

Abstract
The air and water stable π-allyliridium C,O-benzoate modified by (S)-tol-BINAP, (S)-Ir-II, catalyzes highly regio- and enantioselective Tsuji-Trost type aminations of racemic branched alkyl-substituted allylic acetates using primary or secondary (hetero)aromatic amines. Specifically, in the presence of (S)-Ir-II (5 mol%) in DME solvent at 60-70 °C, α-methyl allyl acetate 1a (100 mol%) reacts with primary (hetero)aromatic amines 2a-2l (200 mol%) or secondary (hetero)aromatic amines 3a-3l (200 mol%) to form the branched products of allylic amination 4a-4l and 5a-5l, respectively, as single regioisomers in good to excellent yield with uniformly high levels of enantioselectivity. As illustrated by the conversion of heteroaromatic amine 3m to adducts 6a-6g, excellent levels of regio- and enantioselectivity are retained across diverse branched allylic acetates bearing normal alkyl or secondary alkyl substituents. For reactants 3n-3p, which incorporate both primary and secondary aryl amine moieties, regio- and enantioselective amination occurs with complete site-selectivity to furnish adducts 7a-7c. Mechanistic studies involving amination of the enantiomerically enriched deuterium labelled acetate 1h corroborate C-N bond formation via outer-sphere addition.
Graphical Abstract
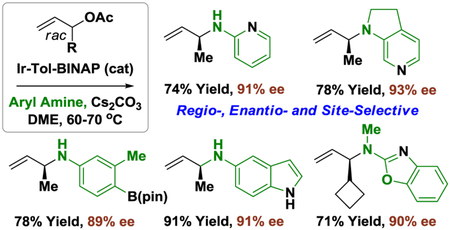
Introduction
Cyclometallated π-allyliridium C,O-benzoate complexes have been shown to catalyze diverse alcohol-mediated carbonyl allylations using allyl carboxylates as pronucleophiles.1 In these umpoled allylations,2 the C,O-benzoate moiety assists in maintaining neutrality and, hence, nucleophilicity of the π-allyliridium intermediate. Neutral iridium complexes that are not cyclometallated also display nucleophilic properties.3 In contrast, as illustrated by enantioselective Tsuji-Trost-type allylic aminations developed by Takeuchi,4,5 Helmchen,6,7 Hartwig,8,9 Carreira10 and You,11,12 cationic π-allyliridium species invariably serve as electrophiles. In this latter context, two distinct classes of iridium catalysts have emerged. Type I catalysts are used under basic conditions in combination with linear allyl proelectrophiles (as branched allyl proelectrophiles react stereospecifically).13 Type II catalysts are used under acidic conditions in combination with branched allyl proelectrophiles, which react in a non-stereospecific fashion, perhaps due to displacement of the π-bond of (σ+π)-allyl (enyl) iridium intermediates by the tethered olefin of the phosphoramidite ligand (Figure 1).14
Figure 1.

Cationic vs neutral chiral iridium complexes for regio- and enantioselective allylic amination.
The present authors recently found that neutral π-allyliridium C,O-benzoate complexes, which behave as nucleophilic allyl donors,1 can also act as electrophiles, representing the first examples of amphiphilic reactivity in the context of transition metal catalysis.15 In the initial communication of these findings, enantioselective allylic aminations of branched allylic acetates bearing linear alkyl groups with primary aliphatic amines were disclosed.15 These aminations proceed with complete branched regioselectivity, overcoming a significant limitation associated with Type I and II catalysts, which display incomplete regioselectivity for π-allyl precursors bearing linear alkyl groups.16,17 With regard to the amine nucleophile, the Type III SEGPHOS-modified π-allyliridium complexes used in our initial study enforced high enantioselectivities in reactions of primary aliphatic amines (Figure 1). Here, we show that the corresponding tol-BINAP-modified iridium catalyst avail a significant expansion in scope, enabling highly enantioselective aminations of branched alkyl-substituted allylic acetates with electronically diverse primary and secondary aryl amines, including site-selective reactions of bis(amine) nucleophiles. Additionally, we report deuterium labelling studies that corroborate C-N bond formation through an outer-sphere mechanism.
