

Abstract
Immunomodulatory drugs (IMiDs) including lenalidomide and pomalidomide bind cereblon (CRBN) and activate the CRL4crbn ubiquitin ligase to trigger proteasomal degradation of the essential transcription factors IKZF1 and IKZF3 and multiple myeloma (MM) cytotoxicity. We have shown that CRBN is also targeted for degradation by SCFFbxo7 ubiquitin ligase. In the current study, we explored the mechanisms underlying sensitivity of MM cells to IMiDs using genome-wide CRISPR-Cas9 screening. We validate that CSN9 signalosome complex, a deactivator of Cullin-RING ubiquitin ligase, inhibits SCFFbxo7 E3 ligase-mediated CRBN degradation, thereby conferring sensitivity to IMiDs; conversely, loss of function of CSN9 signalosome activates SCFFbxo7 complex, thereby enhancing degradation of CRBN and conferring IMiD resistance. Finally, we show that pretreatment with either proteasome inhibitors or NEDD8 activating enzyme (NAE) inhibitors can abrogate degradation and maintain levels of CRBN, thereby enhancing sensitivity to IMiDs. These studies therefore demonstrate that CSN9 signalosome complex regulates sensitivity to IMiDs by modulating CRBN expression.
Introduction
Multiple myeloma (MM) is characterized by the infiltration of abnormal plasma cells in bone marrow and represents 1% of all cancers and 2% of blood cancers. Both genetic and environmental/host factors have been implicated in progression of monoclonal gammopathy of undetermined significance (MGUS) to smoldering MM and active MM requiring therapy. [1, 2] Major improvements in patient outcome have resulted from the development of high dose therapy and autologous stem cell transplantation (ASCT), [3–5] as well as the development of novel agents targeting the tumor in its bone marrow microenvironment including: immunomodulatory drugs (IMiDs) thalidomide, lenalidomide and pomalidomide; [6–8] proteasome inhibitors (PIs) bortezomib, carfilzomib, and ixazomib; [9–12] monoclonal antibodies daratumumab and elotuzumab; [13–15] and histone deacetylase inhibitor panobinostat. [16] Integration of novel therapies as induction before and maintenance after ASCT has achieved the highest extents and frequency of response. [17] In spite of this progress, MM commonly acquires resistance leading to clinical relapse, highlighting the importance of defining mechanisms of sensitivity versus resistance to these agents.
Recent studies have delineated the mechanism of action of IMiDs. The IMiD thalidomide (Thai), which tragically resulted in phocomelia when prescribed as a sedative to treat morning sickness of pregnant women 50 years ago, targets CRBN, [18] a substrate specificity factor of Cullin4 Ring Ligase (CRL4). CRL4crbn ubiquitin ligase targets the large conductance Ca2+-activated potassium (BK) channels [19] and the CLC-1 chloride channels [20] for ubiquitination, and when mutated is associated with autosomal recessive non-syndromic mental retardation. [21] The teratogenicity of Thal may be related to binding to CRBN and inhibition of CRL4crbn ubiquitin ligase activity. [18] Importantly, the IMiDs thalidomide, lenalidomide, and pomalidomide have been shown to target and trigger cytotoxicity against MM cells in their bone marrow microenvironment in preclinical studies, and to have remarkable clinical efficacy when used as initial, salvage, and maintenance therapy in MM. [22, 23] These IMiDs activate the CRL4crbn ubiquitin ligase to target B-cell specific transcription factors IKZF1 and IKZF3, which are essential for MM cell survival, for ubiquitination, and destruction. [24, 25] Similarly, lenalidomide induces ubiquitination and degradation of another substrate casein kinase 1A1 (CK1α) in myelodysplastic syndrome (MDS) with deletion of chromosome 5q (del(5q)). [26] Mechanistically, IMiDs dock to an exposed surface of CRBN distant from CRL4, [27, 28] and the complex then recruits and binds these substrates. [29] We recently showed that CRBN is also a substrate for another ubiquitin ligase SCFFbxo7. [30] Most recently, we have identified p53-related protein kinase as another substrate of IMiDs and promising therapeutic target in MM. [31] To date however, mechanisms regulating CRBN and sensitivity to IMiDs have not been defined.
In the present study, we performed a CRISPR-Cas9 based genome-wide screening in MM cells to identify and delineate mechanisms of IMID sensitivity. We identify and validate mechanisms whereby CSN9 signalosome (CSN) subunits modulate CRBN expression levels and sensitivity to IMiDs in MM. These studies also delineate mechanisms underlying the observed enhanced clinical efficacy of IMiDs when used in combination with PIs.
Materials and methods
CRISPR-Cas9 library screening
Library amplification
The human GeCKOv2 sgRNA libraries were purchased from Addgene and amplified according to the protocol provided by Zhang’s lab (www.genome-engineering.org). Briefly, we conducted 4 electroporations using 8 μl of 50 ng/pL GeCKOv2 A or B sub-library in Lucigen Endura electrocompetent cells, with yield of 2 mg plasmids for each sub-library. SgRNAs from the original and amplified libraries were further amplified using a two-step PCR, followed by a Miseq 150 deep-sequencing for quality control.
