

Abstract
Clostridium difficile is a deadly, opportunistic bacterial pathogen. In the last two decades, C. difficile infections (CDIs) have become a national concern due to the emergence of hypervirulent mutants armed with a higher capability of producing toxins and spores. This has resulted in an increased number of infections and death associated with CDI. The scarcity of anticlostridial drugs has led to unsatisfactory cure rates, elevated recurrence rates and permitted enhanced colonization with other drug-resistant pathogens (such as vancomycin-resistant enterococci), in afflicted patients. Therefore, both patients and physicians are facing an urgent need for more effective therapies to treat CDI. In an effort to find new anticlostridial drugs, we investigated auranofin, an FDA-approved oral antirheumatic drug which has recently been found to also possess antibacterial activity. Auranofin exhibited potent activity against C. difficile isolates inhibiting growth at a concentration of 1 μg/ml against 50% of all the tested isolates. Auranofin inhibited both toxin production and spore formation, a property that is lacking in both vancomycin and metronidazole (the primary agents used to treat CDI). Auranofin had a direct protective activity against C. difficile toxin-mediated inflammation and inhibited the growth of vancomycin-resistant enterococci. Overall, auranofin is a promising candidate that warrants further investigation as a treatment option for C. difficile infections.
Keywords: C. difficile infection, anti-toxin, spores formation, auranofin, antibacterial, repurposing
1. INTRODUCTION
Clostridium difficile is the most common hospital-acquired infectious agent [1]. C. difficile infection (CDI), also known as C. difficile associated diarrhea (CDAD), afflicted nearly half a million patients in the United States alone and was associated with over 29,000 deaths in 2011 resulting in a direct cost that exceeds $5 billion (U.S.) annually [2]. Though first discovered in the 1970s, the incidence and severity of CDI has sharply increased over the past two decades [3]. This upsurge in CDI has been attributed to the emergence of hypervirulent strains (e.g. the North American pulsotype 1 (NAP1), PCR-ribotype 027, and restriction endonuclease analysis (REA) group BI 8). These hypervirulent C. difficile strains exhibit enhanced motility and adherence, increased drug-resistance and production of higher levels of toxins (toxins A and B and binary toxin) [1, 4]. C. difficile toxins are the main virulence factor, and they are essential for the bacteria to cause disease Furthermore, C. difficile forms very resistant spores that can persist in the environment for extensive periods of time. These spores serve as the springboard for disease transmission. Moreover, spores can survive in the gastrointestinal tract of infected patients until the cessation of antibiotic treatment, provoking relapse of CDI [5, 6].
Three primary drugs are used to treat CDI - vancomycin, metronidazole and fidaxomicin. Vancomycin and metronidazole have been used for decades with limited efficacy and high recurrence rates [7]. Fidaxomicin is the only anticlostridial drug approved in the past three decades. However, fidaxomicin treatment is not superior to vancomycin treatment with regards to reducing the recurrence rate or in the occurrence of treatment-emergent adverse events. Additionally, reports about C. difficile resistance or reduced susceptibility to metronidazole, vancomycin and to lesser extent fidaxomicin are starting to emerge worldwide [8, 9]. Altogether, there is a critical and imperative need for new anticlostridial drugs with improved treatment outcomes.
Drug repurposing is a promising approach to find new indications for existing or abandoned drugs. Adopting an old drug with a well-studied safety and pharmacokinetic profile can circumvent some of the pitfalls and costs associated with clinical testing and regulatory approval processes for novel compounds [10–12]. Auranofin [2,3,4,6-tetra-o-acetyl-l-thio-β-d-glycopyranp-sato-S-(triethyl-phosphine)-gold] is a gold-containing anti-inflammatory oral drug that has been used for treatment of rheumatoid arthritis for over 30 years. Auranofin’s safety and pharmacokinetic profile in humans has been well-characterized, which has permitted investigation of auranofin for other clinical indications [13, 14]. The present study evaluates auranofin’s potential to be repurposed as a novel anticlostridial drug to treat CDI.
In the current study, we report that auranofin possesses potent antibacterial activity against a wide panel of C. difficile strains. Additionally, auranofin is capable of inhibiting toxin production, spore formation and protects human gut cells against the inflammation induced by sterile C. difficile toxins. Taken all together, auranofin is a promising candidate to evaluate further to treat CDI.
2. MATERIALS AND METHODS
2.1. Chemicals, media and bacterial strains.
Auranofin, linezolid (Chem-Impex International, Wood Dale, IL), vancomycin hydrochloride (Gold Biotechnology, St. Louis, MO) metronidazole (Beantown Chemical Corporation, Hudson, NH), sodium selenite (MP Biomedicals, Santa Ana, CA), and fidaxomicin (Apexbio, Houston, TX) were procured from commercial vendors. Brain heart infusion (BHI) was purchased from Becton, Dickinson and Company (Cockeysville, MD). Phosphate buffered saline, fetal bovine serum and non-essential amino acids (NEAA) were purchased from Fisher Scientific (Waltham, MA). Yeast extract, L-cysteine, vitamin K, hemin, Dulbecco’s Modified Eagle’s medium (DMEM), and penicillin/streptomycin were obtained from Sigma-Aldrich (St. Louis, MO). C. difficile and enterococcal isolates (Table 1) were obtained from the American Type Culture Collection (ATCC) and Biodefense and Emerging Infections Research Resources Repository (BEI Resources).
Table 1.
