

Abstract
Intratumoral stimulatory dendritic cells (SDCs) play an important role in stimulating cytotoxic T cells and driving immune responses against cancer. Understanding the mechanisms that regulate their abundance in the tumor microenvironment (TME) could unveil new therapeutic opportunities. We find that in human melanoma SDC abundance is associated with intratumoral expression of the gene encoding the cytokine FLT3LG. FLT3LG is predominantly produced by lymphocytes, notably natural killer (NK) cells in mouse and human tumors. NK cells stably form conjugates with SDCs in the mouse TME, and genetic and cellular ablation of NK cells in mice demonstrates their importance in positively regulating SDC abundance in the tumor through production of Flt3L. Although anti-PD-1 “checkpoint” immunotherapy for cancer largely targets T cells, we find that NK cell frequency correlates with protective SDCs in human cancers, with patient responsiveness to anti-PD-1 immunotherapy, and with increased overall survival. Our studies reveal that innate immune SDCs and NK cells cluster together as an excellent prognostic tool for T cell directed immunotherapy and that these innate cells are necessary for enhanced T cell tumor responses, suggesting this axis for novel therapies.
INTRODUCTION
Checkpoint blockade therapies, such as anti-CTLA-4 or anti-PD-1 immunotherapy, have been remarkably effective in reactivating T cell responses to tumors and providing long-lasting protection to patients. However, it is common for upwards of 80% of patients in any given indication to have no objective responses to these treatments1. While the frequency of mutations leading to new T cell epitopes is suggested to be one factor associated with better responses2, other immune parameters and cell types that control responsiveness to these treatments remain to be determined.
We previously identified a rare intratumoral DC subset that is uniquely capable of re-stimulating T cells in the TME3 and is required for adoptive T cell therapy in mouse models3–7. These rare intratumoral type I conventional dendritic cells (cDC1, when taken from tumors referred to as Stimulatory Dendritic Cells; SDC) were defined in the mouse by surface expression of the integrin CD103 and in the human by expression of BDCA3 (also known as CD141)3. Studies in lung have demonstrated that these cells are rarer in tumors as compared to adjacent normal tissues8. In the infrequent cases of WNT/β-catenin pathway mutations in melanoma, decreases in these DCs have been implicated in poor outcome and this has been mapped to defects in chemokine expression patterns in tumors9. Here we find that the relationship of SDC number to outcome is likely more generalized.
In this study, we show that the levels of protective BDCA3+ SDCs in the TME correlate with better overall survival of melanoma patients. We further link the population level of SDCs to the expression of the cDC1 formative cytokine, FLT3LG. Using a novel Flt3lg reporter mouse we identify intratumoral lymphocytes as the producers of Flt3lg in the tumor, with genetic and functional studies demonstrating that natural killer (NK) cells are the integral cell type that produces Flt3lg to control the levels of SDCs in the tumor. We further show that SDCs in human melanoma correlate with levels of intratumoral NK cells and that both innate immune cell types correlate with responsiveness to anti-PD-1 immunotherapy. These findings suggest that NK cells, through the production of FLT3LG in the tumor, control the levels of SDCs in the tumor and further the responsiveness of patients to anti-PD-1 immunotherapy.
RESULTS
BDCA3+ SDC levels in human melanoma correlate with increased overall survival.
Our previous work identified an 8 gene “SDC signature”, derived from direct comparisons of SDCs versus all other myeloid populations within mouse tumors3 (Fig. 1a). We utilized this SDC gene signature to estimate the levels of SDCs across the spectrum of melanoma patient samples with clinical outcome data in a previously published metastatic melanoma dataset10 and found that 6 “SDC signature” genes had a significant individual association with increased overall survival (OS) from the time of metastasis (Supplementary Table 1). Furthermore, the entire signature, binned into ‘high’ or ‘low’ expression with a 66% stringency cutoff significantly correlated with increased OS in Kaplan-Meier analysis (Fig. 1b); similar correlations were observed at 33% and 50% stringency (Supplementary Fig. 1a). The correlation of the SDC gene signature with increased overall survival was recapitulated in the TCGA melanoma dataset (Supplementary Fig. 1b)11. We further found that measures of TIL category were highly correlated with the SDC gene signature (Fig. 1c). Furthermore, a gene signature that uses a ratio of signatures for SDCs and non-stimulatory myeloid cells (NSMs)3, representing the relative abundance of stimulatory and inhibitory myeloid populations, also showed a strong correlation with OS and increased T cell infiltration (Supplementary Fig. 1c-e). These data suggest that the relative levels of SDCs in the tumor correlate with increased overall survival.
Figure 1. BDCA3+ DCs define overall outcome in melanoma patients and predict responsiveness to anti-PD-1 immunotherapy.
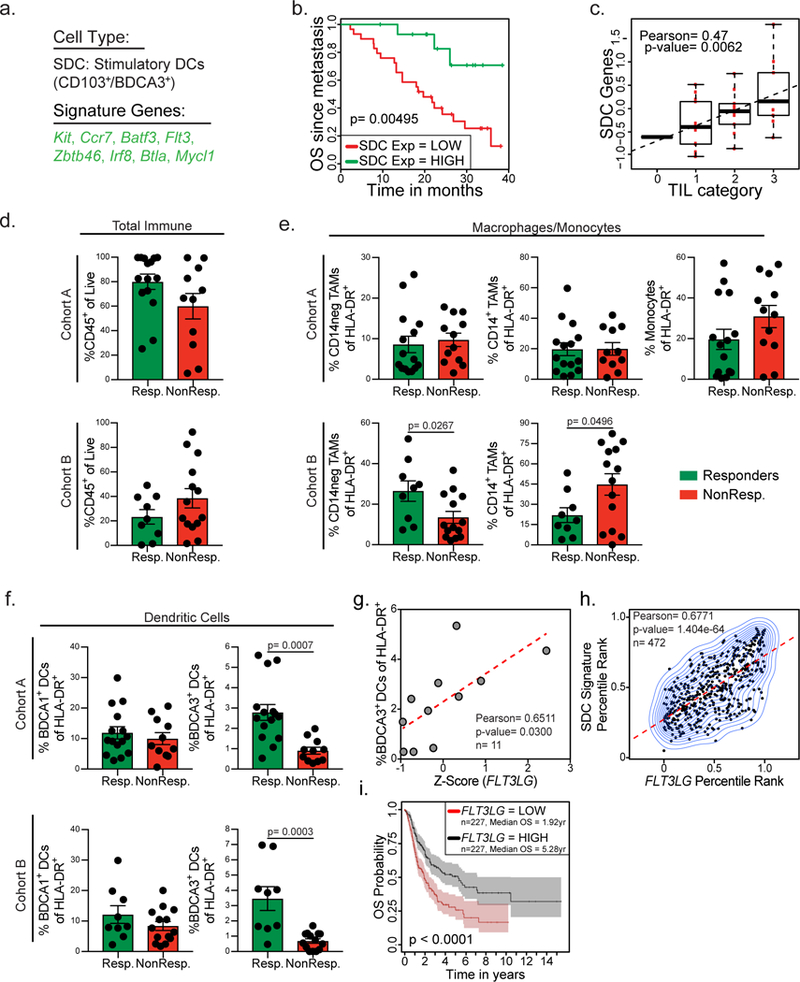
(a) Signature genes identifying SDC (from3). (b) Kaplan-Meier plot for post recurrence survival of metastatic melanoma patients for SDC gene list expression. Data from 10 (n = 44 metastatic melanoma samples from 38 biologically independent patients) are parsed into “high” (green; n=15 metastatic melanoma tumor samples) and “low” (red; n=29 metastatic melanoma tumor samples) bins at 66% stringency threshold for levels of expression of the SDC genes. p-value calculated by log rank test after adjusting for multiple comparisons. (c) Class-based measures of TIL category from 10 plotted versus SDC gene signature (n=33 metastatic melanoma samples). Data plotted as box and whisker plot (box ends= upper and lower quartiles, middle line= median, error bars= maximum and minimum value) with regression line superimposed. TIL categories defined as: 0: 0–5% (n=1), 1: 5–25% (n=13), 2: 25–50% (n=10), 3: 50–100% (n=9). Correlation assessed using the Pearson correlation coefficient and two-tailed p-value. (d-f) Quantified frequency of percentage of CD45+ immune cells of total cells (d), Macrophage/Monocyte populations of HLA-DR+ cells (e), and dendritic cell populations of total HLA-DR+ cells (f) in the tumor of individuals. Patients binned as either responders (green, including partial or complete responses) or non-responders (red, including stable disease and progressive disease). Data were collected from two independent patient cohorts, pooled across patients, and analyzed by two-tailed unpaired parametric t-test, and presented as mean ± S.E.M. with all individual points shown. Cohort A: n=26 samples collected from 24 biologically independent patients (Resp.: n=15 samples from 15 patients, NonResp.: n=11 from 9 patients). Cohort B: n=23 biologically independent patient samples (Resp.: n=9 and NonResp.: n=14 independent patient samples). (g) Z-score of FLT3LG expression in total live cells plotted versus BDCA3+ DC levels of total HLA-DR+ cells in a subset of human melanoma patients in cohort A (n=11 biologically independent metastatic melanoma samples). Data plotted as scatter plot with regression line superimposed (dotted red line); correlation assessed using the Pearson correlation coefficient and two-tailed p-value. (h) Percentile rank of SDC gene expression plotted versus normalized expression of FLT3LG in tumors from the TCGA melanoma dataset11. Correlation of n=472 independent patient samples was assessed using the Pearson correlation coefficient and two-tailed p-value. (i) Kaplan-Meier plot for overall survival of melanoma patients median split for FLT3LG gene expression. Data from 11 are parsed into “high” (black; n=227 biologically independent patient samples) and “low” (red; n=227 biologically independent patient samples) bins at 50% stringency threshold for levels of expression of FLT3LG. Plots and statistics generated by the UZH TCGA Cancer Browser28. Shaded regions identify 95% confidence interval; p-value calculated by log rank test after adjusting for multiple comparisons.