Lentivirus production
To produce lentivirus, 4.5 × 108 293 T cells (Thermo Fisher #R700–07) were harvested and reverse-transfected into twenty-five T-150 flasks. For each flask, 20 μg GeCKOv2 A or B library plasmid, 15 μg psPAX2 (Addgene #12260), and 10 μg pMD2.G (Addgene #12259) diluted in 3 ml Opti-MEM were combined with 150 μL Lipofectamine 2000 diluted in 3 ml Opti-MEM. The mixture was left for 20min and then added to the cells. 12 h after transfection, the media was replaced by fresh culture media. Virus supernatant was collected 72 h post-transfection, followed by centrifugation at 2000 rpm for 10 min to pellet cell debris. Filtration was then performed with a 0.45 pm low protein binding membrane (Millipore #SE1M003M00), followed by ultra-centrifugation at 25,000 rpm at 4 °C for 2 h to pellet virus. The pellets of virus were resuspended and aliquots stored at −80 °C. For virus titration, HCT116 cells were seeded (1 × 105 per well in 6-well plates) one day before transduction. Lentivirus stock was thawed and diluted (10–3 to 10–7) for transduction. Cells were incubated with virus overnight, then in culture media for 24 h followed by puromycin selection for 5 days, before staining by crystal violet.
Cell transduction
Virus was first tested to achieve an MOI of 0.3–0.4 in MM.1S cells, and 75 μl virus was used for large-scale spin-infection of MM.1S cells (3 × 106). A total of 2.1 × 108 MM.1S cells (70 wells per 12-well plate, 10 wells for transfection efficiency control) were transduced with concentrated virus (5.25 ml), attaining transduction efficiency of 25% (700 cells per lenti-CRISPRv2 construct). Puromycin was added to cells 24 h after transduction, and maintained for 7 days. Subsequently, 4×107 cells were collected for base-line genomic DNA analysis. The remaining cells were split for pomalidomide versus vehicle control treatment. The starting drug concentrations were 75 nM GeCKO v2A and 100 nM GeCKO v2B, the IC70 for MM.1S cells, and drug was refreshed every 3 days. On Days 7, 14 and 21 after drug treatment, 4 × 107 cells were removed from either drug group or vehicle control group for analysis.
Genomic DNA sequencing
Genomic DNA extraction (QIAGEN cat#12362) and PCR amplification were performed according to Zhang’s protocol. In brief, for each sample we performed at least 40 separate 50 μl reactions, with 4 μg genomic DNA in each reaction, for the 1st step PCR. The 2nd PCR was done in separate 50 μl reactions, with 5 μl of combined amplicons from the first PCR. The primers for the 1st PCR are the same as published, and the primers for the 2nd PCR are below:
F2: (1–9 bp variable length sequence) tcttgtggaaaggac gaaacaccg;
R2: (1–9 bp variable length sequence) tgtgggcgatgtgc gctct;
Resulting amplicons were purified for sequencing library construction with Illumina kits, followed by Hiseq 2500 Rapid PE75 deep sequencing.
Data processing and initial analysis
Raw reads and normalized reads were obtained as Zhang et al. described. Genes were identified by the algorithm, as described above.
Cell lines
HEK 293T, MM1.S, and NCI-H929 cell lines were obtained from American Type Culture Collection (ATCC). OPM2 cell line was obtained from Leibniz-Institut DSMZ. Cells were cultured in media recommended by ATCC, maintained at 37 °C in 5% CO2 condition, and regularly tested for mycoplasma contamination.
cDNA and plasmids
Full-length CRBN was PCR amplified and sub-cloned into pXF4H expression vector between the XbaI and HindIII restriction sites. Full-length Fbxo7 sequences was sub-cloned into pcDNA3.1(–)-Myc vector between the XbaI and HindIII restriction sites. Full length and truncations of Fbxo7 or CRBN were amplified and sub-cloned into pXF6F expression vector between the XbaI and HindIII restriction sites.
Compounds and antibodies
bortezomib (S1013), lenalidomide (S1029), pomalidomide (S1567), and MLN4924 (S7109) were purchased from Selleck Chem; all were dissolved in DMSO. Anti-Flag (F3165; Sigma-Aldrich), anti-HA (H6908; H3663; Sigma- Aldrich), anti-Myc (2272; Cell Signaling Technology), anti- Cul1 (AP16324b; Abgent), anti-β-actin (5779–1; Epi- tomics), anti-actin (1844–1; Epitomics), anti-TRIP15 (ab155774; Abcam), anti-CSN6 (NBP1–79752), anti-JAB1 (ab182756; Abcam), anti-CSN7B (ab133548; Abcam), anti- CSN7A (AP12810b; Abgent), anti-CSN8 (AP14987a; Abgent), anti-Fbxo7 (ab57037; Abcam), anti-IKZF3 (ab139408; Abcam), anti-IKZF1 (sc-13039; Santa Cruz), anti-Nedd8 (ab81264; Abcam), anti-HA agarose (E6779; Sigma-Aldrich), anti-Myc agarose (E6654; Sigma-Aldrich), and anti-CRBN (SAB045910; Sigma-Aldrich) were purchased and used according to the manufacturers’ recommendations. Secondary antibodies included goat anti-mouse IgG-HRP (sc-2005; Santa Cruz) and Goat anti-rabbit IgG- HRP (sc-2004; Santa Cruz).