Bacterial strains used in this study.
| Bacterial Strain | Alternate Designation | Source | Comments |
|---|---|---|---|
| C. difficile NAP07 (CDC#2007054) | HM-88 | Isolated from human feces. | Reference genome for The Human Microbiome Project (HMP). |
| C. difficile P2 | NR-32883 | Isolated from the fecal material of a human patient with CDI in Western Pennsylvania, USA in 2001. | Toxigenic strain. |
| C. difficile P3 | NR-32884 | It was obtained in 2001 from the fecal material of a human patient with CDI in Western Pennsylvania, USA. | Toxigenic strain. |
| C. difficile P4 | NR-32889 | Isolated from the fecal material of a human patient with a relapsing CDI in Western Pennsylvania, USA in 2001. | Toxigenic strain. |
| C. difficile P5 | NR-32885 | It was obtained in 2001 from the fecal material of a human patient with CDI in Western Pennsylvania, USA. | Toxigenic strain. |
| C. difficile P6 | NR-32886 | Isolated from the fecal material of a human patient with a relapsing CDI in Western Pennsylvania, USA in 2001. | Toxigenic strain. |
| C. difficile P7 | NR-32887 | It was obtained in 2001 from the fecal material of a human patient with CDI in Western Pennsylvania, USA. | Toxigenic strain. |
| C. difficile P8 | NR-32888 | Isolated from the fecal material of a human patient with CDI in Western Pennsylvania, USA in 2001. | Toxigenic strain. |
| C. difficile P13 | NR-32891 | Isolated from the fecal material of a human patient with CDI in Western Pennsylvania, USA in 2005. | Toxigenic strain. |
| C. difficile P15 | NR-32892 | Isolated from the fecal material of a human patient with CDI in Western Pennsylvania, USA in 2005. | Toxigenic strain. |
| C. difficile P19 | NR-32895 | It was obtained in 2005 from the fecal material of a human patient with a relapsing C. difficile infection in Western Pennsylvania, USA. | Toxigenic strain. |
| C. difficile P20 | NR-32896 | Isolated from the fecal material of a human patient with a relapsing C. difficile infection in Western Pennsylvania, USA in 2005. | Toxigenic strain. |
| C. difficile P21 | NR-32897 | Isolated from the fecal material of a human patient with a relapsing C. difficile infection in Western Pennsylvania, USA in 2005. | Toxigenic strain. |
| C. difficile P29 | NR-32903 | Isolated from the fecal material of a human patient with CDI in Western Pennsylvania, USA in 2009. | Toxigenic strain and reported to be co-colonized with non-toxigenic C. difficile. |
| C. difficile P30 | NR-32904 | It was obtained in 2009 from the fecal material of an asymptomatic human patient in Western Pennsylvania, USA. | Non-toxigenic strain. |
| C. difficile Isolate 1 | NR-13427 | It was obtained from a human patient from the Mid-Atlantic region of the United States in 2008/2009. | -- |
| C. difficile Isolate 2 | NR-13428 | It was obtained from a human patient from the Mid-Atlantic region of the United States in 2008/2009. | -- |
| C. difficile Isolate 4 | NR-13430 | It was obtained from a human patient from the Mid-Atlantic region of the United States in 2008/2009. | -- |
| C. difficile Isolate 5 | NR-13431 | It was obtained from a human patient from the Mid-Atlantic region of the United States in 2008/2009. | -- |
| C. difficile Isolate 6 | NR-13432 | It was obtained from a human patient from the Mid-Atlantic region of the United States in 2008/2009. | -- |
| C. difficile Isolate 7 | NR-13433 | It was obtained from a human patient from the Mid-Atlantic region of the United States in 2008/2009. | -- |
| C. difficile Isolate 9 | NR-13435 | It was obtained from a human patient from the Mid-Atlantic region of the United States in 2008/2009. | -- |
| C. difficile Isolate 10 | NR-13436 | It was obtained from a human patient from the Mid-Atlantic region of the United States in 2008/2009. | -- |
| C. difficile Isolate 11 | NR-13437 | It was obtained from a human patient from the Mid-Atlantic region of the United States in 2008/2009. | -- |
| C. difficile Isolate 13 | NR-13553 | It was obtained from a human patient from the Mid-Atlantic region of the United States in 2008/2009. | -- |
| C. difficile 002-P50-2011 | HM-746 | Isolated from the stool of a patient with diarrhea in January 2011. | Reference genome for the Human Microbiome Project (HMP). |
| C. difficile ATCC 700057 | VPI 11186 | -- | Toxinotype tcdA-, tcdB-, Ribotype 038, Nontoxigenic. |
| C. difficile ATCC 43598 | 1470 | Human feces, asymptomatic neonate, Belgium | Presence of tcdB gene confirmed by PCR, Ribotype 017. |
| C. difficile ATCC BAA 1801 | 3232 | Human feces (adult with diarrhea), Belgium. | Nontoxigenic, Ribotype 010. |
| C. difficile ATCC BAA 1870 | 4118 | -- | Presence of binary toxin, tcdA and tcdB genes. Toxinotype IIIb, Ribotype 027. |
| C. difficile Isolate 20100207 | NR-49278 | Obtained from the stool of an elderly adult male patient with a healthcare-associated (HA) C. difficile infection in New York, USA, in 2010. | PCR ribotype 027, NAP11, contains tcdA2, tcdB3 and tcd4 C (with 18 base pair deletion) PaLoc5 operon and the CDT6 |
| C. difficile Isolate 20100502 | NR-49277 | Obtained from the stool of an older adult male patient with a community-associated (CA) C. difficile infection in Colorado, USA, in 2010. | PCR ribotype 019, NAP11, contains tcdA2, tcdB3 and tcd4 C of the PaLoc5 operon, as well as the CDT6 |
| C. difficile Isolate 20110052 | NR-49281 | Obtained from the stool of an elderly male patient with a healthcare-associated (HA) C. difficile infection in northeastern USA in 2010. | PCR ribotype 027, NAP11, contains tcdA2, tcdB3 and tcd4 C (with 18 base pair deletion) PaLoc5 operon and the CDT6 |
| C. difficile Isolate 20110868 | NR-49287 | Obtained from the stool of an elderly female patient with a healthcare-associated (HA) C. difficile infection in southern USA, in 2011. | PCR ribotype 027, NAP11, contains tcdA2, tcdB3 and tcd4 C (with 18 base pair deletion) PaLoc5 operon and the CDT6 |
