

Summary
“Biased” G protein-coupled receptor (GPCR) agonists preferentially activate pathways mediated by G proteins or β-arrestins. Here we use double electron-electron resonance spectroscopy to probe the changes that ligands induce in the conformational distribution of the angiotensin II type I receptor. Monitoring distances between ten pairs of nitroxide labels distributed across the intracellular regions enabled mapping of four underlying sets of conformations. Ligands from different functional classes have distinct, characteristic effects on the conformational heterogeneity of the receptor. Compared to angiotensin II, the endogenous agonist, agonists with enhanced Gq coupling more strongly stabilize an “open” conformation with an accessible transducer-binding site. β-Arrestin-biased agonists deficient in Gq coupling do not stabilize this open conformation but instead favor two more occluded conformations. These data suggest a structural mechanism for biased ligand action at the angiotensin receptor that can be exploited to rationally design GPCR-targeting drugs with greater specificity of action.
Graphical Abstract
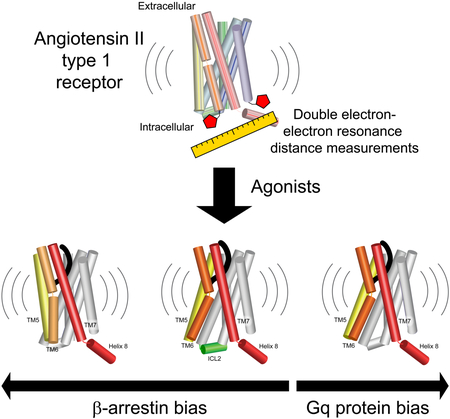
In brief
GPCR ligands that lead preferentially to G protein or β-arrestin signaling promote distinct activated conformations of the receptor.
Introduction
G protein-coupled receptors (GPCRs, or seven transmembrane receptors) are versatile membrane proteins that initiate cascades of intracellular signaling events in response to an extraordinary array of extracellular signals. In the canonical model of GPCR activation (Lefkowitz, 2013), binding of agonists to an extracellular site stabilizes conformational changes in the cytoplasmic G protein-binding pocket, activating heterotrimeric G proteins. Subsequent phosphorylation of agonist-activated GPCRs by GPCR kinases (GRKs) recruits β-arrestins to the receptor. β-Arrestins initiate additional signaling cascades and dampen G protein-mediated signaling by mechanisms that include steric occlusion of the G protein-binding site and facilitation of receptor endocytosis.
The assumption that these transducer-mediated events must transpire in linear succession was challenged by the discovery of “biased” GPCR agonists. Biased agonists preferentially activate particular signaling pathways (e.g., G protein- vs. β-arrestin-mediated signaling) (Rankovic et al., 2016). The angiotensin II type 1 receptor (AT1R) is a particularly compelling model of biased agonism, as two distinct classes of biased ligands have been thoroughly characterized in cellular signaling and in vitro transducer coupling assays (Rajagopal et al., 2011; Strachan et al., 2014). Compared to the endogenous ligand angiotensin II (AngII), “Gq-biased” AT1R agonists are more efficacious in stimulating Gq-mediated cellular signaling and in allosterically promoting Gq coupling, but they have comparable efficacy towards β-arrestin. In contrast, “β-arrestin-biased” ligands are deficient in Gq-mediated cellular signaling and allosteric coupling to Gq, yet they retain efficacy towards β-arrestin. Physiologically, Gq-mediated AT1R signaling increases blood pressure, while β-arrestin signaling increases cardiac contractility and performance (Rajagopal et al., 2006; Violin et al., 2010). For certain heart failure indications, β-arrestin-biased AT1R agonists such as TRV027 have shown promise clinically as alternatives to angiotensin receptor blockers (ARBs), mainstay hypertension treatments that categorically inhibit both signaling arms (Cotter et al., 2018; Ryba et al., 2017).
There is a paucity of information on the structural mechanisms underlying biased ligands’ actions. It is widely believed that biased ligands stabilize distinct receptor conformations that favor coupling to particular transducers. In support of this hypothesis, techniques such as FRET, BRET, chemical labeling, and NMR have identified ligand-specific conformational differences in multiple receptors (Devost et al., 2017; Granier et al., 2007; Kahsai et al., 2011; Liu et al., 2012; Okude et al., 2015; Rahmeh et al., 2012; Zurn et al., 2009). However, these methods lack the resolution to comprehensively describe the conformations they detect. Several crystallographic and cryo-electron microscopy structures have provided snapshots of GPCRs bound to biased ligands (Liang et al., 2018; Wacker et al., 2013; Wacker et al., 2017), but these structures capture only a single, low-energy conformation. The conformational heterogeneity of GPCRs (Latorraca et al., 2017) could play an important role in biased agonists’ mechanisms.
Here we map the conformational changes that biased ligands induce across the intracellular surface of the AT1R using double electron-electron resonance (DEER) spectroscopy. This pulsed electron paramagnetic resonance (EPR) technique measures the distribution of distances between two nitroxide labels 15-80 Å apart (Figure 1A), providing high-resolution information on the conformations present in a protein’s ensemble as well as their populations (Jeschke, 2012). A series of spin label pairs reveals the distinct effects of different functional classes of ligands on the AT1R’s conformational landscape.
Figure 1: DEER Distance Measurements of the AT1R.

(A) Site-directed spin labeling of AT1R intracellular regions with the nitroxide-containing V1 side chain allows the detection of ligand-dependent conformational changes by DEER spectroscopy, as shown schematically for TMs 1 and 6.
(B) AT1R labeling strategy indicating six labeling sites and their ten pairwise combinations (dotted lines), shown on the crystal structure of AT1R bound to the candesartan precursor ZD7155 (PDB: 4YAY).
Results and Discussion
Conformational Heterogeneity of “Inactive” AT1R
To enable site-specific spin labeling through thiol-selective chemistry, we developed a minimal cysteine AT1R, in which the six cysteines not involved in the receptor’s two extracellular disulfide bonds were mutated to alanines. These modifications modestly lower the receptor’s affinity for AngII and its derivatives but otherwise preserve its functionality both in a cellular context and in detergent after purification (Figures S1A-S1C, Table S1). Initially guided by homology models and later by the published structure of inactive AT1R (Zhang et al., 2015), we engineered cysteine residues at the cytoplasmic ends of transmembrane helices (TMs) 1, 5, 6, and 7, and in intracellular loop 2 (ICL2) and helix 8 (H8). Optimal labeling sites were identified by empirical screening of 32 single cysteine constructs with respect to their expression and biochemical properties, efficient labeling of purified protein with (1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl) methanethiosulfonate spin label (MTSSL), and continuous-wave EPR analysis of labels’ mobility. Six labeling sites with favorable properties were combined in ten pairwise combinations (Figure 1B), omitting pairs which showed poor biochemical behavior upon purification and those expected to be outside the distance range of DEER, based on structural information. This extensive cross-referencing of sites increases the likelihood of finding a favorable vantage point for observing all spin label movements, thereby enabling detection of global conformational changes and allowing trilateration (mapping) of all label positions from the combined set of data (Altenbach et al., 2008). We verified that these mutations and spin labeling of the receptor do not substantially alter its biochemical behavior or ability to bind to AngII (Figures S1D-S1F, Table S2). For DEER experiments, we found that labeling with bis-(2,2,5,5-tetramethyl-3-imidazoline-1-oxyl-4-yl)disulfide (IDSL) gave less background labeling of minimal cysteine AT1R than more commonly used spin labels (e.g., MTSSL, IA-PROXYL). In addition, IDSL generates a side chain (V1) with restricted rotameric freedom, providing sharper, more readily interpretable distance distributions (Balo et al., 2016; Warshaviak et al., 2013).
DEER measurements collected on the ten V1-labeled double mutants attest to the inherent conformational heterogeneity of the AT1R in the absence of ligands (Apo state) (Figures 2A, S2 and S3). All experiments were performed using receptor purified in lauryl maltose neopentyl glycol detergent with cholesterol hemisuccinate. The distance distributions of most pairs are multimodal, showing either more than one major peak or a broad distribution of multiple overlapping peaks. Addition of candesartan or olmesartan, inverse agonist ARBs that suppress the AT1R’s low levels of constitutive activity (Van Liefde and Vauquelin, 2009), either negligibly affects the distributions (e.g., TM1-ICL2, ICL2-TM7) or stabilizes certain distances already present (e.g., ICL2-TM5, TM5-H8) (Figure 2A).
Figure 2: DEER Distance Measurements Detect Conformational Changes in the AT1R.

(A) Distance distributions of ligand-free (Apo) AT1R overlaid with those of AT1R bound to the inverse agonist candesartan (Cand) and the endogenous (balanced) agonist AngII.
(B) Spin densities based on the combined DEER distance distributions of candesartan-bound AT1R (colored clouds) are overlaid with the ZD7155-bound AT1R crystal structure (ribbons, PDB: 4YAY) including modeled V1 side chain rotamers.
(C) Conformational heterogeneity of ZD7155-AT1R in MD simulations. When the most probable V1 rotamer is modeled onto hundreds of simulation frames, the nitroxide oxygen positions (small colored spheres) overlap with the DEER spin densities (clouds outlined for clarity) at all spin label positions. This suggests conformations among those captured in simulation can explain the experimentally observed DEER conformations. Each simulation frame represents a snapshot sampled every 10 ns from the last 1 μs of simulation, across four simulations of ZD7155-bound AT1R. The AT1R-ZD7155 structure (PDB: 4YAY) and DEER spin densities (outlined for clarity) shown in panel B are overlaid for comparison.