Q-PCR
Relative levels of CSN4 transcripts was determined with GAPDH as housekeeping gene using SYBR Green (Life Technology). Primers are listed below:
CSN4 forward: TTCGAGCGGCTCTCATAAAGA, reverse: CGAGATCACGAGACT-GACATTCT; GAPDH forward: AGGTGAAGGTCGGAGTCAAC, reverse: CG CTCC-TGGAAGATGGTGAT.
Western blot
Protein lysates were separated by SDS-PAGE and transferred onto PVDF membranes. Blots were blocked in 5% non-fat dry milk, and incubated with primary antibodies in 5% BSA at 4 °C overnight. Blots were then washed 3 times with 1 × TBS-T, prior to incubation with secondary antibodies for 1 h. Detection was performed using chemoluminescent substrates (Thermo Scientific).
Co-immunoprecipitation
HEK 293T or MM cells were transiently transfected with indicated plasmids, and then lysed in lysis buffer supplemented with complete protease inhibitors (Roche). After centrifugation at 13,000 rpm for 20min, super-natants were subjected to immunoprecipitation by incubation with anti-HA Affinity Gel or anti-Myc Affinity Gel (Sigma) for 4 h or overnight at 4 °C. The samples were then washed 3 times with lysis buffer, and the immuno-precipitates were separated by SDS-PAGE and analyzed by Western blot.
Lentiviral sgRNA vectors and infection
Oligonucleotides (Supplementary Table S1) targeting different genes were annealed and sub-cloned into lenti-CRISPRv2 vectors. The sgRNA vectors were then transfected into HEK 293FT cells, together with pVSVg and psPAX2 vectors, using Lipofectamine 2000. Media was replaced at 24 h, and the viral supernatant was collected 72 h post-transfection, with ultra-centrifugation at 25,000 rpm at 4 °C for 2 h.
For viral infection, MM cells were seeded in 12-well plates supplemented with 1 μg/ml Polybrene (Sigma). Cells were centrifuged at 2000 rpm for 2 h at 37 °C; media was then aspirated, and fresh media was added, excluding polybrene.
Viability assay
MM cells were seeded in 96-well plates and treated with bortezomib and lenalidomide or pomalidomde at indicated concentrations and time intervals. Total cellular ATP content was assessed using CellTiter-Glo® Luminescent Cell Viability Assay (Promega), according to the manufacturer’s protocol. Luminescence was detected in a multimode detector DTX880 (Beckman Coulter).
Gene expression correlation analysis
We downloaded the gene expression profiles in 304 patients with newly diagnosed and previously treated MM from the Multiple Myeloma Research Consortium (MMRC). The expression signals have been median centered and log2 normalized. The Pearson correlation coefficient, and its significance for each paired gene was calculated.
Results
Genome-wide CRISPR-Cas9 screen for IMiDs sensitivity
To delineate the mechanism underlying IMiDs sensitivity, we first performed genome-wide knockout screening in MM.1S, an IMiDs-sensitive MM cell line, using a CRISPR-Cas9 GeCKOv2 library [32, 33] containing 6 unique sgRNAs against each of 19,050 genes and 4 sgRNAs against each of 1,864 microRNAs (Supplementary Figures S1a and b). Cells infected with the library were treated with pomalidomide at an IC70 concentration (Supplementary Figure S1c), and then harvested after 1, 2, and 3 weeks (Fig. 1a). Deep sequencing of embedded sgRNA barcodes showed coverage of at least 100 reads for more than 98% sgRNAs in cells prior to pomalidomide treatment (Supplementary Figure S1d), and an average total 1.6 × 108 sgRNA reads scored at each time point (Supplementary Figure S1e). Enrichment of a few sgRNA sequences was observed after 2 weeks pomalidomide treatment (Supplementary Figure S1f).
Fig. 1.