| C. difficile Isolate 20110870 | NR-49288 | Obtained from the stool of a young adult female patient with a healthcare-associated (HA) C. difficile infection in Tennessee, USA, in 2011. | PCR ribotype 027, NAP11, contains tcdA2, tcdB3 and tcd4 C (with 18 base pair deletion) PaLoc5 operon and the CDT6 |
| C. difficile Isolate 20110979 | NR-49285 | Obtained from the stool of an elderly female patient with a community-associated (CA) C. difficile infection in midwestern USA in 2011. | PCR ribotype 027, NAP11, contains tcdA2, tcdB3 and tcd4 C (with 18 base pair deletion) PaLoc5 operon and the CDT6 |
| C. difficile Isolate 20110999 | NR-49286 | Obtained from the stool of an elderly female patient with a healthcare-associated (HA) C. difficile infection in western/midwestern USA in 2011. | PCR ribotype 027, NAP11, contains tcdA2, tcdB3 and tcd4 C (with 18 base pair deletion) PaLoc5 operon and the CDT6 |
| C. difficile Isolate 20120013 | NR-49283 | Obtained from the stool of a young male patient with a community-associated (CA) C. difficile infection in northeastern USA, in 2011. | PCR ribotype 027, NAP11, contains tcdA2, tcdB3 and tcd4 C (with 18 base pair deletion) PaLoc5 operon and the CDT6 |
| C. difficile Isolate 20120184 | NR-49289 | Obtained from the stool of an elderly female patient with a fatal healthcare-associated (HA) C. difficile infection in Tennessee, USA, in 2011. | PCR ribotype 027, NAP11, contains tcdA2, tcdB3 and tcd4 C (with 18 base pair deletion) PaLoc5 operon and the CDT6 |
| C. difficile Isolate 20120187 | NR-49290 | Obtained from the stool of an elderly male patient with a healthcare-associated (HA) C. difficile infection in Tennessee, USA, in 2011. | PCR ribotype 019, NAP11, contains tcdA2, tcdB3 and tcd4 C of the PaLoc5 operon, as well as the CDT6 |
| C. difficile Isolate 20120236 | NR-49291 | Obtained from the stool of an older female patient with a community-associated (CA) C. difficile infection in midwestern, USA, in 2011. | PCR ribotype 027, NAP11, contains tcdA2, tcdB3 and tcd4 C (with 18 base pair deletion) PaLoc5 operon and the CDT6 |
| E. faecium HF50104 | NR-32052 | Isolated from swine feces in Michigan, USA in 2008. | Resistant to erythromycin, tetracycline and vancomycin. |
| E. faecium Patient #3–1 | NR-31912 | Isolated from the stool of a human patient having dominance of vancomycin-resistant Enterococcus in the stool but no bacteremia. | Vancomycin-resistant |
| E. faecium E1071 | NR-28978 | Hospitalized person free of enterococcal infection in the Netherlands in 2000 during a hospital surveillance program. | Non-infectious fecal isolate. Resistant to vancomycin. |
| E. faecium ERV165 | HM-970 | Isolated from human feces in Colombia, in 2008. | Resistant to vancomycin. |
| E. faecium ERV102 | HM-968 | Isolated from human oral sputum in Colombia, in 2006. | Resistant to ampicillin, vancomycin and streptomycin. |
NAP= North American pulsed-field gel electrophoresis type
tcdA= C. difficile toxin A gene
tcdB= C. difficile toxin B gene
tcdC= Anti-sigma factor gene
PaLoc= Pathogenicity locus
CDT= C. difficile binary toxin.
2.2. Minimum inhibitory concentration (MIC) of auranofin against C. difficile.
The broth microdilution assay was employed emulating the Clinical and Laboratory Standards Institute (CLSI) guidelines, with slight modifications [15]. A bacterial suspension equivalent to 0.5 McFarland standard was prepared and subsequently diluted in BHIS broth to ~105 CFU/mL. The bacterial suspension was seeded in 96-well plates containing the required concentrations of auranofin and control antibiotics (vancomycin and metronidazole). Plates were then incubated anaerobically for 48 hours, at 37 °C. MICs reported represent the lowest concentration of each agent that suppressed the visual growth of bacteria. MIC50 and MIC90 are the minimum concentration of each agent that inhibited the visual growth of 50% and 90% of the tested isolates, respectively.
2.3. Effect of auranofin on toxin production from a toxigenic C. difficile strain.
To assess auranofin’s ability to inhibit C. difficile toxin production, total amounts of toxins A and B were measured in the cell free supernatant of a late exponential phase culture of C. difficile ATCC BAA-1870. Toxin levels were compared after the addition of different subinhibitory concentrations of auranofin and control anticlostridial drugs [16, 17]. Briefly, C. difficile ATCC BAA-1870 was grown in BHIS broth, washed twice and aliquoted into 500 µL tubes. Drugs at the required concentrations were added to each tube, in triplicates, then tubes were incubated anaerobically (using BD GasPak™ EZ Container Systems) at 37 °C for six hours. One portion of each suspension was serially diluted, plated on BHIS agar and incubated anaerobically at 37 °C for 24 hours to detect the bacterial count. The second portion was centrifuged at 10,000 rpm for five minutes. The total concentration of C. difficile toxins A and B was measured in the supernatant of each tube using an enzyme linked immunosorbent assay (ELISA) kit (Premier®, Meridian Bioscience, Inc, Cincinnati, OH) following the manufacturer’s instructions. The optical density (450 nm), corresponding to the toxin concentration, was measured and compared for auranofin and the control drugs.
2.4. Effect of auranofin on C. difficile spore formation.
C. difficile HM-88, in stationary phase, was diluted in fresh BHIS broth and incubated anaerobically for 4–6 hours at 37 °C. The bacterial suspension was aliquoted into tubes and drugs were added (in triplicate) at concentrations equivalent to ½ × and 1 × MIC. Tubes were then incubated anaerobically for five days at 37 °C. After the incubation period, each tube was divided into two parts. One part was used to count the total amount of bacteria (vegetative bacteria + spores) through serial dilutions and culturing on BHIS agar plates supplemented with 0.1% taurocholic acid. The second part was centrifuged, media was replaced with PBS and stored at 4 °C overnight. The bacterial suspensions in PBS were shock heated at 70 °C for 20 minutes to kill vegetative cells, then serially diluted and plated to determine heat-resistant spore counts.