We compared the conformations of inverse-agonist-bound AT1R inferred from DEER data to the crystallographic conformation. The full distance distributions collected in the presence of candesartan were used to determine a globally minimized arrangement of all spin labels (Altenbach et al., 2008). Subsequently, the distance distributions were “mapped” in the membrane plane by trilateration (see STAR Methods). Based on previously described methods (Polyhach et al., 2011), spin labels were modeled onto the crystal structure based on the steric constraints of the V1 side chain. The DEER data deviate from the crystal structure slightly at the ICL2 site and more substantially at the TM5, TM7 and helix 8 sites (Figure 2B).
To test whether this discrepancy originated due to either the instability of the crystal structure upon removal of lattice contacts or the intrinsic heterogeneity of inactive AT1R, we performed molecular dynamics (MD) simulations using the crystal structure of the AT1R bound to ZD7155, a precursor of candesartan (Zhang et al., 2015). The ZD7155-bound receptor relaxes away from the crystallographic conformation and exhibits considerable conformational heterogeneity (Figure S4A-S4C). A turn at the base of TM7 straightens, and helix 8 reorients toward an axis nearly perpendicular to the membrane plane. The intracellular end of TM5 moves away from the center of the helical bundle, and the intracellular end of TM4 frequently unwinds from its helical structure. Spin label rotamers modeled onto simulation frames overlap with the DEER clouds at every labeling site, suggesting that the conformational heterogeneity of the AT1R observed in simulation can account for the discrepancies between the crystal structure and the DEER data (Figures 2C, S4D, and S4E).
Activation of AT1R by Angiotensin II
Binding of the endogenous agonist AngII causes major conformational changes throughout the intracellular regions (Figure 2A) that reflect known structural hallmarks of GPCR activation. In the TM1-TM6 pair, AngII depletes the 31 and 34 Å distances, and the majority of the population is stabilized at 42 Å. The outward displacement of TM6 is the largest-scale conformational change in the transmembrane region upon GPCR activation (Manglik and Kruse, 2017). Another conformational change typically associated with GPCR activation is the inward movement of TM7, which is consistent with the peak at 36 Å in the ICL2-TM7 AT1R pair. Besides these well-conserved movements, AngII activation of the AT1R induces several other significant changes. Most dramatic is the appearance of a short distance peak at 26 Å in the TM1-ICL2 distribution. Other distance distributions involving ICL2 also have prominent short distance peaks (e.g., 22 Å in ICL2-TM7), suggesting conformational changes are occurring in the ICL2 region rather than at the TM1 site.
AngII broadens the conformational distribution of the AT1R rather than quantitatively populating a single agonist-specific conformation (Figure 2A). TM6 is in the outward position only in ~60% of the population (TM1-TM6 pair), and for other pairs, an even lower fraction of the population shifts to AngII-stabilized peaks. This is consistent with the loose allosteric coupling between the ligand-binding site and the intracellular regions described in other agonist-bound GPCRs (Manglik et al., 2015; Sounier et al., 2015; Ye et al., 2016). Site-specific effects due to mutations and labeling (Figure S1F) could contribute to the differences in the populations of AngII-stabilized peaks among the panel of spin pairs, but this variability could also indicate that AngII activates conformational changes in different intracellular regions with different efficacies.
Notably, most of the clearly resolved AngII-stabilized peaks (e.g., those in TM1-TM6 and TM1-ICL2) are weakly but detectably populated (<10% of the population) in the Apo state (Figure 2A). The finite population of “active conformations” under basal conditions supports a conformational selection model (Ye et al., 2016), but the low abundance of these active conformations in the Apo state accords well with this receptor’s low constitutive activity in cellular systems (Scragg et al., 2005).
Activation of AT1R by Biased Agonists
We then investigated the conformational changes induced by analogs of the AngII octapeptide, which have substitutions, insertions, or deletions at one to three positions (Figure 3A), when bound to the receptor at saturating concentrations. As summarized in Figure 3A and Table S1, these ligands have been extensively characterized in previous studies not only with regards to their cellular signaling profiles (Rajagopal et al., 2011; Violin et al., 2010) but also in the ability of Gq and β-arrestin fusions to the AT1R to allosterically enhance these ligands’ binding (“molecular efficacy”), a direct measure of the free energy change due to ligand-transducer coupling (Strachan et al., 2014). TRV055 and TRV056 are “gain-of-function” ligands that have >10-fold greater allosteric coupling to Gq than AngII and are also more efficacious in stimulating cellular Gq-mediated signaling (e.g., IP1 generation), but they show similar allosteric coupling to β-arrestin as AngII. Thus we describe TRV055 and TRV056 as “Gq-biased.” In contrast, TRV026, TRV034, TRV023, and TRV027 are “loss-of-function” ligands that have ≥20-fold impairment compared to AngII in allosteric coupling to Gq and in Gq-mediated signaling, but they show relatively minor decreases in allosteric coupling to β-arrestin (<3-fold) and in β-arrestin-mediated cellular activities (e.g., β-arrestin recruitment, receptor internalization, β-arrestin-mediated ERK phosphorylation). Therefore we refer to these ligands as “β-arrestin-biased.” Quantitative measures of bias for the ligands in our panel are provided in Table S1.
Figure 3: AT1R Conformational Changes Induced by Biased Agonists.

(A) Peptide sequences and functional profiles of AngII and its biased derivatives. Bold residues indicate mutations relative to AngII (NMe = N-methyl). Coupling to Gq and β-arrestin is defined relative to AngII, with “+” indicating comparable direct allosteric coupling (molecular efficacy) (Strachan et al., 2014) and effective signaling in cell-based assays (Rajagopal et al., 2011), “++” indicating ≥10-fold increase in allosteric coupling and effective signaling, and “—” indicating ≥10-fold decrease in allosteric coupling and effective signaling. These previously reported values are provided in Table S1.
(B and C) Distance distributions of selected spin label pairs collected for AT1R bound to (B) Gq-biased ligands and (C) β-arrestin-biased ligands. Distributions of Apo and AngII-bound AT1R are shown for reference.
TRV055 and TRV056 lead to distance distributions that are very similar to each other but quite distinct from those induced by AngII (Figures 3B and S3). The Gq-biased agonists promote the outward movement of TM6 (TM1-TM6 pair) in a smaller fraction of the population (~40%) than AngII (~60%). Most conformational changes associated with AngII activation are likewise diminished (e.g., 36 Å in ICL2-TM7) or essentially absent (e.g., 26 Å in TM1-ICL2) upon binding to TRV055 or TRV056. However, in a few pairs, the Gq-biased ligands stabilize particular populations more strongly than AngII, including the distances at 31-33 Å in ICL2-TM5.
Despite having very comparable functional profiles, the four β-arrestin-biased ligands induce diverse conformational signatures. In several pairs TRV026 (Figures 3C and S3) populates the same distances from the distributions of Apo receptor that inverse agonists depopulate (Figure 2A). For example, TRV026 does not promote the outward movement of TM6 but rather stabilizes almost the entire population at an intermediate distance (34 Å, TM1-TM6 pair) that is one of two main peaks in the Apo distribution. However, TRV026 binding enriches peaks in the ICL2-TM7 and ICL2-H8 distributions that are not clearly observed in either Apo or AngII-bound receptor. Given the absence of changes in the TM1-ICL2 pair, these new peaks most likely result from movements of TM7 and H8 rather than ICL2, in contrast to the conformational changes stabilized by AngII.
Distributions for AT1R bound to TRV034 and TRV023 are qualitatively quite similar to those of AngII, with slight quantitative differences (Figures 3C and S3). These ligands promote the outward movement of TM6 to 42 Å in approximately 50% of the population (TM1-TM6), compared to ~60% of the population for AngII. In other spin pairs, they vary relative to AngII in their ability to stabilize particular conformational changes, ranging from being less efficacious (e.g., 36 Å peak in ICL2-TM7) to equally or more efficacious (e.g., 26 Å peak in TM1-ICL2, 22 Å in ICL2-TM7, 25 Å in ICL2-H8). The fourth β-arrestin-biased agonist, TRV027, stabilizes a mixture of the distance distributions that are stabilized by TRV026 and TRV034/TRV023 (Figures 3C and S3). It should be noted that the small differences in the peak center observed in several pairs (e.g., the outward TM1-TM6 distance appears ~0.5 Å longer for AngII than for the β-arrestin-biased agonists) may reflect additional ligand-specific differences that are beyond the resolution DEER can detect with confidence (~0.5 Å).
AT1R Conformational Ensemble Reflects Ligand Functionality
We used non-negative matrix factorization (NNMF) (Paatero and Tapper, 1994) to find correlated changes across the entire DEER data set and to identify the minimum number of conformations required to account for all conformational changes (see STAR Methods). NNMF deconvolutes complex datasets into a given number of salient components (here, conformations) and the magnitude of their contributions (here, the conformations’ populations) to each independent observation (here, each ligand condition). NNMF suggests only four components (denoted as NNMF I, II, III, and IV) with distinct dependencies on bound ligands (Figures 4A, 4B, and S5A-S5C; Table S1). Strikingly, the well-defined NNMF distance distributions and the associated population analysis indicate that NNMF is able to resolve the mixing of conformations that is particularly prevalent in agonist-bound AT1R. The persistent multimodality in some distance distributions likely has two causes. First, it reflects the plasticity of the receptor, especially upon TM6 outward movement. Second, particularly for the less-represented NNMF components III and IV, the influence of structural decoupling and small population shifts due to mutation and spin labeling may cause peaks from more dominant NNMF components to be mixed in.
Figure 4: AT1R Conformational Ensemble Reflects Ligand Functionality.

(A) Non-negative matrix factorization (NNMF) reveals four dominant components (colored lines) contributing to the aggregated DEER dataset. For comparison, the experimentally determined distributions for the ligand with the highest population of each component (gray dashed lines) are overlaid.