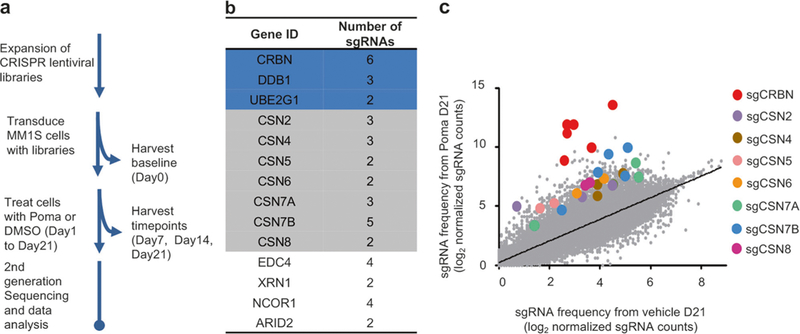
Genome-scale CRISPR-Cas9 screen identifies genes essential for pomalidomide-induced cytotoxicity in MM.1S cells. a Flowchart of pomalidomide resistance screening. Poma pomalidomide. b A sample list of top ranked genes. c Enrichment of specific sgRNAs after Poma treatment. Each dot specifies one sgRNA
Twenty-eight (28) genes and one microRNA (Fig. 1b; Supplementary Table S2) were identified in viable cells based upon the following criteria: (1) time-dependent accumulation; (2) at least a four-fold increase in drug-treated cells compared to vehicle control cells at 21 days; and (3) at least 2 unique sgRNA sequences against each gene meeting criteria 1 and 2. All six sgRNAs targeting CRBN were identified (Fig. 1b, c), consistent with CRBN as the direct target of pomalidomide [28, 29] and required for IMiDs-induced MM cell cytotoxicity. [34, 35]
Degradation of IKZF3 induced by IMiDs is inhibited by CSN knockout
Among the genes identified by CRISPR-based genome-wide knockout screening, 7 of 9 CSN9 signalosome (CSN) subunits [36] were found to be required for MM cell sensitivity to IMiDs (Fig. 1b, c) and are the main focus of this study. To validate the functional importance of these genes mediating pomalidomide and lenalidomide-induced cytotoxicity, sgRNAs against CRBN and these seven genes were individually cloned into the Cas9 lentiviral vector, and then re-introduced into MM.1S cells (Supplementary Figures S2a and b). Importantly, knockout of each CSN gene confers partial resistance to pomalidomide and lenalidomide (Fig. 2a). CSN inactivates the cullin family ubiquitin ligases by removing the ubiquitin-like Nedd8 protein, which is covalently attached to cullins. [37–39] Deletion of these CSN subunits in MM cells led to a dramatic decrease of CRBN protein levels in MM.1S cells (Fig. 2b), except for CSN7A which has a mutually exclusive role with CSN7B. [40] Moreover, pomalidomide-induced IKZF3 degradation was also abrogated by CSN6 knockout (Fig. 2c). We confirmed that deletion of CSN2 or CSN6 in other MM cell lines OPM2 and H929 similarly decreased CRBN levels, decreased IKZF3 degradation, and conferred pomalidomide resistance (Supplementary Figures S3a-c), Finally, CRBN gene expression was positively correlated with expression of CSN subunits in 304 patients with newly diagnosed and previously treated MM (Supplementary Figure S4), indicating potential clinical relevance. Taken together, our CRISPR screening therefore identified CRBN regulation mediated by CSNs as key determinants of IMiDs sensitivity in MM cells.
Fig. 2.
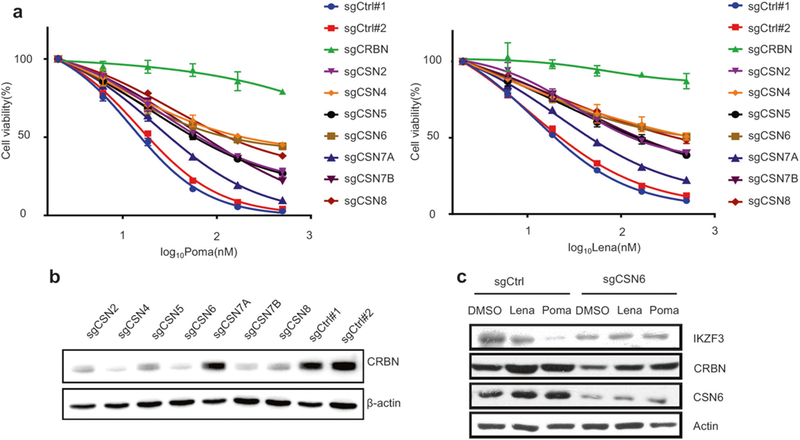
CSN Knockout inhibits the degradation of IKZF3. a Dose-dependent survival of Poma (left panel) and Lena (right panel) treated MM.1S cells infected with individually cloned lentiCRISPR viruses targeting the selected gene candidates. Controls were null-targeting lentiCRISPR viruses. Error bars represent s.e.m. (n = 3). b CRBN levels in each of CSN knockout MM.1S cells. c Immunoblot analysis of IKZF3 and CRBN in CSN6 knockout cells treated with Lena (100 nM) or Poma (25 nM) for 16 h
CSN inhibits the degradation of CRBN by SCFFbxo7
We have previously shown that CRBN can be targeted and degraded by SCFFbxo7 E3 ubiquitin ligase. [30] We here demonstrate that CRBN interacts with CRL4 and SCFFbxo7 at distinct domains (Supplementary Figures S5a-c). We therefore next tested whether CSNs prevent SCFFbxo7 from targeting CRBN for degradation. We found that cul1 neddylation level was significantly increased in CSN2 knockout MM cells (Fig. 3a), consistent with a role of CSNs in negatively regulating SCF ubiquitin ligase activity. [38, 41] Importantly, deletion of either Cul1 (Fig. 3b) or Fbxo7 (Fig. 3c) completely restored CRBN protein levels in CSN2 or CSN6 knockout MM cells to those found in wild type cells; and importantly, re-sensitized these CSN knockout MM cells to pomalidomide-induced cytotoxicity (Fig. 3d, e). These studies indicate a role for CSNs in negatively regulating SCFFbxo7 E3 ligase activity targeting CRBN for degradation.
Fig. 3.