2.5. Protection of human gut cells against the inflammatory effect of C. difficile toxins.
To appraise the anti-inflammatory effect of auranofin against C. difficile toxin-mediated inflammation of human gut cells, a cell rounding assay was utilized [16]. Briefly, C. difficile ATCC BAA-1870 was grown in BHIS broth for 24 hours, centrifuged and the supernatant was sterile filtered and then frozen. Human colorectal epithelial cells (Caco-2) were grown in cell culture medium (DMEM supplemented with 10% FBS, 1 × NEAA, 100 IU/mL penicillin, and 100 μg/mL streptomycin) for five days. Cells were then trypsinized and seeded on a 96-well plate and grown at 37° C + 5% CO2. Once cells reached ~90% confluency, medium was removed, and the bacterial supernatant was added to the cells with or without auranofin (1 and 8 μg/mL) and control anticlostridial drugs (vancomycin, metronidazole and fidaxomicin, 1 – 128 μg/mL). Drugs were incubated with cells for 24 hours at 37° C + 5% CO2. Cells were then observed via a phase contrast microscope for morphological changes (cell rounding) as a result of C. difficile toxin-induced inflammation.
2.6. Reduction of IL-8 release from toxin-treated Caco-2 cells.
In order to further understand the anti-inflammatory activity of auranofin, IL-8 (a key cytokine in the process of C. difficile toxin-induced inflammation of gut cells) was detected in cell supernatants obtained from the cell rounding assay experiment (after 24 hours of incubation with C. difficile toxin with or without auranofin, 1 μg/mL, treatment) [18]. Supernatants were tested for IL-8 concentrations using an ELISA kit (Human IL-8 PicoKine™ ELISA Kit) according to the manufacturer’s instructions.
2.7. Activity of auranofin against vancomycin-resistant enterococci (VRE).
The minimum inhibitory concentrations (MICs) of auranofin and control antibiotics were tested using broth microdilution assay per CLSI guidelines [19]. Briefly, enterococcal isolates were streaked on brain heart infusion (BHI) agar plates and incubated aerobically at 37º C for about 18 hours. Bacterial colonies were scraped off the agar plates and suspended in BHI broth at a concentration of ~105 CFU/mL. Serial dilutions of the drugs were incubated with the bacterial suspensions for 16–20 hours at 37º C. The reported MICs are the lowest concentration of each drug that could inhibit the bacterial growth visually [20, 21].
2.8. Statistical analysis.
GraphPad Prism version 7.0 for Windows (GraphPad Software, La Jolla CA) was utilized for the statistical analyses. One-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test was performed to analyze the IL-8 data from cell supernatants. Two-way ANOVA followed by Dunnett’s post-hoc comparisons test were utilized to analyze the spore inhibition data.
3. RESULTS
3.1. Antibacterial activity of auranofin against C. difficile.
Auranofin’s anticlostridial activity was evaluated against a large panel of C. difficile strains including hypervirulent strains (ribotype 027). As shown in Table 1, auranofin inhibited growth of the 41 tested C. difficile strains at concentrations ranging from 0.25 – 4 μg/mL. Auranofin inhibited 50% of the tested isolates (MIC50) at a concentration of 1 μg/mL and inhibited 90% of the isolates (MIC90) at a concentration of 2 μg/mL. The MIC of auranofin was comparable to values obtained for vancomycin, the drug of choice for treatment of severe CDI. Vancomycin was effective at a range of 0.25 – 2 μg/mL (MIC50 = 0.5 μg/mL and MIC90 = 1 μg/mL). Metronidazole was active at a range of 0.06 – 0.25 μg/mL (MIC50 = 0.0.25 μg/mL and MIC90 = 0.25 μg/mL).
3.2. Auranofin inhibits C. difficile toxin production.
After confirming the potent in vitro anticlostridial activity of auranofin, we next moved to test the inhibitory activity of auranofin against C. difficile toxin production. Bacteria in the late log phase were incubated with subinhibitory concentrations of auranofin and control anticlostridial drugs. Auranofin exhibited a dose-dependent inhibition of C. difficile toxin production, when compared to the untreated control. As depicted in Fig. 1, auranofin at 1/8, 1/4 and 1/2 × MIC inhibited 15.6%, 31.2% and 40% of total toxin production, respectively. Fidaxomicin, an anticlostridial antibiotic known to inhibit toxin production and spore formation [22], was found to inhibit 37.2%, 50.1% and 52.3% of toxin production at 1/8, 1/4 and 1/2 × MIC, respectively. As expected, no toxin inhibition was observed when C. difficile was treated with either vancomycin or metronidazole; on the contrary, the toxin concentration increased at certain concentrations, in agreement with previous reports [17, 22, 23].
Fig. 1. Toxin inhibition activity of auranofin and control anticlostridial drugs (vancomycin, metronidazole and fidaxomicin) against C. difficile.

Drugs, at concentrations of 1/8 ×, 1/4 × and1/2 × MIC were incubated with a hypervirulent, toxigenic strain of C. difficile (strain ATCC BAA-1870). The bacterial counts were determined for each sample, and the toxin levels were assessed in the supernatant using enzyme linked immune fluorescent assay (ELISA). Error bars represent standard deviation values from triplicate samples for each treatment.
3.3. Auranofin’s inhibits C. difficile spore formation.
C. difficile HM-88, in late exponential growth phase, was incubated with subinhibitory concentrations of auranofin and control anticlostridial drugs. Total vegetative cells and heat-resistant spores were counted in each sample. Spores comprised most of the viable count in the untreated control (Fig. 2). Auranofintreated bacteria displayed reduced spore count, ~1.5 log10 at both 1/2 and 1 × MIC. While a similar effect was observed with fidaxomicin, almost no reduction in the spore count was detected with either vancomycin or metronidazole treatment.
Fig. 2. Spore inhibition of activity of auranofin against C. difficile compared to the control anticlostridial drugs, vancomycin and metronidazole.

Drugs (1/2× and 1 × MIC) were incubated with bacteria for five days followed by serial dilution and plating to count both total bacterial count and heat resistant spores. Error bars represent standard deviation values from triplicate samples for each treatment. (*) denotes significant difference between the total and the spore counts.