Intratumoral SDC abundance predicts responsiveness to anti-PD-1 immunotherapy.
Tumoral SDCs were first defined by their ability to cross-present tumor antigens to T cells and stimulate them3,4 and have since been shown to be required for profound anti-PD-1 responses in mouse models5–7. We therefore sought to query whether SDC levels related to the effectiveness of a T cell checkpoint blockade treatment which would be predicted to release more CD8+ T cells for possible tumor control12. To extend this analysis to human melanoma patients and to more broadly determine the immune components required for responsiveness to anti-PD-1 immunotherapy we analyzed tumor biopsies from two independent cohorts (cohort A n=33, all comers, including those already treated with various immunotherapies, cohort B n=23, all pre-anti-PD-1 immunotherapy; Supplementary Table 2) of metastatic melanoma patients’ biopsy or surgical resections. Biopsies were digested into single cell suspensions that were subsequently analyzed by flow cytometry and RNA sequencing while clinical outcome of patients was tracked along with responsiveness to anti-PD-1 immunotherapy. Patients were parsed into groups either as ‘non-responders’, defined as either stable or progressive disease, or ‘responders’, defined as partial or complete responses to anti-PD-1 therapy (see Methods). We designed a comprehensive flow panel to dissect the human immune infiltrates in the tumor (Fig. 1d-f; Supplementary Fig. 2a-b) and although we found heterogeneity in the number of immune infiltrates across melanoma patients there was no significant correlation between total immune infiltrate and responsiveness to anti-PD-1 immunotherapy (Fig. 1d).
In contrast, multiple myeloid populations within the TME trended with response to immunotherapy (Fig. 1e-f). CD14− tumor associated macrophages (TAMs) showed a trend towards increased numbers in anti-PD-1 responding patients, but this was only seen in one cohort (Fig. 1e). CD14+CD16− cells were negatively prognostic for responsiveness to anti-PD-1 immunotherapy in cohort B, with cohort A, where CD14+CD16+ monocytes could be effectively separated from CD14+CD16− TAMs, showing a trend that intratumoral monocyte levels, more than TAMs, act as a negative prognostic value for anti-PD-1 responsiveness (Fig. 1e). Further, BDCA1+ DCs (cDC2s) did not show a significant change upon responder status in either cohort (Fig. 1f). Interestingly, BDCA3+ DCs (which were further confirmed to be cDC1 by staining for CLEC9a; Supplementary Fig. 2c) as a proportion of total APC (gating on CD19−, HLA-DR+ cells) strongly predicted patient responsiveness to anti-PD-1 therapy in both melanoma cohorts (Fig. 1f).
FLT3LG expression correlates with SDC levels in the tumor.
Given the profound association of SDCs with patient outcome and responsiveness to anti-PD-1 immunotherapy, we sought to elicit the cellular and molecular mechanisms controlling the levels of protective myeloid cells in tumors. The formative cytokine for cDC1, which include tumoral SDC, is Flt3L3,13, yet the endogenous source of this cytokine in cancer remains unknown. Using our melanoma dataset containing paired flow cytometry and RNA sequencing of total live cells from the tumor (cohort A; Supplementary Table 2), we found a significant correlation between the levels of BDCA3+ DCs and expression of FLT3LG within the tumor (Fig. 1g). We confirmed this correlation between SDC levels in the tumor and the expression of the cytokine FLT3LG using the SDC gene signature to estimate SDC levels in the public TCGA melanoma dataset11 (Fig. 1h). Consistent with an important role in controlling SDCs in the tumor, TCGA melanoma samples11 with high FLT3LG expression (median split) had significantly increased overall survival (Fig. 1i). These findings suggest that control of the cytokine FLT3LG in the TME can have an important effect on the levels of SDCs, a cell type integrally important for immune responses to cancer and responsiveness to anti-PD-1 immunotherapy.
Lymphocytes are the primary producer of Flt3l in the tumor microenvironment.
The correlation of FLT3LG expression and SDC levels in the tumor introduced the question of which cell type(s) produce FLT3L. We therefore generated a Flt3l-reporter mouse by introducing DNA encoding an inducible Cre and teal fluorescent protein downstream of the endogenous mouse Flt3l locus (Fig. 2a). Mice homozygous for this allele and bearing ectopic B16F10 tumors have similar proportions of conventional DCs and lymphocytes within the TME, despite a small but reproducible decrease in total serum Flt3L, overall suggesting that the protein remains functional to a similar extent within tumors (Supplementary Fig.3a-c). Flt3l-reporter homozygous mice were given ectopic B16F10 tumors and TFP, as a readout of Flt3l expression, was only detected within lymphocytes at two weeks post tumor transplant (Fig. 2b-c). Of the lymphocytes expressing Flt3l, NK cells had the highest expression levels with lower levels in T cells and no significant expression in B cells (Fig. 2b-c; gating strategy Supplementary Fig. 3e-f). These same populations were positive when stained with an anti-Flt3L antibody to detect the protein on cell surfaces (Fig. 2d). Flt3l, measured by TFP expression, was expressed at similar levels in NK cells isolated from the tumor, tumor-draining lymph node (LN), and the non-tumor-draining LN (Supplementary Fig. 3d); suggesting that Flt3l expression is not modulated by the TME. Similar patterns of expression for the reporter allele were found in other tumor models including those that arise spontaneously (e.g. PyMTChOVA14 Supplementary Fig. 4a-b). Interestingly, WT B16F10 tumor-bearing animals have no significant differences in Flt3L levels in the serum, suggesting that local production of Flt3L is important for SDC levels and protection from cancer. (Supplementary Fig. 4c).
Figure 2. Tumor resident lymphocytes produce Flt3l.
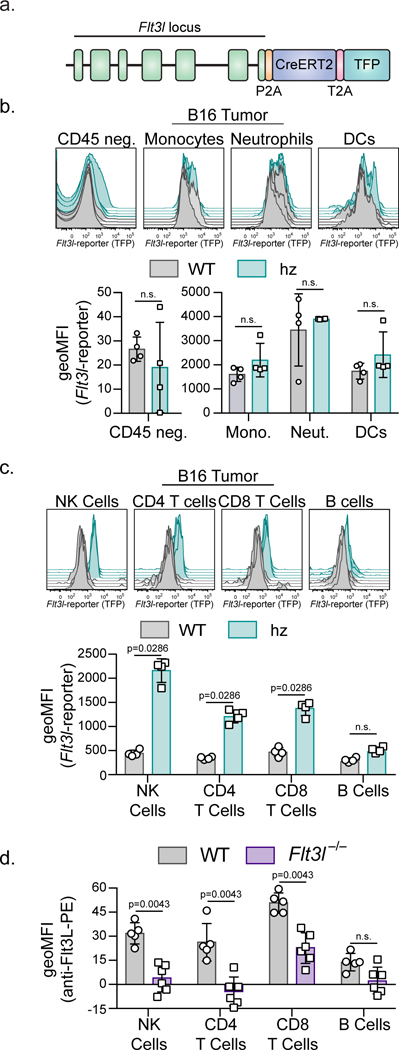
(a) Cartoon diagram of the Flt3l-reporter mouse. (b) Representative flow plots and geometric mean fluorescent intensity (MFI) of Flt3l-reporter expression in CD45 negative and myeloid cells found in two-week-old B16F10 tumors in WT (grey, n=4 biologically independent animals) and Flt3l-reporter homozygous (teal, n=4 biologically independent animals) mice. Monocytes (CD45+, CD11b+, Ly6Chi), neutrophils (CD45+, CD11b+,Ly6Cmid), DCs (CD45+, CD90.2−, CD45R−, Ly6G−, NK1.1−, Ly6C−, F4/80−, MHC-II+, CD24+). (c) Representative flow plots and geometric mean fluorescent intensity (MFI) of Flt3l-reporter expression in NK cells, CD4+ T cells, CD8+ T cells, and B cells (MHC-II+, B220+) found in two-week-old B16F10 tumors in WT (grey; n=4 biologically independent animals) and Flt3l-reporter homozygous (teal; n=4 biologically independent animals) mice. (b-c) Gating strategy depicted in Supplementary Figure 3. (d) Geometric MFI of Flt3L surface protein expression in NK cells, CD4+ T cells, CD8+ T cells, and B cells found in two-week-old B16F10 tumors in WT (grey; n=5 biologically independent animals) or Flt3l-deficient (purple; n=6 biologically independent animals) mice stained with anti-Flt3L antibody. (a-d) Quantification plotted as mean ± S.D and analyzed by Mann-Whitney U Test to generate two-tailed p-values. Data are representative of three (b-c) and two (d) independent replicates.