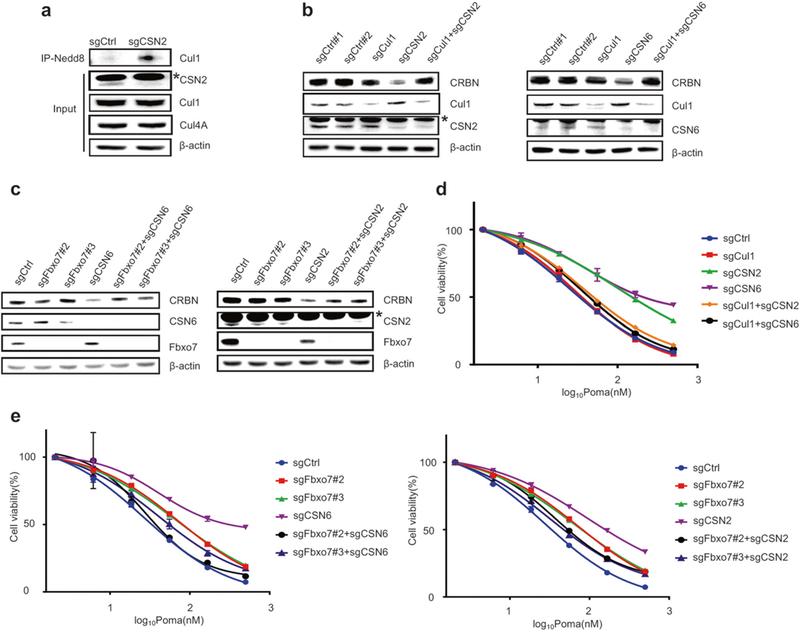
Inactivation of SCFFbxo7 restores CRBN levels and IMiDs sensitivity of CSN knockout MM cells. a Analysis of the levels of neddylated Cul1 and Cul4A in CSN2 deleted MM.1S cells. The asterisk indicates a non-specific band. b, c Immunoblot analysis of CRBN levels in MM.1S cells with CSN2/6 single knockout or CSN2/6-Cul1 (b) or CSN2/6-Fbxo7 (c) double knockout. The asterisk indicates a non-specific band. d, e Dose-dependent survival of MM cells with Cul1 single knockout or CSN2/6-Cul1 double knockout (d) and MM cells with Fbxo7 single knockout or CSN2/6-Fbxo7 double knockout (e) treated with Poma. Error bars represent s.e.m. (n = 3)
Synergy between bortezomib or MLN4924 and IMiDs by sequential treatment
Since CRBN turnover is controlled by a SCF ubiquitin ligase, we next hypothesized that proteasome inhibitor bortezomib, in addition of its inherent anti-MM activity, might stabilize CRBN and thereby enhance IMiDs-induced MM cell cytotoxicity. To address this hypothesis, we pretreated CSN6 knockout cells with bortezomib to prevent SCFFbxo7-mediated CRBN degradation, prior to treatment with pomalidomide. This sequential treatment enhanced CRBN levels, as well as pomalidomide-induced degradation of IKZF3 and cell death (Fig. 4a). We and others have shown that bortezomib and lenalidomide combination therapy mediate synergistic cytotoxicity in preclinical studies, as well as high frequency and extent of response to treat newly diagnosed and relapsed MM. [17] In the clinic, bortezomib is administered to MM patients either twice or once weekly, whereas lenalidomide is taken every day, [42] suggesting that sequential effects of bortezomib followed by lenalidomide may also be operative in the clinic. We confirmed synergistic cytotoxicity in MM.1S and OPM2 MM cells treated sequentially with bortezomib followed by pomalidomide (Fig. 4b). Of note, sequential treatment with MLN4924 followed by pomalidomide similarly increased CRBN levels, decreased IKZF1/3 levels, and triggered synergistic MM cytotoxicity (Fig. 4c).
Fig. 4.