3.4. Protection of human gut cells against inflammatory effect of C. difficile toxins.
Auranofin has been previously shown to have anti-inflammatory activity, an important factor for its use in the treatment of rheumatoid arthritis. We thus moved to investigate if auranofin would also have the ability to protect gut cells from inflammation induced by C. difficile toxins. Human colorectal cells (Caco-2) were treated with sterile-filtered bacterial supernatant, with or without the addition of auranofin or control anticlostridial drugs. As shown in Fig. 4, healthy Caco-2 cells display normal morphologic characteristics of enterocytes. Upon treatment with C. difficile toxins, cell rounding and detachment occurred [24]. The goal was to inspect if drug treatment will preserve the normal Caco-2 morphology, in the presence of C. difficile toxins. Auranofin (1 and 8 μg/mL) protected cells against the inflammatory effect of C. difficile toxins and successfully preserved the normal cell morphology. Similar results were obtained with polarized cells (data not shown). On the other hand, cells treated with vancomycin, metronidazole or fidaxomicin (at a concentration ranging from 1 to 128 μg/mL) all exhibited rounding and detachment after exposure to toxins, indicating these drugs were unable to protect the gut epithelial cells.
Fig. 4. Auranofin mediated IL-8 inhibition from gut cells treated with C. difficile toxins.
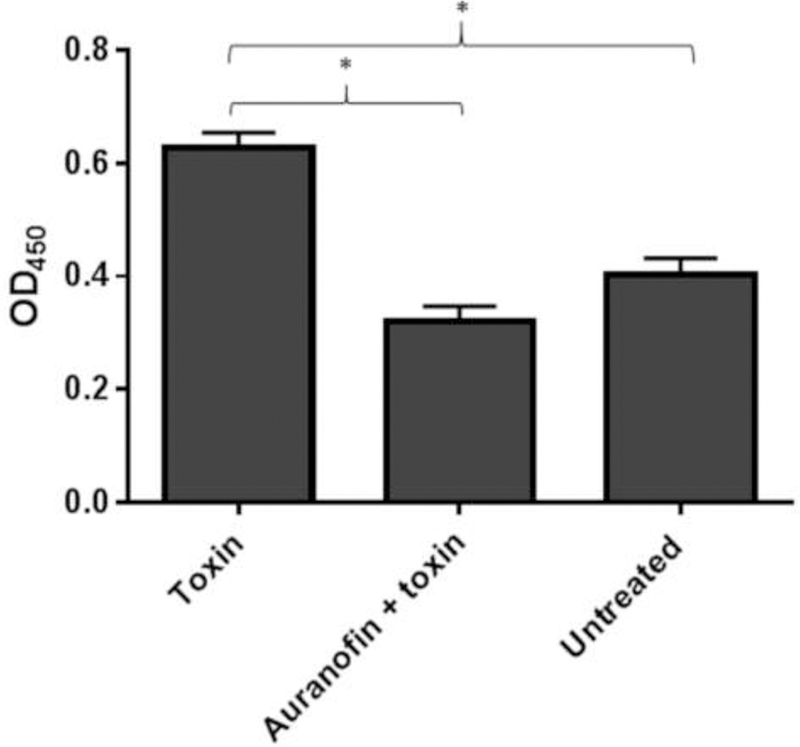
IL-8 level was assessed in Caco-2 cells treated with supernatant containing C. difficile toxins, with or without the addition of auranofin. OD450 coincides with the level of IL-8 in the cell supernatant. Error bars represent standard deviation values from triplicate samples used for each test agent. An asterisk (*) indicates significant difference (P < 0.05) between cells exposed to supernatant containing toxin alone and cells treated with supernatant containing auranofin (1 µg/mL) using one-way (ANOVA) followed by Dunnett’s multiple comparisons test.
3.5. Reduction of IL-8 release from toxin-treated Caco-2 cells.
After confirming auranofin can mitigate C. difficile toxin-mediated inflammation, we investigated if auranofin’s antiinflammatory effect was due to inhibition of IL-8. IL-8 is a major proinflammatory cytokine released upon exposure of gut epithelial cells to C. difficile toxins [18]. IL-8 was measured in the Caco-2 supernatants after incubation with C. difficile toxins with or without auranofin (1 μg/mL). Toxin treatment resulted in a significant increase in IL-8 concentration in the cell supernatant (Fig. 5). However, when cells were treated with toxins along with auranofin, no significant increase inIL-8 was observed. The OD450 is represent to the IL-8 concentration present in the supernatants.
3.6. Activity of auranofin against vancomycin-resistant enterococci (VRE).
To evaluate if auranofin treatment can prompt an overgrowth of VRE, the MIC of auranofin was determined against five VRE strains (Table 3). Auranofin inhibited growth of VRE at concentrations ranging from 0.25 – 0.5 μg/mL). Fidaxomicin also inhibited VRE growth albeit at higher concentrations (MIC range was 1 – 2 μg/mL). In contrast, vancomycin and metronidazole were not effective against all the tested VRE strains.
Table 3.
The minimum inhibitory concentration (MIC, µg/mL) of auranofin and control drugs against vancomycin-resistant Enterococcus faecium isolates.
| Strain | Auranofin | Fidaxomicin | Vancomycin | Metronidazole | Linezolid |
|---|---|---|---|---|---|
| Enterococcus faecium HF50104 NR-32052 | 0.5 | 2 | 256 | >256 | 0.125 |
| Enterococcus faecium Patient #3–1 NR-31912 | 0.5 | 2 | >256 | >256 | 0.25 |
| Enterococcus faecium NR-28978 | 0.5 | 2 | 128 | >256 | 0.5 |
| Enterococcus faecium ERV165 HM-970 | 0.25 | 1 | 256 | >256 | 0.5 |
| Enterococcus faecium ERV102HM-968 | 0.5 | 1 | >256 | >256 | 0.25 |
4. DISCUSSION
Clostridium difficile infections (CDIs) are increasing in morbidity and mortality within the healthcare setting. The U.S. Centers for Disease Control and Prevention (CDC) reported about half a million CDI cases in 2011, an increase from 333,000 cases reported in 2007. More worrisome is the relapse rate was 20% and came at a direct cost of $4.8 billion for acute care facilities alone. Moreover, approximately 29,000 deaths occurred within 30 days of the initial diagnosis, with much higher rates reported among elderly patients [2]. CDI in the healthcare setting is most likely attributed to administration of broad-spectrum antibiotics that subsequently damages the intestinal microbiota permitting C. difficile to expand, attach to epithelial cells and produce toxins. C. difficile toxins afflict the colonic epithelium leading to loss of tight junctions, enhanced mucosal permeability and ultimately intense inflammation and neutrophilic infiltration [25].