Flt3L production by lymphocytes is required for normal SDC levels in the tumor.
We next undertook genetic experiments in mice to test the requirement for lymphocytes and their subsets in controlling the levels of SDCs in the TME. Il2rg-deficient animals, lacking T cells and NK cells (Supplementary Fig. 5a), were injected with ectopic B16F10 melanoma and tumors were analyzed for the levels of myeloid and lymphoid cells. While B16F10 tumors from Il2rg−/− mice had roughly the same tumor area as their WT controls, tumors from Il2rg−/− animals had significantly reduced frequencies of CD103+ SDCs in the TME with no significant change in CD11b+ DCs (Fig. 3a, Supplementary Fig. 5a). To test the role of Flt3L production specifically by lymphocytes in controlling the levels of SDCs we generated mixed bone marrow chimeras with Il2rg−/− bone marrow mixed with WT or Flt3l−/− bone marrow transferred into lethally irradiated Flt3l−/− recipient animals (Fig. 3b). Il2rg−/−:Flt3l−/− mixed bone marrow chimeras, which have a lymphocyte compartment unable to produce Flt3L, have reduced levels of CD103+ SDCs in the tumor as compared to Il2rg−/−:WT mixed bone marrow chimeras, which have a lymphocyte compartment capable of producing Flt3L, or WT animals (Fig. 3b). The differences in CD103+ SDC levels in the tumor are not due a global loss of T or NK cells, as these cells were, if anything, more abundant in the tumor draining and non-draining lymph node (LN) of Il2rg−/−: Flt3l−/− bone marrow chimeras (Supplementary Fig. 5b). CD11b+ DCs in the tumors of Il2rg−/−:Flt3l−/− mixed bone marrow chimeras were not significantly reduced as compared to Il2rg−/−:WT bone marrow chimeras (Fig. 3b); furthermore, in the tumor draining and non-draining skin LN there was no defect in resident or migratory CD11b+ DCs suggesting there is not a global defect in the CD11b+ DC compartment in the Il2rg−/−:Flt3l−/− mixed bone marrow chimeras (Supplementary Fig. 5c). Interestingly, the levels of resident CD8+ DCs and migratory CD103+ DCs in the tumor draining and non-draining LN are also reduced in the Il2rg−/−:Flt3l−/− bone marrow chimeras suggesting that the lymphocyte requirement extends to cDC1 production more generally (Supplementary Fig. 5c).
Figure 3. Flt3L production by NK cells controls the levels of CD103+ DCs in the tumor.
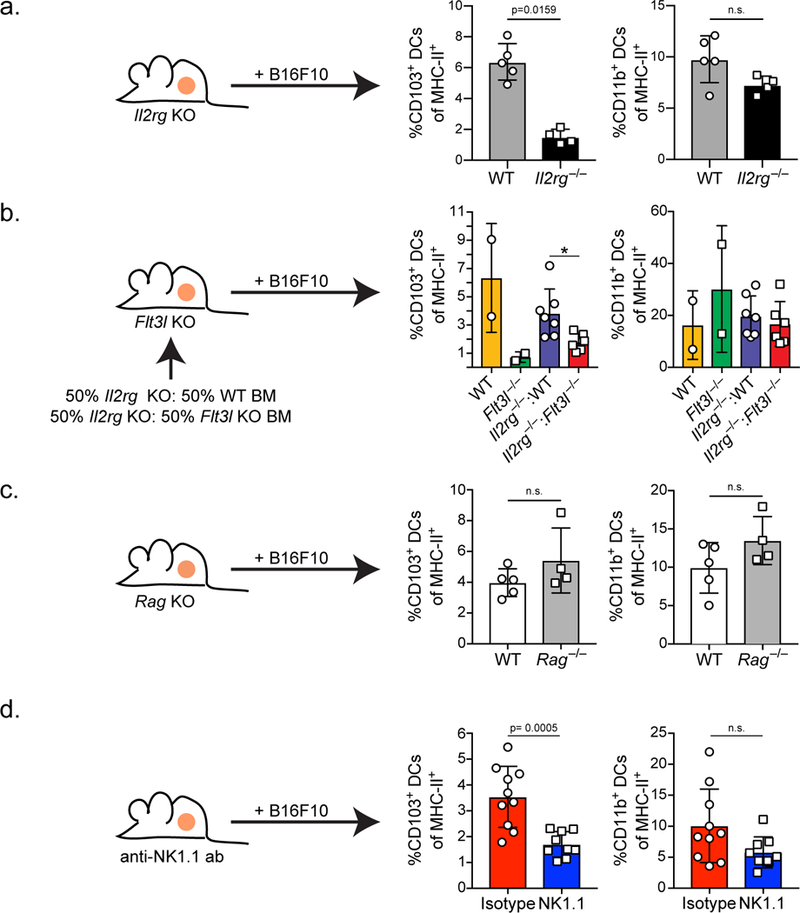
(a) Quantification of CD103+ and CD11b+ DCs of total MHC-II+ cells in two-week-old ectopic B16F10 tumors from WT (grey; n=5 biologically independent animals) or Il2rg−/− (black; n=4 biologically independent animals) mice. (b) Quantification of CD103+ and CD11b+ DCs of total MHC-II+ cells in two-week-old ectopic B16F10 tumors from mixed bone marrow chimeras reconstituted with a 50:50 mixture of Il2rg−/−:WT (blue; n=7 biologically independent animals) or Il2rg−/−:Flt3l−/− (red; n=6 biologically independent animals), and WT (yellow; n=2 biologically independent animals) and Flt3l−/− (green; n= 2 biologically independent animals) controls. * = p=0.0140. Data are combined from 2 independent experimental replicates. (c) Quantification of CD103+ and CD11b+ DCs as a proportion of total MHC-II+ cells in two-week-old B16F10 tumors from WT (white; n=5 biologically independent animals) or Rag−/− (grey; n=4 biologically independent animals) mice. (d) Quantification of CD103+ and CD11b+ DCs as a proportion of total MHC-II+ cells in two-week-old B16F10 tumors from WT mice treated with isotype control (red; n=10 biologically independent animals) or mice treated with anti-NK1.1 antibody (blue; n=9 biologically independent animals) every 3 days, starting 3 days prior to tumor injection. (a-d) Plotted as mean ± S.D and analyzed by Mann-Whitney U Test (a-c) or unpaired parametric test (d) to generate two-tailed p-values. (a-d) Data are representative of two independent experimental replicates.
NK cells, not T cells, are required for SDC levels in the tumor.
To directly test the role of specific types of lymphocytes in controlling CD103+ SDC levels in the tumor we depleted T cells and NK cells, both of which have expression of the Flt3l-reporter, in the mouse B16F10 melanoma model. Mice lacking all T cells as a consequence of a RAG mutation had no defect in CD103+ DC levels in the tumor (Fig. 3c; Supplementary Fig. 5d). Additionally, WT mice depleted of CD4+ or CD8+ T cells with antibodies also displayed normal SDC cellularity (data not shown). To investigate NK cells, mice were treated with an anti-NK1.1 antibody every three days starting three days prior to B16F10 tumor injection, resulting in profound loss of NK cells but limited other changes in lymphocyte cellularity and tumor growth (Supplementary Fig. 5e). However, animals depleted of NK cells had reduced frequencies of CD103+ SDCs in the TME with no significant changes in the levels CD11b+ DCs (Fig. 3d). Consistent with our previous finding that T cell stimulation in tumors relies on tumor-resident CD103+ DCs, animals lacking NK cells show a trend for reduced numbers of activated T cells in the tumor (Supplementary Fig. 5e). Interestingly, depletion of NK cells leads to a small, but significant, decrease in the levels of CD103+ DCs in the tumor draining and non-draining LN (Supplementary Fig. 5f), suggesting again that NK cells may play a more generalized role in controlling CD103+ DC levels as well as in the tumor. Taken as a whole, these findings suggest that while both T cells and NK cells can produce Flt3l in the tumor, the loss of T cells has no effect on tumor CD103+ DC levels, while Flt3l production by NK cells plays an important role in controlling the levels of these protective DCs in the tumor.
NK cells make frequent and stable interactions with SDCs in the TME.