Research Design and Methods
To develop highly regio- and enantioselective allylic aminations mediated by aryl amines, a series of π-allyliridium C,O-benzoate complexes were evaluated in reactions of α methyl allyl acetate (100 mol%) and aniline (200 mol%) under conditions previously optimized for primary aliphatic amines.15 The iridium catalyst modified by tol-BINAP, (S)-Ir-II, delivered the product of allylic amination 4a with significantly higher levels of enantioselectivity than the corresponding SEGPHOS-modified catalyst, (S)-Ir-I, but a lower isolated yield of 4a was observed (eq. 1). Changing the solvent from THF to DME improved the isolated yield of 4a, and by decreasing the reaction temperature from 80 °C to 70 °C 4a could be formed in 82% yield and 89% enantiomeric excess (eq. 1).
 |
(eq. 1) |
Deviation from these reaction parameters did not avail further improvement, and given the low cost of tol-BINAP these conditions were adopted to explore the scope of primary aromatic and heteroaromatic amine nucleophiles 2a-21 in aminations of α-methyl allyl acetate (Table 1). Amine nucleophiles containing a diverse array of functional groups were examined to mirror challenges faced in medicinal chemistry. In each case, the targeted products of allylic amination 4a-4l were formed with complete branched regioselectivity and uniformly high levels of enantioselectivity. As illustrated in the formation of 4d, which incorporates a pinacol boronate moiety, the reaction conditions tolerate rather sensitive functional groups. The tolerance of Ortho-substituted anilines, as demonstrated by the formation of 4e, is also noteworthy. Perhaps the most striking feature, however, is the compatibility of the catalyst with electronically diverse aryl amine partners and the tolerance of Lewis basic N-heterocycles, as illustrated by the formation 4k and 4l The absolute stereochemical assignment of adducts 4b-4l is made in analogy to that determined for compound 4a, which has been prepared in enantiomerically enriched form in two separate reports.8d,18
Table 1.
Iridium-catalyzed amination of α-methyl allyl acetate 1a with primary aromatic and heteroaromatic amines 2a-2l to form enantiomerically enriched allylic amines 4a-41.a
 |
Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
In a further exploration of scope, optimized conditions were applied to the amination of α-methyl allyl acetate using secondary aromatic and heteroaromatic amine nucleophiles 3a-3l (Table 2). Indoline 3a, 6- and 7-aza-indolines 3b and 3c and the 3,3’-spirocyclic indoline 3d each underwent asymmetric allylation with complete branched regioselectivity and high levels of enantioselectivity. Pronounced match-mismatched effects were observed in the conversion of chiral nonracemic (S)- and (R)-2-methyl indolines 3e and 3f to adducts 5e and 5f, respectively, suggesting the potential for kinetic resolution. The amination of 1a using N-methyl aniline 3g and related compounds 3h and 3i bearing electron withdrawing and donating groups at the Para-position proceeded smoothly to form adducts 5g-5i, respectively. Among these three N-methyl aniline derivatives (3g-3i), amination using the more electron rich N-methyl-Para-anisidine 3i occurred with notably higher levels of enantioselectivity. As illustrated by the formation of 5j and 5k, N-methyl anilines containing bromide (3j) and chloride (3k) functional groups are tolerated. Finally, amination of 1a using N-Methyl-2-(methylamino)benzimidazole 31 is remarkably efficient, providing adduct 5l in 94% yield with complete selectivity for allylation of the extranuclear 2-(methylamino) moiety.
Table 2.
Iridium-catalyzed amination of α-methyl allyl acetate 1a with secondary aromatic and heteroaromatic amines 3a-3l to form enantiomerically enriched allylic amines 5a-51.a
 |
Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
To assess how structural variation of the π-allyliridium intermediate impacts reactivity, regio- and stereoselectivity, a set of branched allylic acetates 1a-1g were explored in amination mediated by 2-(methylamino)benzoxazole 3m (Table 3). In addition to α-methyl allyl acetate 1a, linear alkyl-substituted allylic acetates 1b and 1c smoothly underwent amination to form adducts 6a-6c as single regioisomers with high levels of enantiomeric enrichment. Allylic acetates 1d-1f, which incorporate cycloalkyl-substituents, delivered adducts 6d-6f as single regioisomers, although an erosion in enantioselectivity is observed using the larger cyclopentyl-substituted allyl acetate 1f. Finally, using the enantiomeric iridium catalysts (S)-Ir-II and (R)-Ir-II, the (S)-citronellol-derived allylic acetate 1g reacts with 3m to form 6g and iso-6g, respectively, with good levels of catalyst-directed diastereoselectivity. Under these conditions, aryl-substituted allyl acetates gave low yields (<10%) of allylic amination product and linear allylic acetates provided mixtures of allylic amination and hydroamination product in low isolated yield.19
Table 3.