Treatment options for CDI, including for infections caused by hypervirulent strains, are very limited. Only three drugs are currently in use, vancomycin and metronidazole, both discovered in the mid-20th century, and fidaxomicin, approved in 2011. However, there are several limitations with these drugs. For example, it is estimated that about 22% and 14% of patients treated with metronidazole or vancomycin, respectively, will experience treatment failure. Moreover, about 25 – 30% of patients treated with either drug will suffer from CDI recurrence [7, 17]. Unfortunately, treatment outcome remains unsatisfactory even with the introduction of fidaxomicin, where relapsing CDI occurs [26, 27]. This highlights there remains a need to identify new, effective agents to treat CDI.
Antibacterial drug discovery is a very lengthy and expensive process. Repurposing FDA-approved drugs for new indications is a promising approach for drug discovery. Due to extensive preclinical and clinical investigation, key parameters such as the safety profile, pharmacodynamics and pharmacokinetics of these drugs is known, which will undoubtedly reduce both the time and cost associated with drug development [28–31]. Auranofin represents one drug we have been extensively investigating to repurpose as an antibacterial agent. Auranofin is an antirheumatic drug, approved by the FDA in 1985, that has a well-defined safety profile with limited reports of adverse reactions [13]. One of the attractive traits of auranofin is its low oral absorbability as only 15 – 25% of the administered dose is absorbed and about 85% of the drug is excreted in feces [13]. Therefore, auranofin is an attractive drug for development against gut pathogens.
Although Jackson-Rosario, et al. reported auranofin is active in vitro against C. difficile [32], a detailed investigation of auranofin against a wide panel of C. difficile isolates and auranofin’s impact on key virulence factors expressed by C. difficile has yet to be undertaken. In this study, we evaluated auranofin against 41 different C. difficile strains, including hypervirulent (NAP1, ribotype 027) and clinical toxigenic isolates. In agreement with Jackson-Rosario et al.’s study, [32], auranofin inhibited growth of C. difficile at a low concentration (0.25 – 4 µg/mL) which was comparable to the standard anticlostridial drugs, vancomycin and metronidazole.
We next sought to investigate the inhibitory activity of auranofin against C. difficile toxin production. As mentioned above, toxins are crucial for C. difficile to induce inflammation and to provoke disease. As a result, non-toxigenic bacteria are not associated with disease. So it is conjectured that an inhibition of toxin production will contribute to effective treatment of CDI [17, 33]. Additionally, toxin production occurs during stationary phase of bacterial growth where antibiotics are not as effective. Furthermore, toxin production is increased by environmental stress; therefore, some anticlostridial drugs (e.g. vancomycin and metronidazole) induce C. difficile toxin production [34]. Auranofin was previously reported to inhibit protein synthesis, virulence factors and toxin production in Staphylococcus aureus [35, 36]. Therefore, we tested the activity of auranofin at subinhibitory concentrations against a hypervirulent, toxigenic strain of C. difficile. Auranofin inhibited the total toxin production in C. difficile. A similar effect was observed with fidaxomicin but not with vancomycin or metronidazole. We also tested auranofin’s ability to reduce C. difficile toxin-mediated inflammation of the gut epithelial cells, given auranofin exhibits potent anti-inflammatory activity. Exposure of human colonic epithelial cells (Caco-2) to supernatant containing toxins produced by C. difficile to induced cell rounding similar to that was reported previously [18]. Inclusion of a very low concentration of auranofin (1 µg/mL) with the C. difficile culture supernatant protected Caco-2 cells from the deleterious effect of C. difficile toxins (Fig. 4) and suppressed production of the inflammatory cytokine IL-8.
In addition to toxin production, C. difficile utilizes spore formation as a key virulence factor in its pathogenesis. Hypervirulent and epidemic strains of C. difficile are associated with a higher ability to form spores that are resistant to standard disinfection procedures. This accounts for the ability of these strains to spread more efficiently throughout the environment. Furthermore, persistent C. difficile spores can germinate in the intestine, after the conclusion of treatment, leading to relapse [33, 37]. Auranofin was reported to inhibit several major pathways involved in protein biosynthesis in Staphylococcus aureus [35]. Given sporulation requires the synthesis of spore coat proteins, we hypothesized auranofin would be able to inhibit spore formation in C. difficile [38]. As anticipated, auranofin (at ½ × MIC) inhibited C. difficile spore formation; in contrast, neither vancomycin or metronidazole treatment was effective. Auranofin’s ability to interfere with spore formation, may translate into lower recurrence rates of CDI.
One complication of using vancomycin or metronidazole for treating CDI is the promotion of persistent colonization by vancomycin-resistant enterococci (VRE) [39], and avoidance of this problem is one goal for anticlostridial drug development [40]. When investigated against five isolates of VRE, auranofin (MIC ranged from 0.25 – 0.5 µg/mL) was superior to metronidazole, vancomycin, and fidaxomicin. Furthermore, auranofin has been reported to reduce VRE carriage and shedding in a mouse model of VRE colonization [20].
In conclusion, we report auranofin, an FDA-approved antirheumatic drug, possesses potent in vitro antibacterial activity against C. difficile, is capable of inhibiting both toxin production and spore formation. Furthermore, auranofin protected gut epithelial cells from the deleterious effect of C. difficile toxin-mediated inflammation. Additionally, auranofin has dual activity against C. difficile and VRE and should not promote VRE colonization. Further investigation is required to determine the activity of auranofin in animal models of CDI.
Fig. 3. Auranofin’s effect against C. difficile toxin-mediated inflammation of gut epithelial cells.