NK cells are the dominant lymphocyte required for SDC levels in the tumor. However, NK cells and SDCs are quite rare in the tumor, thus we asked: how do NK cells exert control on SDCs in the tumor? SDCs are only sparsely present in the TME compared to other APC and interact infrequently with incoming T cells3,4. In contrast, when we undertook live 2-photon slice imaging of B78 melanoma tumors we found NK cells (marked by Ncr-1-GFP) frequently arrested in close contacts with SDC (marked by anti-XCR1; Fig. 4a, Supplementary Video 1). Undertaking live intravital 2-photon imaging of B78-cherryOVA14 tumors we found approximately 1.9% of imaged NK cells could be found within 5μm of a Xcr1-Venus+ cDC1 while only 0.38% of transferred OT-I T cells can be found within 5μm of a Xcr1-Venus+ cDC1s (Fig. 4b). Further, we observed migration (motion associated with significant displacement) of NK cells greater than 5μm from a XCR1+ cDC1 while NK cells in close contact (<5μm) had reduced motility, consistent with continuous and/or synaptic engagements (Fig. 4c, Supplementary Video 2). These findings suggest that in the tumor NK cells are the most relevant source of Flt3L, which controls SDC levels, likely due to an increased affinity of NK cells for cDC1 DCs. This finding is consistent with recent data showing chemokine/receptor pairing for NK cells and XCR1+ DC in the TME15. Consistent with NK cells playing a direct role in DC survival, when CD103+ DCs were sorted from WT mouse LNs and incubated with media or purified NK cells, CD103+ DCs co-cultured with NK cells showed significantly increased survival over 24hr and 72hrs of incubation (Fig. 4d). These studies suggest that NK cells form stable interactions with SDCs in the tumor, more often than T cells, and suggest that targeting NK cell levels may be a novel therapy to increase Flt3L production and subsequently the levels or survival of SDCs in the tumor. We note that while we see evidence that NK cells provide increased survival to CD103+ DCs in the tumor, it is possible that NK cells may also be acting on DC precursors to control these SDCs.
Figure 4. NK cells make frequent, stable interactions with XCR-1+ DCs and provide pro-survival signals.
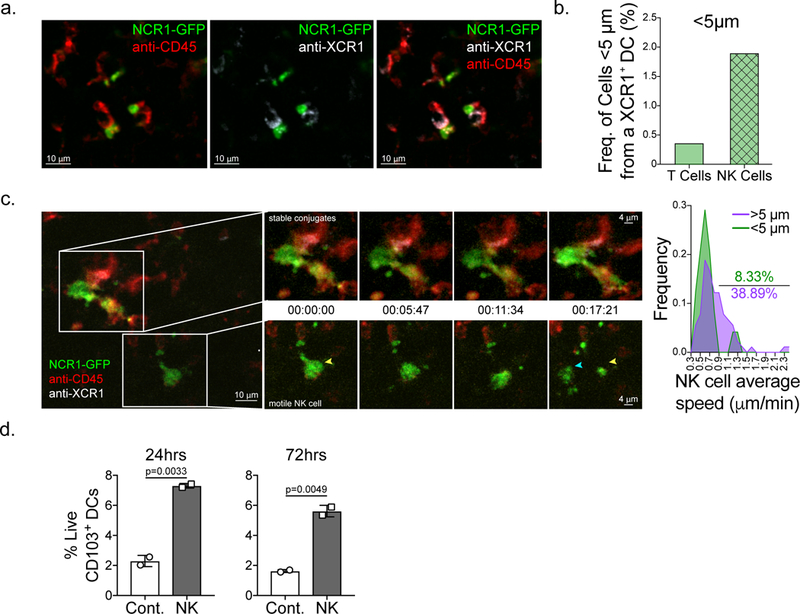
(a-b) Still images from live 2-photon imaging of ectopic B78 melanoma tumor slices from Ncr1-GFP (green) mice stained with anti-XCR1 (white) and anti-CD45 (red) antibodies. (a) Example of static interactions between NK cells and XCR1+ DCs. Representative images from three biologically independent tumors. See also Supplementary Video S1. (b) Quantification of the number of transferred ubiquitin-CFP OT-I CD8+ T cells and endogenous NK cells less than 5μm from a XCR1+ DC in B78cherryOVA tumors in a triple reporter mouse expressing Ncr1-GFP, Xcr1-venus, and CD11c-mCherry. Still images from two biologically independent tumors were used for calculations. (c) Still images of dynamic NK cell-DC interactions. Insets show a time series of two regions of interest where NK cells are in close proximity (< 5μm) to XCR1+ DCs and making stable interactions (top) and a NK cell greater than 5 μm from a XCR1+ DC (bottom) with much more motility. Yellow arrow head, NK cell starting point. Blue arrow head, NK cell ending point. NK cell speeds were quantified (n=114 individual cells) and NK cells were parsed by proximity to XCR1+ DCs (< 5μm or > 5μm from an XCR1+ DC), speeds binned into 22 bins ranging from 0.3–2.4 μm/min, and plotted as an XY plot of Frequency of NK cells by speed. Images are representative of three independent biological replicates. See also Supplementary Video 2. (d) CD103+ DCs from pooled steady state mouse lymph nodes were sorted and incubated with WT NK cells and survival was measured by staining with viability dye at 24hrs and 72hrs of incubation (n=3 technical replicates per treatment group). Data are representative of three biological replicates.
Human NK cell abundance in the tumor correlates with FLT3LG expression and BDCA3+ SDCs
Our mouse studies show that NK cells produce Flt3L and control the levels of SDCs in the tumor. Given these findings, we interrogated human datasets for the abundance of NK cells by generating a NK cell gene signature based on previously published datasets and expression profiling of specific NK cell genes16,17 (Fig. 5a, Supplementary Fig. 6). Using the NK cell gene signature as an estimate of NK cell abundance in samples from the TCGA melanoma dataset11 we found a significant correlation between the levels of NK cells in the tumor and FLT3LG expression (Fig. 5b), consistent with intratumoral NK cells in humans being a source of FLT3LG. NCR1 expression alone, a NK cell receptor expressed specifically in NK cells (Supplementary Fig. 6), showed a significant correlation with FLT3LG levels in the tumor (Supplementary Fig. 7a). Further consistent with our mouse data, using gene signatures for SDCs (Fig. 1a) and NK cells (Fig. 6a) to estimate cellular abundance we found a significant correlation between the levels of NK cells and SDCs in human melanoma patients (Fig. 5c). NCR1, a NK-specific gene, alone significantly correlates with the SDC signature (Supplementary Fig. 7b). To directly compare SDC and NK cell numbers, human melanoma biopsy samples were collected, digested into single cell suspension, and analyzed by flow cytometry (cohort A; Supplementary Table 2; Supplementary Fig. 2). Direct analysis of NK cell and SDC populations in the tumor demonstrated a significant correlation between the levels of NK cells and BDCA3+ DCs in the tumor (Fig. 5d). BDCA3+ DCs in the tumor did not correlate with CD4+ Thelper cells, CD8+ T cells, or CD45 negative cells in the tumor (Supplementary Fig. 7c-e), suggesting this is a specific correlation between NK cells and BDCA3+ DCs. The correlation between NK cells and BDCA3+ DCs was also found in the TME of other cancer types, notably in head and neck squamous cell carcinoma (Fig. 5e), suggesting this innate immune cell relationship may be broader than a single indication or sub-indication.
Figure 5. BDCA3+ DC levels correlate with levels of NK cells in the human melanoma tumor microenvironment.
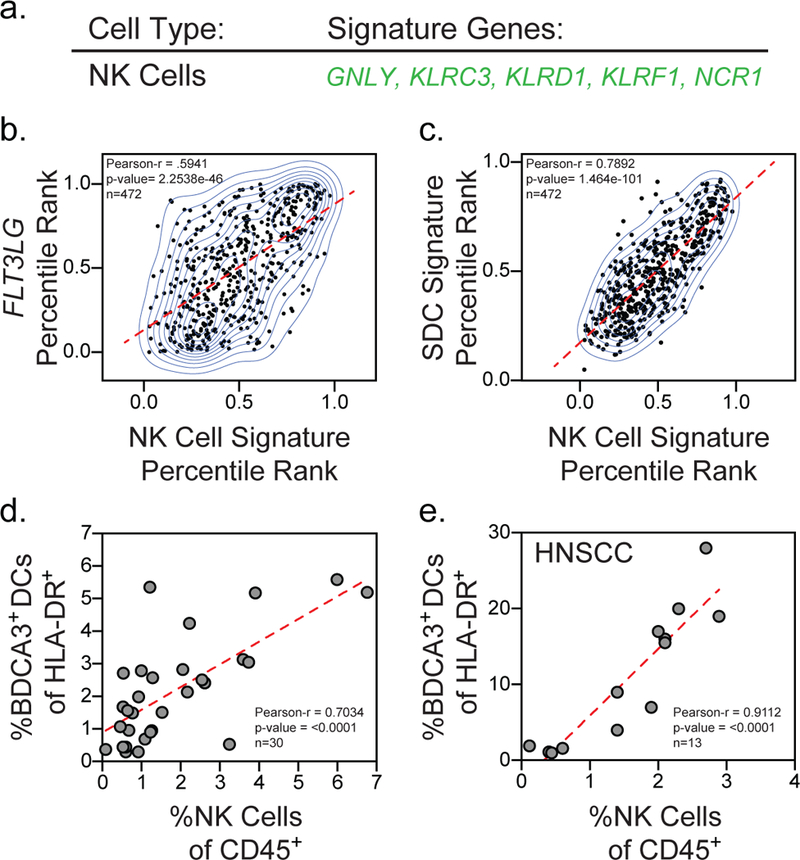
(a) NK cell gene signature. (b) Percentile rank normalization of FLT3LG expression plotted versus the percentile rank of the NK cell gene signature for individual patients in the TCGA melanoma dataset11 (n=472 biologically independent melanoma tumor samples). (c) Percentile rank normalization of the SDC gene signature (Fig. 1a) plotted versus the percentile rank of the NK cell gene signature for individual patients in the TCGA melanoma dataset11 (n=472 biologically independent melanoma tumor samples). (d) Quantification of NK cells of total CD45+ cells plotted versus quantification of BDCA3+ DC of total HLA-DR+ cells for individual melanoma patients in cohort A (n=30 tumor samples from 29 patients). (e) Quantification of NK cells of total CD45+ cells plotted versus quantification of BDCA3+ DC of total HLA-DR+ cells for individual patients in a cohort of head and neck squamous cell carcinoma patient samples (n=13 biologically independent tumor samples). (a-d) Data plotted as scatter plot with regression line superimposed (dotted red line); correlation assessed using the Pearson correlation coefficient and two-tailed p-value.
