Potential | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Position | P6 | P5 | P4 | P3 | P2 | P1 | P1’ | P2’ | P3’ | P4’ | P5’ | pH 7.4 | (mM) | Mean (mV) ± Std. dev |
| P/A | X1 | G | L | X2 | G | |||||||||
| 1AK | F | F | A | L | G | L | A | G | K | K | +2 | 0.4 | 11.5 ± 0.8 | |
| 1AD | F | F | A | L | G | L | A | G | D | D | −2 | 0.6 | −34.9 ± 1.1 | |
| 1PK | F | F | P | L | G | L | A | G | K | K | +2 | 0.3 | 15.4 ± 0.3 | |
| 1PD | F | F | P | L | G | L | A | G | D | D | −2 | 0.5 | −38.0 ± 1.7 | |
| 2AK | F | F | G | A | L | G | L | K | G | K | +2 | 0.4 | 8.2 ± 2.8 | |
| 2AD | F | F | G | A | L | G | L | D | G | D | −2 | 0.6 | −30.3 ± 1.4 | |
| 2PK | F | F | G | P | L | G | L | K | G | K | +2 | 0.3 | 4.1 ± 1.0 | |
| 2PD | F | F | G | P | L | G | L | D | G | D | −2 | 0.7 | −41.2 ± 2.3 | |
| 3AK | F | F | G | A | A | G | L | K | G | K | +2 | 0.8 | 1.3 ± 0.6 | |
| 3AD | F | F | G | A | A | G | L | D | G | D | −2 | 0.5 | −7.5 ± 1.8 | |
| 3PK | F | F | G | P | A | G | L | K | G | K | +2 | 0.7 | 1.1 ± 1.5 | |
| 3PD | F | F | G | P | A | G | L | D | G | D | −2 | 0.5 | −28.2 ± 0.7 | |
CAC: critical aggregation concentration
To achieve MMP-9 specificity, and to program the distinct morphologies of the peptide nanostructures and the resulting enzymatic products, we designed the cleavable segment based on data from the MEROPS database,32 which suggests PX1G↓LX2G where ↓ represents the scissile bond, and X1 and X2 represent positions where there are no significant preferences for a single amino acid. The amino acid positions are designated starting from the scissile bond (red dash line in Table 1) and are labeled P1 through P6 towards the N terminal, and P1’ and P5’ towards the C terminal. Peptides 1-3 PK/PD contain Pro in P3 which is prevalent in substrates of most MMPs,22 however, Pro is known to disrupt assembly of secondary structures like α-helices and β-sheets in proteins.33 Therefore, in order to promote fiber formation of the post-cleavage enzymatic products, we substituted Pro for Ala in P3 in 1-3 AK/AD which is also found in natural substrates of MMP-9.34 In addition, we varied P2 with small aliphatic residues to observe differences in enzyme specificity, and inserted Gly in P4 to change the self-assembling behavior of peptides and observe the consequent changes in enzyme kinetics.
Lastly, to introduce the hydrophobic segment of our amphiphilic peptides, we used the well-known self-assembling sequence diphenylalanine35,36 on the first two positions of the N termini to drive self-assembly of nanostructures prior to enzyme action via hydrophobic and aromatic interactions, and to form nanofibers in a subset of sequences after enzyme action (1-3 AK/AD). These 12 sequences represent a modular design in which the rate and morphology of the enzymatic response can be customized for desired applications.
Characterization of the peptide nanostructures.
The 12 peptides were synthesized using Fmoc-based solid phase peptide synthesis (SPPS), purified on high-performance liquid chromatography (HPLC) using C18 column, lyophilized in water, and identified by high resolution liquid chromatography-mass spectrometry (HRMS) and 1H nuclear magnetic resonance (NMR) spectroscopy (Figure S1-2 in SI). The critical aggregation concentrations of the peptides were determined using pyrene as a fluorescent probe and ranged between 0.3-0.8 mM for cationic peptides and 0.5-0.7 mM for anionic peptides (Table 1 and Figure S9 in SI). The zeta potentials of the peptide assemblies confirmed the presence of the expected charges (Table 1). Higher zeta potentials were observed for the anionic peptides which have negatively charged Asp residues on the free carboxyl termini of the peptides, in comparison to the cationic peptides in which the positive charge of Lys residues is negated by the free C termini (Figure 1A). We note that measurements were made at high concentrations (5 mM) due to the limited light scattering properties of the peptide particles that may have contributed to aggregation which results in overall lower zeta potential values (Figure S8 in SI).