Iridium-catalyzed amination of α-substituted allyl acetates 1a-1g with secondary heteroaromatic amine 3m to form enantiomerically enriched allylic amines 6a-6g.a
 |
Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
Having established the ability to functionalize both primary and secondary aromatic amines, the site-selective modification of reactants 3n-3p, which incorporate both primary and secondary aromatic amines, was attempted using the branched allylic acetate 1a (eq. 2-4). Upon exposure to standard conditions, 5-aminoindole 3n undergoes completely chemoselective functionalization at the primary amine to form adduct 7a as a single constitutional isomer with excellent levels of enantioselectivity (eq. 2). Similarly, in the conversion of 3o to adduct 7b, complete control of regio- and site-selectivity is accompanied by high levels of enantioselectivity (eq. 3). The structure of adduct 7b was verified by single crystal X-ray diffraction analysis, further corroborating the absolute stereochemical assignment of adducts 4a-4l, 5a-5l and 6a-6g. Finally, N-cyclohexyl-1,2-diaminobenzene 3p reacts with 1a to deliver adduct 7c, which is modified exclusively at the primary amine (eq. 4). The ability to engage diamines in site-selective regio- and enantioselective amination enhances step economy by avoiding manipulations devoted to N-protection-deprotection.
 |
(eq. 2) |
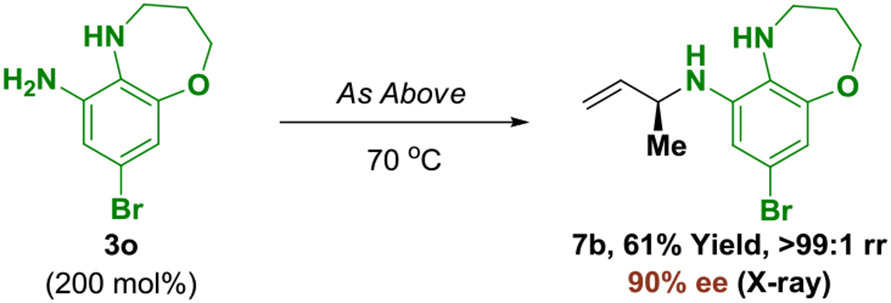 |
(eq. 3) |
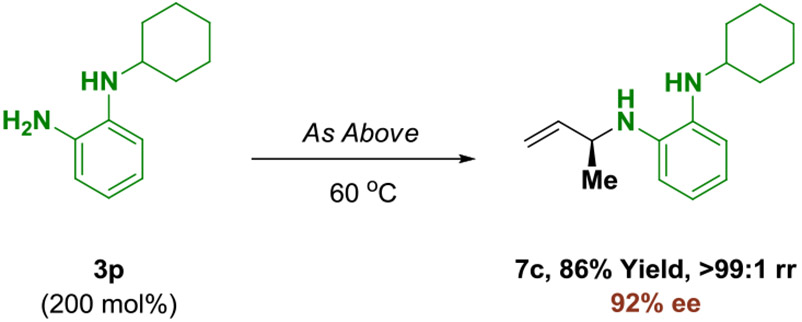 |
(eq. 4) |
Finally, to better understand the nature of the C-N bond forming event, asymmetric amination of the enantiomerically enriched (Z)-deuterated allylic acetate 1h was conducted under standard conditions using (0S)-Ir-II (Scheme 1, eq. 5).20 Compound 8a is formed with complete alkene (Z)-stereoselectivity, as determined by 1H NMR. Assuming formation of the π-allyliridium occurs with inversion of stereochemistry, as established in analogous iridium phosphoramidite catalyzed processes,4-12 the stereochemistry of the amination product 8a is consistent with outer-sphere addition of the nitrogen nucleophile. To corroborate the veracity of this experiment, the amination of allylic acetate 1h was conducted using the enantiomeric iridium catalyst, (R)-Ir-II (Scheme 1, eq. 6). The amination product 8b is formed with complete alkene (E)-stereoselectivity, as determined by 1H NMR. The stereochemistry of 8a is again consistent with outer-sphere C-N bond formation.
Scheme 1.

Iridium-catalyzed amination of enantiomerically enriched deuterated allylic acetate 1h with the enantiomeric catalysts (S)-Ir-II and (R)-Ir-II.a
a Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
Based on the collective data, a general catalytic mechanism and stereochemical model were proposed (Scheme 2). The π-allyliridium(I) complex I is subject to outer-sphere amine addition to form the C-N bond and the zwitterionic iridium(I) olefin complex II. Deprotonation of ammonium moiety of complex II mediated by cesium carbonate generates the anionic iridium(I) species III. Alkene exchange with the allylic acetate releases the product of allylic amination and forms the olefin complex IV. Loss of acetate ion regenerates the π- allyliridium(I) complex I to close the catalytic cycle. The indicated stereochemical model accounts for the observed sense of absolute stereoinduction for outer sphere addition of a nucleophile to the neutral iridium π-allyl complex I. This model is based upon the coordination mode revealed in closely related crystal structures.21 Orientation of the π-allyl is controlled through alleviation of steric clashes between the naphthyl and tolyl substituents of the phosphine ligand with the R-group of the resulting π-allyl, as illustrated in the disfavored mode of addition (Scheme 2).
Scheme 2.

General catalytic mechanism and stereochemical model for enantioselective iridium-catalyzed allylic amination.