(B) The population (amplitude factor) of each NNMF component for each ligand condition. Values are tabulated in Table S1. (Cand = candesartan, Olm = olmesartan)
(C) Correlation between agonist molecular efficacy in Gq coupling (αGq) (Strachan et al., 2014) and the population of NNMF component IV. TRV027 was not included in the previous study of molecular efficacy. Error bars representing standard error of the mean for α values are smaller than the markers.
Clear relationships are seen between ligand functionality and the NNMF component populations (Figure 4B). NNMF I, which captures features such as the shortest TM1-TM6 distance (31 Å), dominates the Apo state, and inverse agonists further increase its population. β-Arrestin-biased ligands stabilize various mixtures of two main components, NNMF II (including features such as the intermediate 34 Å TM1-TM6 distance) and NNMF III (including both the fully outward 42 Å TM1-TM6 distance and the short 26 Å TM1-ICL2 distance), with TRV026 at one extreme and TRV034 at the other. Compared to TRV034, AngII stabilizes a higher percentage of the population in NNMF IV, which has the outward TM1-TM6 distance (42 Å) but a rather heterogeneous TM1-ICL2 distribution disfavoring the 26 Å distance. Gq-biased ligands have an even higher NNMF IV population (~40%) than AngII and a conspicuous absence of the NNMF III component. The NNMF IV population correlates remarkably well with agonists’ efficacy in promoting Gq coupling (Figure 4C). In contrast, efficacy towards β-arrestin is associated with population of any of the three components NNMF II, III, and IV (Figure S5D).
Models of Biased Agonist-Stabilized Conformations
We next modeled the global arrangement of spin labels for the four sets of conformations detected by NNMF. Since the high degree of heterogeneity precludes the application of triangulation techniques to the full distributions (as in Figure 2B), we selected the dominant or most characteristic ligand-stabilized peak from each distribution and used those distances to determine the globally refined distribution of spin label positions (see Figure S5A and STAR Methods for details). It is important to note that due to the plasticity of the AT1R, each map describes only a dominant conformation capturing the characteristic features for each component, not the full ensemble explored by the receptor.
As expected, the model based on NNMF I (Figure 5A) is very similar to that generated based on the full candesartan distributions (cf. Figure 2B). We refer to this modeled conformation as Aclosed since it stabilizes the most inward TM6 position observed. Compared to Aclosed, the map derived from NNMF II (Figure 5B), dominant upon TRV026 binding, is characterized by a slight outward movement of TM6, the inward movement of TM5, a slight shift of TM7, and a larger movement of H8. We refer to this model as Aoccl1 since it suggests the central transducer-binding pocket remains inaccessible despite being stabilized by ligands with some biological activity. The changes in helix 8 are consistent with the label moving from lying parallel to more perpendicular to the membrane, as MD simulations suggest is possible (Figure S4C). However, greater uncertainty is associated with assigning coordinates out of the membrane plane, where the other labels are located.
Figure 5: Model of Biased Ligand Activation of the AT1R.

(A-D) Distance mapping of AT1R conformations based on the four NNMF components: (A) Aclosed (NNMF I), (B) Aoccl1 (NNMF II), (C) Aoccl2 (NNMF III), and (D) Aopen (NNMF IV). Colored surfaces reflect spin densities based on major distance peaks selected from the NNMF distributions. A representative frame from molecular dynamics simulations of ZD7155-bound AT1R is overlaid in gray. Colored arrows represent changes in label positions relative to the Aclosed map (light gray surfaces in (B), (C), and (D)).
(E) Superposition of the spin densities of the Aopen conformation and the crystal structure of the AT1R activated by a conformation-specific nanobody (PDB: 6DO1).
(F) The relationship between ligands’ biological activities and the characteristic receptor populations they stabilize suggest a model of receptor activation where (i) Aclosed represents the inactive conformation (stabilized by inverse agonists), (ii) Aoccl1 and Aoccl2 are capable of triggering β-arrestin but not Gq protein signaling (stabilized by β-arrestin-biased agonists), and (iii) Aopen represents a fully activated conformation able to couple to both β-arrestin and G protein with the highest efficiency (stabilized by AngII and even more by Gq-biased ligands).
See also Figure S5.
The map based on NNMF III, most prevalent in the presence of the β-arrestin-biased agonist TRV034, shows the pronounced inward movement of the ICL2 label as well as the large-scale outward displacement of TM6 (Figure 5C), along with an inward shift of TM7 and outward movement of TM5. The map from NNMF IV (Figure 5D), stabilized most by the Gq-biased ligands, displays an outward tilt of TM6, the subtle inward shift of TM7, and a larger movement of TM5 toward TM6, which would be consistent with the rotation of TM5 observed in multiple activated GPCR structures (Manglik and Kruse, 2017). We designate the models derived from NNMF III and IV as Aoccl2 and Aopen, respectively. While both Aoccl2 and Aopen show a similar translocation of TM6, the more substantial movement of TM5 and the absence of rearrangements in ICL2 in Aopen suggest a more accessible transducer-binding pocket overall. The configuration of labels in Aopen agrees remarkably well with the conformation observed in our nanobody-stabilized active AT1R structure (Wingler et al., co-submitted) (Figure 5E).
Implications for Mechanisms of AT1R Biased Agonists
There is a striking correlation between the functional profiles of AT1R ligands and the conformations they stabilize, arguing the receptor’s equilibrium conformational distribution can account for the ligands’ biological activities (Figure 5F). The best predictor of a ligand’s efficacy towards Gq is the population in NNMF IV (Aopen) (Figures 4B, 4C and S5D). This conformation is most prevalent upon AT1R binding to Gq-biased ligands, rationalizing their augmented activity compared to AngII. Interestingly, over half of the receptor remains in “inactive” conformations when bound to TRV055 or TRV056, suggesting AT1R “super-agonists” even more efficacious in Gq signaling could be developed. AngII stabilizes NNMF IV (Aopen) more than any β-arrestin-biased agonist (Figures 4B and 4C), accounting for the inability of the latter class to activate Gq. In contrast, β-arrestin can allosterically couple not only to ligands stabilizing NNMF IV (Aopen) but also those stabilizing any mixture of NNMF components II (Aoccl1) and III (Aoccl2) (Figures 4B, 5F, and S5D). As a result, ligands that promote very different conformational ensembles (e.g., TRV026 and TRV034) produce very similar functional outputs (Rajagopal et al., 2011; Strachan et al., 2014). However, Gq-biased ligands promote coupling to β-arrestin with slightly higher efficacy than the β-arrestin-biased ligands, despite having a lower combined population in the three agonist-stabilized conformations (Figure S5D). This suggests that β-arrestin couples to Aopen more efficiently than to either Aoccl1 or Aoccl2.
We cannot exclude the possibility that the detergent environment influences the absolute populations of conformations (Van Eps et al., 2017) or that the kinetics of conformational transitions (Manglik et al., 2015) and ligand binding (Lane et al., 2017) contribute to these ligands’ bias. However, our overall model of biased ligand action at the AT1R is consistent with independent lines of work on other GPCRs. NMR data on the μ-opioid receptor shows G-biased ligands favor more open intracellular conformations, while agonist activation of a β-arrestin-biased mutant promotes more closed conformations (Okude et al., 2015). It further accords with work demonstrating the visual arrestin finger loop, the key interaction motif, binds two distinct conformations of active rhodopsin (Elgeti et al., 2018). Since β-arrestin can bind to GPCRs through both a higher affinity interaction with the GRK-phosphorylated carboxy terminus and a lower affinity interaction with the TM core (Latorraca et al., 2018; Shukla et al., 2014), the enforced proximity of β-arrestin resulting from the carboxy terminal interaction could facilitate its binding to occluded conformations or to very low populations of open conformations.
The outward displacement of TM6 is considered the principal marker of GPCR activation and has often correlated with agonists’ efficacy in G protein activation (Ghanouni et al., 2001; Gregorio et al., 2017). However, movement of TM6 in the presence of the β-arrestin-biased agonists such as TRV034 indicates this change alone is not sufficient for Gq activation. The Aoccl2 and Aopen models (expected to be inactive and active towards Gq, respectively) differ primarily in the position of TM5 and the large inward movement of the ICL2 label in Aoccl2, though there may be additional distinguishing features that our current spin label pairs are not positioned to detect. Interestingly, recent structural data and MD simulations have highlighted the role of ICLs in transducers’ binding mechanisms (Ho et al., 2018; Latorraca et al., 2018).
Notably, β-arrestin-biased agonists such as TRV034 do not stabilize a conformation unique to this functional class. The AngII and TRV034 distributions differ only in each conformation’s relative population, with TRV034 favoring conformations lacking the full complement of features associated with “active” receptors. Spectroscopic data and MD simulations of other GPCRs have suggested that partial agonists similarly drive the equilibrium towards activation intermediates instead of the canonical fully active conformation (Dror et al., 2011; Solt et al., 2017; Ye et al., 2016). It is possible that activation intermediates of some GPCRs may have partial activity towards all transducers, while analogous intermediate conformations of other receptors only interact with certain transducers. This would inherently predispose certain GPCRs to biased signaling.
The absence of outwardly displaced TM6 in the distributions of TRV026 (Figure 3C) suggests it is not absolutely required to initiate β-arrestin recruitment and coupling. While this is surprising since TM6 is displaced in GPCRs fully engaged with arrestins (Kang et al., 2015), it is consistent with previous evidence that GRK- and β-arrestin-mediated events can occur without this conformational change (Pulvermuller et al., 1993; Vilardaga et al., 2001). The primary features differentiating the TRV026 and Apo conformational ensembles are the shift of TM7 and the associated inward movement of helix 8, regions implicated in biased agonists’ effects on other receptors (Fay and Farrens, 2015; Liu et al., 2012; Rahmeh et al., 2012). While depletion of the most inaccessible conformations (Aclosed) could contribute to TRV026’s biological activity, it is interesting to speculate that helix 8 assists in “flagging down” GRKs and/or β-arrestins in the absence of G protein-dependent mechanisms for their mobilization (Pack et al., 2018; Pitcher et al., 1992).