Atomic force microscopy (AFM) images show that AK/AD sequences form one-dimensional nanostructures and PK/PD sequences form spherical nanostructures with the exception of 1 PK/PD (Figure 2A-B). Analysis of Fourier-transform infrared spectroscopy (FTIR) spectra reveal that the self-assembly of most of the peptide sequences are not majorly driven by highly ordered hydrogen bonding of the peptide backbones. Instead, the major peaks in the amide I region absorb between 1640-1650 cm−1, indicative of disordered hydrogen bonds (shaded green in Figure 2C-D).38 Thus, the major contribution in the formation of these spherical micelles (2-3 PK/PD) or worm-like micelles (1 PK/PD and 2-3 AK/AD) are driven by the formation of diphenylalanine hydrophobic core solubilized by the hydrophilic lysine or aspartic acid tail.39 In contrast, 1 AK/AD show prominent characteristics of anti-parallel β-sheet arrangement of the peptide backbone which absorbs at 1620 and 1687 cm−1 for 1 AK and at 1623 and 1696 cm−1 for 1 AD (shaded blue in Figure 2C-D). In addition, the carboxylate peak which absorbs in 1580-1590 cm−1 (shaded red in Figure 2C-D) is red shifted to 1541 cm−1 in 1 AD, indicative of cation complexation which suggests intermolecular salt bridge formation between the C and N terminal (or aspartic acid residues next to the C terminal) which contributes to the long-range order of the peptide assembly.37 We speculate that these highly ordered nanofibers are caused by the assembly of linear and rigid backbone of 1 AK/AD in P6-P1 (FFALG), and this is clearly evident in 1 PK/PD in which a single amino acid substitution of Ala to Pro in P3 (FFPLG) creates a kink in the peptide backbone and significantly disrupts the anti-parallel β-sheet formation. Furthermore, the addition of Gly in P4 (FFGAX1G) in 2-3 AK/AD adds flexibility to the peptide backbone40 which hinders long range order of the peptide assembly and thus forms smaller nanofibers.
Figure 2.

AFM images and FTIR spectra of self-assembled peptide nanostructures. (A-B) 1 AK/AD form large fibers organized in anti-parallel β-sheet arrangement, 2-3 AK/AD and 1 PK/PD form elongated worm-like micelles, and 2-3 PK/PD form spherical micelles. Scale bar 500 nm. Additional AFM images can be found in Figure S5 in SI. (C-D) FTIR spectra of the peptides in amide I region. Red shade highlights the carboxylate groups (1580-1590 cm−1) which can red shift to lower wavenumber (1541 cm−1 for 1 AD) upon cation complexation,37 blue shade highlights parallel β-sheet (near 1620 cm−1) and anti-parallel β-sheet (additional peak at1688 cm−1) hydrogen bonding of the peptide backbones, and the green shade highlights disordered hydrogen bonds (1640-1650 cm−1). Peaks are listed in Table S2 in SI.
Controlling enzyme kinetics.
Next, we measured the rate of MMP-9 cleavage of the 12 peptides. Lyophilized peptides were dissolved in phosphate-buffered saline (PBS) supplemented with 1 mM CaCl2 and 55 μM ZnCl2 to be compatible with MMP-9 (a metalloproteinase with zinc and calcium dependent catalytic domain), the pH adjusted to 7.4, and sonicated for 10 mins to achieve 1 mM of peptide solutions. 100 ng/mL MMP-9 was incubated with peptides at 37°C and the reaction was monitored up to 96 hrs using LC-MS to identify and quantify the enzymatic products by calculating the area of the product peak over the initial peptide peak (Figure S3-4 in SI).
Electrostatic recruitment or repulsion of MMP-9.
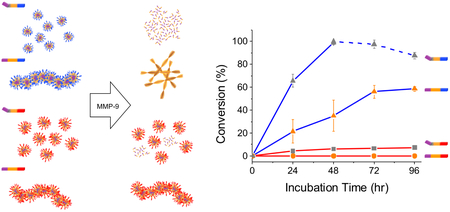
Comparing the P1↓P1’ enzymatic products, the cationic peptides (Figure 3A) were preferentially cleaved by (anionic) MMP-9. For example, 2 PK had a 12-fold higher conversion in comparison to 2 PD (Figure 3B). This stark difference resulting from preferential hydrolysis of nanostructures by oppositely charged enzymes has also been observed by Wooley et al.24 using polymeric micelles.
Figure 3.

1 mM of (A) 1-3 AK/PK and (B) 1-3 AD/PD incubated with 100 ng/mL of MMP-9 at 37°C. Peptides with Pro in P3 are marked grey, and Ala with orange. Average % conversion of peptides to the post-enzymatic products (P1↓P1’) from two separate trials. 1-3 PK showed the highest conversion and further degradation of the post-enzymatic product was observed for 2 PK (blue dash line). Over 50% of 2 AK was cleaved, while less than 10% of 3 AK and 1-2 PD was converted, and the rest were not responsive to MMP-9.
Interestingly, the major cleavage site for the cationic peptides was between G↓L in P1↓P1’ as anticipated, whereas the major cleavage site for the anionic peptides was between F↓F in P6↓P5 with inconsistent results between two separate trials (Table S1 in SI). It has been reported that MMP-9 cleavage site in peptides of similar length can shift to P2↓P1 or P1’↓P2’,41 but this drastic shift to P6↓P5 suggests that the anionic peptides do not meet MMP-9 specificity. By electrostatic (mis) matching, we are able to recruit or repel MMP-9 to trigger the desired response in the nanomaterial.