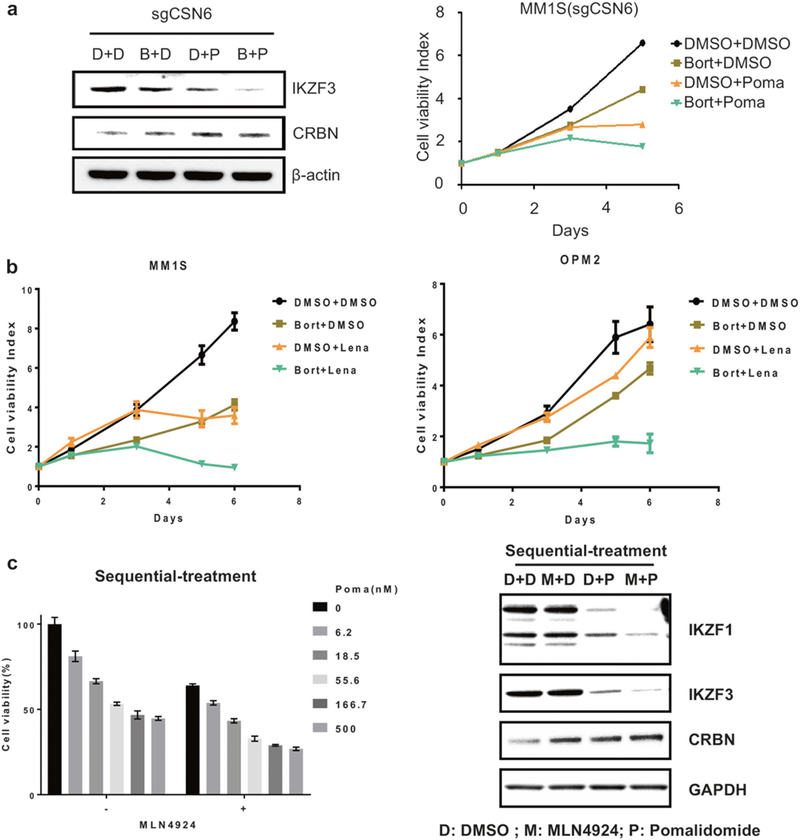
Bortezomib or MLN4924 potentiates IMiDs-induced MM cell cytotoxicity by inhibiting SCFFbox7 -mediated degradation of CRBN. a Immunoblot analysis of IKZF3 and CRBN levels in CSN6 knockout MM.1S cells. Cells were treated with Poma (P, 10 nM) for 4 h after incubating with bortezomib (B, 5 nM) for 24 h or DMSO (d) (left panel). Proliferation of CSN6 knockout MM.1S cells treated with Poma (20 nM) for 5 days after incubating with or bortezomib (1.5 nM) or DMSO for 24 h. Error bars represent s.e.m. (n = 3) (right panel). b Proliferation of MM.1S or OPM2 cells treated with Lena (10 nM) for 5 days after incubating with bortezomib (1.5 nM) or DMSO for 24 h. Error bars represent s.e.m. (n = 3). c Cell viability of MM.1S cells treated with indicated concentrations of Poma for 3 days after incubating with MLN4924 (0.2 μM) for 36 h. Error bars represent s.e.m. (n = 3) (left panel). Immunoblot analysis of IKZF1/3 and CRBN levels in MM.1S cells treated with Poma (P, 10 nM) for 16 h, after incubating with MLN4924 (M, 0.2 μM) or DMSO (d) for 48 h (right panel)
Taken together, these data indicate that CSN regulated CRBN turnover mediated by SCFFbxo7 critically regulates CRBN degradation and pomalidomide sensitivity in MM cells. Moreover, either bortezomib or MLN4924 maintains CRBN levels and confers sensitivity to subsequent IMiDs treatment, which may underlie synergistic preclinical cytotoxicity and clinical efficacy of these combination therapies in MM. [43]
Discussion
RNA-guided CRISPR-Cas9 has been a powerful tool to introduce mutations at specific sites in the genome. [44, 45]. Recently, this method has been applied to interrogate gene function on a genome-wide scale, with several advantages relative to RNA interference technology. [33, 46] In the current study, we explored the mechanisms underlying sensitivity versus resistance of MM cells to IMiDs using this genome-wide knockout screen, and identified many genes that are critically required for sensitivity to pomalidomide in MM cells. From our screening, we showed that inactivation of seven of nine CSN subunits confer IMiDs resistance. CSN subunits negatively regulate the activities of all cullin ubiquitin ligases by deneddylating cullins, [37, 38] and are deregulated in many types of cancer. [47] Importantly, here we validated that knockout of 6 CSNs decrease CRBN level and confers resistance to IMiDs-induced MM cytotoxicity.
SCF E3 ligases are the largest family of ubiquitin ligases, and regulate many biological process including signal transduction, cell proliferation, and genomic stability. [48] The F-box proteins, as adapters of the SCF complex, determine substrate specific of these E3 ligases, which have an important role in cancer. [49] Importantly, CRBN is targeted and degraded by SCFFbxo7 E3 ligase. [30] In the current study, we show that CSN knockout increases the activity of SCFFbxo7, thereby enhancing degradation of CRBN and conferring resistance to IMiDs. Our results highlight the importance of evaluating the impact of novel IMiDs and derivatives on SCFFbxo7 activities.
Combination treatment with proteasome inhibitor borte- zomib or neddylation inhibitor MLN4924 with IMiDs has demonstrated increased MM cytotoxicity in preclinical studies, as well as remarkable extent and frequency of clinical responses. Our study has begun to identify mechanisms underlying these enhanced activities. Here we show that pretreatment with either PI bortezomib or MLN4924 maintains increased levels of CRBN by counteracting SCFFbxo7-targeted CRBN degradation, thereby conferring sensitivity to subsequent IMiDs (Poma or Lena) induced cytotoxicity.
Taken together, our genome-scale CRISPR-Cas9 screening has identified the novel molecular mechanisms regulating sensitivity of MM cells to IMiDs (Fig. 5). CSN9 signalosome complex inhibits the activity of SCFFbxo7 E3 ligase-related CRBN degradation; conversely, loss of function of CSN9 signalosome activates SCFFbxo7 complex, enhances degradation of CRBN, and confers IMiD resistance. Finally, proteasome inhibitors or NAE inhibitors can suppress degradation and maintain levels of CRBN, thereby enhancing sensitivity to IMiDs. These studies identify targets for novel therapeutics to enhance sensitivity to or overcome resistance to IMiDs.
Fig. 5.