Conclusions
Previously reported enantioselective allylic aminations are largely restricted to chiral iridium-phosphoramidite catalysts.4-12 In the initial communication of our work,15 we have shown that the air and water stable π-allyliridium C,O-benzoate modified by SEGPFIOS, which is well-known for its ability to catalyze nucleophilic carbonyl allylation,1,2 also promotes highly regio- and enantioselective electrophilic allylation of primary aliphatic amines. As demonstrated here, the corresponding tol-BINAP-modified complex is a significantly more effective catalyst for allylic amination, which has enabled use of primary and secondary aromatic or heteroaromatic amine nucleophiles. These π-allyliridium C,O-benzoate catalyzed processes overcome a longstanding limitation associated with all known catalytic systems for asymmetric allylic amination - the ability to promote enantioselective aminations of racemic branched allylic acetates bearing n-alkyl groups with complete levels of regioselectivitity.4-12,16 Another notable feature of these catalysts involves the ability to promote site-selective N-allylations of reactants that incorporate both primary and secondary aromatic amines. As demonstrated by mechanistic studies involving amination of the enantiomerically enriched deuterated allylic acetate 1h, an outer-sphere mechanism for C-N bond formation is operative. This work, along with our prior studies,15 significantly expands the scope of catalytic asymmetric allylic amination methodology, broadening access to diverse chiral α-stereogenic amines. Future work under the aegis of this academic-industrial collaboration will focus on expanding the range of amine nucleophiles for π-allyliridium C,O-benzoate catalyzed allylic amination, in particular, the use of secondary aliphatic amines and amide, and the development of related methods for asymmetric C-N bond formation.
Supplementary Material
ACKNOWLEDGMENT
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM069445) are acknowledged for partial support of this research. Genentech is acknowledged for summer predoctoral internship support (S. W. K. & L. A. S.). We would like to thank Kewei Xu and Yanzhou Liu for analytical data and Amber Guillen and Mengling Wong for assistance with chiral SFC analyses.
Footnotes
Supporting Information. Experimental procedures and spectroscopic data for all new compounds (1H NMR, 13C NMR, IR, HRMS), including images of NMR spectra and HPLC traces for racemic and enantiomerically enriched compounds. Single crystal X-ray diffraction data for compound 7b. This material is available free of charge via the internet at http://pubs.acs.org
Notes.
The authors declare no competing financial interest.
REFERENCES
- (1).(a) For selected reviews on alcohol-mediated carbonyl allylation, see: Han SB; Kim IS; Krische MJ Enantioselective iridium-catalyzed carbonyl allylation from the alcohol oxidation level via transfer hydrogenation: minimizing pre-activation for synthetic efficiency. Chem. Commun 2009, 7278. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ketcham JM; Shin I; Montgomery TP; Krische MJ Catalytic Enantioselective C−H Functionalization of Alcohols by Redox-Triggered Carbonyl Addition: Borrowing Hydrogen, Returning Carbon. Angew. Chem. Int. Ed 2014, 53, 9142. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim SW; Zhang W; Krische MJ Catalytic Enantioselective Carbonyl Allylation and Propargylation via Alcohol-Mediated Hydrogen Transfer: Merging the Chemistry of Grignard and Sabatier. Acc. Chem. Res 2017, 50, 2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).For a recent review on “umpoled allylations”, see: Spielmann K; Niel G; de Figueiredo RM; Campagne J-M Catalytic nucleophilic ‘umpoled’ π-allyl reagents. Chem. Soc. Rev 2018, 47, 1159. [DOI] [PubMed] [Google Scholar]