Increasing or limiting accessibility to enzymatic hydrolysis.
In addition to the dramatically different conversion rates (and cleavage sites) resulting from electrostatic interactions, we can further fine-tune the rate of enzymatic hydrolysis by achieving enzyme specificity, and controlling the degree of order in the supramolecular peptide nanostructures. Of the cationic peptides, sequences with Pro in P3 (1-3 PK) were almost completely digested by MMP-9 by 96 hrs. In particular, 1 PK, which forms the smallest micelles of the three, was completely converted to the enzyme product, FFPLG, in 48 hrs, and continued to be further hydrolyzed between F↓F (dash blue line in Figure 3A). This demonstrates the biodegradability of peptide nanoparticles, which may be useful for applications where (di) phenylalanine generated amyloid-like toxic fibrils are of concern.42 Overall, 1-3 PK were cleaved to completion at a similar rate due to the preferred Pro residue in P3, and the subtle differences in the peptide sequences did not affect the enzymatic hydrolysis rates.
In the case of 1-3 AK, significant differences were observed due to the degree of supramolecular organization of the nanostructures. 3 AK which has the highest CAC value of 0.8mM had a low conversion (below 10% by 96 hrs), whereas, 2 AK with a lower CAC value of 0.4 mM had the highest conversion (up to 60% by 96 hrs), and no cleavage was observed for 1 AK (CAC value 0.4 mM). These data suggest that the kinetics of the enzyme hydrolysis are determined by the supramolecular order of the peptide assemblies rather than the hydrolysis of unassembled peptide monomers. The slight difference of introducing a glycine residue in the sequences of 2 AK and 1 AK (between diphenylalanine and alanine) contributes to a major change in the morphology of the two nanostructures. 2 AK, which has a more flexible peptide backbone due to the insertion of Gly residue in P4, forms disordered hydrogen bonds (Figure 2C) that leads to the formation of smaller fibrils (Figure 2A and Figure S5A in SI) and is susceptible to MMP-9 hydrolysis. However, without the Gly residue, the rigid peptide backbone of 1 AK in P6-P1 arranges in an anti-parallel β-sheet configuration (Figure 2C) and form fibers that are microns in length (Figure 2A and Figure S5A in SI). These highly organized fibers are less susceptible to enzyme degradation, especially for the endopeptidase MMP-9, which has been observed to bind to type I fibril collagen, a large extra cellular matrix component, but cannot digest it until it is in the denatured, gelatin form.43 Concluding from this set of 12 peptides, it is clear that we can dramatically control the rate and specificity of MMP-9 hydrolysis through systematic exchange of amino acids.
Biocompatibility of peptide nanostructures in vitro.
Moving forward, we chose to test the biocompatibility of the two peptides that were active against MMP-9, 2 AK and 3 AK, and their Pro analogous peptides 2 PK and 3 PK. We chose a human clear cell renal cell carcinoma line, Caki-1, because tissues from patients with this type of kidney cancer show significantly higher expression of MMP-9 mRNA, with increasingly higher expression in advanced tumor stages.44 As a control, we chose a non-cancerous human lung fibroblast cell line, IMR90, which was reported to have negligible expression of MMP-9 mRNA.45 1 mM of the peptides were incubated with Caki-1 and IMR90 cells at 37°C for 72hrs and the cell viability was determined using the Presto Blue assay (Figure 4A-B). The peptides are non-toxic to both cell lines with cell viabilities over 90%, except for 3 AK which forms β-sheet fiber post-cleavage and decreased the cell viability of Caki-1 cells to 66%, despite the observed low conversion (Figure 3A).
Figure 4.

Biocompatibility of peptide nanostructures. Cell viability of human renal cancer cell line, Caki-1, with high expression of MMP-9 (A), and non-cancerous human fibroblast, IMR90, with normal expression of the enzyme (B), incubated with 1 mM peptides for 72 hrs. The peptides are non-toxic to both cell lines, except for 3 AK which decreased the cell viability of Caki-1 to 66%. (C) FTIR spectra of the post-cleavage peptides show that only 3 A forms anti-parallel β-sheet peptide fibers which causes the cytotoxic effects in Caki-1 only. (D) TEM images of 3 AK and 2 PK show formation of worm-like and spherical micelles, respectively, and the post-enzymatic products, 3 A and 2 P which form rigid fibers or random aggregates, respectively. Scale bar 100nm.
Nanofibers induce selective toxicity in cancer cells.
