

Abstract
Neuroprotective agents administered post cerebral ischemia have failed so far in the clinic to promote significant recovery. Thus, numerous efforts were redirected towards prophylactic approaches such as preconditioning as an alternative therapeutic strategy. Our lab has revealed a novel long-term window of cerebral ischemic tolerance mediated by resveratrol preconditioning (RPC) that lasts for two weeks in mice. To identify its mediators, we conducted an RNA-seq experiment on the cortex of mice two weeks post RPC, which revealed 136 differentially expressed genes. The majority of genes (116/136) were downregulated upon RPC and clustered into biological processes involved in transcription, synaptic signaling, and neurotransmission. The downregulation in these processes was reminiscent of metabolic depression, an adaptation used by hibernating animals to survive severe ischemic states by downregulating energy-consuming pathways. Thus to assess metabolism, we used a neuronal-astrocytic co-culture model and measured the cellular respiration rate at the long-term window post RPC. Remarkably, we observed an increase in glycolysis and mitochondrial respiration efficiency upon RPC. We also observed an increase in the expression of genes involved in pyruvate uptake, TCA cycle, and oxidative phosphorylation, all of which indicated an increased reliance on energy-producing pathways. We then revealed that these nuclear and mitochondrial adaptations, which reduce the reliance on energy-consuming pathways and increase the reliance on energy-producing pathways, are epigenetically coupled through acetyl-CoA metabolism and ultimately increase baseline ATP levels. This increase in ATP would then allow the brain, a highly metabolic organ, to endure prolonged durations of energy deprivation caused by cerebral ischemia.
Keywords: Resveratrol, cerebral ischemia, preconditioning, brain metabolism, brain transcriptomic, acetyl-CoA
Introduction
The brain only constitutes around 2% of body weight, yet it consumes around 20% of total body energy [1]. This high energy demand renders it highly susceptible to ischemic injuries that result from the prolonged interruption of blood flow to the brain caused by either a hemorrhagic or ischemic stroke or a cardiac arrest [1, 2]. Despite decades of preclinical and clinical research, cerebral ischemic injuries remain one of the leading causes of death and adult disability in the United States and worldwide, where stroke ranks as the fifth leading cause of death and cardiovascular disorders rank as the first [2]. The restriction of blood flow to the brain deprives cells of sufficient oxygen and glucose needed to maintain ATP (adenosine triphosphate) production. This causes an energy failure in the brain, which affects several energy-demanding processes including the sodium-potassium ATPase. The failure of this pump causes neurons to depolarize and release excessive glutamate into the synaptic cleft, which then activates post-synaptic glutamate receptors thus initiating a complex cascade of excitotoxic events that can ultimately lead to cell death [3]. To date, numerous neuroprotective agents targeting the excitotoxic cascade have failed in the clinic to promote significant recovery [4, 5]. Currently, the tissue plasminogen activator (tPA) and mechanical thrombectomy are the only two clinically-approved therapeutic approaches [2]. Yet both involve the physical removal of the clot itself rather than promote direct neuroprotection by targeting cellular or molecular processes [2]. Additionally, these therapeutic approaches can only benefit a small percentage of acute ischemic stroke patients leaving the majority of cerebral ischemic patients with no treatment [2, 6].
The failure of neuroprotective agents in the clinic is thought to result mainly from the rapid and complex cascade of events that initiate immediately after an ischemic insult thus making it difficult for a therapeutic agent to be capable of halting this complex cascade of events within a clinically relevant time window. Hence, numerous efforts have been redirected towards prophylactic approaches such as preconditioning as an alternative therapeutic strategy for the treatment of cerebral ischemia. [5, 7]. Ischemic preconditioning (IPC) is defined as a phenomenon whereby a non-injurious ischemic insult protects against a subsequent injurious one by activating endogenous mechanisms of ischemic protection [7]. This phenomenon could be mimicked by the administration of pharmacological compounds such as resveratrol (3,5,4′-trihydroxystilbene), a natural polyphenol found on the skin of grapes and berries as well as in red wine [8]. Our group and others have observed ischemic tolerance with resveratrol preconditioning (RPC) using several in vitro and in vivo models of cerebral ischemia [8–13].
It was previously thought that the long-term window (LTW) of ischemic tolerance can last for a maximum of one week after preconditioning [7]. In an attempt to extend this window, Stowe et al. in 2011 exposed mice to repetitive hypoxic stimuli where they were able to prolong the window of ischemic tolerance to a period of 8 weeks [14]. Similarly, our lab tested different paradigms of RPC in an attempt to extend the window of ischemic tolerance mediated by pharmacological preconditioning. The results of the study remarkably revealed a novel window of ischemic tolerance that lasted for at least two weeks in vivo after a single administration of resveratrol; making it the longest window of ischemic tolerance discovered to date by a single administration of a pharmacological agent [9]. This finding led us to further investigate its underlying mechanism in our current study.
In an attempt to identify candidate genes and adaptations that promote this long-term window of ischemic tolerance, we first aimed to characterize the global transcriptomic changes induced in the brain upon RPC within the two-week window. Previous studies have aimed to characterize these changes within the 24–72 hour time-window and using preconditioning stimuli that were mostly hypoxic or ischemic [15]. The results of our transcriptomic dataset has redirected our focus towards metabolism. Taken together, the genomic and metabolic results revealed a novel endogenous mechanism of long-term cerebral ischemic tolerance characterized by bioenergetic efficiency in the brain. This adaptive mechanism involves nuclear and mitochondrial adaptations epigenetically coupled through acetyl-CoA metabolism. These coupled adaptations increased the cells’ reliance on energy producing pathways and reduced their reliance on energy consuming pathways. This, in turn, increases baseline ATP levels allowing the brain, a highly metabolic organ, to endure prolonged durations of energy deprivation caused by cerebral ischemia. Knowing that the initial trigger behind the excitotoxic injury is an energy failure in the brain, our study should foster future efforts to focus on activating these long-lasting endogenous mechanisms to promote ischemic tolerance rather than only aim to halt or reverse the excitotoxic pathway after its initiation, an attempt that has failed in the clinic to promote recovery.
The translation of resveratrol among other preconditioning agents to the clinic would be of therapeutic value to the general population as a whole and in particular to patients with a high proclivity to stroke. These patients include those with comorbidities such as diabetes, hypertension, and cardiovascular disorders as well as patients undergoing long invasive surgeries that subjects their brains to temporary states of ischemia [2, 16]. Additionally, knowing that individuals above the age of 55 have a lifetime risk of stroke of 1 in 6, preconditioning would also be of great therapeutic potential to our aging population in general [17].
Results
RPC induces a global downregulation in gene expression within the cortex during the LTW of ischemic tolerance.
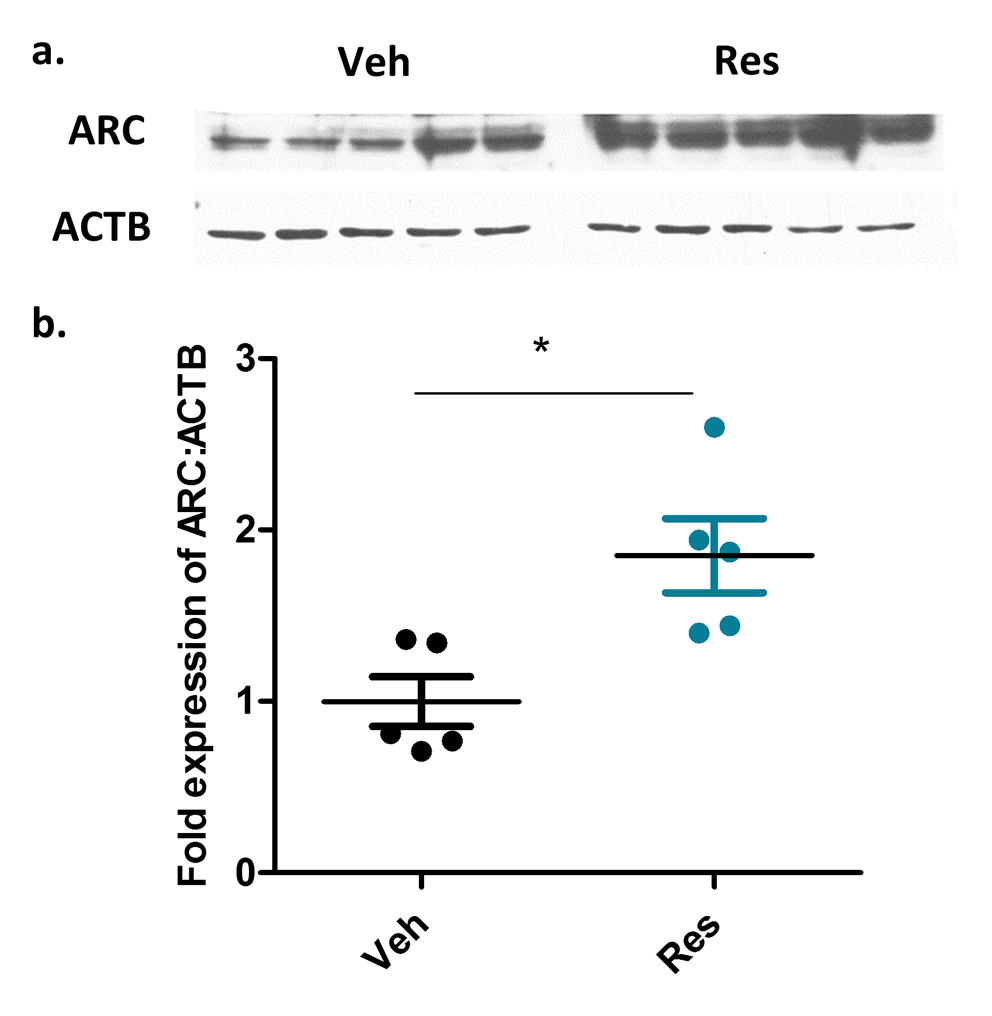
In order to identify candidate genes and adaptations that promote the RPC-induced long-term cerebral ischemic tolerance, we performed an unbiased transcriptomic analysis on the cortex of mice two weeks following an i.p. injection of resveratrol (Res) or vehicle (Veh) (10mg/kg) (Fig.1a). We chose the cortex since we have previously reported cortical neuroprotection promoted by RPC using a mouse model of middle cerebral artery occlusion (MCAo) [9]. Using RNA sequencing (RNA-seq) analysis, we identified 136 differentially expressed genes (False discovery rate (FDR)<0.1) within the two-week window following RPC. The majority of these genes consisting of 116/136 (85%) were downregulated upon RPC while only 20/136 (15%) were upregulated (Fig.1b) (the complete list of genes is provided in Supplementary Table 1). In order to validate the results of our RNA-seq experiment, we measured the expression of randomly selected differentially expressed genes using real-time PCR analysis. The results showed a high correlation (R-square 0.95) between the changes in gene expression revealed by the RNA-seq experiment compared to real-time PCR (Fig.1c). We then performed pathway using the Genomatix Pathway System (GePS). The results revealed several biological processes altered upon RPC including transcription from RNA-polymerase II, synaptic signaling, regulation of ion transmembrane transport, regulation of neurotransmitter levels, and histone acetylation (Fig.1d). The majority of the genes clustered in these pathways exhibited a trend of downregulation, an interesting observation that we speculated may reflect a reduced state of cellular activity induced by RPC. Despite the global downregulation in gene expression, we observed a significant increase in the mRNA levels of the immediate early genes (IEGs) Arc (activity-regulated cytoskeleton associated protein), Fos (Fos proto-oncogene), JunB (JunB proto-oncogene), Npas4 (Neuronal PAS Domain Protein 4), and Nr4a1 (Nuclear Receptor Subfamily 4 Group A Member 1) (Fig.1c&d). We also observed a significant increase in the protein levels of the immediate early gene ARC in the two-week window post-RPC (Supplementary Fig.1).