- (3).(a) For selected examples of alcohol-mediated carbonyl allylation involving non-cyclometallated iridium catalysts, see:Bower JF; Skucas E; Patman RL; Krische MJ Catalytic C–C Coupling via Transfer Hydrogenation: Reverse Prenylation, Crotylation, and Allylation from the Alcohol or Aldehyde Oxidation Level. J. Am. Chem. Soc 2007, 129, 15134. [DOI] [PubMed] [Google Scholar]; (b) Bower JF; Patman RL; Krische MJ Iridium-Catalyzed C–C Coupling via Transfer Hydrogenation: Carbonyl Addition from the Alcohol or Aldehyde Oxidation Level Employing 1,3-Cyclohexadiene. Org. Lett 2008, 10, 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nguyen KD; Herkommer D; Krische MJ Enantioselective Formation of All-Carbon Quaternary Centers via C–H Functionalization of Methanol: Iridium-Catalyzed Diene Hydrohydroxymethylation. J. Am. Chem. Soc 2016, 138, 14210. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Holmes MT; Nguyen KD; Schwartz LA; Luong T; Krische MJ Enantioselective Formation of CF3-Bearing All-Carbon Quaternary Stereocenters via C–H Functionalization of Methanol: Iridium Catalyzed Allene Hydrohydroxymethylation. J. Am. Chem. Soc 2017, 139, 8114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Takeuchi R; Ue N; Tanabe K; Yamashita K; Shiga N Iridium Complex-Catalyzed Allylic Amination of Allylic Esters. J. Am. Chem. Soc 2001, 123, 9525. [DOI] [PubMed] [Google Scholar]; (b) Onodera G; Watabe K; Matsubara M; Oda K; Kezuka S; Takeuchi R Iridium-Catalyzed Enantioselective Allylic Alkylation using Chiral Phosphoramidite Ligand Bearing an Amide Moiety. Adv. Synth. Catal. 2008, 2725. [Google Scholar]
- (5).(a) Reviews:Takeuchi R Iridium Complex-Catalyzed Highly Selective Organic Synthesis. Synlett 2002, 1954. [Google Scholar]; (b) Takeuchi R; Kezuka S Iridium-Catalyzed Formation of Carbon-Carbon and Carbon-Heteroatom Bonds. Synthesis 2006, 3349. [Google Scholar]
- (6).(a) Bartels B; García-Yebra C; Rominger F; Helmchen G Iridium-Catalysed Allylic Substitution: Stereochemical Aspects and Isolation of IrIII Complexes Related to the Catalytic Cycle. Eur. J. Inorg. Chem 2002, 2569. [Google Scholar]; (b) Lipowsky G; Helmchen G Regio- and enantioselective iridium-catalysed allylic aminations and alkylations of dienyl esters. Chem. Commun 2004, 116. [DOI] [PubMed] [Google Scholar]; (c) Welter C; Koch O; Lipowsky G; Helmchen G First intramolecular enantioselective iridium-catalysed allylic aminations. Chem. Commun 2004, 896. [DOI] [PubMed] [Google Scholar]; (d) Welter C; Dahnz A; Brunner B; Streiff S; Dübon P; Helmchen G Highly Enantioselective Syntheses of Heterocycles via Intramolecular Ir-Catalyzed Allylic Amination and Etherification. Org. Lett 2005, 7, 1239. [DOI] [PubMed] [Google Scholar]; (e) Weihofen R; Dahnz A; Tverskoy O; Helmchen G Highly enantioselective iridium-catalysed allylic aminations with anionic N-nucleophiles. Chem. Commun 2005, 3541. [DOI] [PubMed] [Google Scholar]; (f) Weihofen R; Tverskoy O; Helmchen G. Salt-Free Iridium-Catalyzed Asymmetric Allylic Aminations with N,N-Diacylamines and ortho-Nosylamide as Ammonia Equivalents. Angew. Chem. Int. Ed. 2006, 45, 5546. [DOI] [PubMed] [Google Scholar]; (g) Speiss S; Berthold C; Weihofen R, Helmchen G. Synthesis of α,β-unsaturated γ-lactams via asymmetric iridium-catalysed allylic substitution. Org. Biomol. Chem. 2007, 5, 2357. [DOI] [PubMed] [Google Scholar]; (h) Speiss S; Raska-tov JA; Gnamm C; Brödner K; Helmchen G. Ir-Catalyzed Asymmetric Allylic Substitutions with (Phosphoramidite)Ir Complexes—Resting States, Synthesis, and Characterization of Catalytically Active (π-Allyl)Ir Complexes. Chem. Eur. J. 2009, 15, 11087. [DOI] [PubMed] [Google Scholar]; (i) Gärtner M; Jäkel M; Achatz M; SonnenSchein C; Tverskoy O; Helmchen G Enantioselective Iridium-Catalyzed Allylic Substitutions with Hydroxamic Acid Derivatives as N-Nucleophiles. Org. Lett. 2011, 13, 2810. [DOI] [PubMed] [Google Scholar]