In order to understand the effects of the post-cleavage products in vitro, we synthesized the hydrophobic N terminal fragments of the resulting enzymatic products (herein referred as 2-3 A/P) by SPPS and studied their self-assembly behaviors using FTIR, AFM, and TEM. The full characterization of 1-3 A/P can be found in supporting information (HRMS, 1H NMR, AFM, FTIR, and additional TEM). Distinct differences can be observed by peptides with Ala in P3 (2-3 A) which formed ordered structures, and peptides with Pro in P3 (2-3 P) which was expected not to self-assemble due to the Pro disrupting formation of ordered hydrogen bonds. FTIR spectra of the non-assembling peptides (Figure 4C gray lines) show absorptions at 1643 and 1653 cm−1 for 2 P and 3 P, respectively, which is observed for amides of peptide backbones with disordered hydrogen bonds. A mixture of parallel β-sheet (1628 cm−1) and disordered (1643 cm−1) hydrogen bonds were observed for 2 A. In contrast, 3 A absorbs at 1624 and 1688 cm−1, which are distinct characteristic peaks of anti-parallel β-sheet hydrogen bonds (Figure 4C orange lines). TEM images (Figure 4D) show that the precursor 2 PK forms spherical particles which, when converted to 2 P, disassemble and remain as random aggregates. In comparison, precursor 3 AK forms flexible fibers and the post-enzymatic product, 3 A, forms ordered rigid β-sheet fibers. Enzyme-triggered fibers which form on or near the cell surface of cancer cells that produce the enzyme stimuli are known to be cytotoxic to the cells.46 Likewise, the observed decrease in viability of Caki-1 cells treated with 3 AK is most likely due to the formation of toxic anti-parallel β-sheet nanofibers (3 A) that are selectively triggered to form by the overexpressed MMP-9 in the diseased cells.16 This effect is not observed in the control cell line, IMR90, where overexpression of MMP-9 is not expected.
CONCLUSION
In conclusion, we have created a modular system of self-assembling peptide nanostructures to customize surface charge and supramolecular order to control enzyme kinetics and response action. These peptide nanostructures are non-toxic and bio-degradable, and after enzyme action, the non-assembling products remain non-toxic while the β-sheet nanofiber forming products can selectively kill cancer cells. The kinetics of nanoparticles is important in biomedical applications in which the rate of enzyme responsiveness should be regulated (i.e. predetermined drug release profile, degradation rate of hydrogels, etc.). In addition, the morphology of the nanoparticles should be logically designed to achieve optimal function of the precursors (i.e. manipulation of cellular uptake, bio-distribution, etc.) and of the post-enzymatic product (i.e. biodegradability or therapeutic nanofiber/drug depots, etc.).
METHODS
Solid Phase Peptide Synthesis (SPPS).
Fluorenylmethyloxycarbonyl (Fmoc)-protected amino acids and pre-loaded Wang resins were purchased from Bachem. Peptides were synthesized on CEM Liberty Blue microwave assisted solid phase peptide synthesizer using ~1:5 resin to amino acid ratio and excess of diisopropylcarbodiimide (DIC), Oxyma (Ethyl(hydroxyimino)cyanoacetate), and 20% piperidine in dimethylformamide (DMF). The complete peptide-loaded resins were washed three times in dichloromethane, followed by three washes in diethyl ether on a filtration column. The peptides were cleaved from the resins and side chain protecting groups were removed by reacting with TFA cocktail (95% trifluoroacetic acid, 2.5% triisopropyl silane, and 2.5% water) for 2hr. The cleaved peptides were recovered by removing TFA cocktail, followed by precipitation in cold diethyl ether. Peptides were washed three times in cold diethyl ether, using centrifuge to decant the supernatant. The crude peptides were dissolved in Milli-Q (MQ) water and lyophilized.
Preparatory High-Performance Liquid Chromatography (HPLC).
Lyophilized crude peptides were dissolved in 50% acetonitrile in water containing 0.1% TFA and purified through a preparatory C18 column on the Thermo Scientific Dionex Ultimate 3000. Acetonitrile was removed from the collected fragments on a rotary evaporator before lyophilization. Purified peptides were dissolved in 10mM HCl solution to make 1mM peptide solution and lyophilized to remove residual TFA salts.
High Resolution Mass Spectrometry (HRMS).
HRMS data were obtained on an Agilent 6550 QToF, with a dual sprayer ESI source, coupled to an Agilent 1290 Infinity LC system. Samples were analyzed by FIA (flow injection analysis) using a mobile phase of 50% acetonitrile in water (0.1% formic acid) with a flow rate of 0.4 mL/min.
1H Nuclear Magnetic Resonance (NMR) Spectroscopy
NMR spectra were recorded in a Bruker AV400 at 400 MHz. Chemical shifts (δ) are given in ppm using D2O as solvent.
Assay to determine the rate of MMP-9 cleavage of peptide sequences
MMP-9 (catalytic domain) (human), (recombinant, E. coli) was purchased from Enzo. The purchased enzyme solution was defrosted and aliquots made for storage in −80°C. 1mM peptides were prepared in PBS (2.7mM KCl, 137mM NaCl, supplemented with 1mM CaCl2, 55μM ZnCl2, pH=7.4) and the pH adjusted to 7.4 using 0.5M NaOH or 0.5M HCl. Peptides were sonicated for 10minutes to facilitate self-assembly and then incubated at 37°C in a stationary heat block. Peptide samples for t=0 analysis were reserved and 100ng/mL of MMP-9 (stock aliquot was diluted to 10,000ng/mL in PBS) was gently mixed into the 1mM peptide solution. Reaction samples were taken every 24hr for LC-MS analysis.