Fig. 1. Identifying the transcriptomic changes induced in the cortex by RPC within the long-term window of ischemic tolerance.

a. Schematic showing the experimental design followed. (n=3 per group). b. Volcano plot displaying the expression level and significance value for the genes identified from the RNA-seq analysis. Differentially expressed genes (FDR<0.1) are displayed in color (red is upregulated, green is downregulated). c. Validating the expression of differentially expressed genes identified from the RNA-seq analysis using real-time PCR. d. Pathway analysis performed on the differentially expressed genes using Genomatix Pathway System (GePS) (red is upregulated, green is downregulated).
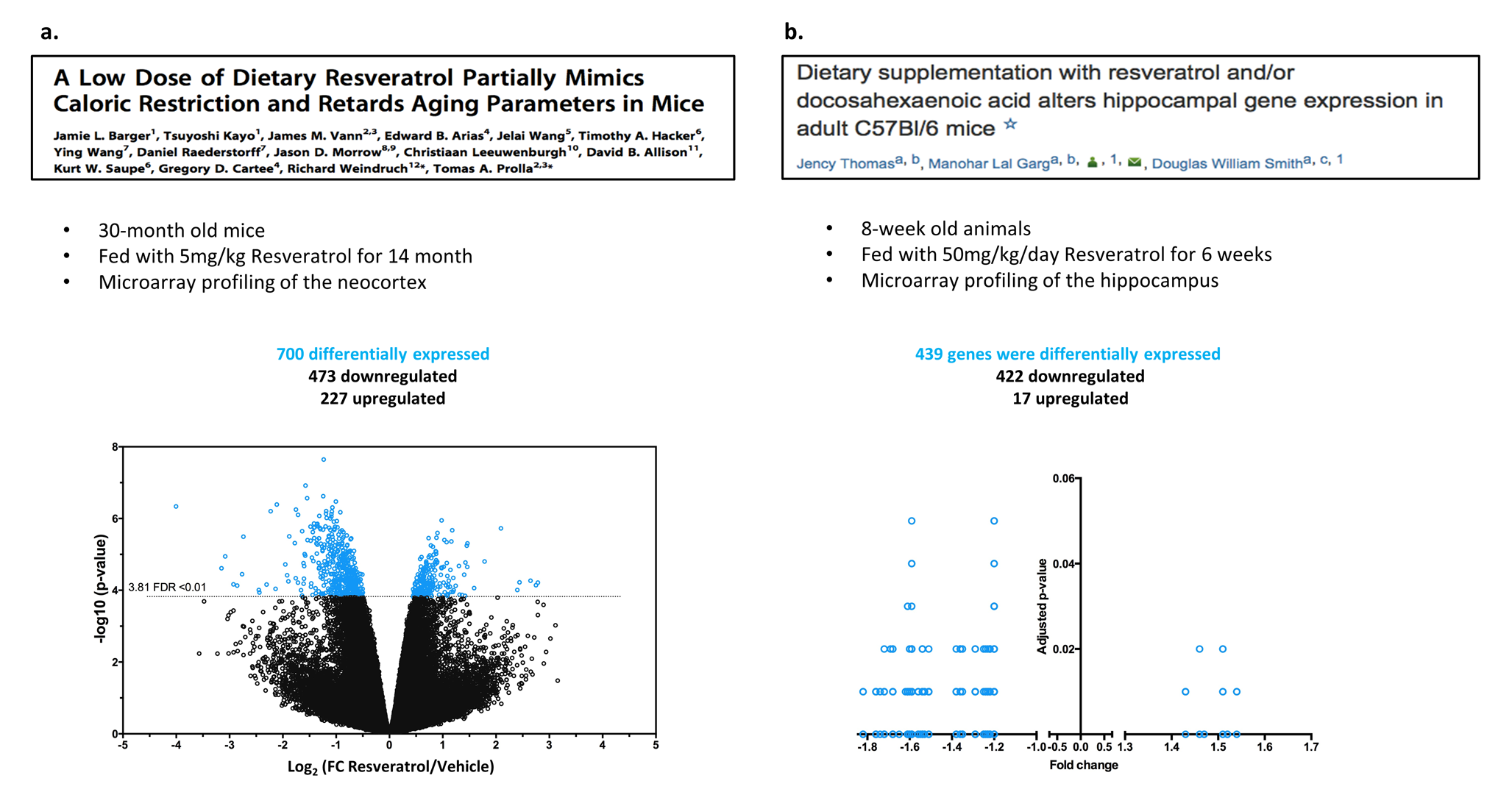
Furthermore, we analyzed the results of two publically available datasets that were conducted by Barger et al. in 2008 and Thomas et al. in 2013 both of which involved the transcriptomic analysis of mouse brains after resveratrol administration [18, 19]. Consistent with our results, the two studies also revealed a global downregulation in gene expression in the brains of mice upon resveratrol treatment (Supplementary Fig.2 and Tables2&3).
RPC induces long-term ischemic tolerance in a model of neuronal-astrocytic co-cultures.
To determine whether the global downregulation in gene expression actually reflects a reduced state of metabolism as suggested by previous studies [20, 21], we aimed to measure the rate of metabolism using an in vitro model of long-term ischemic tolerance. It was previously reported that resveratrol has short-term protective effects against oxygen and glucose deprivation (OGD) in primary cortical cultures [22–24]. Thus to develop an in vitro model of long-term RPC, we used primary neuronal-astrocytic co-cultures to better represent the cellular complexity of the cortex (Supplementary Fig.3). We exposed the 8 DIV (days in vitro) neuronal-astrocytic co-cultures to a single treatment of Res/Veh (25–100uM) and maintained them for 2, 4, 6, or 8 days post-treatment. In order to mimic an ischemic insult, we exposed the cells to an OGD at the respective time points (Fig.2a). Cell death was then assessed 24hrs post OGD using lactate dehydrogenase assay (LDH) and propidium iodide (PI) staining. The results of these experiments revealed that 100μM of Res promoted the highest level of protection that lasted for 8 days post-treatment (Fig.2b). Lower doses of Res such as the 75μM were also protective starting days 4 to 8 while the 50μM dose was only protective at day 6 (Fig.2b). Even though we observed ischemic tolerance at 8 days post RPC, we chose the 6-day time-point as our LTW since the overall health of the cultures declined past that time point. To further validate our results, we stained the cultures with PI and Hoechst 24hrs post-OGD at the six-day time-point (Fig.2c). The results revealed a significant (p<0.001) reduction in the PI:Hoechst stained cells in the Res group (9.8%) compared to Veh (35.9%) 24-hrs post OGD which further supported the LDH assay results (Fig.2d). Remarkably, there was no difference (p>0.05) in cell death of the Res group exposed to OGD (9.8%) compared to sham wells (Veh 6.7%; Res 11.4%), which reflected the robustness of the protection induced by RPC (Fig.2d).
Fig. 2. Identifying the long-term window of cerebral ischemic tolerance mediated by resveratrol preconditioning in vitro using primary neuronal-astrocytic co-cultures.

a. Schematic showing the experimental paradigm followed. b. Bar graph showing the level of cell death as revealed by the LDH activity in the supernatant of the cultures 24hrs post OGD normalized to a non-treated sham control wells. (Two-way ANOVA, post-hoc Bonferroni matched values (n=3–4); ns not significant; * p<0.05, ** p<0.01, *** p<0.001). c. Representative images of the propidium iodide (PI) and Hoechst stained cells 6d post RPC and 24hrs post OGD/sham. d. Bar graph showing the percentage of cell death revealed by the PI to Hoechst staining. (One-way ANOVA, Bonferroni multiple comparison posttest (n=6–9); with averages of at least eight images per well; ns not significant; *** p<0.001).
The in vitro model of RPC-induced long-term cerebral ischemic tolerance recapitulates in vivo transcriptomic changes.
To determine if RPC induces similar transcriptomic changes in vitro using the primary neuronal-astrocytic co-culture model compared to the in vivo model, we measured the expression of several genes identified from the RNA-seq analysis using real-time PCR analysis at the six-day time-point (Fig.3a–b). The genes measured were selected based on their involvement in gene expression regulation: Polr2a (RNA polymerase II subunit A), vesicular transport and neurotransmitter release: Wasf1 (was protein family member 1), Vamp2 (vesicle associated membrane protein 2), Doc2B (double c2 domain beta), Shank1 (sh3 And multiple ankyrin repeat domains 1), and Smap2 (small ArfGAP2), or act as ion channels: Scn9a (sodium voltage-gated channel alpha subunit 9). Strikingly, we observed high similarities in the trend of expression of several of these genes between the in vitro and in vivo models. With the in vitro model, we observed a significant downregulation in the Polr2a (77%), Wasf1 (79%), Doc2B (64%), and Scn9a (71%) genes as revealed by the percent expression compared to vehicle (Fig.3b). Similarly, with the in vivo model, we also observed a downregulation in these genes upon RPC Polr2a (70%), Wasf1 (60%), Doc2B (64%), and Scn9a (57%) (Fig.3a). Together, the observed consistency in gene expression between the two models further adds validity to the use of our in vitro model.