The eighth and final residue of AT1R peptide ligands appears to be the most critical determinant of the AT1R’s intracellular conformational ensemble (Figures 3A and S5E), just as it is for Gq activation (Domazet et al., 2015; Holloway et al., 2002). Extensive structure-activity studies have indicated AngII’s terminal carboxylate directly triggers conformational changes activating Gq, while the bulky Phe side chain properly orients the carboxylate (Aumelas et al., 1985; Noda et al., 1995). Our structure of activated AT1R bound to the partial agonist Sar1,Ile8-AngII confirms the terminal carboxylate ion pairs with AT1R Lys199, and the Ile8 side chain fits snugly in a hydrophobic cavity at the pocket’s base (Wingler et al., co-submitted). The larger Phe8 of AngII and Gq-biased ligands would require additional conformational rearrangements, which must promote Aopen. Decreasing side chain bulk (Ala8, TRV034 and TRV023) reduces the population of Aopen but not Aoccl2, while reorienting the side chain outside the hydrophobic cavity (D-Ala8, TRV027) begins favoring Aoccl1. Only Aoccl1 is observed upon outright deletion of residue 8 (TRV026), pointing to a categorical requirement for this residue for outward TM6 movement. Higher in the ligand-binding pocket, the network of interactions involving Arg2 (Feng et al., 1995; Wingler et al., co-submitted), a position conserved in β-arrestin-biased but not Gq-biased ligands, may be required along with residue 8’s interactions to stabilize Aoccl2, which is not detectable upon Gq-biased agonist binding (Figure S5E). The extended ligand-binding pockets of peptide receptors could make them particularly amenable to biased ligand development.
Conclusions
Despite tremendous advances in GPCR structural biology, a detailed understanding of how biased ligands modulate receptors to achieve diverse patterns of transducer activation remains elusive. An increasing number of atomic-resolution crystallographic and cryo-electron microscopy structures of GPCRs in activated conformations or bound to biased ligands are becoming available (Manglik and Kruse, 2017), but these snapshots lose information about the conformational flexibility explored and utilized by receptors. Spectroscopic methods have demonstrated that GPCRs are conformationally heterogeneous (Liu et al., 2012; Manglik et al., 2015; Okude et al., 2015; Ye et al., 2016), but they generally deliver lower-resolution information on specific regions of proteins. DEER marries many advantages of both types of approaches, providing Angstrom-resolution measurements and quantitative information on the composition of the conformational ensemble (Van Eps et al., 2017). This study is by far the most detailed mapping of a GPCR’s different activation states by DEER reported to date.
Our data highlight the importance of taking a global view of receptor activation that encompasses both the inherent plasticity of GPCRs and conformational changes throughout the protein. Numerous vantage points are required to resolve the multiple “activated” receptor conformations stabilized by functionally diverse ligands. The correlation between biased ligands’ functional profiles and conformational effects are consistent with a model in which Gq has relatively strict conformational requirements for binding, while β-arrestin is more promiscuous, initiating binding either to occluded conformations or a minute population of the most open conformations (Figure 5F). As biophysical data emerge on other biased ligands, it will be fascinating to discover the common themes and receptor-specific features of the underlying mechanisms. The next challenge will be to harness this information to design ligands, and ultimately therapeutics, with tailored pharmacological profiles.
STAR Methods
Contact for Reagent and Resource Sharing
Requests for further information or reagents may be directed to the Lead Contact, Robert Lefkowitz (lefko001@receptor-biol.duke.edu).
Experimental Model and Subject Details
Expi293F cells were grown in suspension culture in Expi293F expression media at 37 °C in a humidified environment with 8% CO2 shaking at 110 rpm. The derivative line with the stably transfected tetracycline repressor (pcDNA/TR) was maintained in the presence of 10 μg/mL blasticidin, which was removed prior to transfection. The activated endocytosis U2OS parental cell line was grown at 37 °C in a humidified enviro nment with 5% CO2 in MEM media supplemented with 10% FBS, 1% penicillin-streptomycin, 250 μg/mL hygromycin, 0.875 μg/mL puromycin, and 2 mM glutamine. All cell lines are female.
Method Details
AT1R Expression and Purification
Full-length wild-type human AT1R with an N-terminal hemagglutinin membrane insertion signal sequence and FLAG tag (Strachan et al., 2014) was cloned into pACMV-tetO (Reeves et al., 2002). The six native cysteine residues not involved in extracellular disulfide bonds (C76, C121, C149, C289, C296, and C355) were mutated to alanine to generate AT1R minimal cysteine, and engineered cysteines (F55C, R139C, K220C, D236C, F304C, R311C) were introduced in pairwise combinations. All mutagenesis was performed using the Quikchange Lightning Multi Site-Directed Mutagenesis Kit (Agilent). Constructs were transfected into a derivative of the HEK293 suspension cell line Expi293F (Thermo) stably expressing the tetracycline repressor (Staus et al., 2018) using the Expifectamine transfection kit (Thermo) according to the manufacturer’s recommendations. Two days post-transfection, AT1R expression was induced with doxycycline (4 μg/mL, 5 mM sodium butyrate) in the presence of the low-affinity AT1R antagonist losartan (1 μM). Cells were harvested approximately 30 hours post-transfection, washed with cold phosphate-buffered saline containing 1 μM losartan, flash frozen with liquid nitrogen, and stored at −80 °C.
Cell pellets were subjected to hypotonic lysis (10 mM Tris pH 7.4, 2 mM EDTA, 10 mM MgCl2, 1 μM losartan, 2.5 U/mL benzonase (Sigma), protease inhibitors benzamidine and leupeptin) before solubilization with lauryl maltose neopentyl glycol (MNG) (0.5%, 0.05% cholesterol hemisuccinate (CHS), 20 mM HEPES pH 7.4, 500 mM NaCl, 10 mM MgCl2, 1 μM losartan, benzonase, benzamidine, leupeptin) for two hours at 4 °C. Following the addition of 2 mM CaCl2, the solubilized material was loaded onto M1 anti-FLAG resin over the course of 1-2 hours at 4 °C. The resin was washed with 20 column volumes of cold wash buffer (20 mM HEPES pH 7.4, 500 mM NaCl, 0.01% MNG, 0.01% CHS, benzamidine, leupeptin) supplemented with 2mM CaCl2 and eluted in the same buffer supplemented with 5 mM EDTA and 0.2 mg/mL FLAG peptide. For DEER experiments, receptor was eluted from M1 resin with pH 6.8 buffer and labeled with a 10-fold molar excess of bis-(2,2,5,5-tetramethyl-3-imidazoline-1-oxyl-4-yl)disulfide (Enzo) relative to the number of free cysteine residues for 3 hours at room temperature. The receptor was then purified away from free spin label and any aggregated material and exchanged into HNM buffer (20 mM HEPES pH 7.4, 100 mM NaCl, 0.01% MNG, and 0.001% CHS) using size exclusion chromatography (SEC) (Superdex 200 Increase 10/300 GL column, GE).
DEER experiments
For DEER experiments, SEC buffer was prepared with D2O (Cambridge Isotopes) and a higher concentration of HEPES (40 mM), and the spin-labeled AT1R was concentrated to approximately 250 μM. Binding reactions consisted of 150-200 μM AT1R, 1 mM ligand (peptide ligands dissolved in water, candesartan and olmesartan dissolved in DMSO), 20% d8-glycerol (Cambridge Isotopes), and 2% DMSO (added to all samples achieve the same final buffer composition) in a final volume of 12 μL. After a one-hour incubation at room temperature, samples were flash frozen with liquid nitrogen in 1.4/1.7 mm (i.d./o.d.) borosilicate capillaries (VitroCom, Mountain Lakes, NJ) and stored at −80 °C.
Q-band four pulse DEER measurements were conducted on an Elexsys E580 pulsed EPR spectrometer equipped with an ER5106QT-2 cavity resonator (Bruker, Billerica, MA). During experiments sample temperature was kept at 50 K using a recirculating helium cryo-cooling system (ColdEdge Technologies, Allentown, PA). Observe pulse lengths were: 18-22 ns (π/2) and 36-44 ns (π), as determined by an echo nutation experiment. A linear frequency-swept (chirp) pulse of 50 MHz half-width, generated by an arbitrary waveform generator (Bruker) and applied 70 MHz below the observer frequency, served as pump pulse. All pulses were further amplified via an external 150 W TWT amplifier (Applied Engineering Systems, Fort Worth, TX).
Background correction and model free analysis of dipolar evolution data was performed using LongDistances (version 590 or newer). The program is available via http://www.biochemistry.ucla.edu/biochem/Faculty/Hubbell/software.html. A single smoothness parameter for each spin pair across all ligand conditions was chosen by L-curve criterion.
DiscoverX assays
β-Arrestin endocytosis was measured by the active endocytosis assay (DiscoverX) as described (Strachan et al., 2014) with slight modifications. Wild-type or minimal cysteine versions of pACMV-tetO-Flag-AT1R were transiently transfected into U2OS cells stably expressing β-arrestin2 with an Enzyme Acceptor tag and endosome-localized ProLink tag protein. On the following day, transfected cells were plated at a density of 35,000 cells per well. Two days post-transfection, cells were treated with a serial dilution of AngII in duplicate for three hours at 37 °C. Following the addition of reagents from the PathHunter Detection kit (DiscoverX) and an additional one hour incubation at 27 °C, chemiluminescence arising from the complementation of β-galactosidase fragments (Enzyme Acceptor and ProLink) within endosomes due to β-arrestin endocytosis was detected on a NOVOstar plate reader (BMG Labtech).