Liquid Chromatography-Mass Spectrometry (LC-MS).
30μL of the MMP-9 reaction solution (or peptide only solution at t=0) was directly added to 50% acetonitrile in water containing 0.1% TFA. Samples were analyzed on an LCMS system comprised of an Agilent 1200 LC system coupled to an Agilent 6340 ion trap mass spectrometer. Samples were injected onto an Agilent Zorbax column (SB-C8, 5μM, 2.1×50mm) using a gradient of 2-50% acetonitrile in water (0.1% formic acid) at a flow rate of 200μL/min over 10minutes followed by a 2min wash step with 95% acetonitrile.
Atomic Force Microscopy (AFM).
AFM images were taken on Bruker Dimension FastScan using FASTSCAN-B tip on fast scan mode. 1mM of peptide solution was prepared in phosphate buffer (pH 7.4), sonicated for 10minutes and drop casted on freshly cleaved mica and allowed to dry for 48hr before imaging.
Transmission Electron Microscopy (TEM).
TEM images were taken on FEI Titan Halo 80-300 microscope. 1mM of peptide solution was prepared in 10mM phosphate buffer (pH 7.4) sonicated for 10minutes and 5μL of the solution was drop casted on a carbon film grid (400 mesh, copper) and dried completely. To the dry grid, 5μL of MilliQ water was drop casted and quickly blotted to wash away the phosphate salts and dried completely. Finally, 5μL of methylamine vanadate based negative stain (NanoVan® by Nanoprobes) was drop casted, blotted away, and dried completely.
Fourier Transform Infrared Spectroscopy (FTIR).
Absorbance spectra were taken from 4000cm−1 to 800cm−1 with 64 scans at 4cm−1 resolution on the Bruker Vertex 70 spectrometer. 20mM peptide solutions was prepared in deuterated phosphate buffer (pH=8), pH was adjusted to 7.4 using 0.5M NaOH or 0.5M HCl, and sonicated for 10minutes. 5μL of sample solutions were drop casted between two CaF2 cells with PTFE spacers (12μm thickness × 13mm diameter). For analysis, deuterated phosphate buffer absorbance spectra was subtracted from the sample absorbance and normalized from 1560 to 1655cm−1.
Zeta Potential.
Zeta potential measurements were made on Anton Paar Litesizer 500 Particle Analyzer. 5mM of peptide samples were prepared in 2%PBS and the pH was adjusted to 7.4 using dilute NaOH and HCl. 50uL of samples were pipetted into Univette low volume cuvette and three series of measurements were made at 25°C using Smoluchowski approximation.
Critical Aggregation Concentration (CAC).
1mM of peptides were prepared in PBS and the pH adjusted to 7.4 using 0.5M NaOH or 0.5M HCl, and serially diluted in PBS with thorough vortexing. Peptide solutions were incubated in 50°C for 15 minutes, then 2μL of stock pyrene solution (100μM in methanol) was added to 100μL of each peptide solution, gently mixed, and incubated for 5 minutes at 50°C, then finally cooled down to room temperature to co-assemble the peptides with pyrene molecules. Pyrene emission spectra was measured from 350-450nm (λex=310nm) in a micro fluorescence cuvette (3mm path length) on Jasco FP-8500 Spectrofluorometer (measurement parameter: 20nm excitation and 1nm emission bandwidth, 0.2sec response, medium sensitivity, 0.2nm data interval, at 200nm/min). The CAC was determined by plotting the ratio between intensities of the 3rd to 1st peak of the pyrene emission spectra. Increasing peptide concentrations were measured in 0.1mM increments until the slope of the plot had changed, and simultaneously the 3rd peak shifted from 382.4nm to 384.4nm and the 1st peak shifted from 371.8nm to 373.4nm.
Cell lines.
Human clear cell renal cell carcinoma line Caki-1 was newly obtained for these studies from the American Type Culture Collection (ATCC) (Manassas, Virginia, USA) and cultured in Roswell Park Memorial Institute (RPMI-1640) (Mediatech Inc., Manassas, VA) media containing 10% Fetal Bovine Serum, certified, heat inactivated, US origin (FBS) (Gibco, Life Technologies, US), 1% Minimum Essential Media (MEM) nonessential amino acids (NEAA, Mediatech) and 1% penicillin–streptomycin (PenStrep, Mediatech). IMR90 (human fetal lung fibroblast) cells were purchased from ATCC (Manassas, Virginia, USA) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Mediatech) supplemented with 10% FBS, 1% NEAA and 1% Penicillin Streptomycin.
Cell viability assay.