Fig. 3. The in vitro model of RPC-induced long-term ischemic tolerance recapitulates in vivo transcriptomic changes.

a. The percent mRNA levels for the differentially expressed genes as revealed by RNA-seq analysis performed on the cortex of mice 2 weeks post RPC (n=3). b. The percent mRNA levels for the same genes as revealed by real-time PCR analysis performed at 6 days post RPC using the in vitro rat neuronal-astrocytic co-culture model. (n=7; paired Student’s t.test; ns not significant; * p<0.05).
RPC alters the cellular respiration rate within the LTW of cerebral ischemic tolerance.
To measure the rate of metabolism of the neuronal-astrocytic co-cultures, we performed live cell metabolic flux analysis using the Seahorse Bioscience Technology. This assay allows the dynamic measurement of metabolic parameters of glycolysis and oxidative phosphorylation in real-time. We first assessed the oxygen consumption rate (OCR) of the neuronal-astrocytic co-cultures at the 6-day time-point post RPC as a measure of oxidative phosphorylation (Fig.4a). The results revealed a significant (p<0.05) decrease in the OCR used for both basal and maximal ATP production in the Res group (3.97 fmol/min/cell; 5.5 fmol/min/cell) compared to Veh (4.60 fmol/min/cell; 6.7 fmol/min/cell) (Fig.4b). Similarly, the OCR used by the proton leak was also significantly reduced upon RPC (Res 0.82 fmol/min/cell, Veh 0.92 fmol/min/cell) (p<0.05) (Fig.4b). We then assessed the extracellular acidification rate (ECAR) as a measure of glycolysis (Fig.4c). Interestingly, the results revealed a significant increase (p<0.05) in both basal and maximal rates of glycolysis in the Res group (1.3 μpH/min/cell; 2.7 μpH/min/cell) compared to Veh (1.1 μpH/min/cell; 2.4 μpH/min/cell), with no significant changes (p>0.05) in the cells’ glycolytic reserve (Fig.4d). To confirm that the changes in cellular respiration are not due to an alteration in the proportion of neurons to astrocytes, we fixed the cells 6 days post-RPC and stained them with NeuN and DAPI. We noticed no significant changes (p>0.05) in the percentage of neurons in either the Res group (41%) or Veh (35%) (Supplementary Fig.4), thus suggesting that the observed changes in metabolic parameters actually reflect intrinsic changes in metabolism. Furthermore, the immunostaining results also showed that the co-culture model consists of approximately 40% neurons at the LTW which further provides additional validation to the use of our in vitro model knowing that the cortex of young rodent animals possesses a similar ratio of neurons in vivo [25].
Fig. 4. The effect of RPC on the respiration rates of the neuronal-astrocytic co-cultures within the long-term window of ischemic tolerance.

a. Oxygen consumption rates (OCR) of the neuronal-astrocytic co-cultures measured using the Seahorse Biosciences Technology at the six day time-point post RPC before and after the sequential injection of the mitochondrial stressors (Oligomycin, FCCP, Rotenone/Antimycin A). b. Bar graphs showing the calculated OCR used to produce ATP at the basal and maximal level and OCR consumed by the mitochondrial proton leak (see methods). c. Extracellular acidification rates (ECAR) of the neuronal-astrocytic co-cultures measured using the Seahorse Biosciences Technology at the six day time-point post RPC in the absence of glucose and after the sequential injection of glucose, oligomycin, and 2-deoxyglucose (2-DG). d. Bar graphs showing the calculated basal rates of glycolysis, glycolytic capacity, and glycolytic reserve of the cells (see methods). (n=9 biological replicates with 3 technical replicates; paired Student’s t.test; ns not significant, * p<0.05).
RPC increases mitochondrial abundance within the LTW of ischemic tolerance in vitro.
Since we observed reduced basal and maximal OCR at the LTW, we wanted to determine if this reduction was due to a decrease in mitochondrial abundance. Thus at the 6-day time-point post RPC, we measured the relative abundance of mitochondrial to nuclear DNA using real-time PCR analysis with two sets of primers that exclusively detect mitochondrial Atp6 (mitochondrially encoded ATP synthase membrane subunit 6); Nd1 (mitochondrially encoded NADH:Ubiquinone oxidoreductase core subunit 1) and nuclear DNA Actb (Actin Beta); Neb (nebulin). Remarkably, with both sets of primers, we observed a significant increase in the mitochondrial to nuclear DNA ratio in the Res group compared to Veh. (Atp6:Actb, Res 166, Veh 100, p<0.05; Nd1:Neb Res 246, Veh 128, p<0.001) (Fig.5a). To further confirm the increase in mitochondrial abundance, we measured the mRNA levels of mitochondrial-encoded genes with real-time PCR analysis using primers that amplify the minimum number of nuclear pseudogenes. We also observed a significant increase in the mRNA levels of mitochondrial-encoded genes involved in oxidative phosphorylation Mt-co2 (mitochondrially encoded cytochrome c oxidase II) (139%), Mt-co3 (mitochondrially encoded cytochrome c oxidase III) (147%), Mt-nd4 (mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 4) (137%), Mt-nd5 (mitochondrially encoded NADH:Ubiquinone oxidoreductase core subunit 5) (109%), and Mt-atp6 (mitochondrially encoded ATP synthase membrane subunit 6) (133%) (Fig.5b). We also measured the expression of nuclear-encoded mitochondrial genes involved in diverse mitochondrial functions (Fig.5b). We assessed genes involved in: 1) protein import into the mitochondria Tim22 (translocase of inner mitochondrial membrane 22) and Tom22 (translocase of outer mitochondrial membrane 22), 2) mitochondrial biogenesis Pgc1α (pparg coactivator 1 alpha) and Tfam (transcription factor a, mitochondrial), and 3) oxidative phosphorylation Atp5s (ATP synthase, H+ transporting, mitochondrial Fo complex subunit S (Factor B) and Cox4i1 (cytochrome c oxidase subunit 4I1). Among these nuclear-encoded mitochondrial genes, we did not observe any significant alterations upon RPC (Fig.5b). This result was consistent with our RNA-seq experiment results where no significant changes in nuclear-encoded mitochondrial genes was observed. To further assess mitochondrial abundance, we stained the cells with the fluorescent Mitotracker Green probe, which binds the mitochondria independent of membrane potential (Fig.5c). By normalizing the fluorescence intensity of the cells to cell number, we also observed a significant increase in mitochondrial abundance in the Res group (0.00150 A.U/cell) compared to Veh (0.00057 A.U/cell) (Fig.5d). Thus, taken together although the mixed neuronal-astrocytic cultures have significantly reduced basal and maximal OCR in response to RPC, they exhibit an increase in mitochondrial abundance within the LTW of ischemic tolerance.
Fig. 5. RPC increases the mitochondrial abundance of the rat neuronal-astrocytic co-cultures within the long-term window of ischemic tolerance.

a. Measuring the mitochondrial to nuclear DNA ratio using real-time PCR analysis with two rat-specific primer sets that uniquely amplify mitochondrial (Atp6, Nd1) or nuclear (Actb, Neb) DNA at the six day time point post RPC (n=7; paired Student’s t.test; * p<0.05, *** p<0.001). b. Measuring the mRNA levels of mitochondrial- or nuclear- encoded genes involved in oxidative phosphorylation (Mt-co2, Mt-co3, Mt-nd4, Mt-nd5, Atp5s, Mt-Atp6, Cox4i1), import of proteins to the mitochondria (Tim22, Tom22), or mitochondrial biogenesis (Pgc1α, Tfam) (n=5–7; paired t-test; ns not significant, * p<0.05, ** p<0.01, *** p<0.001). c. Representative fluorescent images of live neuronal-astrocytic co-cultures stained with Mitotracker Green and Hoechst at the six day time point post-RPC (40X magnification). d. Quantification of the fluorescence intensity of the Mitotracker green stained cells normalized to cell number (n=3; paired Student’s t-test; * p<0.05).
RPC increases mitochondrial abundance within the LTW of ischemic tolerance in vivo.
We then wanted to determine if the increase in mitochondrial abundance in response to RPC is also occurring in vivo. Since the analysis of the RNA-seq experiment did not involve mitochondrial-encoded mRNA (see methods), we measured the expression of mitochondrial-encoded genes in the cortex of mice 2 weeks post-RPC using real-time PCR analysis with primers that amplify the minimum number of nuclear pseudogenes. Similar to our in vitro model, we also observed a significant increase in the mRNA levels of the three mitochondrial-encoded genes Mt-co2 (115%), Mt-co3 (113%), Mt-nd4 (119%) upon RPC with no significant changes in the levels of Mt-nd5 (106%) (Fig.6a). Additionally, we assessed the relative abundance of the mitochondrial to nuclear DNA using two sets of mitochondrial (Rnr1 (RNA, ribosomal 45s cluster 1), Nd1) and nuclear primers (ActB, Neb) with real-time PCR analysis which also revealed an increase in the mitochondrial:nuclear DNA levels (Fig.6b). Using the Mt-rnr1:ActB we observed a significant (p<0.05) increase in the Res group (121) compared to vehicle (86) while using Nd1:Neb we observed a non-significant (p-0.16) increase in the Res group (113) compared to vehicle (96). We then performed western blot analysis on the cortex of mice within the two-week window where we observed a significant increase in the protein levels of COXIV (cytochrome c oxidase subunit 4) (185%, p<0.05) and VDAC (voltage dependent anion channel) levels (137%, p<0.001) in the Res group compared to Veh, with no significant (p>0.05) changes in the ATPB (atp synthase subunit beta) protein levels (Fig.6c&d). Together, these results supported the observation that RPC also increases mitochondrial abundance within the LTW of ischemic tolerance in vivo.
Fig. 6. RPC increases mitochondrial abundance in the mouse cortex within the long-term window of ischemic tolerance.