Figure 6. NK cell and BDCA3+ DC levels uniquely correlate with anti-PD-1 responsiveness in melanoma patients.
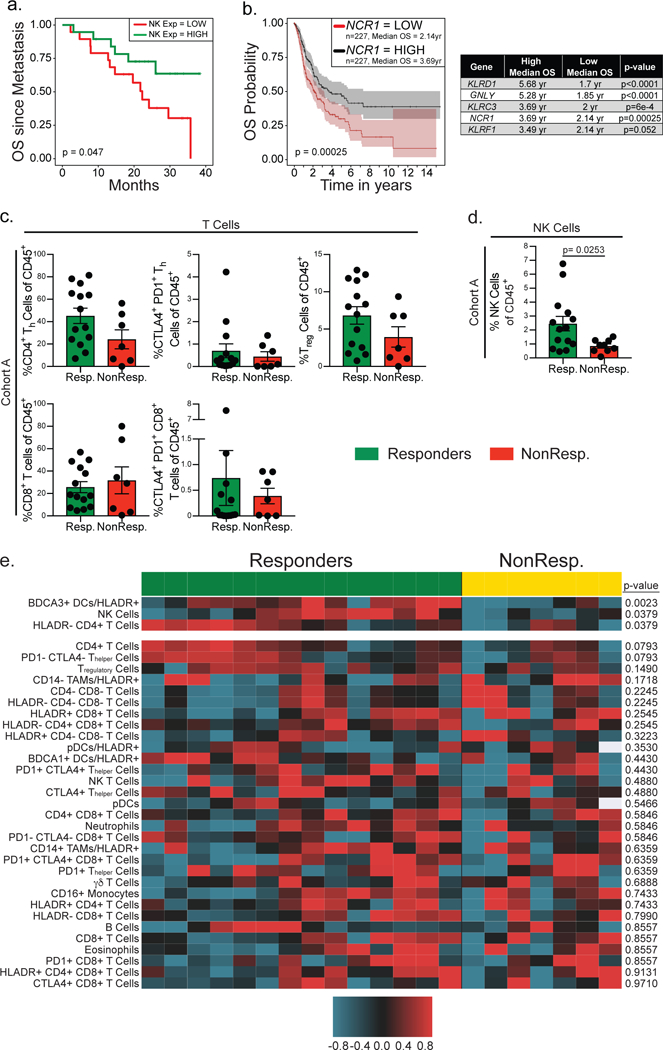
(a) Kaplan-Meier plot of overall survival of metastatic melanoma patients. Data (from 10, n = 38 biologically independent primary metastatic melanoma samples) are parsed into “high” (green; n=19 biologically independent metastatic melanoma samples) and “low” (red; n=19 biologically independent metastatic melanoma samples) bins at 50% (median) stringency thresholds for levels of expression of the NK cell genes. Two-tailed p-value calculated by log rank test after adjusting for multiple comparisons. (b) Kaplan-Meier plot for overall survival of melanoma patients median split for the gene expression of the NK cell specific gene NCR1 and individual survival statistics for each gene in the NK cell gene signature. Data from 11 are parsed into “high” (black; n=227 biologically independent melanoma samples) and “low” (red; n=227 biologically independent melanoma samples) bins at 50% stringency threshold for levels of expression of NCR1, KLRD1, GNLY, KLRC3, or KLRF1. Plots and statistics generated by the UZH TCGA Cancer Broswer28. n=454 biologically independent melanoma patient samples; shaded regions identify 95% confidence interval; two-tailed p-value calculated by log rank test after adjusting for multiple comparisons. (c) Frequency of T cell populations (CD4+ Thelper, CD4+ Tregulatory, and CD8+ T cells) of CD45+ cells in the tumor of individuals. Patients binned as either responders (green, including partial or complete responses; n=14 biologically independent metastatic melanoma tumor samples) or non-responders (red, including stable disease and progressive disease; n=7 biologically independent metastatic melanoma tumor samples). (d) Frequency of NK cells of total CD45+ cells in the tumor of individuals. Patients binned as either responders (green, including partial or complete responses; n=14 biologically independent metastatic melanoma tumor samples) or non-responders (red, including stable disease and progressive disease; n=9 biologically independent metastatic melanoma tumor samples). (c-d) Data were analyzed by two-tailed unpaired parametric t-test, and presented as mean ± S.E.M. with all individual points shown. (e) Heat map of 33 cell populations defined from flow cytometric analysis of a subset of melanoma samples from cohort A (n=21 biologically independent metastatic melanoma tumor samples from 20 patients) separated by anti-PD1 responders (green; partial or complete responses; n=14 biologically independent samples) and non-responder (yellow; stable and progressive disease; n=7 samples from 6 patients). For each population two-tailed p-values were calculated with the Wilcoxon rank-sum test. All populations are a fraction of total CD45+ immune cells unless otherwise noted. Data for each row were logged and mean-centered.
NK cells in human melanoma correlate with increased overall survival.
A positive correlation of SDC and NK cells would logically predict that NK numbers should also predict survival. The NK cell gene signature (Fig. 5a), normalized in each patient as a z-score, was used to bin patients with ‘low’ or ‘high’ NK cell numbers based on a median split (50% stringency). This demonstrated that patients with high NK cell numbers have a significantly increased OS in a Kaplan-Meier plot analysis for two independent datasets (Fig. 6a; Supplementary Fig. 7f11). NCR1 expression alone is significantly correlated with increased overall survival in the TCGA melanoma dataset11 and further, 4 of the 5 genes in the NK cell signature are individually associated with increased overall survival (Fig. 6b). These findings suggest, as we have shown in mouse models, that NK cells produce FLT3LG in human melanoma patients, controlling the levels of BDCA3+ stimulatory DCs in the tumor, and leading to increased patient survival. We note that these findings do not exclude a more traditional role of NK cells in tumor rejection, namely direct tumor cytolysis.
NK cells predict responsiveness to anti-PD-1 immunotherapy in melanoma patients.
Given the important role for NK cells in producing FLT3LG and controlling the levels of SDCs in the tumor, which predict responsiveness to anti-PD-1 immunotherapy, we asked if NK cells and/or T cells correlate with response to immunotherapy. We found no significant correlation with responsiveness to anti-PD-1 immunotherapy with T regulatory cells, CD4+ T helper cells, CD8+ T cells, and T cells double positive for the markers PD-1 and CTLA-4 (Fig. 6c), although there were weak trends with some of these populations. In particular, we did not recapitulate previous data that suggested that a PD-1+CTLA-4+ CD8+ “exhausted” T cell signature was predictive of outcome (see Discussion). However, NK cells were significantly enriched in the tumors of patients that respond to anti-PD-1 immunotherapy (Fig. 6d). These findings are consistent with our finding in mice that NK cells, Flt3L, and SDC form a cluster of determinants for outcome, likely at least in part driven by NK enhancement of SDC through production of Flt3L.
The NK-SDC axis is uniquely associated with responsiveness to anti-PD-1 immunotherapy.
The comprehensive flow cytometry panels used for melanoma cohort A allowed for the quantification of 33 immune populations in the TME (Supplementary Fig. 2, Supplementary Table 2). To ask if NK cells and SDC levels in the TME are uniquely associated with responsiveness to anti-PD-1 immunotherapy we assigned z-scores to population fractions in each sample, and found that, of identified immune cells in the TME, BDCA3+ DCs and NK cells significantly correlate with responsiveness to anti-PD-1 immunotherapy (Fig. 6e, Supplementary Table 3). Furthermore, when viewed this way, it appears that very high frequencies of HLADR− CD4+ T cells significantly correlate with anti-PD-1 responses and may represent an alternative prognostic feature of responsiveness in patients where BDCA3/NK frequencies were not numerous. This may indicate that PD-1 blockade can be supported by more than one type of immune infiltrate although further studies are required to confirm this.