Human fetal lung fibroblast (IMR90) and human clear cell renal cell carcinoma cells (Caki-1) were seeded in 96-well flat bottom microplate (BioLite Microwell Plate, Fisher Scientific, Waltham, MA). For IMR90 we used 6.0 X 103 cell per well and for Caki-1 we used 5.6 X 103 cells per well in 90μL of complete phenol red free cell culture media. The cells were allowed to grow for 24hours at 37°C and 5% CO2 in a humidified incubator. 10mM peptide solutions were prepared in phosphate buffer saline, pH adjusted to 7.4 using NaOH or HCl and sonicated for 10minutes. Then 10μL of each sample was added into wells containing 90μL of media (in triplicate). Following the administration of the peptides, cells were incubated for 72 hours at 37°C under 5% CO2. After each period of incubation, Presto Blue (Life Technologies, Carlsbad, CA) was used as an indicator of cellular toxicity; 11μL of presto blue was added to each well and incubated for 1 hours at 37°C under 5% CO2. The 96-wells plate was then analyzed using a multi-mode plate-reader BioTek Microplate Reader (BioTek U.S., Winooski, VT) at 530nm and 590nm wavelength. The percentage of surviving cells was calculated as a normalized ratio of the fluorescence intensity between cells treated with media and phosphate buffer saline alone.
Supplementary Material
ACKNOWLEDGMENT
LC-MS analysis was performed at Hunter Mass Spectrometry, supported by the City University of New York. We thank the Tow Foundation Graduate Fellowship from the MSKCC Center for Molecular Imaging and Nanotechnology (J.S.), the SEED Grant CUNY-Advanced Science Research Center Program (M.C.), and the National Cancer Institute (NCI) and the National Institute for General Medical Sciences (NIGMS) grants 1SC1CA182844 and 2SC1 GM127278-05A1 (M.C.).
Footnotes
Supporting Information. The Supporting Information (PDF document) is available free of charge on the ACS Publications website (MMP-9 cleavage studies by LC-MS, and characterization of peptides using HRMS, 1H NMR, AFM, TEM, FTIR, zeta potential, and CAC studies).
REFERENCES
- (1).Kinnear C; Moore TL; Rodriguez-Lorenzo L; Rothen-Rutishauser B; Petri-Fink A Form Follows Function: Nanoparticle Shape and Its Implications for Nanomedicine. Chem. Rev. 2017, 117, 11476–11521. [DOI] [PubMed] [Google Scholar]
- (2).Blum AP; Kammeyer JK; Rush AM; Callmann CE; Hahn ME; Gianneschi NC Stimuli-Responsive Nanomaterials for Biomedical Applications. J. Am. Chem. Soc. 2015, 137, 2140–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Mura S; Nicolas J; Couvreur P Stimuli-Responsive Nanocarriers for Drug Delivery. Nat. Mater. 2013, 12, 991–1003. [DOI] [PubMed] [Google Scholar]
- (4).Chen G; Roy I; Yang C; Prasad PN Nanochemistry and Nanomedicine for Nanoparticle-Based Diagnostics and Therapy. Chem. Rev. 2016, 116, 2826–2885. [DOI] [PubMed] [Google Scholar]
- (5).Soukasene S; Toft DJ; Moyer TJ; Lu H; Lee H-K; Standley SM; Cryns VL; Stupp SI Antitumor Activity of Peptide Amphiphile Nanofiber-Encapsulated Camptothecin. ACS Nano 2011, 5, 9113–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zelzer M; Todd SJ; Hirst AR; McDonald TO; Ulijn RV Enzyme Responsive Materials: Design Strategies and Future Developments. Biomater Sci 2013, 1, 11–39. [DOI] [PubMed] [Google Scholar]
- (7).Christian DA; Cai S; Garbuzenko OB; Harada T; Zajac AL; Minko T; Discher DE Flexible Filaments for in Vivo Imaging and Delivery. Mol. Pharm. 2009, 6, 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Visse R; Nagase H Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases: Structure, Function, and Biochemistry. Circ. Res. 2003, 92, 827–839. [DOI] [PubMed] [Google Scholar]
- (9).Overall CM; Kleifeld O Tumour Microenvironment - Opinion: Validating Matrix Metalloproteinases as Drug Targets and Anti-Targets for Cancer Therapy. Nat. Rev. Cancer 2006, 6, 227–239. [DOI] [PubMed] [Google Scholar]
- (10).Kai H; Ikeda H; Yasukawa H; Kai M; Seki Y; Kuwahara F; Ueno T; Sugi K; Imaizumi T Peripheral Blood Levels of Matrix Metalloproteases-2 and −9 Are Elevated in Patients with Acute Coronary Syndromes. J. Am. Coll. Cardiol. 1998, 32, 368–372. [DOI] [PubMed] [Google Scholar]