a. Measuring the mRNA levels of mitochondrial-encoded genes involved in oxidative phosphorylation (Mt-co2, Mt-co3, Mt-nd4, Mt-nd5) in the mouse cortex two weeks post RPC (n=10; paired Student’s t-test; ns not significant, * p<0.05). b. Measuring the mitochondrial to nuclear DNA ratio using real-time PCR analysis with two mouse-specific primer sets that uniquely amplifies the mitochondrial (Mt-rnr1, Nd1) or nuclear (Actb, Neb) DNA at the two-week window post RPC (n=10; paired Student’s t.test; * p<0.05). c. Western blot analysis showing the protein levels of COXIV, VDAC, ATPB, and ACTB in the mouse cortex two weeks post RPC. d. Quantification of the western blot results of COXIV, VDAC, and ATPB relative to ACTB (n=4–5; Student’s t-test; ns not significant, * p<0.05, ** p<0.01).
RPC alters acetyl-coenzymeA (acetyl-CoA) metabolism within the LTW of ischemic tolerance.
Knowing that cells couple changes in gene expression to nutrient availability, we wanted to determine if the concomitant nuclear and mitochondrial adaptations observed at the LTW were epigenetically coupled. We chose to focus on acetyl-CoA regulation since we observed a significant reduction in several genes involved in the regulation of histone acetylation upon RPC (Fig.1d,7a). We postulated that RPC increases acetyl-CoA availability in the mitochondria in order to maximize mitochondria ATP production, while its export to the nucleus is reduced in order to reduce its availability to epigenetic enzymes that promote transcriptional activation. To test this, we first measured the mRNA levels of several genes involved in acetyl-CoA metabolism at the 6-day time point post RPC using the neuronal-astrocytic culture model. The genes included those involved in the import of pyruvate to the mitochondria (Mpc1 (mitochondrial pyruvate carrier 1), Mpc2 (mitochondrial pyruvate carrier 2)), the conversion of pyruvate into acetyl-CoA Pdha1 (pyruvate dehydrogenase E1 alpha 1 subunit), Dlat (dihydrolipoamide s-acetyltransferase), and Dld (dihydrolipoamide dehydrogenase). We also measured the enzyme involved in the condensation of acetyl-CoA with oxaloacetate to produce citrate Cs (citrate synthase), the mitochondrial carrier involved in citrate transport to the cytosol Slc25a1 (solute carrier family 25 member 1), and the enzyme that breaks down citrate into acetyl-CoA in the nucleus and cytosol Acly (atp citrate lyase). As expected, we observed a significant increase in the enzymes involved in the import of pyruvate into the mitochondria Mpc2 (152%) (p<0.001) and entry of acetyl-CoA into the TCA cycle Cs (139%) (p<0.01) (Fig.7b). Additionally, we observed a significant reduction in the Acly (70%) gene which codes for an enzyme involved in the breakdown of citrate into acetyl-CoA in the cytosol and nucleus (Fig.7b).
Fig. 7. RPC alters acetyl-CoA metabolism and ATP levels within the long-term window of ischemic tolerance.

a. Schematic depicting the catabolic pathways that breakdown glucose into acetyl-coA and the two subsequent paths for acetyl-CoA metabolism involving either the TCA cycle or its extra-mitochondrial transport in the form of citrate. (Circles with (+) sign indicates enzymes are upregulated at the mRNA level; circle with (–) sign indicates downregulation, circle with no sign indicate no change). b. Bar graph showing the mRNA levels of enzymes involved in acetyl-coA metabolism at the six day time-point post RPC (normalized to geometric mean of six nuclear endogenous controls) (n=7; paired Student’s t-test; * p<0.05, ** p<0.01, *** p<0.001). c. Western blot analysis for histone lysine acetylation marks H3K9ac and H4K16ac relative to total H3 and H4. Bar graphs representing the percent expression of proteins based on the western blot analysis (n=3–4; Mann-Whitney; * p<0.05). d. Bar graph representing ATP levels per cell measured at the six day time point post RPC. E. mRNA levels of the uncoupling protein (Ucp) measured at the six day time point post RPC (normalized to the geometric mean of six endogenous controls) (n=7; paired Student’s t-test; ns not significant, * p<0.05).
Since acetyl-CoA in the nucleus serves the substrate for histone acetyltransferases (HATs), we measured total levels of histone 3 lysine 9 acetylation (H3K9ac) and histone 4 lysine 16 acetylation (H4K16ac) using western blot analysis. Both H3K9ac and H4K16ac are well-characterized epigenetic marks that highly correlate with transcriptional activation. Consistent with the observed reduction in the enzyme that produces nuclear acetyl-CoA (Acly), we found a significant decrease (p<0.05) in the abundance of H3K9ac (42%) and H4K16ac (not detectable) at the LTW post RPC (Fig.7c), which further supported the observed global downregulation in gene expression mediated by RPC.
Since we observed increased reliance on energy-producing pathways (glycolysis, pyruvate uptake, citrate synthesis, oxidative phosphorylation) along with reduced reliance on energy-consuming pathways (reduced transcriptional activity, epigenetic marks), we wanted to determine if these coupled adaptations affect basal ATP levels at the LTW. To test this, we used the ATP bioluminescence kit at the 6-day time point where we observed a significant (p<0.05) increase in the ATP levels of the Res group (8.5 × 10−16 nmols/cell) compared to vehicle (9.0 × 10−17 nmols/cell) which consisted of a remarkable nine fold increase in basal ATP levels (Fig.7d). This result strongly supported our observation of altered metabolism and enhanced bioenergetic efficiency upon RPC.
Despite the significant increase in mitochondrial abundance (Fig.5), and the expression of enzymes involved in pyruvate uptake, citrate synthesis, and oxidative metabolism (Fig.5b&7b) along with the increase in basal ATP levels (Fig.7d), both basal and maximal OCR were significantly reduced upon RPC. In an attempt to explain this reduction, we wanted to determine if the expression of uncoupling proteins (UCPs) is altered upon RPC (Fig.7e). Uncoupling proteins are found on the inner mitochondrial membrane and allow the movement of protons across the inner membrane thus dissipating the proton gradient used to power the ATP-synthase. This, in turn, reduces the efficiency of ATP synthesis and increases oxygen consumption [26]. Thus we measured the mRNA levels of Ucp1–5 at the 6 days where we observed a significant (p<0.05) decrease in Ucp4 (73.9%) and a non-significant (p-0.11) decrease in Ucp2 (69.7%). Ucp1 and Ucp3 were not detected in our cellular model and Ucp5 (104.9%) was not altered (Fig.7e). The reduction in Ucp2 and Ucp4 suggested reduced proton leaks across the inner mitochondrial membrane and as a consequence enhanced ATP synthesis efficiency due to increased coupling between oxygen reduction and ATP production (Fig.4e).
Acute treatment with resveratrol inhibits mitochondrial respiration in the neuronal-astrocytic co-cultures.
In an attempt to identify the initial trigger that may cause this enhanced bioenergetic efficiency within the LTW, we wanted to determine if the acute exposure to resveratrol affect the cells’ ability to produce energy. Despite its well-known health benefits, resveratrol in nature is found on the skin of grapes and berries and is thought to protect the plant from fungal infections by acting as a mitochondrial respiration inhibitor [27–29]. Thus, to test if the preconditioning dose of Res (100μM) inhibits cellular respiration, we measured oxygen consumption (OCR) and extracellular acidification rates (ECAR) immediately following acute resveratrol application. When normalized to baseline rates, the acute treatment with Res (100μM) caused a small but significant (p<0.01) drop in OCR (90.8%) compared to Veh (98.1%). This was concomitant with a small but non-significant (p-value 0.16) increase in ECAR in the Res group (114.0%) compared to Veh (109.7%) (Fig.8a&b). This data suggests that resveratrol acutely inhibits mitochondrial respiration, in turn, allowing more substrates to be available for lactic acid production. To confirm this observation, we used a higher dose of Res (500μM) where we observed a more pronounced and significant (p<0.01) drop in OCR rates in the Res group (86.0%) compared to Veh (97.35%) concomitant with a significant increase (p<0.01) in ECAR rates in the Res group (120.4%) compared to Veh (106.2%) (Fig.8c&d). Together, the results of this experiment further supported previous finding suggesting that resveratrol acutely inhibits mitochondrial respiration.
Fig. 8. Acute treatment with resveratrol inhibits mitochondrial respiration.

a&c. Oxygen consumption rates (OCR) measured in non-treated cells before and after the sequential injection of Veh/Res (A:100μM; C: 500μM), FCCP, and Rotenone/Antimycin A. Bar graphs represent the average of the three time points measured after the acute injection of Veh/Res using either the 100μM or 500μM Res. b&d. Extracellular acidification rates (ECAR) measured in non-treated cells before and after the sequential injection of Veh/Res (B:100μM; D:500μM), FCCP, and Rotenone/Antimycin A. Bar graph representing the average of the three time points measured after the acute injection of Veh/Res using either the 100μM or 500μM Res. (n=3 biological replicates; paired Student’s t-test; ns not significant, * p<0.05, ** p<0.01).
Discussion
Our study has revealed a previously uncharacterized endogenous mechanism of ischemic tolerance induced in the brain by RPC that leads to bioenergetic efficiency (Fig.9) The components of this adaptive response involves epigenetically-coupled nuclear and mitochondrial adaptations involving acetyl-CoA metabolism and histone acetylation. These coupled responses reduce the cells’ reliance on energy consuming pathways while increasing their reliance on energy producing pathways thus leading to an increase in baseline ATP levels, allowing the cells to endure prolonged durations of energy deprivation caused by cerebral ischemia.
Fig. 9. Schematic depicting the genomic and metabolic adaptation induced by RPC within the long-term window of cerebral ischemic tolerance leading to bioenergetic efficiency.