DISCUSSION
Here we have identified a dendritic cell-NK cell contexture that defines the responsiveness of a patient to anti-PD1 immunotherapy. The idea that the non-T cell compartment would have a profound effect upon overall survival and T cell therapy is not in itself novel, but this work specifically identifies an axis that connects one innate cell type (NK) to another antigen presenting cell subset (SDC). This work also extends very recent work that shows NK cells produce cytokines in tumors that attract XCR1+ DCs15. However, it takes this significantly further insofar as it provides the additional important fact that NK cells make the formative cytokine for SDC, namely FLT3LG. In our study, exhausted T cell frequencies themselves did not specifically predict response to therapy as it did in previous studies18. At present, the source of this discrepancy is unclear but may result from differences in digestion protocols, those previous studies relying on long overnight digestion that may differentially liberate or preserve these cells or raise these markers on activating T cells. In any case, given the findings that exhausted T cells are insensitive to PD1 restoration of function19, it is not immediately obvious how truly exhausted cells in vivo would then predict responsiveness; it is possible that the phenotype masquerades as exhaustion in some way. It is clear that more work is needed in this area, but both mouse and human data support the role for NK cells in making Flt3L, with the mouse studies unequivocally showing that NK cells play a major role in enhancing the frequencies of SDCs in the tumor. Whether this also occurs via interaction of NK cells with incoming pre-cDC precursors also remains to be determined. Such experiments are expected to be difficult due to the low frequencies of each of the populations as well as the lack of available single-channel imaging reagents for live-imaging pre-cDC.
It also remains to be determined if the NK-DC axis in the tumor that defines responsiveness to anti-PD-1 immunotherapy have surrogates in the peripheral blood that may provide another, more accessible, readout of this population. A recent study has shown one marker to be upregulated in the peripheral blood of metastatic melanoma patients that respond to anti-PD-1 immunotherapy is CD56, a marker associated with NK and NKT cells, and further suggest that a myeloid population in the blood may be a good prognostic tool for identifying responsive patients20. Further studies are required to determine how these cell types relate to NK-DC axis found in the tumor microenvironment but suggests that there may be prognostic cell types in the blood that represent a tumor microenvironment receptive to immunotherapy.
Both NK cell and SDC populations are routinely ‘rare’ at less than a few percent of total immune populations, yet this is an important dynamic range that predicts both overall survival as well as responsiveness to checkpoint blockade. Previous studies have implicated DC-NK interactions in activating NK cells in vitro leading to better tumor responses in vivo21,22. In this study we move our understanding of NK-DC interactions further and show direct NK-DC interactions in the tumor in vivo. On the one hand this may result from the aforementioned chemokine attraction between NK cells and SDCs15, but may also imply that they live in special anatomical locations, perhaps akin to tertiary lymphoid structures. While our imaging suggests much smaller ‘clusters’ of SDC with NK, it is formally possible that these clusters have additional features that we have not yet seen with imaging to date. We also note that T cells, while more abundant than NK and apparently similarly loaded with Flt3L do not appear to be capable of driving SDC numbers and this raises the question as to whether they might be blocked for production of the protein or simply do not have ample opportunity to interact with SDCs.
Prior to this work, it has been long thought that the predominant source of Flt3L is epithelial cells. This idea is traced to studies of in vitro elicted Human vascular endothelial cells (HUVEC)23 which likely are a uniquely elicited in vitro population whose in vivo correlate may not be obvious. While our work does not negate that Flt3L may be produced by epithelial cells, in tumors at least, the CD45− compartment typically shows no significant reporter (e.g. RNA) expression as compared to lymphocytes. It is also possible that epithelial cells in other tissue sites are more formidable sources of this cytokine. Consistent with this idea, work in mouse models suggests that nonhematopoietic cells are required to produce Flt3L in nonlymphoid tissues (lung, kidney, pancreas, and liver)24. Along similar lines, although NK cells are critical Flt3L producers in the TME, T cells may well be more important in other tissues or contexts.
It has long been acknowledged that NK cells can have a greater effect on tumors than their ability to kill cancer cells alone21,25 but their role as critical producers of Flt3L and the formation of an apparently non-lytic interaction was not known. Our studies suggest that targeting NK cells by either increasing the numbers in the TME or activating these cells should increase the levels of FLT3LG in the tumor and in turn increase the levels of protective SDCs in the TME, either through skewed differentiation of precursor DCs in the tissue or prolonged survival of differentiated SDCs, providing increased responses to anti-PD-1 immunotherapy. Consistent with this idea, blocking CD96, an inhibitory receptor on NK cells and T cells26,27, has been shown to increase NK cell activity and has recently been shown to work synergistically with anti-PD-1 and anti-CTLA-4 checkpoint blockade26. We predict that therapies aimed at recruiting and activating NK cells in tumors, may prove to have orthogonal benefits to those provided by checkpoint inhibition.
Online Methods
Melanoma bioinformatics analysis
The melanoma data set GEO accession GSE19234 (n = 44)10 was pre-processed by quantile normalizing in the R environment prior to evaluating the signature and assessing associations with survival (Cox proportional hazards). For NK gene signature analysis only, 6 secondary tumors were removed from the analysis of GSE19234, leaving 38 samples for analysis. The TCGA melanoma dataset downloaded from fireBrowse (n=469)11 with RSEM normalized expression data was further normalized by taking Log2(Exp+1) and z-scoring across genes in R prior to evaluating the signature. SDC/NSM ratio signature is calculated as the log of the mean expression of SDC genes divided by the mean expression of the NSM genes, followed by z-score standardization (mean = 0, s.d. = 1). Survival analyses were performed using Cox Proportional Hazards modeling. Log rank p-values are used to assess significance after adjusting for multiple comparisons using Benjamini-Hochberg method29. Kaplan-Meier survival plots were generated using the Survival package in R and we classified each sample as ‘high’ or ‘low’ using the 33%, 50% (Median), or 66% value of the SDC or SDC/NSM ratio signature. Kaplan-Meier survival plots were generated for the NK cell gene signature using the Survival package in R and classified as ‘high’ or ‘low’ using a median split. The NK cell gene signature (Figure 5A) was based on previously published differential expression analysis in peripheral lymphocytes16,17 and further refined by selecting genes that were predominantly expressed in NK cells using the Primary Cell Atlas database from BioGPS30 (Fig S6). Survival analysis with the NK cell gene signature was undertaken on median normalized datasets (TCGA melanoma and metastatic melanoma). The NK cell gene signature was extracted from the dataset and used to calculate a median-based z-score for each gene of the NK cell gene signature. Z-scores were then averaged across all signature genes for each patient, generating a vector of means, one for each patient, that was used to assess association with survival (Cox proportional hazards). Kaplan-Meier survival plots were generated using the Survival package in R and we classified each sample as ‘high’ or ‘low’ using a median split (50 % stringency), with gene signatures above the median classified as ‘high’ and below the median classified as ‘low’.
Patients and samples
Patients were enrolled in this study if they had histologically confirmed stage IV or III unresectable metastatic melanoma. Patients were consented for tissue collection under a UCSF IRB approved protocol (UCSF CHR #13–12246). The study enrollment period started from December 2012 to November 2016, and the sample size was determined by the availability of specimens throughout this period. Patients were treated with the following anti-PD-1/PDL-1 axis blocking monoclonal antibodies: pembrolizumab (Keynote 001, 002, 006 or expanded access program or commercial supply) or nivolumab (commercial supply).
Melanoma cohort A patient selection and sample collection:
Cutaneous/subcutaneous metastatic melanomas (including metastases to the lymph node) were biopsied with either a punch (4 mm or 6 mm), a surgical excision, or core biopsies (16 g or 18 g) exclusively. Patients in cohort A (n = 21 patients with n = 23 samples) were selected without regard to prior treatment with immunotherapies, therefore in this cohort there is a range of patients from immunotherapy naïve to patients having been treated with anti-PD-1, ipilimumab, and/or IL-12 (Supplementary Table 2). Samples suspected to be the primary melanoma lesion are noted in Supplementary Table 2.
Melanoma cohort B patient selection and sample collection:
Cutaneous/subcutaneous tumors were biopsied with either a punch (4 mm or 6 mm), a surgical excision (sample K10), and all other tumor biopsies were with core biopsies (16 g or 18 g) exclusively. An additional sample was sent for pathology evaluation. Biopsies (n = 23) were collected prior to infusion of anti-PD-1 (Supplementary Table 2).
The fresh biopsy samples (cohort A and B) were immediately placed in a sterile container on saline soaked gauze (or submerged in PBS) and placed in a container of wet ice for transport to the laboratory for evaluation. Patient samples were coded and flow analysis and outcome data was scored by separate individuals prior to data agglomeration. All samples were processed and analyzed by flow cytometry, but only those with at least 1,000 live CD45+ cell events were included in the analysis.