- (11).S Burrage P; Mix K; Brinckerhoff C Matrix Metalloproteinases: Role in Arthritis. Front Biosci. 2006, 11, 529–543. [DOI] [PubMed] [Google Scholar]
- (12).West JL; Hubbell JA Polymeric Biomaterials with Degradation Sites for Proteases Involved in Cell Migration. Macromolecules 1999, 32, 241–244. [Google Scholar]
- (13).Lutolf MP; Hubbell JA Synthetic Biomaterials as Instructive Extracellular Microenvironments for Morphogenesis in Tissue Engineering. Nat. Biotechnol. 2005, 23, 47–55. [DOI] [PubMed] [Google Scholar]
- (14).Patterson J; Hubbell JA Enhanced Proteolytic Degradation of Molecularly Engineered PEG Hydrogels in Response to MMP-1 and MMP-2. Biomaterials 2010, 31, 7836–7845. [DOI] [PubMed] [Google Scholar]
- (15).Tanaka A; Fukuoka Y; Morimoto Y; Honjo T; Koda D; Goto M; Maruyama T Cancer Cell Death Induced by the Intracellular Self-Assembly of an Enzyme-Responsive Supramolecular Gelator. J. Am. Chem. Soc. 2015, 137, 770–775. [DOI] [PubMed] [Google Scholar]
- (16).Zhou J; Du X; Yamagata N; Xu B Enzyme-Instructed Self-Assembly of Small D-Peptides as a Multiple-Step Process for Selectively Killing Cancer Cells. J. Am. Chem. Soc. 2016, 138, 3813–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Callmann CE; Barback CV; Thompson MP; Hall DJ; Mattrey RF; Gianneschi NC Therapeutic Enzyme-Responsive Nanoparticles for Targeted Delivery and Accumulation in Tumors. Adv. Mater. 2015, 27, 4611–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lin Y-A; Ou Y-C; Cheetham AG; Cui H Rational Design of MMP Degradable Peptide-Based Supramolecular Filaments. Biomacromolecules 2014, 15, 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kulkarni PS; Haldar MK; Nahire RR; Katti P; Ambre AH; Muhonen WW; Shabb JB; Padi SKR; Singh RK; Borowicz PP; Shrivastava DK; Katti KS; Reindl K; Guo B; Mallik S MMP-9 Responsive PEG Cleavable Nanovesicles for Efficient Delivery of Chemotherapeutics to Pancreatic Cancer. Mol. Pharm. 2014, 11, 2390–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kim K; Bae B; Kang YJ; Nam J-M; Kang S; Ryu J-H Natural Polypeptide-Based Supramolecular Nanogels for Stable Noncovalent Encapsulation. Biomacromolecules 2013, 14, 3515–3522. [DOI] [PubMed] [Google Scholar]
- (21).Kalafatovic D; Nobis M; Son J; Anderson KI; Ulijn RV MMP-9 Triggered Self-Assembly of Doxorubicin Nanofiber Depots Halts Tumor Growth. Biomaterials 2016, 98, 192–202. [DOI] [PubMed] [Google Scholar]
- (22).Eckhard U; Huesgen PF; Schilling O; Bellac CL; Butler GS; Cox JH; Dufour A; Goebeler V; Kappelhoff R; dem Keller UA; Klein T; Lange PF; Marino G; Morrison CJ; Prudova A; Rodriguez D; Starr AE; Wang Y; Overall CM Active Site Specificity Profiling of the Matrix Metalloproteinase Family: Proteomic Identification of 4300 Cleavage Sites by Nine MMPs Explored with Structural and Synthetic Peptide Cleavage Analyses. Matrix Biol. J. Int. Soc. Matrix Biol. 2016, 49, 37–60. [DOI] [PubMed] [Google Scholar]
- (23).Thornton PD; Mart RJ; Webb SJ; Ulijn RV Enzyme-Responsive Hydrogel Particles for the Controlled Release of Proteins: Designing Peptide Actuators to Match Payload. Soft Matter 2008, 4, 821–827. [DOI] [PubMed] [Google Scholar]
- (24).Samarajeewa S; Zentay RP; Jhurry ND; Li A; Seetho K; Zou J; Wooley KL Programmed Hydrolysis of Nanoassemblies by Electrostatic Interaction-Mediated Enzymatic-Degradation. Chem. Commun. Camb. Engl. 2014, 50, 968–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Pires RA; Abul-Haija YM; Costa DS; Novoa-Carballal R; Reis RL; Ulijn RV; Pashkuleva I Controlling Cancer Cell Fate Using Localized Biocatalytic Self-Assembly of an Aromatic Carbohydrate Amphiphile. J. Am. Chem. Soc. 2015, 137, 576–579. [DOI] [PubMed] [Google Scholar]