The acute treatment with resveratrol mildly inhibits mitochondrial respiration. As a long-term adaptation to this stressful response, cells of the cortex develop a bioenergetically efficient phenotype characterized by increased reliance on energy-producing pathways and reduced reliance on energy-consuming pathways. The nuclear adaptations to this stressful response involve a reduction in the abundance of epigenetic marks that promote transcriptional activation (H3K9ac, H4K16ac). This correlates with reduced global gene expression levels specifically in genes involved in the regulation of membrane potential, neurotransmitter regulation, and gene expression all of which are energy-demanding biological processes. While the long-term metabolic adaptations to RPC involve an increase in the glycolytic rates, expression of enzymes involved in pyruvate uptake, citrate synthesis, and oxidative phosphorylation, as well as an increase in mitochondrial abundance and respiration efficiency all of which indicate increased investment in energy production. Additionally, the RPC-induced mitochondrial and genomic adaptations are coupled through epigenetic mechanisms involving acetyl-CoA metabolism ultimately increasing basal ATP levels, which in turn would delay the initiation of the excitotoxic injury caused by cerebral ischemia. (Circles with a (+) sign indicate increased abundance or activity; Circles with a (–) sign indicate downregulation or reduced activity).
Metabolic depression is an adaptation used in nature by hibernating animals and hypoxia-tolerant organisms to survive severe states of hypoxia and hypoglycemia [30, 31]. By depressing their rate of metabolism such that their ATP consumption rate matches their ATP production rate, animals survive these ischemic states with no detrimental damages to their organs including the brain [32]. In 2003 and 2004, two studies by Stenzel-Poore et al. have suggested that IPC induces a similar state of metabolic depression in the brains of ischemic preconditioned mice that creates the ischemic tolerant phenotype [20, 21]. Subsequently, other studies have also reported similar findings [7, 33–35]. Our study further supports these observations; as we also noted a global downregulation in gene expression upon RPC in genes involved in the transcription from RNA-polymerase II promoter, synaptic signaling, regulation of ion transmembrane transport, and regulation of neurotransmitter levels, all of which are energy-demanding processes. We further reported this observation using our in vitro model of primary neuronalastrocytic co-cultures. Although this model does not completely recapitulate the cellular complexity found in vivo including oligodendrocytes, microglia, and endothelial cells, it suggests that many of these adaptations are intrinsic to neurons and/or astrocytes. The downregulation in these energy-demanding processes suggested a reduced state of cellular and electrical activity. Consistent with this observation, our lab previously reported a reduction in the electrical activity of organotypic hippocampal slices in response to ischemic preconditioning and to another form of pharmacological preconditioning involving Protein kinase C epsilon (PKCɛ) activation [36, 37]. Remarkably, despite the global downregulation in gene expression, we observed a significant increase in the expression IEGs at the LTW despite their known transient and dynamic nature of expression. IEGs are well-known mediators of neuroprotection seen in hibernating animals [38]. Additionally, our lab previously reported that the IEG Arc is needed to promote the cerebral ischemic tolerance mediated PKCɛ activation [39]. By internalizing glutamate receptors Arc delays the initiation of anoxic depolarization [39]. Thus, the observed increase in Arc levels in response to RPC further supports our observation of reduced electrical activity upon preconditioning. It is important to note that the term metabolic depression may lead to confusing interpretations as it may suggest both reduced energy-producing and -consuming activities. Previous studies have suggested that metabolic depression including both reduced energy-producing and -consuming pathways is the protective mechanism behind the observed ischemic tolerance [7, 33–35]. As opposed to these studies, the comprehensive results from our study suggest that in response to RPC although we observe a reduction in energy-consuming activities, this does not correlate with a reduction in energy-producing activities as we further discuss below.
To determine whether RPC affects the rate of metabolism, we measured the rate of cellular respiration of the neuronal-astrocytic co-cultures at the 6-day time point post RPC (100μM). Interestingly, we observed a reduction in OCR along with an increase in glycolytic rates upon RPC. Initially, we assumed this observation reflects reduced reliance on oxidative phosphorylation along with increased reliance on glycolysis for ATP production, an observation termed Warburg’s effect and used by cancer cells to survive hypoxic environments. Yet, further analysis showed that this was not the case with RPC. Although the cells have reduced OCR, they exhibited significantly increased mitochondrial abundance as revealed by the mitochondrial to nuclear DNA levels and Mitotracker green staining. The cells also exhibited increased expression of genes involved in pyruvate uptake, citrate synthesis, and oxidative phosphorylation all of which suggested increased reliance on mitochondrial activity for ATP production. Additionally, we observed a significant decrease in Ucp4 suggesting reduced proton leak and increased coupling between ATP synthesis and oxygen consumption. Thus, all these observations combined, support an increased reliance on mitochondrial activity for ATP production and suggest that the reduced OCR does not reflect reduced oxidative phosphorylation rates but rather more efficient respiration. A similar observation has been made previously by our lab in a study that showed RPC reduced UCP2 levels in rat brain and increased the ATP synthesis ratio per oxygen molecule (ADP/O rates) 2 days post RPC [13]. This enhanced respiration efficiency induced by RPC would be crucial for the cells to survive hypoxic and ischemic states as it would allow them to produce ATP using fewer oxygen molecules. Additionally, our study sheds light on the importance of using caution when interpreting OCR as a measure of oxidative phosphorylation or cellular metabolism since these parameters may not always correlate [40].
Since cells couple changes in gene expression to nutrient availability by sensing intermediate metabolites of the mitochondria, we wanted to determine if the genomic and mitochondrial adaptions induced by RPC were epigenetically coupled [41]. We chose to focus on acetyl-CoA metabolism since several of the differentially expressed genes affected by RPC were involved in histone acetylation. Acetyl-CoA is the product of pyruvate breakdown in the mitochondria and the substrate for the TCA cycle. When present in limited amounts, such as during calorie restriction, acetyl-CoA remains in the mitochondria and is used for maximum ATP production [41]. Yet when present in excess, acetyl-CoA gets shuttled to the nucleus and cytosol in the form of citrate where it is broken down back to acetyl-CoA. In the nucleus, acetyl-CoA serves as the substrate for histone acetyl-transferases (HATs) that add epigenetic marks to the chromatin leading to transcriptional activation [41]. Supporting our observation of increased reliance on energy production, we observed an increased expression of genes involved in pyruvate uptake and citrate synthesis upon RPC. Additionally, we observed a reduction in the expression of the enzyme citrate lyase (Acly) suggesting reduced nucleo-cytoplasmic levels of acetyl-CoA. Since Acly indirectly regulates histone acetylation [42, 43], we measured the abundance of the histone posttranslational marks H3K9ac and H4K16ac. We chose these epigenetic marks since they highly correlate with transcriptional activation [44]. Consistent with the reduction in Acly, we observed a significant reduction in the levels of H3K9ac:H3 and H4K16ac:H4 thus further supporting the global downregulation in gene expression. Thus, RPC seems to mimic the effect of calorie restriction on acetyl-CoA regulation, where acetyl-CoA levels in the mitochondria is increased to maximize ATP production, while its export to the nucleus is reduced which limits its availability for histone acetylation. This observation further supports previous findings suggesting resveratrol is a mimetic of calorie restriction [18, 45, 46].
Consistent with the increased reliance on energy producing pathways and reduced reliance on energy consuming pathways, we observed a striking 9-fold increase in basal ATP levels at the LTW. This substantial increase in ATP levels would allow the cells to endure prolonged durations of energy deprivation encountered during a cerebral ischemia and thus delay the initiation of the excitotoxic injury.
The question remained as to why does resveratrol elicit such a pronounced metabolic change in neuronalastrocytic co-cultures. The answer to this seems to be on the acute effect of resveratrol on cells. Despite its numerous reported health benefits, resveratrol in nature it is thought to act as an antifungal agent that protects the skin of grapes and berries from fungal infections by inhibiting mitochondrial respiration [27–29]. Consistent with these reports, we observed a significant drop in OCR upon acute Res (100μM) treatment. The drop in OCR was dose-dependent as evidenced by the use of the 500μM dose. Furthermore, upon the inhibition of OCR, we observed an increase in the rate of glycolysis further suggesting that respiration is halted and additional substrates are available for glycolysis. Thus, in support of previous findings [47], our study further shows the inhibitory effects of resveratrol on oxidative phosphorylation. Since IPC is defined as a non-injurious ischemic insult that protects against a subsequent injurious one by sub-lethally inhibiting brain metabolism, it stands to reason that RPC acts in a similar fashion by mildly inhibiting mitochondrial respiration.
Resveratrol was originally discovered in a screen as a direct activator of sirtuin1 (SIRT1), a mediator of longevity in several model organisms [46]. Yet subsequent studies have refuted its direct effect on SIRT1 [48, 49]. Some of these studies have suggested that it acts indirectly by inhibiting mitochondrial respiration which in turn increases the AMP (adenosine monophosphate)/ATP levels thus activating AMPK (AMP-activated protein kinase), an upstream activator of SIRT1 [50–52]. Supporting the fact that SIRT1 is indeed a mediator of resveratrol, our lab recently reported using an inducible neuronal-specific SIRT1 transgenic mouse model, that SIRT1 is required to promote the neuroprotective effects of RPC [10]. Yet the results from our current study shows that the acute effect of resveratrol involves the inhibition of mitochondrial respiration. Thus, taken together our studies suggest that resveratrol indirectly activates SIRT1 by inhibition of mitochondrial respiration thus supporting previously made predictions [52, 53].
Pharmacological agents that act as mitochondrial inhibitors have been approved by the FDA for the treatment of several disorders such as the drug metformin, which is used for the treatment of type II diabetes [54]. Metformin acts by inhibiting mitochondrial complex I which leads to the activation AMPK that then promotes favorable outcomes in patients [54]. Interestingly, recent studies have reported neuroprotective effects of metformin against ischemic injuries using in vitro and in vivo models of cerebral ischemia [55]. Also, epidemiological reports have suggested that metformin reduces the incidence of stroke independent of its glucose-lowering effects [54–57]. Thus, knowing that the FDA-approved drug metformin acts by mildly inhibiting oxidative phosphorylation yet promotes neuroprotective effects, this may pave the way for the preconditioning agent resveratrol to reach clinical trials for the treatment of cerebral ischemia. Additional mitochondrial inhibitors have also been reported to exhibit preconditioning effects [58, 59]. Moreover, insults to the mitochondria in the form of oxygen deprivation have been shown to protect cells carrying defects in mitochondrial respiration [60, 61]. In these cells, oxygen deprivation activates the hypoxic response allowing cells to adapt to limited oxygen availability by reducing their reliance on oxidative phosphorylation for energy production [60–62]. Thus, mild form of stressors to the mitochondria are protective to cells as they activate endogenous survival mechanisms allowing the cells to adapt to the corresponding insult.