Radioligand binding assays
Purified AT1R (~50 ng) was incubated with varying concentrations of cold competitor ligand in duplicate and [3H]-olmesartan (American Radiolabeled Chemicals) in HNM buffer containing 0.1% bovine serum albumin (BSA) in for 90 minutes at room temperature. The 200 μL binding reactions were harvested onto GF/B filters and rapidly washed with cold 20 mM HEPES pH 7.4, 100 mM NaCl. Alternatively, for experiments performed with the scintillation proximity assay, 20 μL of Protein A yttrium silicate beads (PerkinElmer) coated with M1 anti-FLAG antibody in 20 mM HEPES pH 7.4, 100 mM NaCl, 12 mM CaCl2 were added to 100 μL reactions, which were agitated for 15 minutes at room temperature, centrifuged for 1 minute at 1500 g, and counted on a MicroBeta counter (PerkinElmer). Data were analyzed in GraphPad Prism.
Molecular dynamics simulations
We performed simulations of inactive-state AT1R bound to the crystallographic ligand ZD7155. The structure was prepared by first removing the co-crystallized apocytochrome BRIL from the ZD7155-bound crystal structure (PDB: 4YAY). Prime (Schrödinger, Inc.) was used to model in missing side-chains and missing extracellular and intracellular loops. Hydrogen atoms were added, and protein chain termini were capped with neutral acetyl and methylamide groups. In simulations, titratable residues were left in their dominant protonation state at pH 7.0. Asp74 and Asp125 remained deprotonated, in accordance with the observation that in other GPCRs, the residues may be deprotonated in their inactive state and protonated in their active state (Ghanouni et al., 2000). The engineered, shortened amino terminus, present in the crystal structure, was maintained in simulations. Finally, the Dowser program was used to hydrate pockets within the GPCR (Hermans et al., 2003). The ZD7155 tetrazole moiety was deprotonated in both cases, in accordance with its low pKa of 4.9.
The prepared protein structure was aligned on the transmembrane helices to the Orientation of Proteins in Membranes (OPM; (Lomize et al., 2006)) structure of PDB entry 4YAY and then inserted into a pre-equilibrated palmitoyl-oleoyl-phosphatidylcholine (POPC) bilayer using Dabble (Betz, 2017, August 1). Final system dimensions were 77.9 × 79.4 × 92.8 Å, including 130 lipids, ~11,200 waters, 17 sodium ions, and 30 chloride ions.
We used the CHARMM36 parameter set for protein molecules, lipids, and ions, and the CHARMM TIP3P water model for waters (Huang and MacKerell, 2013; Klauda et al., 2010). Parameters for ZD7155 were generated using the CHARMM General Force Field (CGenFF) with the ParamChem server (Vanommeslaeghe and MacKerell, 2012). Full parameter sets are available upon request. Because CGenFF lacked atom types for the deprotonated form of tetrazole, we obtained partial charges for the nitrogen and carbon atoms within the tetrazole ring, as well as the carbon extending from the tetrazole ring to the biphenyl linker, from SwissParam (Zoete et al., 2011). We confirmed that the final ZD7155 net charge was −1. Across all multi-microsecond simulations, the ligand remained stably bound within the binding pocket and formed persistent contacts with surrounding residues.
Simulations were performed on GPUs using the CUDA version of Particle Mesh Ewald Molecular Dynamics (PMEMD) in Amber16 (Case, 2016; Salomon-Ferrer et al., 2013). Prepared systems were minimized. Equilibration was performed by heating the system with the Langevin thermostat from 0 to 100 K in the NVT ensemble over 12.5 ps with harmonic restraints of 10.0 kcal·mol−1·Å−2 on the non-hydrogen atoms of lipid, protein, and ligand, and initial velocities sampled from the Boltzman distribution. The system was then heated to 310 K over 125 ps in the NPT ensemble with anisotropic pressure coupling. Further equilibration was performed at 310 K with harmonic restraints on the protein and ligand starting at 5.0 kcal·mol−1·Å−2 and reduced by 1.0 kcal·mol−1·Å−2 in a stepwise fashion every 2 ns for 10 ns and then by 0.1 kcal·mol−1·Å−2 in a stepwise fashion every 2 ns for 20 ns, for a total of 30 ns of additional restrained equilibration. Production simulations, each of 5.0 μs in length, were performed at 310 K and 1 bar in the NPT ensemble using the Langevin thermostat and Monte Carlo barostat.
Simulations were performed with hydrogen mass partitioning (Hopkins et al., 2015) and a time step of 4.0 fs. All bond lengths were constrained by SHAKE. Non-bonded interactions were cut off at 9.0 Å, and long-range electrostatic interactions were computed using the particle mesh Ewald (PME) method with an Ewald coefficient β of approximately 0.31 Å and B-spline interpolation of order 4. The FFT grid size was chosen such that the width of a grid cell was approximately 1 Å.
Snapshots from each trajectory were saved every 200 ps during the production phase of each simulation and visualized with VMD (Humphrey et al., 1996). To generate Figure 2C, we sampled frames every 10 ns during the last 1 μs of simulation for four of the five simulations. We omitted one simulation in which helix 8 exhibited extremely heterogeneous behavior. We used the MMM software to attach V1 spin labels to each of the six sites in every sampled simulation frame, allowing us to predict the distribution of spin labels based on MD simulation. Rotamer libraries like the one used here overestimate the number of conformations the spin label might adopt, and we display only the most probable spin label nitroxide oxygen position in Figure 2C, with the caveat that we do not have experimental data on which rotameric state occurs most frequently in our particular samples. To generate Figure S4E, we used Chimera’s fitmap command using fixed atom positions to calculate the overlap between the DEER cloud generated through triangulation and each of the three highest probability V1 rotamers, A–C, modeled onto each site on the crystal structure and a subset of simulation frames (sampled every 100 ns during the last 1 μs of simulation for the same simulations displayed in Figure 2C). For each site in Figure S4D, the probability of the three highest probability spin label rotamers are as follows. TM1: A (45%), B (9%), C (6%); ICL2: A (18%), B (12%), C (8%); TM5: A (21%), B (14%), C (8%); TM6: A (96%), B (4%); TM7: A (63%), B (14%), C (11%); H8: A (21%), B (20%), C (10%). Despite the fact that the simulations may not have explored the full conformational space available to the protein, they populate conformations in which attached V1 spin labels sample positions that could explain the locations of the labels predicted by DEER. All simulation trajectories are available upon request.
NNMF analysis
NNMF analysis was performed on the complete data set of DEER distance distributions using the Matlab built-in routines (1000 replicates). The NNMF components and amplitudes were normalized for comparison. For distance mapping we generally selected the most intense peak from the NNMF-determined distance distributions (cf. Figure S5A). Due to partial structural decoupling upon TM6 outward movement and potential unequal effects of site-specific mutations and spin labeling on the conformational equilibrium (Kusnetzow et al., 2006), higher order NNMF components in particular may be incompletely separated and thus contain considerable admixtures of other NNMF component peaks. During mapping of the NNMF III and IV conformations three distance distributions gave internally inconsistent results when the most intense peak was selected for mapping. In those cases each of the distance peaks was picked one at a time, and the χ2 value of the resulting conformation (involving all 10 distance distributions) was calculated. The peak with the lowest χ2 value (in all three cases the second most intense peak) was used for mapping of the internally consistent conformation.
Mapping of AT1R conformations
To visualize and to be able compare the different conformations we calculated actual spin density clouds by trilateration using the recorded distance distributions in LabVIEW (Altenbach et al., 2008). Briefly, the procedure comprises three steps:
1) Spin label starting positions: The native residues at position 55, 139, 220, 236, 304 and 311 of two reference structures (4YAY or selected MD simulation frames, see above) were exchanged for a cysteine with attached V1 spin label side chain using the program MMM (http://www.epr.ethz.ch/software.html). For each labeling site, one of the three highest probability MMM-generated rotamers was picked so that the inter-nitroxide distances approximately reproduced the main distance peaks of the candesartan distance distributions.
2) Refinement of spin label positions: An optimization of the nitroxide coordinates was performed to achieve better agreement with the DEER distance distributions. During this step, the positions 55V1 (TM1) and 236V1 (TM6) were kept in place since MD simulations indicated the least deviation from the crystal structure at those sites. All other label positions were allowed to move freely in the membrane plane to match the dominant inter-spin distance from the respective distance distribution. After the first refinement the best fit coordinates were used as starting coordinates for a second refinement, leaving 311V1 free to move in all three dimensions.
The MD simulation label positions refined for the candesartan distance distributions were used as starting coordinates for NNMF I, NNMF II and NNMF IV with 55V1 and 139V1 fixed. Due to the large change at site 139V1, we used 55V1 and 236V1 of the NNMF IV refined coordinates as starting coordinates for NNMF III refinement.
3) Spin density mapping: The spin density cloud of a specific spin label is the product of all probability distributions involving this label, keeping the coordinates of each second spin label fixed (after refinement). A two-dimensional map was generated and visualized in NGL viewer (Rose and Hildebrand, 2015) using surface or slice representation (z-coordinate of the refined label position).
For the comparison to the Nb-stabilized AT1R crystal structure (Figure 5E) (Wingler et al., co-submitted), the Aopen map was manually aligned in the membrane plane until it matched the position of MMM-generated, high probability V1 rotamers.
Quantification and Statistical Analysis
Radioligand binding data were fit to a one-site model in GraphPad Prism. Error is reported or shown as the standard error of the mean of the values calculated in at least three independent experiments. β-Arrestin endocytosis data were fit to a sigmoidal dose response curve in GraphPad Prism; the standard error of the mean of the values calculated in at least three independent experiments is shown.