- (26).Wang H; Feng Z; Wang Y; Zhou R; Yang Z; Xu B Integrating Enzymatic Self-Assembly and Mitochondria Targeting for Selectively Killing Cancer Cells without Acquired Drug Resistance. J. Am. Chem. Soc. 2016, 138, 16046–16055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Feng Z; Wang H; Wang S; Zhang Q; Zhang X; Rodal AA; Xu B Enzymatic Assemblies Disrupt the Membrane and Target Endoplasmic Reticulum for Selective Cancer Cell Death. J. Am. Chem. Soc. 2018, 140, 9566–9573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Feng Z; Wang H; Chen X; Xu B Self-Assembling Ability Determines the Activity of Enzyme-Instructed Self-Assembly for Inhibiting Cancer Cells. J. Am. Chem. Soc. 2017, 139, 15377–15384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhang S Fabrication of Novel Biomaterials through Molecular Self-Assembly. Nat. Biotechnol. 2003, 21, 1171–1178. [DOI] [PubMed] [Google Scholar]
- (30).Kalafatovic D; Nobis M; Javid N; Frederix PWJM; Anderson KI; Saunders BR; Ulijn RV MMP-9 Triggered Micelle-to-Fibre Transitions for Slow Release of Doxorubicin. Biomater. Sci. 2015, 3, 246–249. [DOI] [PubMed] [Google Scholar]
- (31).Jaiswal A; Chhabra A; Malhotra U; Kohli S; Rani V Comparative Analysis of Human Matrix Metalloproteinases: Emerging Therapeutic Targets in Diseases. Bioinformation 2011, 6, 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Rawlings ND; Barrett AJ; Finn R Twenty Years of the MEROPS Database of Proteolytic Enzymes, Their Substrates and Inhibitors. Nucleic Acids Res. 2016, 44, 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Woolfson DN; Williams DH The Influence of Proline Residues on α-Helical Structure. FEBS Lett. 1990, 277, 185–188. [DOI] [PubMed] [Google Scholar]
- (34).Kridel SJ; Chen E; Kotra LP; Howard EW; Mobashery S; Smith JW Substrate Hydrolysis by Matrix Metalloproteinase-9. J. Biol. Chem. 2001, 276, 20572–20578. [DOI] [PubMed] [Google Scholar]
- (35).Reches M; Gazit E Casting Metal Nanowires Within Discrete Self-Assembled Peptide Nanotubes. Science 2003, 300, 625–627. [DOI] [PubMed] [Google Scholar]
- (36).Gazit E Self-Assembled Peptide Nanostructures: The Design of Molecular Building Blocks and Their Technological Utilization. Chem. Soc. Rev. 2007, 36, 1263–1269. [DOI] [PubMed] [Google Scholar]
- (37).Barth A The Infrared Absorption of Amino Acid Side Chains. Prog. Biophys. Mol. Biol. 2000, 74, 141–173. [DOI] [PubMed] [Google Scholar]
- (38).Fleming S; Ulijn RV Design of Nanostructures Based on Aromatic Peptide Amphiphiles. Chem. Soc. Rev. 2014, 43, 8150–8177. [DOI] [PubMed] [Google Scholar]
- (39).Shimada T; Lee S; Bates FS; Hotta A; Tirrell M Wormlike Micelle Formation in Peptide-Lipid Conjugates Driven by Secondary Structure Transformation of the Headgroups. J. Phys. Chem. B 2009, 113, 13711–13714. [DOI] [PubMed] [Google Scholar]
- (40).Pace CN; Scholtz JM A Helix Propensity Scale Based on Experimental Studies of Peptides and Proteins. Biophys. J. 1998, 75, 422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Huang Y; Shi J; Yuan D; Zhou N; Xu B Length-Dependent Proteolytic Cleavage of Short Oligopeptides Catalyzed by Matrix Metalloprotease-9: Proteolytic Cleavage of Short Oligopeptides by MMPs. Biopolymers 2013, 100, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Adler-Abramovich L; Vaks L; Carny O; Trudler D; Magno A; Caflisch A; Frenkel D; Gazit E Phenylalanine Assembly into Toxic Fibrils Suggests Amyloid Etiology in Phenylketonuria. Nat. Chem. Biol. 2012, 8, 701–706. [DOI] [PubMed] [Google Scholar]
- (43).Collier IE; Legant W; Marmer B; Lubman O; Saffarian S; Wakatsuki T; Elson E; Goldberg GI Diffusion of MMPs on the Surface of Collagen Fibrils: The Mobile Cell Surface – Collagen Substratum Interface. PLoS ONE 2011, 6, 24029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Qiao Z; Li Y; Lu H; Wang K; Xu W Expression of Tissue Levels of Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Renal Cell Carcinoma. World J. Surg. Oncol. 2013, 11, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Giambernardi TA; Grant GM; Taylor GP; Hay RJ; Maher VM; McCormick JJ; Klebe RJ Overview of Matrix Metalloproteinase Expression in Cultured Human Cells. Matrix Biol. J. Int. Soc. Matrix Biol. 1998, 16, 483–496. [DOI] [PubMed] [Google Scholar]
- (46).Zhou J; Xu B Enzyme-Instructed Self-Assembly: A Multistep Process for Potential Cancer Therapy. Bioconjug. Chem 2015, 26, 987–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.