In summary, our study revealed a novel endogenous mechanism of long-term cerebral ischemic tolerance characterized by metabolic and bioenergetic efficiency in the brain (Fig.9). We showed that the initial trigger behind the preconditioning effect of resveratrol may be the inhibition of mitochondrial respiration upon acute treatment. As a consequence, the cells adapt by developing epigenetically coupled mitochondrial and nuclear adaptations through acetyl-CoA metabolism that ultimately increase baseline ATP levels. This increase in baseline energy levels would allow the brain, a highly metabolic organ, to endure prolonged durations of energy deprivation and as such delay the initiation of the excitotoxic injury. Thus, as opposed to previous studies, our study suggests that enhanced bioenergetic efficiency rather than solely metabolic depression is the underlying mechanism behind the observed ischemic tolerance, an observation that has not been previously reported in the field of cerebral ischemia.
Our discovery of this novel mechanism of neuroprotection constitutes an advancement to the field of cerebral ischemia. It should promote future therapeutic efforts to focus on its activation as an attempt to promote long-term ischemic tolerance rather than solely target the excitotoxic pathways post-ischemia, the efforts of which has failed so far in the clinic. Additionally, knowing that other neurologic and neurodegenerative disorders converge on similar mechanisms of cell death, this endogenous mechanism of neuroprotection may also be of therapeutic value to these disorders as well.
Methods
Animals
All animal usage and experimentation was approved by the Institutional Animal Care and Use Committee at the University of Miami and was in accordance with the US Public Health Service’s Policy on Humane Care and Use of Laboratory Animals. Animals were supervised and housed in facilities of the division of veterinary resources with free access to food and water. Animals were allowed to acclimate for one week before the conduction of any experiment or treatment.
Primary neuronal-astrocytic co-cultures
The primary neuronal-astrocytic co-cultures were prepared from embryonic day E19–20 rats obtained from time-pregnant Sprague-Dawley rats which were acquired from Charles River (Wilmington, MA). Embryos were harvested and decapitated, and their brains were maintained in dissection media on ice during the procedure until dissociated into single cells. Briefly, the cortices of multiple embryos was collected and pooled and then incubated with Trypsin and DNase for 15min at 37°C. The tissues were then washed and replaced with plating medium (minimum essential media, 1% Glutamax, 5% fetal bovine serum, and 15mmol/L glucose). Using a pipette, the brains were dissociated into single cells and filtered using a 70μm cell strainer. The cells were then cultured in the plating medium on poly-D lysine coated tissue culture plates. Cultures underwent half media changes every third day and were allowed to mature for more than 8 days in vitro (DIV) before any treatment to allow the maturation of neurons and astrocytes. A representative immunofluorescence staining of the neuronal-astrocytic co-cultures is presented in Supplementary Fig.3. Cells were plated at a density of 106 cells per well in a 24-well plate for the OGD (oxygen and glucose deprivation) and staining experiments. For the Seahorse experiments cells were plated at a density of 8 × 104 cells per well using the Seahorse Extracellular Flux Analyzer Miniplate (Agilent, Santa Clara, CA).
Drug Preparation and Treatments
Resveratrol (Sigma-Aldrich) was dissolved in DMSO at a concentration of 65mg/ml and stored in aliquots of 11.54 μl in the −20°C. Immediately before the treatment, 738.46 μl of saline (0.9% NaCl) was added to the resveratrol/vehicle (Res/Veh) aliquots and vortexed thoroughly and then used to treat the cells or mice based on the following concentrations. For the in vitro experiments, the 8DIV mixed neuronal-astrocytic cultures were treated with a single dose of Res/Veh ranging in concentration from 25–100μM. The mixed neuronal-astrocytic cultures were then exposed to a half-media change 24hours post treatment and then every third day. The cells were left in culture for 2–8 days post-treatment. For the in vivo experiments, 8-week-old male mice (Jackson Laboratories) were treated with a single intraperitoneal injection (i.p.) of 10mg/kg of Res/Veh. Two weeks post treatment, the mice were anesthetized with 3% isofluorane in 30% oxygen and 70% nitrous oxide, and their cortices were collected and snap frozen in liquid nitrogen for further processing.
Oxygen and Glucose Deprivation (OGD)
The OGD experiments were performed at least 10 days post culturing. Briefly, the cells were washed twice with OGD media and then exposed to two and a half hours of OGD by incubating the cells in an anaerobic chamber (Coy Lab Products, Grass Lake MI, USA) flushed with 90% N2 5% CO2 5% H2 gas at 37°C. The mixed cultures were then refed with the plating media but lacking FBS (due to the interference of the FBS with the LDH assay) and returned to the normoxic incubator for 24 hours. (The OGD media is constituted of 1.26mM CaCl2, 5.37mM KCl, 0.44mM KH2PO3, 0.49mM MgCl2, 0.41mM MgSO4, 136.9mM NaCl, 4.17mM NaHCO3, 0.34mM Na2HPO4, 20mM sucrose, and 10mM HEPES adjusted to pH 7.4)
Lactate Dehydrogenase (LDH) Assay
The LDH assay was performed using the Cytotoxicity detection kit LDH (Roche). Briefly, 100ul was collected from the supernatant of the mixed cultures 24 hours after their exposure to an OGD or sham OGD and used along with the kit reagents to perform the LDH assay in a 96-well plate format. The absorbance was then read using a plate reader at 490nm. The fold change in LDH released after OGD was normalized to the LDH levels released from sham wells, which are cells only exposed to a complete media change without OGD.
Propidium iodide (PI) to Hoechst staining
Propidium iodide staining was performed 24 hours after the exposure of the mixed culture to an OGD or sham OGD. The cells were supplemented with a final concentration of 2ug/ml PI and Hoechst and incubated at 37°C for 5min. Images were then captured at 20X magnification using the EVOS™ inverted microscope (CKX41/CKX31). The percent cell death was then quantified by normalizing the number of PI positive cells to Hoechst using ImageJ analysis.
RNA extraction, cDNA synthesis, real-time PCR analysis
As described previously RNA was extracted and purified using a combination of TRIzol reagent and QIAGEN RNeasy columns [63]. Also as previously described one microgram of RNA was then reverse transcribed into cDNA using the qScript cDNA Synthesis Kit (Quantabio). The cDNA was then diluted 5 times and used as template for real-time PCR analysis using the Power SYBR Green PCR Master Mix on the QuantStudioTM 6 Flex Real-Time PCR System [64]. For all RT-qPCR reactions the average of three technical replicates were used. The SYBR green primers were designed using Primer3 (version 0.4.0) to span an exon-exon junction in order to avoid any DNA amplifications. The complete list of primers and sequences used is found in Supplementary Table 4. For the in vitro experiments, the relative expression of genes was normalized to the geometric mean of six endogenous controls (ActB, Gapdh, Cypa, Pgk-1, B2m, 18srRNA).
RNA-seq: Library preparation, RNA-seq analysis, and Pathway Analysis
RNA was extracted from the mice’s right cortex two weeks following RPC. A staring input material of 500ng was used for library preparation using the TruSeq Stranded Total RNA Sample Preparation guide (15031048 E) with Ribozero Gold. Sequencing was then conducted on the Illumina Hiseq 2500 to generate 125bp paired-end reads. RNA-seq analysis was performed on the cloud-based genomics analysis platform called BaseSpace Sequence Hub from Illumina https://basespace.illumina.com/home/index. Briefly, paired end reads were trimmed and aligned to the Mus musculus reference genome mm9 using STAR v1.1.1. with the option of no novel transcript assembly. Cufflinks v2.1.0 was then used to assess transcripts abundance (FPKM) and to conduct differential expression. A value of 0.1 was used as a cutoff for the false discovery rate (FDR). (The RNA-seq analysis pipeline described here filters out mitochondrial RNA to avoid misinterpretations due to misalignment to nuclear pseudogenes, thus mitochondrial-encoded genes are not revealed by the RNA-seq analysis). The data discussed in this publication was deposited in NCBI’s Gene Expression Omnibus and accessible through GEO Series accession number GSE113298 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113298). For pathway analysis we used Genomatix Pathway System (GePS) v.1.0.0. within BaseSpace.
Mitochondrial to nuclear DNA analysis
DNA was extracted from the mixed neuronal-astrocytic cultures and anterior region of the cortex using the DNeasy Blood and Tissue Kit (Qiagen). The DNA was diluted to 10ng/ul and used for real-time PCR analysis as described above to measure the relative abundance of mitochondrial and nuclear DNA. The primer names and sequences used to detect rat and mice mitochondrial nuclear DNA is found in the Supplementary Table 4.
Seahorse Biosciences
Neuronal-astrocytic co-cultures were plated on poly-D-lysine coated Seahorse miniplates and treated with Res/Veh (100μM) 6 days prior to the experiments. The glycolytic and oxidative rates were measured using the Seahorse Extracellular Flux Analyzer (Agilent) according to the manufacturer’s instructions. To measure the glycolytic rates, cells were washed 3x with seahorse media (XF Base Medium and 2 mM glutamine) and incubated in a CO2-free incubator at 37°C for 1hr prior to the plate run. Extracellular acidification rate (ECAR) was measured prior and after the injection of glucose, oligomycin, and 2-deoxyglucose resulting in a final well concentration of 20mM, 4μM, and 50mM respectively. The glycolytic rates were then calculated as: Glycolysis=ECARGlucose-ECAR2-DG; Glycolytic capacity= ECARoligomycin-ECAR2-DG; Glycolytic reserve=ECAROligomycin-ECARGlucose. To measure the rate of oxidative phosphorylation, the cells were washed 3x with seahorse media (XF Base Medium-Minimum Dulbecco’s Modified Eagle’s Medium, 20 mM glucose, 1 mM sodium pyruvate, and 2 mM glutamine) and incubated in a CO2-free incubator at 37°C for 1hr before plate run. Oxygen consumption rate (OCR) was measured prior and after the injection of oligomycin, FCCP, and a mix of rotenone and antimycin A resulting in a final well concentration of 4μM, 0.5μM, and 0.5μM respectively. The OCR rates were then calculated as: OCR for Basal ATP Production=OCROligomycin-OCRBasal; OCR for Maximal ATP Production=OCRFCCP-OCRRot/Ant; OCR used by Proton Leak=OCROligomycin-OCRRot/Ant. ECAR and OCR rates were normalized to cell count as measured by Hoechst staining, followed by image aquisition using the EVOS™ inverted fluorescent microscope (CKX41/CKX31), and quantified with ImageJ analysis. For the experiments involving the acute treatment with Res/Veh. The cells were not exposed to any prior treatments. On the day of the experiment same steps to measure the oxidative phosphorylation mentioned above were followed with the exception that oligomycin was replaced with Res such the final concentration in the well reaches 100μM or 500μM.