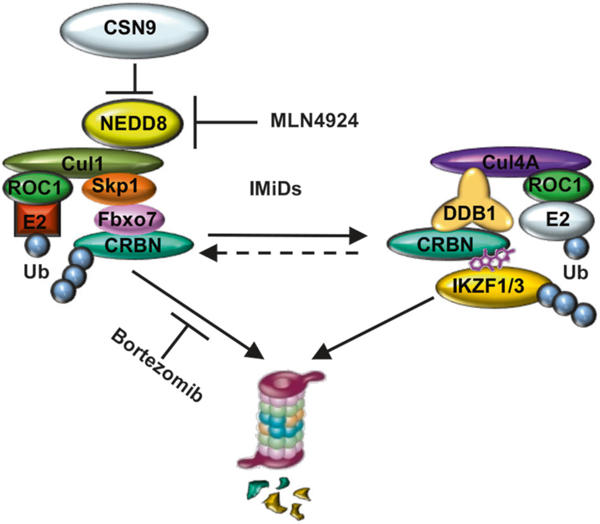
CSN9-mediated CRBN degradation in MM. CSN9 block the function of SCFFbxo7 thereby inhibiting proteasomal degradation of CRBN. Proteasome inhibitor bortezomib or NAE inhibitor MLN4924 treatment stabilizes CRBN level, and enhances IMiDs-induced MM cytotoxicity
Supplementary Material
Acknowledgements
We thank X.H. Feng and Z.P. Xia for expression vectors. We thank the staff from the hematologic neoplasia core facilities at Dana-Farber Cancer Institute for technical assistance. We also thank the Genome Center of WuXi AppTec Inc. for the initial data analysis of the CRISPR screening.
Funding This study was supported by the National Institute of Health Grant; SP0RE-P50100707 (KCA), P01-CA078378 (K.C.A.), R01- CA050947 (K.C.A), and R01-CA178264 (T.H. and K.C.A.). K.C.A. is an American Cancer Society Clinical Research Professor. This study was also supported in part by funds from the National 973 Plan for Basic Research (2015CB553803), National Natural Science Foundation of China (31671334), Fundamental Research Funds for the Central Universities, and Key Construction Program of the National ‘985’ Project.
Footnotes
Compliance with ethical standards
Conflict of interest K.C.A. serves on advisory boards to Celgene and Millennium. All other authors declare no competing financial interests.
Electronic supplementary material The online version of this article (https://doi.org/10.1038/s41375–018-0205-y) contains supplementary material, which is available to authorized users.
References
- 1.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364:1046–60. [DOI] [PubMed] [Google Scholar]
- 2.Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer. 2002;2:175–87. [DOI] [PubMed] [Google Scholar]
- 3.Fermand JP, Ravaud P, Chevret S, Divine M, Leblond V, Belanger C, et al. High-dose therapy and autologous peripheral blood stem cell transplantation in multiple myeloma: up-front or rescue treatment? Results of a multicenter sequential randomized clinical trial. Blood. 1998;92:3131–6. [PubMed] [Google Scholar]
- 4.Lenhoff S, Hjorth M, Holmberg E, Turesson I, Westin J, Nielsen JL, et al. Impact on survival of high-dose therapy with autologous stem cell support in patients younger than 60 years with newly diagnosed multiple myeloma: a population-based study. Nordic Myeloma Study Group. Blood. 2000;95:7–11. [PubMed] [Google Scholar]
- 5.Fermand JP, Levy Y, Gerota J, Benbunan M, Cosset JM, Castaigne S, et al. Treatment of aggressive multiple myeloma by high-dose chemotherapy and total body irradiation followed by blood stem cells autologous graft. Blood. 1989;73:20–23. [PubMed] [Google Scholar]
- 6.Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341:1565–71. [DOI] [PubMed] [Google Scholar]
- 7.McCarthy PL, Owzar K, Hofmeister CC, Hurd DD, Hassoun H, Richardson PG, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366:1770–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lacy MQ, Hayman SR, Gertz MA, Dispenzieri A, Buadi F, Kumar S, et al. Pomalidomide (CC4047) plus low-dose dex-amethasone as therapy for relapsed multiple myeloma. J Clin Oncol. 2009;27:5008–14. [DOI] [PubMed] [Google Scholar]
- 9.Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352:2487–98. [DOI] [PubMed] [Google Scholar]
- 10.Herndon TM, Deisseroth A, Kaminskas E, Kane RC, Koti KM, Rothmann MD, et al. U.S. Food and Drug Administration approval: carfilzomib for the treatment of multiple myeloma. Clin Cancer Res. 2013;19:4559–63. [DOI] [PubMed] [Google Scholar]
- 11.Shirley M Ixazomib: First Global Approval. Drugs. 2016;76: 405–11. [DOI] [PubMed] [Google Scholar]
- 12.Gandolfi S, Laubach JP, Hideshima T, Chauhan D, Anderson KC, Richardson PG. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastas- Rev. 2017;36:561–84. [DOI] [PubMed] [Google Scholar]
- 13.McKeage K Daratumumab: first global approval. Drugs. 2016;76: 275–81. [DOI] [PubMed] [Google Scholar]
- 14.Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373:1207–19. [DOI] [PubMed] [Google Scholar]
- 15.Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373:621–31. [DOI] [PubMed] [Google Scholar]
- 16.Laubach JP, Moreau P, San-Miguel JF, Richardson PG. Panobi- nostat for the treatment of multiple myeloma. Clin Cancer Res. 2015;21:4767–73. [DOI] [PubMed] [Google Scholar]
- 17.Attal M, Lauwers-Cances V, Hulin C, Leleu X, Caillot D, Escoffre M, et al. Lenalidomide, bortezomib, and dexamethasone with transplantation for myeloma. N Engl J Med. 2017;376: 1311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–50. [DOI] [PubMed] [Google Scholar]