Evaluating Responses to Therapy
All response evaluation was with radiologic imaging and limited to best overall response using the Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST). Complete Response was defined as a complete regression of all target and non-target lesions, Partial Response was defined as regression of target lesions > 30 % with no new lesions appearing, Stable disease was defined as ≤ 30 % decrease or ≤ 20 % increase in the size of target lesions. Progressive disease was defined as increases in target lesions ≥ 20 % or appearance of new lesions > 1 cm in size. Patients with complete or partial response were classified as “responders” while those with stable disease or progressive disease were classified as “non-responders”.
Associated Clinical trial registration numbers (NCT Numbers):
Protocol 01: 01295827
Protocol 02: 01704287
Protocol 06: 01866319
EAP: 02083484
MTB: Noneà UCSF CHR Number: 13–12246
Human tissue digestion
Cohort A tissue was vigorously minced with surgical scissors and transferred to a GenleMACs™ C Tube (Miltenyi Biotec) with 100 μg/mL Liberase TL (Roche) and 200 μg/mL DNase I (Roche) at 3mL per gram of tissue. C Tubes were incubated in the GentleMACS™ octo dissociator (Miltenyi Biotec) with heaters following the manufacturers dissociation protocol (Miltenyi Biotec Tumor Dissociation Kit). 10mL of sort buffer (PBS + 2% fetal calf Serum (FCS) + 2mM EDTA) was added to samples and filtered through a 100 μm filter, spun down, and red blood cells lysed with 175mM ammonium chloride. Cohort B tissue was vigorously minced with surgical scissors and transferred to a 50 mL conical with 20 μL/mL Liberase TL (at 5 mg/mL, Roche) and 50 U/ml DNase I (Roche) per 0.3 g of tissue for 30 minutes at 37° C with 5 % CO2 with constant agitation. Samples were then filtered through a 70 μm filter, spun down, and re-suspended for staining31.
Antibodies against human antigens
CD45 clone Hl30 (eBioscience 47–0459-42), CD3e clone OKT3 (eBioscience 46–0037-42), HLA-DR clone L243 (eBioscience 48–9952-42), CD56 clone CMSSB (eBioscience 46–0567-42), CD56 clone HCD65 (BioLegend 318304), CD19 clone H1B19 (eBioscience 46–0198-42 and 56–0199-42), CD14 clone 61D3 (Invitrogen Q10056; Cohort 1), CD14 clone M5E2 (BioLegend 301836; Cohort 2), CD16 clone 3G8 (BioLegend 302040), CD11c clone 3.9 (eBioscience 56–0116-42), CD85g clone 17G10.2 (eBioscience 12–5179-42), BDCA1 clone L161 (BioLegend 331516), BDCA3 clone AD5–14H12 (Miltenyi 130–090-513), CD4 clone RPA-T4 (BioLegend 300550), CD8 clone RPA-T8 (BioLegend 301040), CD25 clone BC96 (BioLegend 302612), CD25 clone 2A3 (eBioscience 17–0259-42), FoxP3 clone PCH101 (eBioscience 77–5776-40), FoxP3 clone 236A/E7 (eBioscience 25–4777-42), PD-1 clone EH-12 (BioLegend 329930), CTLA-4 clone BNI3 (BioLegend 369606), γδ TCR clone B1.1 (BioLegend 331212), Flt3L (Abcam ab9688).
Flow cytometry
All antibodies were purchased from BD Pharmingen, eBioscience, Invitrogen, BioLegend, the UCSF hybridoma core, R&D Systems, Abcam, or were produced in the Krummel Lab. For surface staining, cells were incubated with Fc receptor blocking solution (Human: BioLegend Human FcX, Mouse: anti-CD16/32 antibody clone 2.4G2) and stained with antibodies in PBS + 2% FCS + 2 mM EDTA for 30 min on ice. Viability was assessed by staining with Zombie Aqua fixable viability dye (BioLegend, 423102) or Zombie NIR fixable viability dye (BioLegend 423106). All human samples were fixed with BD Cytofix prior to analysis following the manufacturer’s protocol. All mouse samples were run on the cytometer without fixation. For intracellular staining samples were first stained for surface markers, as described above, and then fixed with Fixation/Permeabilization solution and washed with permeabilization buffer following the manufacturer’s suggested protocol (eBioscience 00–5523-00). Intracellular staining was undertaken in the presence of 2% FCS and an Fc receptor blocking solution (BioLegend Human FcX). Flow cytometry was performed on a BD Fortessa or Aria Fusion flow cytometer. Analysis of flow cytometry data was done using FlowJo (Treestar) software. It should be noted that while CD85g is a strong marker of pDCs in the peripheral blood, it can provide low signal on tissue pDCs, meaning that the use of this marker to define pDCs in the tumor may underrepresent the pDC population in the tumor.
Bioinformatic analysis of human flow cytometry data
Myeloid and lymphoid populations in the tumor were defined as shown in Supplementary Fig. S2.
For each population, the Wilcoxon rank-sum test statistic, p-value and q-value was calculated between responders and non-responders. Wilcoxon rank-sum test statistics and p-values were calculated using the wilcox.exact function from the exactRankTests R package and q-values were calculated using the q-value function in the Bioconductor R package. A heatmap of population frequency out of total CD45+ or HLA-DR+ cells (as indicated) was made using the clustermap method from the seaborn python module. The population frequency data was logged and mean-centered. Row and column clustering were suppressed when generating the heatmap and instead rows were ordered by the calculated p-value and the columns were ordered by responder status.
RNAseq analysis of human melanoma samples
With a subset of cohort A melanoma patient samples, 50,000 total live cells, determined by staining with the Zombie Aqua fixable viability dye (BioLegend, 423102), were directly sorted into lysis buffer by fluorescence activated cell sorting (FACS) to isolate RNA. RNA was isolated using the Dynabeads mRNA DIRECT purification kit (ThermoFisher Scientific 61011) following the manufacturer’s suggested protocol. Isolated RNA was converted into amplified cDNA using the Ovation RNA-Seq System V2 kit (NuGen 7102-A01) and cDNA was converted into RNAseq libraries using the Nextera XT DNA Library Prep kit (Illumina FC-131–2001), following the manufacturer’s suggested protocol. RNAseq library quality was checked using a Bioanalyzer HS DNA chip (Agilent) and pooled. Pooled libraries were then sequenced via a single-read 50bp MiSeq (Illumina) run and libraries containing >10% of reads aligned to coding regions and >1000 unique reads in the total library were selected for further sequencing. RNAseq libraries that met the previous quality control criteria were pooled and submitted to the University of California, San Francisco Center for Advance Technology for paired-end, 100bp (PE100) sequencing on the HiSeq4000 (Illumina). Using the Burrows-Wheeler Aligner32, sequenced RNAseq library reads were then aligned against a reference of ribosomal RNA (rRNA) and mitochondrial RNA (mtRNA) to deplete the dataset of rRNA and mtRNA. The remaining reads were then aligned to the Ensembl GrCh38.85 transcriptome build using the software package RSEM33 with the alignment tool Bowtie 2 34. RNAseq libraries with >1×106 protein-coding, non-rRNA read counts were used to analyze RNA expression. Transcripts per million (TPM) for individual genes were used for analyses and in all cases TPMs were normalized by z-score. When gene signatures were calculated from these RNAseq datasets the TPM of each individual gene in the signature was normalized by calculating a z-score and then the z-scores were averaged for the entire signature to give the final gene signature z-score.
Mouse strains
All mice were housed and bred at the University of California, San Francisco and maintained under specific pathogen-free conditions in accordance with the regulatory standards of the NIH and American Association of Laboratory Animal Care standards, and are consistent with the UCSF Institution of Animal Care and Use Committee (IACUC approval: AN170208–01B). C57Bl/6 (WT) mice were purchased from The Jackson laboratory or Simonsen Laboratories or bred in house. Age matched and sex matched animals were used for all experiments. Ectopic tumor experiments utilized 6–8 week old animals. The Flt3l−/−35, Il2rg−/− 36, and Ncr1-GFP 37 mouse strains were purchased from The Jackson Laboratory. Ncr1-GFP mice were used as heterozygous animals as the knock-in disrupts gene expression. The Flt3l-reporter mice were generated, by contract, by the company Biocytogen. The P2A-iCreERT2-T2A-TFP sequence was inserted between exon 8 and the 3’-UTR of Flt3l at the endogenous locus using CRISPR-Cas9 targeting. Proper insertion of the reporter construct was confirmed by PCR and DNA sequencing. Genotyping of Flt3l-reporter mice was done by PCR using a common forward primer that binds in the Flt3l locus: (5’-AAGGAGTCCCATAGCCCTAGAAGCC-3’), a WT reverse primer that binds in the Flt3l gene (5’-CTCCACAGCTTACGATTGTCGGAGC-3’), and a knock-in reverse primer specific to the iCreERT2 knock-in (5’-CCTGTCCCTGAACATGTCCATCAG-3’). Flt3l-reporter mice were crossed onto the PyMT-mCherry-OVA spontaneous tumor model background 14 and offspring were screened for the PyMT-ChOVA transgene and the Flt3l-reporter knock-in by PCR. Female PyMT-ChOVA; Flt3l-reporter animals were monitored for tumors and used at 20 to 30 weeks of age.