To avoid overfitting our data during NNMF analysis we determined the corrected Akaike information criterion (AICc) using one to six NNMF factors. The AICc values showed a broad minimum at 3 to 4 components (Figure S5B). Inspection of the reconstructed data set revealed that four components were required to recreate the characteristic bands specific to G protein-biased ligands (Figure S5C). Using only three components clearly failed to account for these features.
Data and Software Availability
The accession number for the raw DEER spectroscopy data reported in this paper is Mendeley Data: (https://doi.org/10.17632/xnv5k5vb85.1).
Supplementary Material
Figure S1. Characterization of Minimal Cysteine and Spin-labeled AT1R Constructs. Related to Figure 1, Table S1, and Table S2.
(A) PathHunter β-arrestin endocytosis assay with wild-type (log EC50 = −8.37 ± 0.06) and minimal cysteine AT1R (log EC50 = −7.57 ± 0.09). Data are normalized to the maximum AngII-stimulated signal, and error bars represent the standard error of the mean from three independent experiments.
(B and C) Competition [3H]-olmesartan radioligand binding with purified wild-type and minimal cysteine AT1R captured on GF/B filters. (B) AngII competition binding curves. (C) Log Ki values for the biased ligand panel show the minimal cysteine AT1R mutations affect the binding of all ligands proportionally. The solid line is a linear regression of the data, and the dashed line is the line of unity. Error bars represent the standard error of the mean of three independent experiments. Log Ki values are tabulated in Table S1.
(D and E) Representative purifications of two spin-labeled AT1R constructs, (C) 139V1 311V1 (ICL2-H8) and (D) 55V1 236V1 (TM1-TM6). Coomassie-stained gels showing receptor purity at each step and size exclusion chromatography traces are shown. The broad migration of purified AT1R is due to its three N-linked glycosylation sites.
(F) Competition [3H]-olmesartan radioligand binding (scintillation proximity assay) with the ten purified, spin-labeled AT1R constructs used for DEER measurements. Error bars represent the standard error of the mean of at least three independent experiments. Log IC50 values are provided in Table S2.
Figure S2. DEER Data. Related to Figure 2.
Complete panel of dipolar evolutions for all spin label pairs and ligand conditions. The raw (background corrected) data are shown in gray underneath the colored best fit lines (model-free fit smoothness parameter was 3 for all fits except for ICL2-TM5, where 1 was chosen to account for all features in the dipolar evolution).
Figure S3. DEER Data Analysis. Related to Figure 2 and Figure 3.
Distance distributions for the complete panel of spin label pairs and ligand conditions, calculated from the model-free fits of DEER data shown in Figure S2
Figure S4. Conformational Heterogeneity of the AT1R based on MD simulations. Related to Figure 2.
(A-C) Molecular dynamics simulations of the AT1R bound to ZD7155, starting with the 4YAY crystal structure (gray). Simulation frames sampled every 0.1 μs during the last 1 μs of simulation are aligned on TMs 1-4, and the conformations of native residue side chains located at the spin label sites used in this study (cf. Figure 1B) are displayed as sticks. Each image corresponds to data from a single, representative simulation.
(A) In the course of these simulations, the intracellular end of TM4 loses helicity, and ICL2 shows a great deal of conformational heterogeneity. (Loss of helicity seen in two out of five simulations with ZD7155.)
(B) The top of TM5 sometimes moves inward to contact the ligand, while the base of TM5 moves away from the helical bundle. (Intracellular outward motion of TM5 occurred in four out of five simulations with ZD7155.)
(C) A helical turn in TM7 (near residues 299-303) straightens, while helix 8 populates multiple states. (These motions occurred in four out of five simulations with ZD7155.) For (A-C), once these changes took place, they persisted on time scales of several hundred nanoseconds to microseconds.
(D) High probability V1 spin label rotamers modeled onto the crystal structure reveal only minimal overlap with DEER-generated clouds at several labeling sites. Spheres indicate the position of the nitroxide oxygen atoms for each of the three highest probability rotamers attached to each site. Left: the DEER clouds displayed at a relaxed, inclusive contour. Right: DEER clouds displayed at a tightened contour, corresponding to the darker shaded regions of higher probability density in Figures 2B and 2C.
(E) Fraction of MD simulation frames (n = 40) in which modeled V1 spin label rotamers overlap with either the relaxed (darker bars) or tightened (lighter bars) DEER clouds at each site. The left three bars correspond to the fraction of frames containing either rotamers A, B or C, while the right-most bar corresponds to the fraction of frames containing any one of those three rotamers. For all sites, at least a subset of simulation frames possesses a modeled V1 spin label rotamer whose nitroxide oxygen atom is contained within the volume of the DEER-generated cloud, indicating that conformations among those captured in simulation can explain the experimentally observed DEER conformations.
Figure S5. Non-Negative Matrix Factorization (NNMF) Analysis of AT1R DEER Data. Related to Figure 4, Figure 5, and Table S1.
(A) Calculated distance distributions for the four NNMF components. Gaussian peaks indicate distances employed for mapping of the representative AT1R conformations.
(B) Plot of Akaike information criterion (corrected for small sample size, AICc) values for NNMF analysis of the complete data set of DEER distance distributions.
(C) Reconstruction of the indicated distance distributions for Gq-biased ligands using either 3 or 4 components for NNMF.
(D) Relationship between ligand molecular efficacy in Gq coupling (αGq) and β-arrestin coupling (αβarr) (Strachan et al., 2014) and NNMF component populations (see Figure 4B). TRV027 was not included in the previous study of molecular efficacy. Error bars represent standard error of α values determined in three independent experiments.
(E) Relationship between the structures of AngII analogs used in this study (shown as points, cf. Figure 3A) and the intracellular AT1R conformations stabilized (shown in colored regions, cf. Figures 4B and 5A-5D). AngII has an arginine at position 2 and phenylalanine at position 8 and stabilizes a mixture of Aoccl2 and Aopen. When Phe8 is retained, repositioning Arg2 by an insertion mutation (TRV056) or replacing Arg2 with glycine (TRV055) stabilizes only Aopen. In ligands with Arg2, reducing the size of residue 8’s side chain to a methyl stabilizes only Aoccl2 (TRV034 and TRV023), inverting its stereochemistry stabilizes a mixture of Aoccl1 and Aoccl2 (TRV027), and deleting residue 8 stabilizes Aoccl1 exclusively (TRV026).
Highlights.
Intracellular conformational changes of the angiotensin receptor monitored by DEER.
Different functional classes of ligands stabilize distinct sets of conformations.
Gq- and β-arrestin-biased ligands have opposing effects relative to angiotensin II.
Maps of conformations suggest a structural basis for ligands’ biased signaling.
Acknowledgments
We would like to thank Victoria Ronk and Quivetta Lennon for administrative assistance; Aashish Manglik, Henry Chiou, and Jon Zmuda for helpful discussion; Christian Altenbach for his help with distance mapping; and Raphael Townshend, Scott Hollingsworth, Evan Antoniuk, and A.J. Venkatakrishnan for assistance with MD simulations and analysis. We thank Gunnar Jeschke and Yevhen Polyhach for the development of the V1 rotamer library and its implementation into the MMM software package. Support was provided by the National Institutes of Health grants R01GM127359 (R.O.D), R01EY005216 (W.L.H), P30EY00331 (W.L.H), and R01HL16037 (R.J.L.); German Research Foundation (DFG) research fellowship EL779 (M.E.); the German Academic Exchange Service (DAAD) (D.H.); the Jules Stein Professorship Endowment (W.L.H.); and the Mandel Center for Hypertension and Atherosclerosis at Duke (R.J.L.). R.J.L. is an investigator with the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
B.K.K. is a cofounder of and consultant for ConfometRx, Inc. R.J.L. is a founder and stockholder of Trevena, Inc., and is a director of Lexicon Pharmaceuticals.