- (7).(a) Reviews:Helmchen G; Dahnz A; Dübon P; Schelwies M; Weihofen R Iridium-catalysed asymmetricallylic substitutions Chem. Commun. 2007, 675. [DOI] [PubMed] [Google Scholar]; (b) Helmchen G In Iridium Complexes in Organic Synthesis; Oro LA, Claver C, Eds.; Wiley-VCH: Weinheim, 2009; pp 211–250. [Google Scholar]; (c) Qu J; Helmchen G Applications of Iridium-Catalyzed Asymmetric Allylic Substitution Reactions in Target-Oriented Synthesis Acc. Chem. Res. 2017, 50, 2539. [DOI] [PubMed] [Google Scholar]
- (8).(a) Ohmura T; Hartwig JF Regio- and Enantioselective Allylic Amination of Achiral Allylic Esters Catalyzed by an Iridium—Phosphoramidite Complex. J. Am. Chem. Soc 2002, 124, 15164. [DOI] [PubMed] [Google Scholar]; (b) Kiener CA; Shu C; Incarvito C; Hartwig JF Identification of an Activated Catalyst in the Iridium-Catalyzed Allylic Amination and Etherification. Increased Rates, Scope, and Selectivity. J. Am. Chem. Soc 2003, 125, 14272. [DOI] [PubMed] [Google Scholar]; (c) Leitner A; Shu C; Hartwig JF Editing the stereochemical elements in an iridium catalyst for enantioselective allylic amination. Proc. Nat. Acad. Sci 2004, 101, 5830. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shu C; Leitner A; Hartwig JF Enantioselective Allylation of Aromatic Amines after In Situ Generation of an Activated Cyclometalated Iridium Catalyst. Angew. Chem. Int. Ed 2004, 43, 4797. [DOI] [PubMed] [Google Scholar]; (e) Leitner A; Shekhar S; Pouy MJ; Hartwig JF A Simple Iridium Catalyst with a Single Resolved Stereocenter for Enantioselective Allylic Amination. Catalyst Selection from Mechanistic Analysis. J. Am. Chem. Soc 2005, 127, 15506. [DOI] [PubMed] [Google Scholar]; (f) Leitner A; Shu C; Hartwig JF Effects of Catalyst Activation and Ligand Steric Properties on the Enantioselective Allylation of Amines and Phenoxides. Org. Lett 2005, 7, 1093. [DOI] [PubMed] [Google Scholar]; (g) Shekhar S; Trantow B; Leitner A; Hartwig JF Sequential Catalytic Isomerization and Allylic Substitution. Conversion of Racemic Branched Allylic Carbonates to Enantioenriched Allylic Substitution Products. J. Am. Chem. Soc 2006, 128, 11770. [DOI] [PubMed] [Google Scholar]; (h) Yamashita Y; Gopalarathnam A; Hartwig JF Iridium-Catalyzed, Asymmetric Amination of Allylic Alcohols Activated by Lewis Acids. J. Am. Chem. Soc 2007, 129, 7508. [DOI] [PubMed] [Google Scholar]; (i) Markovic D; Hartwig JF Resting State and Kinetic Studies on the Asymmetric Allylic Substitutions Catalyzed by Iridium—Phosphoramidite Complexes. J. Am. Chem. Soc 2007, 129, 11680. [DOI] [PubMed] [Google Scholar]; (j) Pouy MJ; Leitner A; Weix DJ; Ueno S; Hartwig JF Enantioselective Iridium-Catalyzed Allylic Amination of Ammonia and Convenient Ammonia Surrogates. Org. Lett 2007, 9, 3949. [DOI] [PubMed] [Google Scholar]
- (9).(a) Reviews:Hartwig JF; Stanley LM Mechanistically Driven Development of Iridium Catalysts for Asymmetric Allylic Substitution. Acc. Chem. Res 2010, 43, 1461. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hartwig JF; Pouy MJ Top. Organomet. Chem 2011, 38, 169. [Google Scholar]
- (10).(a) Defieber C; Ariger MA; Moriel P; Carreira EM Iridium-Catalyzed Synthesis of Primary Allylic Amines from Allylic Alcohols: Sulfamic Acid as Ammonia Equivalent. Angew. Chem. Int. Ed 2007, 46, 3139. [DOI] [PubMed] [Google Scholar]; (b) Roggen M; Carreira EM Stereospecific Substitution of Allylic Alcohols To Give Optically Active Primary Allylic Amines: Unique Reactivity of a (P,alkene)Ir Complex Modulated by Iodide. J. Am. Chem. Soc 2010, 132, 11917. [DOI] [PubMed] [Google Scholar]; (c) Lafrance M; Roggen M; Carreira EM Direct, Enantioselective Iridium-Catalyzed Allylic Amination of Racemic Allylic Alcohols. Angew. Chem. Int. Ed 2012, 51, 3470. [DOI] [PubMed] [Google Scholar]; (d) Rossler SL; Krautwald S; Carreira EM Study of Intermediates in Iridium—(Phosphoramidite,Olefin)-Catalyzed Enantioselective Allylic Substitution. J. Am. Chem. Soc 2017, 139, 3603. [DOI] [PubMed] [Google Scholar]