- 19.Liu J, Ye J, Zou X, Xu Z, Feng Y, Chen Z, et al. CRL4A(CRBN) E3 ubiquitin ligase restricts BK channel activity and prevents epileptogenesis. Nat Commun. 2014;5:3924. [DOI] [PubMed] [Google Scholar]
- 20.Chen YA, Peng YJ, Hu MC, Huang JJ, Chien YC, Wu JT, et al. The Cullin 4A/B-DDB1-cereblon E3 ubiquitin ligase complex mediates the degradation of CLC-1 chloride channels. Sci Rep. 2015;5:10667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Higgins JJ, Pucilowska J, Lombardi RQ, Rooney JP. A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology. 2004;63:1927–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7:585–98. [DOI] [PubMed] [Google Scholar]
- 23.Hideshima T, Chauhan D, Podar K, Schlossman RL, Richardson P, Anderson KC. Novel therapies targeting the myeloma cell and its bone marrow microenvironment. Semin Oncol. 2001;28: 607–12. [DOI] [PubMed] [Google Scholar]
- 24.Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343: 301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343: 305–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kronke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature. 2015;523: 183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fischer ES, Bohm K, Lydeard JR, Yang H, Stadler MB, Cavadini S,et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512:49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chamberlain PP, Lopez-Girona A, Miller K, Carmel G, Pagarigan B, Chie-Leon B, et al. Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat Struct Mol Biol. 2014;21:803–9. [DOI] [PubMed] [Google Scholar]
- 29.Petzold G, Fischer ES, Thoma NH. Structural basis of lenalidomide-induced CK1alpha degradation by the CRL4 (CRBN) ubiquitin ligase. Nature. 2016;532:127–30. [DOI] [PubMed] [Google Scholar]
- 30.Song T, Liang S, Liu J, Zhang T, Yin Y, Geng C, et al. CRL4 antagonizes SCF(Fbxo7)-mediated turnover of cereblon and BK channel to regulate learning and memory. PLoS Genet. 2017;14:e1007165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hideshima T, Cottini F, Nozawa Y, Seo HS, Ohguchi H, Samur MK, et al. p53-related protein kinase confers poor prognosis and represents a novel therapeutic target in multiple myeloma. Blood. 2017;129:1308–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome- wide libraries for CRISPR screening. Nat Methods. 2014;11: 783–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu YX, Braggio E, Shi CX, Bruins LA, Schmidt JE, Van Wier S, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2011;118: 4771–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, Kang J, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012;26:2326–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lingaraju GM, Bunker RD, Cavadini S, Hess D, Hassiepen U, Renatus M, et al. Crystal structure of the human COP9 signalo- some. Nature. 2014;512:161–5. [DOI] [PubMed] [Google Scholar]
- 37.Dubiel D, Rockel B, Naumann M, Dubiel W. Diversity of COP9 signalosome structures and functional consequences. FEBS Lett. 2015;589(19 Pt A):2507–13. [DOI] [PubMed] [Google Scholar]
- 38.Cope GA, Deshaies RJ. COP9 signalosome: a multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell. 2003;114:663–71. [DOI] [PubMed] [Google Scholar]
- 39.Lyapina S, Cope G, Shevchenko A, Serino G, Tsuge T, Zhou C, et al. Promotion of NEDD-CUL1 conjugate cleavage by COP9 signalosome. Science. 2001;292:1382–5. [DOI] [PubMed] [Google Scholar]
- 40.Olma MH, Roy M, Le Bihan T, Sumara I, Maerki S, Larsen B, et al. An interaction network of the mammalian COP9 signalo- some identifies Dda1 as a core subunit of multiple Cul4-based E3 ligases. J Cell Sci. 2009;122(Pt 7):1035–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–6. [DOI] [PubMed] [Google Scholar]
- 42.Richardson PG, Xie W, Jagannath S, Jakubowiak A, Lonial S, Raje NS, et al. A phase 2 trial of lenalidomide, bortezomib, and dexamethasone in patients with relapsed and relapsed/refractory myeloma. Blood. 2014;123:1461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Avigan D, Rosenblatt J. Current treatment for multiple myeloma. N Engl J Med. 2014;371:961–2. [DOI] [PubMed] [Google Scholar]
- 44.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343: 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee MH, Zhao R, Phan L, Yeung SC. Roles of COP9 signalosome in cancer. Cell Cycle. 2011;10:3057–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee EK, Diehl JA. SCFs in the new millennium. Oncogene. 2014;33:2011–8. [DOI] [PubMed] [Google Scholar]
- 49.Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol. 2013;14:369–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.