Mitotracker Green
To measure the absolute fluorescence of the Mitotracker Green probe, mixed neuronal-astrocytic cultures were plated in a 96-well plate format and exposed to a single treatment of Res/Veh. Live staining was performed 6 days post-treatment using the MitoTracker™ Green FM (Life Technology M7514). Briefly, cells were washed twice with the plating media lacking FBS and then incubated with 600nM of the probe diluted in plating media with no FBS for 25min at 37°C. The cells were then washed twice and fluorescence was read using the plate reader using the excitation/emission wavelength of 490/516nm. The average of three technical replicates was used per experiment. Cell count was determined by staining the cells with Hoechst, followed by imaging using the EVOS™ inverted fluorescent microscope (CKX41/CKX31), and quantified with ImageJ analysis. The results were reported as the average fluorescence per cell number. The representative images shown were taken from the mixed neuronalastrocytic cultures plated on poly-D lysine coated coverslips (Warner, CS-12R15). Images were taken using the EVOS™ inverted fluorescent microscope at 40x magnification.
Immunostaining
Immunostaining was performed as previously described by Magistri et al. 2016 [64]. Antibodies used: rabbit anti-NEUN (1:1000) (Cell Signaling, D3S3I); rabbit anti-MAP2 (1:200) (Millipore AB5622). NeuN:DAPI cell count performed using ImageJ analysis.
Western Blot Analysis
Total cell and tissue lysate was homogenized using RIPA buffer followed by western blot analysis as previously described by Cohan et al. 2017 [39]. In order to detect histone H3K9ac and H3, cells were lysed using RIPA buffer containing additional 2% SDS. To detect histone H4K16ac and H4, acid extraction was performed from the cells using the histone extraction (abcam, ab221031). Antibodies used: Rabbit anti-COXIV (Cell Signaling, #4850) (1:1000); Rabbit anti-VDAC (D73D12) (1:1000) (Cell Signaling, #4661); Mouse anti-ACTB (8H10D10) (Cell Signaling, #3700) (1:25,000); Mouse anti-ATPB [3D5] (Abcam, ab14730) (1:1000); Rabbit anti-H4K16ac (Active motif, 39168) (1:1000); Rabbit anti-H4 (Active motif, 39270) (1:1000); Rabbit anti-H3K9ac (Abcam, abcam4441) (1:400); Rabbit anti-H3 (Cell Signaling, #9715S) (1:1000); and Rabbit anti-ARC (h-300) (Santa Cruz, sc-15325) (1:1000).
ATP measurement
The neuronal-astrocytic co-cultures were plated in 24-well plate at a density of 106 cell/well. The day of the experiment, the cells were washed twice with PBS, incubated with 100μl of 1x reaction buffer, and then scarped. The cell lysate was then homogenized using dounce homogenizer to increase lysis efficiency. ATP measurement was performed using the StayBrite™ Highly Stable ATP Bioluminescence Assay Kit (Biovision) following the manufacturer’s recommendations. The results were normalized to cell count as measured by Hoechst staining as described above.
Statistical Analysis and Blinding Procedures
Mice were randomly assigned to groups throughout. Data acquisition and analyses were conducted in a blinded manner when possible. Statistical analysis was performed using Prism6 software (GraphPad, San Diego, CA). Data are presented as mean ± SEM. The respective statistical test for each experiment is indicated in the figure legends. Significance is denoted in each figure. Sample size was determined for each experiment depending on prior experiments conducted by the laboratory and power analysis to detect an effect size of 0.8 using G*power 3.1 software. Statistical tests meet necessary assumptions as measured in Prism6.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We would like to acknowledge Claudia Sofia Pereira and Tania Arguello for their help with the Seahorse Biosciences experiments and Isabel Saul for her help with animal blinding and injections.
Funding: This work was supported by the NIH/NINDS grants NS45676, NS054147, NS097658, and NS34773 (to M.A.P.P), the American Heart Association (AHA) predoctoral award 16PRE29170004 (to N.K.), the NIH F31 predoctoral award NS089356–01A1 (to K.B.K.), and the Lois Pope LIFE Foundation Fellowship (to N.K. and K.B.K.)
References
- 1.Clarke DD and Sokoloff L, Circulation and Energy Metabolism of the Brain, in Basic Neurochemistry, 6th edition Molecular, Cellular and Medical Aspects, Siegel GJ, Editor-in-Chief, et al. , Editors. 1999, Lippincott-Raven: Philadelphia: p. 637–669. [Google Scholar]
- 2.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American C Heart Association Statistics, and S. Stroke Statistics, Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation, 2017. 135(10): p. e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sims NR and Muyderman H, Mitochondria, oxidative metabolism and cell death in stroke. Biochim Biophys Acta, 2010. 1802(1): p. 80–91. [DOI] [PubMed] [Google Scholar]
- 4.Kikuchi K, Tanaka E, Murai Y, and Tancharoen S, Clinical trials in acute ischemic stroke. CNS Drugs, 2014. 28(10): p. 929–38. [DOI] [PubMed] [Google Scholar]
- 5.Minnerup J, Sutherland BA, Buchan AM, and Kleinschnitz C, Neuroprotection for stroke: current status and future perspectives. Int J Mol Sci, 2012. 13(9): p. 11753–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lansberg MG, Bluhmki E, and Thijs VN, Efficacy and safety of tissue plasminogen activator 3 to 4.5 hours after acute ischemic stroke: a metaanalysis. Stroke, 2009. 40(7): p. 2438–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khoury N, Koronowski KB, and Perez-Pinzon MA, Long-term window of ischemic tolerance: An evolutionarily conserved form of metabolic plasticity regulated by epigenetic modifications? J Neurol Neuromedicine, 2016. 1(2): p. 6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raval AP, Dave KR, and Perez-Pinzon MA, Resveratrol mimics ischemic preconditioning in the brain. J Cereb Blood Flow Metab, 2006. 26(9): p. 1141–7. [DOI] [PubMed] [Google Scholar]
- 9.Koronowski KB, Dave KR, Saul I, Camarena V, Thompson JW, Neumann JT, Young JI, and Perez-Pinzon MA, Resveratrol Preconditioning Induces a Novel Extended Window of Ischemic Tolerance in the Mouse Brain. Stroke, 2015. 46(8): p. 2293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koronowski KB, Khoury N, Saul I, Loris ZB, Cohan CH, Stradecki-Cohan HM, Dave KR, Young JI, and Perez-Pinzon MA, Neuronal SIRT1 (Silent Information Regulator 2 Homologue 1) Regulates Glycolysis and Mediates Resveratrol-Induced Ischemic Tolerance. Stroke, 2017. 48(11): p. 3117–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Narayanan SV, Dave KR, Saul I, and Perez-Pinzon MA, Resveratrol Preconditioning Protects Against Cerebral Ischemic Injury via Nuclear Erythroid 2-Related Factor 2. Stroke, 2015. 46(6): p. 1626–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris-Blanco KC, Cohan CH, Neumann JT, Sick TJ, and Perez-Pinzon MA, Protein kinase C epsilon regulates mitochondrial pools of Nampt and NAD following resveratrol and ischemic preconditioning in the rat cortex. J Cereb Blood Flow Metab, 2014. 34(6): p. 1024–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Della-Morte D, Dave KR, DeFazio RA, Bao YC, Raval AP, and Perez-Pinzon MA, Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience, 2009. 159(3): p. 993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stowe AM, Altay T, Freie AB, and Gidday JM, Repetitive hypoxia extends endogenous neurovascular protection for stroke. Ann Neurol, 2011. 69(6): p. 975–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cox-Limpens KE, Gavilanes AW, Zimmermann LJ, and Vles JS, Endogenous brain protection: what the cerebral transcriptome teaches us. Brain Res, 2014. 1564: p. 85–100. [DOI] [PubMed] [Google Scholar]
- 16.Narayanan SV, Dave KR, and Perez-Pinzon MA, Ischemic preconditioning and clinical scenarios. Curr Opin Neurol, 2013. 26(1): p. 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seshadri S, Beiser A, Kelly-Hayes M, Kase CS, Au R, Kannel WB, and Wolf PA, The lifetime risk of stroke: estimates from the Framingham Study. Stroke, 2006. 37(2): p. 345–50. [DOI] [PubMed] [Google Scholar]
- 18.Barger JL, Kayo T, Vann JM, Arias EB, Wang J, Hacker TA, Wang Y, Raederstorff D, Morrow JD, Leeuwenburgh C, Allison DB, Saupe KW, Cartee GD, Weindruch R, and Prolla TA, A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS One, 2008. 3(6): p. e2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas J, Garg ML, and Smith DW, Dietary supplementation with resveratrol and/or docosahexaenoic acid alters hippocampal gene expression in adult C57Bl/6 mice. J Nutr Biochem, 2013. 24(10): p. 1735–40. [DOI] [PubMed] [Google Scholar]
- 20.Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, Meller R, Rosenzweig HL, Tobar E, Shaw TE, Chu X, and Simon RP, Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet, 2003. 362(9389): p. 1028–37. [DOI] [PubMed] [Google Scholar]
- 21.Stenzel-Poore MP, Stevens SL, and Simon RP, Genomics of preconditioning. Stroke, 2004. 35(11 Suppl 1): p. 2683–6. [DOI] [PubMed] [Google Scholar]
- 22.Gao D, Huang T, Jiang X, Hu S, Zhang L, and Fei Z, Resveratrol protects primary cortical neuron cultures from transient oxygen-glucose deprivation by inhibiting MMP-9. Mol Med Rep, 2014. 9(6): p. 2197–204. [DOI] [PubMed] [Google Scholar]
- 23.Huang T, Gao D, Jiang X, Hu S, Zhang L, and Fei Z, Resveratrol inhibits oxygen-glucose deprivation-induced MMP-3 expression and cell apoptosis in primary cortical cells via the NF-kappaB pathway. Mol Med Rep, 2014. 10(2): p. 1065–71. [DOI] [PubMed] [Google Scholar]