- (11).(a) Ye K-Y; Dai L-X; You S-L Enantioselective synthesis of 2,5-dihydrobenzo[b]azepine derivatives via iridium-catalyzed asymmetric allylic amination with 2-allylanilines and ring-closing-metathesis reaction. Org. Biomol. Chem 2012, 10, 5932. [DOI] [PubMed] [Google Scholar]; (b) Liu WB; Zhang X; Dai L-X; You S-L Asymmetric N-Allylation of Indoles Through the Iridium-Catalyzed Allylic Alkylation/Oxidation of Indolines. Angew. Chem. Int. Ed 2012, 51, 5183. [DOI] [PubMed] [Google Scholar]; (c) Ye K-Y; Zhao Z-A; Lai Z-W; Dai L-X; You S-L Highly Regioselective Allylic Substitution Reactions Catalyzed by an Air-Stable (π- Allyl)iridium Complex Derived from Dinaphthocyclooctatetraene and a Phosphoramidite Ligand. Synthesis 2013, 2109. [Google Scholar]; (d) Ye K-Y; Dai L-X; You S-L Regio- and Enantioselective Synthesis of N-Allylindoles by Iridium-Catalyzed Allylic Amination/Transition-Metal-Catalyzed Cyclization Reactions. Chem. Eur. J 2014, 20, 3040. [DOI] [PubMed] [Google Scholar]; (e) Yang Z-P; Wu Q-F; You S-L Direct Asymmetric Dearomatization of Pyridines and Pyrazines by Iridium-Catalyzed Allylic Amination Reactions. Angew. Chem. Int. Ed 2014, 53, 6986. [DOI] [PubMed] [Google Scholar]; (f) Zhang X; Yang Z-P; Huang L; You S-L Highly Regio- and Enantioselective Synthesis of N-Substituted 2-Pyridones: Iridium-Catalyzed In-termolecular Asymmetric Allylic Amination. Angew. Chem. Int. Ed 2015, 54, 1873. [DOI] [PubMed] [Google Scholar]; (g) Yang Z-P; Wu Q-F; Shao W; You S-L Iridium-Catalyzed Intramolecular Asymmetric Allylic Dearomatization Reaction of Pyridines, Pyrazines, Quinolines, and Isoquinolines. J. Am. Chem. Soc 2015, 137, 15899. [DOI] [PubMed] [Google Scholar]; (h) Ye K-Y; Cheng Q; Zhuo C-X; Dai L-X; You S-L An Iridium(I) N-Heterocyclic Carbene Complex Catalyzes Asymmetric Intramolecular Allylic Amination Reactions. Angew.Chem. Int. Ed 2016, 55, 8113. [DOI] [PubMed] [Google Scholar]; (i) Zhang X; Liu W-B; Cheng Q; You S-L Iridium-Catalyzed Asymmetric Allylic Amination Reactions with N-Aryl Phosphoramidite Ligands. Organ-ometallics 2016, 35, 2467. [Google Scholar]; (j) Yang Z-P; Zheng C; Huang L; Qian C; You S-L Iridium-Catalyzed Intramolecular Asymmetric Allylic Dearomatization Reaction of Benzoxazoles, Benzothiazoles, and Benzimidazoles. Angew. Chem. Int. Ed 2017, 56, 1530. [DOI] [PubMed] [Google Scholar]
- (12).(a) Liu W-B; Xia J-B; You S-L Iridium-Catalyzed Asymmetric Allylic Substitutions. Top. Organomet. Chem 2011, 38, 155. [Google Scholar]; (b) Zhou C-X; Zhang W; You S-L Catalytic Asymmetric Dearomatization Reactions. Angew. Chem. Int. Ed 2012, 51, 12662. [DOI] [PubMed] [Google Scholar]; (c) Wu W-T; Zhang L; You S-L Catalytic asymmetric dearomatization (CADA) reactions of phenol and aniline derivatives. Chem. Soc. Rev 2015, 45, 1570. [DOI] [PubMed] [Google Scholar]
- (13).Bartels B; Helmchen G Ir-catalysed allylic substitution: mechanistic aspects and asymmetric synthesis with phosphorus amidites as ligands. Chem. Commun 1999, 741. [Google Scholar]
- (14).(a) For spectroscopic and crystallographic evidence of a stable rhodium enyl complex and its role in enabling regio- and stereospecific rhodium catalyzed allylic substitution, respectively, see:Lawson DN; Osborn JA; Wilkinson G Interaction of tris(triphenylphosphine)chlororhodium(I) with iodomethane, methyl-allyl, and allyl chloride. J. Chem. Soc. A 1966, 1733. [Google Scholar]; (b) Tanaka I; Jin-no N; Kushida T; Tsutsui N; Ashida T; Suzuki H; Sakurai H; Moro-oka Y; Ikawa T Crystal Structures of (1,5-Cyclooctadiene)di-μ-methoxo-dirhodium(I) and Tetrakis(η3-allyl)di-μ-hydroxo-dirhodium(III). Bull. Chem. Soc. Jpn 1983, 56, 657. [Google Scholar]; (c) Evans PA; Nelson JD Conservation of Absolute Configuration in the Acyclic Rhodium-Catalyzed Allylic Alkylation Reaction: Evidence for an Enyl (σ + π) Organorhodium Intermediate. J. Am. Chem. Soc 1998, 120, 5581. [Google Scholar]