References
- Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, and Hubbell WL (2008). High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc. Natl. Acad. Sci. U. S. A 105, 7439–7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aumelas A, Sakarellos C, Lintner K, Fermandjian S, Khosla MC, Smeby RR, and Bumpus FM (1985). Studies on angiotensin II and analogs: impact of substitution in position 8 on conformation and activity. Proc. Natl. Acad. Sci. U. S. A 82, 1881–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balo AR, Feyrer H, and Ernst OP (2016). Toward precise interpretation of DEER-based distance distributions: insights from structural characterization of V1 spin-labeled side chains. Biochemistry 55, 5256–5263. [DOI] [PubMed] [Google Scholar]
- Betz RM (2017, August 1). Dabble (Version v2.6.3). Zenodo. 10.5281/zenodo.836914.. [DOI] [Google Scholar]
- Case DA, et al. (2016). AMBER 2016 (University of California, San Francisco: ). [Google Scholar]
- Cotter G, Davison BA, Butler J, Collins SP, Ezekowitz JA, Felker GM, Filippatos G, Levy PD, Metra M, Ponikowski P, et al. (2018). Relationship between baseline systolic blood pressure and long-term outcomes in acute heart failure patients treated with TRV027: an exploratory subgroup analysis of BLAST-AHF. Clin. Res. Cardiol 107, 170–181. [DOI] [PubMed] [Google Scholar]
- Devost D, Sleno R, Petrin D, Zhang A, Shinjo Y, Okde R, Aoki J, Inoue A, and Hebert TE (2017). Conformational profiling of the AT1 angiotensin II receptor reflects biased agonism, G protein coupling, and cellular context. J. Biol. Chem 292, 5443–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domazet I, Holleran BJ, Richard A, Vandenberghe C, Lavigne P, Escher E, Leduc R, and Guillemette G (2015). Characterization of angiotensin II molecular determinants involved in AT1 receptor functional selectivity. Mol. Pharmacol 87, 982–995. [DOI] [PubMed] [Google Scholar]
- Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, Borhani DW, and Shaw DE (2011). Activation mechanism of the beta2-adrenergic receptor. Proc. Natl. Acad. Sci. U. S. A 108, 18684–18689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgeti M, Kazmin R, Rose AS, Szczepek M, Hildebrand PW, Bartl FJ, Scheerer P, and Hofmann KP (2018). The arrestin-1 finger loop interacts with two distinct conformations of active rhodopsin. J. Biol. Chem 293, 4403–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JF, and Farrens DL (2015). Structural dynamics and energetics underlying allosteric inactivation of the cannabinoid receptor CB1. Proc. Natl. Acad. Sci. U. S. A 112, 8469–8474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng YH, Noda K, Saad Y, Liu XP, Husain A, and Karnik SS (1995). The docking of Arg2 of angiotensin II with Asp281 of AT1 receptor is essential for full agonism. J. Biol. Chem 270, 12846–12850. [DOI] [PubMed] [Google Scholar]
- Ghanouni P, Schambye H, Seifert R, Lee TW, Rasmussen SG, Gether U, and Kobilka BK (2000). The effect of pH on beta(2) adrenoceptor function. Evidence for protonation-dependent activation. J. Biol. Chem 275, 3121–3127. [DOI] [PubMed] [Google Scholar]
- Ghanouni P, Steenhuis JJ, Farrens DL, and Kobilka BK (2001). Agonist-induced conformational changes in the G-protein-coupling domain of the beta 2 adrenergic receptor. Proc. Natl. Acad. Sci. U. S. A 98, 5997–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier S, Kim S, Shafer AM, Ratnala VR, Fung JJ, Zare RN, and Kobilka B (2007). Structure and conformational changes in the C-terminal domain of the beta2-adrenoceptor: insights from fluorescence resonance energy transfer studies. J. Biol. Chem 282, 13895–13905. [DOI] [PubMed] [Google Scholar]
- Gregorio GG, Masureel M, Hilger D, Terry DS, Juette M, Zhao H, Zhou Z, Perez-Aguilar JM, Hauge M, Mathiasen S, et al. (2017). Single-molecule analysis of ligand efficacy in beta2AR-G-protein activation. Nature 547, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans J, Xia X, Zhang L, Cavanaugh, and Dowser D (2003). Dowser program. [Google Scholar]
- Ho JD, Chau B, Rodgers L, Lu F, Wilbur KL, Otto KA, Chen Y, Song M, Riley JP, Yang H-C, et al. (2018). Structural basis for GPR40 allosteric agonism and incretin stimulation. Nat. Commun 9, 1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway AC, Qian H, Pipolo L, Ziogas J, Miura S, Karnik S, Southwell BR, Lew MJ, and Thomas WG (2002). Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol. Pharmacol 61, 768–777. [DOI] [PubMed] [Google Scholar]
- Hopkins CW, Le Grand S, Walker RC, and Roitberg AE (2015). Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput 11, 1864–1874. [DOI] [PubMed] [Google Scholar]
- Huang J, and MacKerell AD Jr. (2013). CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: visual molecular dynamics. J. Mol. Graph 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Jeschke G (2012). DEER distance measurements on proteins. Annu. Rev. Phys. Chem 63, 419–446. [DOI] [PubMed] [Google Scholar]
- Kahsai AW, Xiao K, Rajagopal S, Ahn S, Shukla AK, Sun J, Oas TG, and Lefkowitz RJ (2011). Multiple ligand-specific conformations of the beta2-adrenergic receptor. Nat. Chem. Biol 7, 692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, et al. (2015). Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O'Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr., and Pastor RW (2010). Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusnetzow AK, Altenbach C, and Hubbell WL (2006). Conformational states and dynamics of rhodopsin in micelles and bilayers. Biochemistry 45, 5538–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane JR, May LT, Parton RG, Sexton PM, and Christopoulos A (2017). A kinetic view of GPCR allostery and biased agonism. Nat. Chem. Biol 13, 929–937. [DOI] [PubMed] [Google Scholar]
- Latorraca NR, Venkatakrishnan AJ, and Dror RO (2017). GPCR dynamics: structures in motion. Chem. Rev 117, 139–155. [DOI] [PubMed] [Google Scholar]
- Latorraca NR, Wang JK, Bauer B, Townshend RJL, Hollingsworth SA, Olivieri JE, Xu HE, Sommer ME, and Dror RO (2018). Molecular mechanism of GPCR-mediated arrestin activation. Nature 557, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ (2013). A brief history of G-protein coupled receptors (Nobel Lecture). Angew. Chem. Int. Ed. Engl 52, 6366–6378. [DOI] [PubMed] [Google Scholar]
- Liang YL, Khoshouei M, Glukhova A, Furness SGB, Zhao P, Clydesdale L, Koole C, Truong TT, Thal DM, Lei S, et al. (2018). Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature 555, 121–125. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Horst R, Katritch V, Stevens RC, and Wuthrich K (2012). Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science 335, 1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize MA, Lomize AL, Pogozheva ID, and Mosberg HI (2006). OPM: orientations of proteins in membranes database. Bioinformatics 22, 623–625. [DOI] [PubMed] [Google Scholar]
- Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, et al. (2015). Structural insights into the dynamic process of beta2-adrenergic receptor signaling. Cell 161, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, and Kruse AC (2017). Structural basis for G protein-coupled receptor activation. Biochemistry 56, 5628–5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda K, Saad Y, Kinoshita A, Boyle TP, Graham RM, Husain A, and Karnik SS (1995). Tetrazole and carboxylate groups of angiotensin receptor antagonists bind to the same subsite by different mechanisms. J. Biol. Chem 270, 2284–2289. [DOI] [PubMed] [Google Scholar]
- Okude J, Ueda T, Kofuku Y, Sato M, Nobuyama N, Kondo K, Shiraishi Y, Mizumura T, Onishi K, Natsume M, et al. (2015). Identification of a conformational equilibrium that determines the efficacy and functional selectivity of the mu-opioid receptor. Angew. Chem. Int. Ed. Engl 54, 15771–15776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paatero P, and Tapper U (1994). Positive matrix factorization - a nonnegative factor model with optimal utilization of error-estimates of data values. Environmetrics 5, 111–126. [Google Scholar]
- Pack TF, Orlen MI, Ray C, Peterson SM, and Caron MG (2018). The dopamine D2 receptor can directly recruit and activate GRK2 without G protein activation. J. Biol. Chem 293, 6161–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher JA, Inglese J, Higgins JB, Arriza JL, Casey PJ, Kim C, Benovic JL, Kwatra MM, Caron MG, and Lefkowitz RJ (1992). Role of beta gamma subunits of G proteins in targeting the beta-adrenergic receptor kinase to membrane-bound receptors. Science 257, 1264–1267. [DOI] [PubMed] [Google Scholar]
- Polyhach Y, Bordignon E, and Jeschke G (2011). Rotamer libraries of spin labelled cysteines for protein studies. Phys. Chem. Chem. Phys 13, 2356–2366. [DOI] [PubMed] [Google Scholar]
- Pulvermuller A, Palczewski K, and Hofmann KP (1993). Interaction between photoactivated rhodopsin and its kinase: stability and kinetics of complex formation. Biochemistry 32, 14082–14088. [DOI] [PubMed] [Google Scholar]
- Rahmeh R, Damian M, Cottet M, Orcel H, Mendre C, Durroux T, Sharma KS, Durand G, Pucci B, Trinquet E, et al. (2012). Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc. Natl. Acad. Sci. U. S. A 109, 6733–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal K, Whalen EJ, Violin JD, Stiber JA, Rosenberg PB, Premont RT, Coffman TM, Rockman HA, and Lefkowitz RJ (2006). Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc. Natl. Acad. Sci. U. S. A 103, 16284–16289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S, Ahn S, Rominger DH, Gowen-MacDonald W, Lam CM, Dewire SM, Violin JD, and Lefkowitz RJ (2011). Quantifying ligand bias at seven-transmembrane receptors. Mol. Pharmacol 80, 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankovic Z, Brust TF, and Bohn LM (2016). Biased agonism: an emerging paradigm in GPCR drug discovery. Bioorg. Med. Chem. Lett 26, 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves PJ, Kim JM, and Khorana HG (2002). Structure and function in rhodopsin: a tetracycline-inducible system in stable mammalian cell lines for high-level expression of opsin mutants. Proc. Natl. Acad. Sci. U. S. A 99, 13413–13418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AS, and Hildebrand PW (2015). NGL Viewer: a web application for molecular visualization. Nucleic Acids Res. 43, W576–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba DM, Li J, Cowan CL, Russell B, Wolska BM, and Solaro RJ (2017). Long-term biased beta-arrestin signaling improves cardiac structure and function in dilated cardiomyopathy. Circulation 135, 1056–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon-Ferrer R, Gotz AW, Poole D, Le Grand S, and Walker RC (2013). Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh ewald. J. Chem. Theory Comput 9, 3878–3888. [DOI] [PubMed] [Google Scholar]