- 24.Tang F, Guo S, Liao H, Yu P, Wang L, Song X, Chen J, and Yang Q, Resveratrol Enhances Neurite Outgrowth and Synaptogenesis Via Sonic Hedgehog Signaling Following Oxygen-Glucose Deprivation/Reoxygenation Injury. Cell Physiol Biochem, 2017. 43(2): p. 852–869. [DOI] [PubMed] [Google Scholar]
- 25.Herculano-Houzel S, Mota B, and Lent R, Cellular scaling rules for rodent brains. Proc Natl Acad Sci U S A, 2006. 103(32): p. 12138–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim-Han JS and Dugan LL, Mitochondrial uncoupling proteins in the central nervous system. Antioxid Redox Signal, 2005. 7(9–10): p. 1173–81. [DOI] [PubMed] [Google Scholar]
- 27.Caruso F, Mendoza L, Castro P, Cotoras M, Aguirre M, Matsuhiro B, Isaacs M, Rossi M, Viglianti A, and Antonioletti R, Antifungal activity of resveratrol against Botrytis cinerea is improved using 2-furyl derivatives. PLoS One, 2011. 6(10): p. e25421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gledhill JR, Montgomery MG, Leslie AG, and Walker JE, Mechanism of inhibition of bovine F1-ATPase by resveratrol and related polyphenols. Proc Natl Acad Sci U S A, 2007. 104(34): p. 13632–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng J and Ramirez VD, Inhibition of mitochondrial proton F0F1-ATPase/ATP synthase by polyphenolic phytochemicals. Br J Pharmacol, 2000. 130(5): p. 1115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dave KR, Christian SL, Perez-Pinzon MA, and Drew KL, Neuroprotection: lessons from hibernators. Comp Biochem Physiol B Biochem Mol Biol, 2012. 162(1–3): p. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lutz PL, Nilsson GE, and Perez-Pinzon MA, Anoxia tolerant animals from a neurobiological perspective. Comp Biochem Physiol B Biochem Mol Biol, 1996. 113(1): p. 3–13. [DOI] [PubMed] [Google Scholar]
- 32.Storey KB and Storey JM, Metabolic rate depression: the biochemistry of mammalian hibernation. Adv Clin Chem, 2010. 52: p. 77–108. [PubMed] [Google Scholar]
- 33.Scornavacca G, Gesuete R, Orsini F, Pastorelli R, Fanelli R, de Simoni MG, and Airoldi L, Proteomic analysis of mouse brain cortex identifies metabolic down-regulation as a general feature of ischemic pre-conditioning. J Neurochem, 2012. 122(6): p. 1219–29. [DOI] [PubMed] [Google Scholar]
- 34.Navarro G, Martinez-Pinilla E, Sanchez-Melgar A, Ortiz R, Noe V, Martin M, Ciudad C, and Franco R, A genomics approach identifies selective effects of trans-resveratrol in cerebral cortex neuron and glia gene expression. PLoS One, 2017. 12(4): p. e0176067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stapels M, Piper C, Yang T, Li M, Stowell C, Xiong ZG, Saugstad J, Simon RP, Geromanos S, Langridge J, Lan JQ, and Zhou A, Polycomb group proteins as epigenetic mediators of neuroprotection in ischemic tolerance. Sci Signal, 2010. 3(111): p. ra15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neumann JT, Thompson JW, Raval AP, Cohan CH, Koronowski KB, and Perez-Pinzon MA, Increased BDNF protein expression after ischemic or PKC epsilon preconditioning promotes electrophysiologic changes that lead to neuroprotection. J Cereb Blood Flow Metab, 2015. 35(1): p. 121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeFazio RA, Raval AP, Lin HW, Dave KR, Della-Morte D, and Perez-Pinzon MA, GABA synapses mediate neuroprotection after ischemic and epsilonPKC preconditioning in rat hippocampal slice cultures. J Cereb Blood Flow Metab, 2009. 29(2): p. 375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lei M, Dong D, Mu S, Pan YH, and Zhang S, Comparison of brain transcriptome of the greater horseshoe bats (Rhinolophus ferrumequinum) in active and torpid episodes. PLoS One, 2014. 9(9): p. e107746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohan CH, Stradecki-Cohan HM, Morris-Blanco KC, Khoury N, Koronowski KB, Youbi M, Wright CB, and Perez-Pinzon MA, Protein kinase C epsilon delays latency until anoxic depolarization through arc expression and GluR2 internalization. J Cereb Blood Flow Metab, 2017. 37(12): p. 3774–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salin K, Auer SK, Rey B, Selman C, and Metcalfe NB, Variation in the link between oxygen consumption and ATP production, and its relevance for animal performance. Proc Biol Sci, 2015. 282(1812): p. 20151028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi L and Tu BP, Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol, 2015. 33: p. 125–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CB, ATP-citrate lyase links cellular metabolism to histone acetylation. Science, 2009. 324(5930): p. 1076–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, Carrer A, Sengupta A, Campbell SL, Kuo YM, Frey AJ, Meurs N, Viola JM, Blair IA, Weljie AM, Metallo CM, Snyder NW, Andrews AJ, and Wellen KE, ATP-Citrate Lyase Controls a Glucose-to-Acetate Metabolic Switch. Cell Rep, 2016. 17(4): p. 1037–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choudhary C, Weinert BT, Nishida Y, Verdin E, and Mann M, The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol, 2014. 15(8): p. 536–50. [DOI] [PubMed] [Google Scholar]
- 45.Chung JH, Manganiello V, and Dyck JR, Resveratrol as a calorie restriction mimetic: therapeutic implications. Trends Cell Biol, 2012. 22(10): p. 546–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, and Sinclair DA, Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature, 2003. 425(6954): p. 191–6. [DOI] [PubMed] [Google Scholar]
- 47.de Oliveira MR, Nabavi SF, Manayi A, Daglia M, Hajheydari Z, and Nabavi SM, Resveratrol and the mitochondria: From triggering the intrinsic apoptotic pathway to inducing mitochondrial biogenesis, a mechanistic view. Biochim Biophys Acta, 2016. 1860(4): p. 727–45. [DOI] [PubMed] [Google Scholar]
- 48.Beher D, Wu J, Cumine S, Kim KW, Lu SC, Atangan L, and Wang M, Resveratrol is not a direct activator of SIRT1 enzyme activity. Chem Biol Drug Des, 2009. 74(6): p. 619–24. [DOI] [PubMed] [Google Scholar]
- 49.Kaeberlein M, McDonagh T, Heltweg B, Hixon J, Westman EA, Caldwell SD, Napper A, Curtis R, DiStefano PS, Fields S, Bedalov A, and Kennedy BK, Substrate-specific activation of sirtuins by resveratrol. J Biol Chem, 2005. 280(17): p. 17038–45. [DOI] [PubMed] [Google Scholar]
- 50.Dasgupta B and Milbrandt J, Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A, 2007. 104(17): p. 7217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, and Auwerx J, AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature, 2009. 458(7241): p. 1056–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madrigal-Perez LA and Ramos-Gomez M, Resveratrol Inhibition of Cellular Respiration: New Paradigm for an Old Mechanism. Int J Mol Sci, 2016. 17(3): p. 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tang BL, Resveratrol is neuroprotective because it is not a direct activator of Sirt1-A hypothesis. Brain Res Bull, 2010. 81(4–5): p. 359–61. [DOI] [PubMed] [Google Scholar]
- 54.Adak T, Samadi A, Unal AZ, and Sabuncuoglu S, A reappraisal on metformin. Regul Toxicol Pharmacol, 2017. [DOI] [PubMed] [Google Scholar]
- 55.Arbelaez-Quintero I and Palacios M, To Use or Not to Use Metformin in Cerebral Ischemia: A Review of the Application of Metformin in Stroke Rodents. Stroke Res Treat, 2017. 2017: p. 9756429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng YY, Leu HB, Chen TJ, Chen CL, Kuo CH, Lee SD, and Kao CL, Metformin-inclusive therapy reduces the risk of stroke in patients with diabetes: a 4-year follow-up study. J Stroke Cerebrovasc Dis, 2014. 23(2): p. e99–105. [DOI] [PubMed] [Google Scholar]
- 57.Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet, 1998. 352(9131): p. 854–65. [PubMed] [Google Scholar]
- 58.Riepe MW, Esclaire F, Kasischke K, Schreiber S, Nakase H, Kempski O, Ludolph AC, Dirnagl U, and Hugon J, Increased hypoxic tolerance by chemical inhibition of oxidative phosphorylation: “chemical preconditioning”. J Cereb Blood Flow Metab, 1997. 17(3): p. 257–64. [DOI] [PubMed] [Google Scholar]
- 59.Busija DW, Gaspar T, Domoki F, Katakam PV, and Bari F, Mitochondrial-mediated suppression of ROS production upon exposure of neurons to lethal stress: mitochondrial targeted preconditioning. Adv Drug Deliv Rev, 2008. 60(13–14): p. 1471–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, Goldberger O, Peng J, Shalem O, Sanjana NE, Zhang F, Goessling W, Zapol WM, and Mootha VK, Hypoxia as a therapy for mitochondrial disease. Science, 2016. 352(6281): p. 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferrari M, Jain IH, Goldberger O, Rezoagli E, Thoonen R, Cheng KH, Sosnovik DE, Scherrer-Crosbie M, Mootha VK, and Zapol WM, Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc Natl Acad Sci U S A, 2017. 114(21): p. E4241–E4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arroyo JD, Jourdain AA, Calvo SE, Ballarano CA, Doench JG, Root DE, and Mootha VK, A Genome-wide CRISPR Death Screen Identifies Genes Essential for Oxidative Phosphorylation. Cell Metab, 2016. 24(6): p. 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Velmeshev D, Magistri M, and Faghihi MA, Expression of non-protein-coding antisense RNAs in genomic regions related to autism spectrum disorders. Mol Autism, 2013. 4(1): p. 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Magistri M, Khoury N, Mazza EM, Velmeshev D, Lee JK, Bicciato S, Tsoulfas P, and Faghihi MA, A comparative transcriptomic analysis of astrocytes differentiation from human neural progenitor cells. Eur J Neurosci, 2016. 44(10): p. 2858–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.