- (15).Meza AT; Wurm T; Smith L; Kim SW; Zbieg JR; Stivala CE; Krische MJ Amphiphilic π-Allyliridium C,O-Benzoates Enable Regio- and Enantioselective Amination of Branched Allylic Acetates Bearing Linear Alkyl Groups. J. Am. Chem. Soc 2018, 140, 1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).(a) For selected examples of catalytic enantioselective allylic aminations beyond iridium, see:Cooke ML; Xu K; Breit B Enantioselective Rhodium-Catalyzed Synthesis of Branched Allylic Amines by In-termolecular Hydroamination of Terminal Allenes. Angew. Chem. Int. Ed 2012, 51, 10876. [DOI] [PubMed] [Google Scholar]; (b) Arnold JS; Nguyen HM Rhodium-Catalyzed Dynamic Kinetic Asymmetric Transformations of Racemic Tertiary Allylic Trichloroacetimidates with Anilines. J. Am. Chem. Soc. 2012, 134, 8380. [DOI] [PubMed] [Google Scholar]; (c) Chen Q-A; Chen Z; Dong VM Rhodium-Catalyzed Enantioselective Hydroamination of Alkynes with Indolines. J. Am. Chem. Soc 2015, 137, 8392. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Xu K; Wang Y-H; Khakyzadeh V; Breit B Asymmetric synthesis of allylic amines via hydroamination of allenes with benzophenone imine. Chem. Sci 2016, 7, 3313. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Mwenda ET; Nguyen HM Enantioselective Synthesis of 1,2-Diamines Containing Tertiary and Quaternary Centers through Rhodium-Catalyzed DYKAT of Racemic Allylic Trichloroacetimidates. Org. Lett 2017, 19, 4814. [DOI] [PubMed] [Google Scholar]; (f) Guo W; Cai A; Xie J; Kleij AW Asymmetric Synthesis of α,α-Disubstituted Allylic Amines through Palladium-Catalyzed Allylic Substitution. Angew. Chem. Int. Ed 2017, 56, 11797. [DOI] [PubMed] [Google Scholar]
- (17).(a) For selected reviews on the catalytic enantioselective synthesis of allylic amines, see:Cannon JS; Overman LE Palladium(II)-Catalyzed Enantioselective Reactions Using COP Catalysts. Ace. Chem. Res 2016, 49, 2220. [DOI] [PubMed] [Google Scholar]; (b) Grange RL; Clizbe EA; Evans PA Recent Developments in Asymmetric Allylic Amination Reactions. Synthesis 2016, 2911. [Google Scholar]; (c) Mailyan AK; Eickhoff JA; Minakova AS; Gu Z; Lu P; Zakarian A Cutting-Edge and Time-Honored Strategies for Stereoselective Construction of C–N Bonds in Total Synthesis. Chem. Rev 2016, 116, 4441. [DOI] [PubMed] [Google Scholar]; (d) Fernandes Rodney A.; Kattanguru Pullaiah; Gholap Sachin P.; Chaudhari Dipali A. Recent advances in the Overman rearrangement: synthesis of natural products and valuable compounds. Org. Biomol. Chem 2017, 15, 2672. [DOI] [PubMed] [Google Scholar]
- (18).Madrahimov ST; Markovic D; Hartwig JF The Allyl Intermediate in Regioselective and Enantioselective Iridium-Catalyzed Asymmetric Allylic Substitution Reactions. J. Am. Chem. Soc 2009, 131, 7228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Remarkably, primary aliphatic amine react with linear allylic acetates under similar conditions to form products of hydroamination with complete levels of acetate-directed 1,3-regioselectivity: Kim SW; Wurm T; Brito GA; Jung W-O; Zbieg JR; Stivala CE; Krische MJ Hydroamination versus Allylic Amination in Iridium Catalyzed Reactions of Allylic Acetates with Amines:1,3-Aminoalcohols via Ester-Directed Regioselectivity. J. Am. Chem. Soc 2018, 140, 9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) For related deuterium labelling experiments, see:Zhang P; Brozek LA; Morken JP Pd-Catalyzed Enantioselective Allyl–Allyl Cross-Coupling. J. Am. Chem. Soc 2010, 132, 10686. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen J-P; Peng Q; Lei B-L; Hou X-L; Wu Y-D Chemo- and Regioselectivity-Tunable Pd-Catalyzed Allylic Alkylation of Imines. J. Am. Chem. Soc 2011, 133, 14180. [DOI] [PubMed] [Google Scholar]
- (21).For a comparison of 4 closely related π-allyliridium C,O-benzoate complexes, see: Schmitt DC; Dechert-Schmitt A-MR; Krische MJ Iridium-Catalyzed Allylation of Chiral β-Stereogenic Alcohols: Bypassing Discrete Formation of Epimerizable Aldehydes. Org. Lett 2012, 14, 6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.