- Scragg JL, Warburton P, Ball SG, and Balmforth AJ (2005). Constitutive activity of human angiotensin II type-1 receptors by Gq overexpression. Biochem. Biophys. Res. Commun 334, 134–139. [DOI] [PubMed] [Google Scholar]
- Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, et al. (2014). Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 512, 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solt AS, Bostock MJ, Shrestha B, Kumar P, Warne T, Tate CG, and Nietlispach D (2017). Insight into partial agonism by observing multiple equilibria for ligand-bound and Gs-mimetic nanobody-bound beta1-adrenergic receptor. Nat. Commun 8, 1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sounier R, Mas C, Steyaert J, Laeremans T, Manglik A, Huang W, Kobilka BK, Demene H, and Granier S (2015). Propagation of conformational changes during mu-opioid receptor activation. Nature 524, 375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Wingler LM, Choi M, Pani B, Manglik A, Kruse AC, and Lefkowitz RJ (2018). Sortase ligation enables homogeneous GPCR phosphorylation to reveal diversity in beta-arrestin coupling. Proc. Natl. Acad. Sci. U. S. A 115, 3834–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan RT, Sun JP, Rominger DH, Violin JD, Ahn S, Rojas Bie Thomsen A, Zhu X, Kleist A, Costa T, and Lefkowitz RJ (2014). Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR). J. Biol. Chem 289, 14211–14224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eps N, Caro LN, Morizumi T, Kusnetzow AK, Szczepek M, Hofmann KP, Bayburt TH, Sligar SG, Ernst OP, and Hubbell WL (2017). Conformational equilibria of light-activated rhodopsin in nanodiscs. Proc. Natl. Acad. Sci. U. S. A 114, E3268–E3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Liefde I, and Vauquelin G (2009). Sartan-AT1 receptor interactions: in vitro evidence for insurmountable antagonism and inverse agonism. Mol. Cell. Endocrinol 302, 237–243. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K, and MacKerell AD Jr. (2012). Automation of the CHARMM general force field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model 52, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardaga JP, Frank M, Krasel C, Dees C, Nissenson RA, and Lohse MJ (2001). Differential conformational requirements for activation of G proteins and the regulatory proteins arrestin and G protein-coupled receptor kinase in the G protein-coupled receptor for parathyroid hormone (PTH)/PTH-related protein. J. Biol. Chem 276, 33435–33443. [DOI] [PubMed] [Google Scholar]
- Violin JD, DeWire SM, Yamashita D, Rominger DH, Nguyen L, Schiller K, Whalen EJ, Gowen M, and Lark MW (2010). Selectively engaging beta-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J. Pharmacol. Exp. Ther 335, 572–579. [DOI] [PubMed] [Google Scholar]
- Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, et al. (2013). Structural features for functional selectivity at serotonin receptors. Science 340, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Wang S, McCorvy JD, Betz RM, Venkatakrishnan AJ, Levit A, Lansu K, Schools ZL, Che T, Nichols DE, et al. (2017). Crystal structure of an LSD-bound human serotonin receptor. Cell 168, 377–389 e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshaviak DT, Khramtsov VV, Cascio D, Altenbach C, and Hubbell WL (2013). Structure and dynamics of an imidazoline nitroxide side chain with strongly hindered internal motion in proteins. J. Magn. Reson 232, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingler LM, McMahon C, Staus DP, Lefkowitz RJ, and Kruse AC (2019) Distinctive angiotensin receptor activation mechanism revealed by synthetic nanobody. Cell 176, 479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Van Eps N, Zimmer M, Ernst OP, and Prosser RS (2016). Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 533, 265–268. [DOI] [PubMed] [Google Scholar]
- Zhang H, Unal H, Gati C, Han GW, Liu W, Zatsepin NA, James D, Wang D, Nelson G, Weierstall U, et al. (2015). Structure of the angiotensin receptor revealed by serial femtosecond crystallography. Cell 161, 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoete V, Cuendet MA, Grosdidier A, and Michielin O (2011). SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem 32, 2359–2368. [DOI] [PubMed] [Google Scholar]
- Zurn A, Zabel U, Vilardaga JP, Schindelin H, Lohse MJ, and Hoffmann C (2009). Fluorescence resonance energy transfer analysis of alpha 2a-adrenergic receptor activation reveals distinct agonist-specific conformational changes. Mol. Pharmacol 75, 534–541. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Characterization of Minimal Cysteine and Spin-labeled AT1R Constructs. Related to Figure 1, Table S1, and Table S2.
(A) PathHunter β-arrestin endocytosis assay with wild-type (log EC50 = −8.37 ± 0.06) and minimal cysteine AT1R (log EC50 = −7.57 ± 0.09). Data are normalized to the maximum AngII-stimulated signal, and error bars represent the standard error of the mean from three independent experiments.
(B and C) Competition [3H]-olmesartan radioligand binding with purified wild-type and minimal cysteine AT1R captured on GF/B filters. (B) AngII competition binding curves. (C) Log Ki values for the biased ligand panel show the minimal cysteine AT1R mutations affect the binding of all ligands proportionally. The solid line is a linear regression of the data, and the dashed line is the line of unity. Error bars represent the standard error of the mean of three independent experiments. Log Ki values are tabulated in Table S1.
(D and E) Representative purifications of two spin-labeled AT1R constructs, (C) 139V1 311V1 (ICL2-H8) and (D) 55V1 236V1 (TM1-TM6). Coomassie-stained gels showing receptor purity at each step and size exclusion chromatography traces are shown. The broad migration of purified AT1R is due to its three N-linked glycosylation sites.
(F) Competition [3H]-olmesartan radioligand binding (scintillation proximity assay) with the ten purified, spin-labeled AT1R constructs used for DEER measurements. Error bars represent the standard error of the mean of at least three independent experiments. Log IC50 values are provided in Table S2.
Figure S2. DEER Data. Related to Figure 2.
Complete panel of dipolar evolutions for all spin label pairs and ligand conditions. The raw (background corrected) data are shown in gray underneath the colored best fit lines (model-free fit smoothness parameter was 3 for all fits except for ICL2-TM5, where 1 was chosen to account for all features in the dipolar evolution).
Figure S3. DEER Data Analysis. Related to Figure 2 and Figure 3.
Distance distributions for the complete panel of spin label pairs and ligand conditions, calculated from the model-free fits of DEER data shown in Figure S2
Figure S4. Conformational Heterogeneity of the AT1R based on MD simulations. Related to Figure 2.
(A-C) Molecular dynamics simulations of the AT1R bound to ZD7155, starting with the 4YAY crystal structure (gray). Simulation frames sampled every 0.1 μs during the last 1 μs of simulation are aligned on TMs 1-4, and the conformations of native residue side chains located at the spin label sites used in this study (cf. Figure 1B) are displayed as sticks. Each image corresponds to data from a single, representative simulation.
(A) In the course of these simulations, the intracellular end of TM4 loses helicity, and ICL2 shows a great deal of conformational heterogeneity. (Loss of helicity seen in two out of five simulations with ZD7155.)
(B) The top of TM5 sometimes moves inward to contact the ligand, while the base of TM5 moves away from the helical bundle. (Intracellular outward motion of TM5 occurred in four out of five simulations with ZD7155.)
(C) A helical turn in TM7 (near residues 299-303) straightens, while helix 8 populates multiple states. (These motions occurred in four out of five simulations with ZD7155.) For (A-C), once these changes took place, they persisted on time scales of several hundred nanoseconds to microseconds.
(D) High probability V1 spin label rotamers modeled onto the crystal structure reveal only minimal overlap with DEER-generated clouds at several labeling sites. Spheres indicate the position of the nitroxide oxygen atoms for each of the three highest probability rotamers attached to each site. Left: the DEER clouds displayed at a relaxed, inclusive contour. Right: DEER clouds displayed at a tightened contour, corresponding to the darker shaded regions of higher probability density in Figures 2B and 2C.
(E) Fraction of MD simulation frames (n = 40) in which modeled V1 spin label rotamers overlap with either the relaxed (darker bars) or tightened (lighter bars) DEER clouds at each site. The left three bars correspond to the fraction of frames containing either rotamers A, B or C, while the right-most bar corresponds to the fraction of frames containing any one of those three rotamers. For all sites, at least a subset of simulation frames possesses a modeled V1 spin label rotamer whose nitroxide oxygen atom is contained within the volume of the DEER-generated cloud, indicating that conformations among those captured in simulation can explain the experimentally observed DEER conformations.
Figure S5. Non-Negative Matrix Factorization (NNMF) Analysis of AT1R DEER Data. Related to Figure 4, Figure 5, and Table S1.
(A) Calculated distance distributions for the four NNMF components. Gaussian peaks indicate distances employed for mapping of the representative AT1R conformations.
(B) Plot of Akaike information criterion (corrected for small sample size, AICc) values for NNMF analysis of the complete data set of DEER distance distributions.
(C) Reconstruction of the indicated distance distributions for Gq-biased ligands using either 3 or 4 components for NNMF.
(D) Relationship between ligand molecular efficacy in Gq coupling (αGq) and β-arrestin coupling (αβarr) (Strachan et al., 2014) and NNMF component populations (see Figure 4B). TRV027 was not included in the previous study of molecular efficacy. Error bars represent standard error of α values determined in three independent experiments.
(E) Relationship between the structures of AngII analogs used in this study (shown as points, cf. Figure 3A) and the intracellular AT1R conformations stabilized (shown in colored regions, cf. Figures 4B and 5A-5D). AngII has an arginine at position 2 and phenylalanine at position 8 and stabilizes a mixture of Aoccl2 and Aopen. When Phe8 is retained, repositioning Arg2 by an insertion mutation (TRV056) or replacing Arg2 with glycine (TRV055) stabilizes only Aopen. In ligands with Arg2, reducing the size of residue 8’s side chain to a methyl stabilizes only Aoccl2 (TRV034 and TRV023), inverting its stereochemistry stabilizes a mixture of Aoccl1 and Aoccl2 (TRV027), and deleting residue 8 stabilizes Aoccl1 exclusively (TRV026).
Data Availability Statement
The accession number for the raw DEER spectroscopy data reported in this paper is Mendeley Data: (https://doi.org/10.17632/xnv5k5vb85.1).