Human colorectal (Caco-2) epithelial cells were incubated with filtered C. difficile culture supernatant plus B. DMSO, 2.5%, C. vancomycin, 128 μg/mL, D. metronidazole, 128 μg/mL, fidaxomicin, 128 μg/mL, F. auranofin, 1 μg/mL or G. auranofin, 8 μg/mL for 24 hours and observed under the microscope. Reference wells were not treated with C. difficile supernatant but still treated with 2.5% DMSO (panel A). Cell rounding is an indication of inflammation.
Table 2.
The minimum inhibitory concentration (MIC, µg/mL) of auranofin and control anticlostridial drugs against C. difficile isolates.
| C. difficile Strain | ID number | Auranofin | Vancomycin | Metronidazole |
|---|---|---|---|---|
| P2 | NR-32883 | 0.5 | 0.25 | 0.06 |
| P3 | NR-32884 | 4 | 1 | 0.25 |
| P4 | NR-32889 | 1 | 2 | 0.125 |
| P5 | NR-32885 | 0.5 | 1 | 0.125 |
| P6 | NR-32886 | 2 | 1 | 0.125 |
| P7 | NR-32887 | 0.5 | 1 | 0.125 |
| P8 | NR-32888 | 1 | 0.5 | 0.125 |
| P13 | NR-32891 | 1 | 0.5 | 0.125 |
| P15 | NR-32892 | 1 | 0.5 | 0.06 |
| P19 | NR-32895 | 1 | 1 | 0.25 |
| P20 | NR-32896 | 4 | 1 | 0.25 |
| P21 | NR-32897 | 1 | 0.25 | 0.125 |
| P29 | NR-32903 | 1 | 0.25 | 0.06 |
| P30 | NR-32904 | 1 | 0.5 | 0.25 |
| Isolate 1 | NR-13427 | 1 | 1 | 0.25 |
| Isolate 2 | NR-13428 | 1 | 1 | 0.06 |
| Isolate 4 | NR-13430 | 2 | 0.25 | 0.06 |
| Isolate 5 | NR-13431 | 2 | 0.5 | 0.25 |
| Isolate 6 | NR-13432 | 1 | 0.5 | 0.125 |
| Isolate 7 | NR-13433 | 0.5 | 0.5 | 0.25 |
| Isolate 9 | NR-13435 | 0.25 | 0.5 | 0.06 |
| Isolate 10 | NR-13436 | 1 | 0.5 | 0.125 |
| Isolate 11 | NR-13437 | 2 | 1 | 0.25 |
| Isolate 13 | NR-13553 | 4 | 1 | 0.25 |
| NAP07 | HM-88 | 1 | 0.5 | 0.25 |
| 002-P50–2011 | HM-746 | 0.25 | 0.25 | 0.125 |
| ATCC 700057 | VPI 11186 | 1 | 0.5 | 0.25 |
| ATCC 43598 | 1470 | 0.5 | 0.5 | 0.25 |
| ATCC BAA 1801 | 3232 | 1 | 0.5 | 0.125 |
| ATCC BAA 1870 | 4118 | 0.5 | 1 | 0.25 |
| Isolate 20100502 | NR-49277 | 0.25 | 0.5 | 0.125 |
| Isolate 20100207 | NR-49278 | 0.25 | 0.25 | 1 |
| Isolate 20110052 | NR-49281 | 0.25 | 0.25 | 0.125 |
| Isolate 20110868 | NR-49287 | 0.25 | 0.25 | 0.25 |
| Isolate 20110870 | NR-49288 | 0.5 | 0.5 | 0.25 |
| Isolate 20110979 | NR-49285 | 0.25 | 0.25 | 0.25 |
| Isolate 20110999 | NR-49286 | 0.25 | 0.25 | 0.25 |
| Isolate 20120013 | NR-49283 | 0.25 | 0.25 | 0.125 |
| Isolate 20120184 | NR-49289 | 0.5 | 0.25 | 0.25 |
| Isolate 20120187 | NR-49290 | 0.5 | 0.5 | 0.25 |
| Isolate 20120236 | NR-49291 | 0.25 | 0.5 | 0.25 |
| MIC50 | 1 | 0.5 | 0.25 | |
| MIC90 | 2 | 1 | 0.25 | |
Highlights.
Auranofin is a potent inhibitor of Clostridium difficile growth
Auranofin, not vancomycin or metronidazole, blocked C. difficile toxin production
Auranofin also inhibited C. difficile spore formation
Auranofin protected gut cells against the C. difficile toxins’ effect in vitro
Auranofin halted vancomycin-resistant enterococci more than anticlostridial drugs
Acknowledgments
Funding: This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R01AI130186.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests: There are not any Conflicts of Interest for all authors.
Ethical Approval: Not required
References
- 1.Vindigni SM and Surawicz CM, C. difficile Infection: Changing Epidemiology and Management Paradigms. Clin Transl Gastroenterol, 2015. 6: p. e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lessa FC, et al. , Burden of Clostridium difficile infection in the United States. N Engl J Med, 2015. 372(9): p. 825–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Depestel DD and Aronoff DM, Epidemiology of Clostridium difficile infection. J Pharm Pract, 2013. 26(5): p. 464–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warny M, et al. , Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet, 2005. 366(9491): p. 1079–84. [DOI] [PubMed] [Google Scholar]
- 5.Kuehne SA, Cartman ST, and Minton NP, Both, toxin A and toxin B, are important in Clostridium difficile infection. Gut Microbes, 2011. 2(4): p. 252–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burns DA and Minton NP, Sporulation studies in Clostridium difficile. J Microbiol Methods, 2011. 87(2): p. 133–8. [DOI] [PubMed] [Google Scholar]
- 7.Vardakas KZ, et al. , Treatment failure and recurrence of Clostridium difficile infection following treatment with vancomycin or metronidazole: a systematic review of the evidence. Int J Antimicrob Agents, 2012. 40(1): p. 1–8. [DOI] [PubMed] [Google Scholar]
- 8.Cornely OA, et al. , Treatment of first recurrence of Clostridium difficile infection: fidaxomicin versus vancomycin. Clin Infect Dis, 2012. 55 Suppl 2: p. S154–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baines SD and Wilcox MH, Antimicrobial Resistance and Reduced Susceptibility in Clostridium difficile: Potential Consequences for Induction, Treatment, and Recurrence of C. difficile Infection. Antibiotics (Basel), 2015. 4(3): p. 267–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thangamani S, Younis W, and Seleem MN, Repurposing ebselen for treatment of multidrug-resistant staphylococcal infections. Sci Rep, 2015. 5: p. 11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thangamani S, Younis W, and Seleem MN, Repurposing celecoxib as a topical antimicrobial agent. Front Microbiol, 2015. 6: p. 750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Younis W, Thangamani S, and Seleem MN, Repurposing Non-Antimicrobial Drugs and Clinical Molecules to Treat Bacterial Infections. Curr Pharm Des, 2015. 21(28): p. 4106–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roder C and Thomson MJ, Auranofin: repurposing an old drug for a golden new age. Drugs R D, 2015. 15(1): p. 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walz DT, et al. , Biologic actions and pharmacokinetic studies of auranofin. Am J Med, 1983. 75(6A): p. 90–108. [DOI] [PubMed] [Google Scholar]
- 15.(CLSI), C.a.L.S.I., Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria, 8th Edition. M11–A8. 2012.
- 16.Ochsner UA, et al. , Inhibitory effect of REP3123 on toxin and spore formation in Clostridium difficile, and in vivo efficacy in a hamster gastrointestinal infection model. J Antimicrob Chemother, 2009. 63(5): p. 964–71. [DOI] [PubMed] [Google Scholar]
- 17.Locher HH, et al. , In vitro and in vivo antibacterial evaluation of cadazolid, a new antibiotic for treatment of Clostridium difficile infections. Antimicrob Agents Chemother, 2014. 58(2): p. 892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahida YR, et al. , Effect of Clostridium difficile toxin A on human intestinal epithelial cells: induction of interleukin 8 production and apoptosis after cell detachment. Gut, 1996. 38(3): p. 337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Institute C.a.L.S., Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Ninth Edition, M07–A9. 2012.
- 20.AbdelKhalek A, et al. , Repurposing auranofin as an intestinal decolonizing agent for vancomycin-resistant enterococci. Sci Rep, 2018. 8(1): p. 8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohammad H, et al. , Phenylthiazole Antibacterial Agents Targeting Cell Wall Synthesis Exhibit Potent Activity in Vitro and in Vivo against Vancomycin-Resistant Enterococci. J Med Chem, 2017. [DOI] [PMC free article] [PubMed]
- 22.Babakhani F, et al. , Fidaxomicin inhibits toxin production in Clostridium difficile. J Antimicrob Chemother, 2013. 68(3): p. 515–22. [DOI] [PubMed] [Google Scholar]
- 23.Endres BT, et al. , Evaluating the Effects of Surotomycin Treatment on Clostridium difficile Toxin A and B Production, Immune Response, and Morphological Changes. Antimicrob Agents Chemother, 2016. 60(6): p. 3519–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jafari NV, Allan E, and Bajaj-Elliott M, Human intestinal epithelial response(s) to Clostridium difficile. Methods Mol Biol, 2010. 646: p. 135–46. [DOI] [PubMed] [Google Scholar]
- 25.Bien J, Palagani V, and Bozko P, The intestinal microbiota dysbiosis and Clostridium difficile infection: is there a relationship with inflammatory bowel disease? Therap Adv Gastroenterol, 2013. 6(1): p. 53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orenstein R, Fidaxomicin failures in recurrent Clostridium difficile infection: a problem of timing. Clin Infect Dis, 2012. 55(4): p. 613–4. [DOI] [PubMed] [Google Scholar]
- 27.Louie TJ, et al. , Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med, 2011. 364(5): p. 422–31. [DOI] [PubMed] [Google Scholar]
- 28.Younis W, et al. , In Vitro Screening of an FDA-Approved Library Against ESKAPE Pathogens. Curr Pharm Des, 2017. 23(14): p. 2147–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mohammad H, et al. , Repurposing niclosamide for intestinal decolonization of vancomycin-resistant enterococci. Int J Antimicrob Agents, 2018. 51(6): p. 897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thangamani S, et al. , Exploring simvastatin, an antihyperlipidemic drug, as a potential topical antibacterial agent. Sci Rep, 2015. 5: p. 16407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thangamani S, et al. , Repurposing Approach Identifies Auranofin with Broad Spectrum Antifungal Activity That Targets Mia40-Erv1 Pathway. Front Cell Infect Microbiol, 2017. 7: p. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson-Rosario S, et al. , Auranofin disrupts selenium metabolism in Clostridium difficile by forming a stable Au-Se adduct. J Biol Inorg Chem, 2009. 14(4): p. 507–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vedantam G, et al. , Clostridium difficile infection: toxins and non-toxin virulence factors, and their contributions to disease establishment and host response. Gut Microbes, 2012. 3(2): p. 121–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Onderdonk AB, Lowe BR, and Bartlett JG, Effect of environmental stress on Clostridium difficile toxin levels during continuous cultivation. Appl Environ Microbiol, 1979. 38(4): p. 637–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thangamani S, et al. , Antibacterial activity and mechanism of action of auranofin against multi-drug resistant bacterial pathogens. Scientific Reports, 2016. 6: p. 22571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thangamani S, et al. , Repurposing auranofin for the treatment of cutaneous staphylococcal infections. Int J Antimicrob Agents, 2016. 47(3): p. 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamboj M, et al. , Relapse versus reinfection: surveillance of Clostridium difficile infection. Clin Infect Dis, 2011. 53(10): p. 1003–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paredes-Sabja D, Shen A, and Sorg JA, Clostridium difficile spore biology: sporulation, germination, and spore structural proteins. Trends Microbiol, 2014. 22(7): p. 406–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Al-Nassir WN, et al. , Both oral metronidazole and oral vancomycin promote persistent overgrowth of vancomycin-resistant enterococci during treatment of Clostridium difficile associated disease. Antimicrob Agents Chemother, 2008. 52(7): p. 2403–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seiler P, et al. , Cadazolid Does Not Promote Intestinal Colonization of Vancomycin-Resistant Enterococci in Mice. Antimicrob Agents Chemother, 2016. 60(1): p. 628–31. [DOI] [PMC free article] [PubMed] [Google Scholar]