Determination of Flt3L serum levels
Blood from Flt3l-reporter homozygous mice, wildtype C57Bl/6 mice, or C57Bl/6 mice bearing 2-week old B16F10 tumors was collected from the tail vein or by cardiac puncture and allowed to sit for 2 hours at room temperature to clot. Blood was centrifuged for 20 minutes at 2000g and serum was collected. Serum was diluted 1:1 with PBS and Flt3L levels were determined by ELISA using the Quantikine ELISA kit (R&D Systems) following the manufacturer’s suggested protocol.
Cell lines and cell culture
The following tumor cell lines were cultured under standard conditions prior to injection into mice: B16-F10 38, B78-parental 39, B78-ChOVA (B78 parental cells transfected with a Cherry-OVA fusion construct identical to that used in the PyMTChOVA mouse strain)14, and MC38 40. Cells were cultured at 37° C and 5% CO2 in DMEM plus 10 % FCS with 1% Penicillin/Streptomycin/Glutamine on tissue-culture treated plastic plates and split every other day.
Ectopic tumor injection
Tumor cell lines were harvested and washed 3 times with PBS, then mixed at a 1:1 ratio with growth factor-reduced Matrigel Matrix (BD Biosciences) for a final injection volume of 50 μL. 1.5×105 B16F10 and B78 tumor cells or 5×105 MC38 tumor cells were injected in the left flank of shaved mice subcutaneously and allowed to grow for 14 days before use, unless otherwise stated.
Mouse tissue digestion
Tumors were dissected from mice and total weight of removed tumor tissue was determined. Tumors were then minced using scalpels and digested with 500 U/mL Collagenase IV (Sigma), 100 U/mL Collagenase I (Worthington) and 200 μg/mL DNAse I (Roche) per 0.3 grams of tumor weight for 30 minutes in a 50 mL conical tube at 37° C with constant shaking. After 30 minutes, 7 mL of PBS + 2% FCS + 2 mM EDTA was added to tubes and tumors were passed through a 100 μm cell strainer to remove large pieces of tumor. Large pieces of tumor were dissociated by pushing the cells through the 70 μm cell strainer with the plunger of a 5 mL syringe. To digest lymph nodes (LNs), LNs were carefully dissected from mice and cleaned of fat. Inguinal LN were taken as tumor draining. LNs were pierced and torn with sharp forceps in 24-well plates and incubated for 15 min at 37° C in 1 ml of digestion buffer (100 U/ml collagenase I, 500 U/ml collagenase IV, and 200 μg/ml DNAse I in RPMI-1640). After the first 15 min of incubation, cells were pipetted up and down repeatedly, then returned for a second 15-min incubation at 37° C. After digestion, LNs were washed with PBS + 2% FCS +2 mM EDTA and filtered through a 100-μm cell strainer before staining for flow cytometry.
Antibodies against mouse antigens
CD3 clone 17A2 (BioLegend 100237 and 100244), CD4 clone RM4–5 (BioLegend 100545 and 100510), CD4 clone GK13 (BioLegend 100403), CD8 clone 53–6.7 (BioLegend 100734), CD11b clone M1/70 (BioLegend 101257), CD11c clone N418 (BioLegend 117339 and 117338), CD19 clone 6D5 (BioLegend 115522), CD24 clone M1/69 (BioLegend 101822), CD45 clone 30-F11 (BioLegend 103139, 103132, and 103114; eBioscience 56–0451-82), CD45R clone RA3–6B2 (BioLegend 103247, 103246, and 103226), CD69 clone H1.2F3 (eBioscience 25–0691-81), CD90.2 clone 30-H12 (BioLegend 105331), CD103 clone 2E7 (BioLegend 121406 and 121414), F4/80 clone BM8 (BioLegend 123108), Flt3L (R&D Systems AF427), Ly6C clone HK1.4 (BioLegend 128037), Ly6G clone 1A8 (BioLegend 127645), MHC-II clone M5/114.15.2 (BioLegend 707631), NK1.1 clone PK136 (BioLegend 108707, 108720, and 108749), Streptavidin-Brilliant Violet 650 (BioLegend 405231), Streptavidin-APC (eBioscience 17–4317-82). Depleting antibodies: NK1.1 clone PK136 (BioXCell BE0036), and IgG2a isotype control (BioXCell BE0085).
Isolation of CD103+ DCs/NK cells and in vitro co-culture
To isolate CD103+ DCs, all non-gut draining lymph nodes were harvested from wildtype C57Bl/6J mice and dissociated into a single cell culture by digesting at 37° C in 1mL of 500 U/mL Collagenase IV (Sigma), 100 U/mL Collagenase I (Worthington) and 200 μg/mL DNAse I (Roche) for 30 minutes with agitation every 15 minutes. Single cell LN suspensions were stained with a biotin-conjugated anti-CD2 antibody (BioLegend 100104) and negative selection was performed with an EasySep Biotin Selection Kit following the manufacturer’s instructions before staining. CD103+ DCs were sorted using the gating scheme described in Supplementary Fig. 3e. 1×104 CD103+ DCs were incubated with 1×105 NK cells isolated using the EasySep Mouse NK Cell Isolation Kit following the manufacturer’s instructions. NK cell and CD103+ DCs were co-cultured in RPMI +10% FCS + 1% Penicillin/Streptomycin/Glutamine + 1x non-essential amino acids + 50 μM beta-mercaptoethanol + 7.5ng/mL recombinant murine GM-CSF. Cells were isolated at 24 or 72 hrs post-incubation and viability was assessed by staining with Zombie NIR fixable viability dye (BioLegend 423106). CD103+ DCs were gated as described in Supplementary Fig. 3e.
Ex vivo tumor slice staining and multiphoton imaging
All Imaging was performed using a custom-built two-photon setup equipped with two infrared lasers (MaiTai: Spectra Physics, Chameleon: Coherent). Emitted light was detected using a 25× 1.2NA water lens (Zeiss) coupled to a 6-color detector array (custom; utilizing Hamamatsu H9433MOD detectors), alternating laser excitation was used to yield 12 detection channels. Emission filters used were: violet 417/50, blue 475/23, green 510/42, yellow 542/27, red 607/70, far red 675/67. The microscope was controlled by the MicroManager software suite41, z-stack images were acquired with 20-fold averaging and z-depths of 5 μm. Data analysis was performed using the Imaris software suite (Bitplane).
Ncr1-GFP animals with 2-week-old ectopic melanoma tumors were euthanized and tumors were harvested. Obstructing fat was removed and tumors were embedded in 4 % low-melting agarose in PBS (SeaPlaque, Lonza). 300 μm thick sections were prepared using a Compresstome VF-200 (Precisionary Instruments Inc.) tissue slicer. Slices were stained with Brilliant Violet 421 anti-XCR1 antibody clone ZET (BioLegend 148216) and Brilliant Violet 605 anti-CD45 antibody clone 30-F11 (BioLegend 103139) in RPMI-1640 supplemented with 5 % rat serum for 2 hours at 37° C with 5% CO2. Slices were washed in RPMI-1640 and attached to plastic coverslips using Vetbond (3M). Slices were kept at 37°C with constant flow-over of RPMI-1640 (without phenol red) with CO2 bubbled in throughout imaging. The MaiTai laser was tuned to 800 nm for excitation of the BV421 and BV605 fluorophores. The chameleon laser excitation was tuned to 910 nm for excitation of GFP.
For quantification of T cell and NK cell distances to XCR1+ DCs Xcr1-venus; Ncr1-GFP;Cd11c-mCherry triple reporter mice were injected with 2e4 ubiquitin-CFP OT-I CD8+ T cells into the tail vein one day prior to tumor injection. Animals were injected with150k B78 chOVA tumors in the absence of growth factor-reduced Matrigel Matrix and intravital imaging of tumors was performed 2 weeks later. Still 3D projectionsof tumors were used to calculate distances. T cells-DC and NK cell-DC distances were measured using the Imaris software suite (Bitplane). Briefly, the center of each cell type was determined and the smallest distance betweenthe cell types of interest (T cell-DC or NK cell-DC) was calculated using the equation:
With R being the distance between the two cells and x, y, z representing the coordinates of the cells in the 3D space. 3 separate image frames from 2 independent tumors were used to calculate the percent of T cells and NK cells within 5μm of a XCR1+ DC. For imaging, the MaiTai laser was tuned to 810 nm for excitation of CFP and the chameleon laser excitation was tuned to 980 nm for excitation of Venus, mCherry, and GFP. Z-stack images were acquired with 15-fold averaging and z-depths of 4μm.
Bone Marrow chimera generation
To generate BM chimeras Flt3l−/− mice were lethally irradiated by exposure to 1,100 rads of irradiation in 2 doses, 3 hours apart. 2–5 ×106 BM cells, consisting of 50% Il2rg−/− and 50% WT or Flt3l−/− BM were injected retro-orbitally to reconstitute irradiated mice. Chimeric mice were injected with subcutaneous B16F10 tumor cells 8 weeks after reconstitution.
Tumor measurements
For tumor measurements, tumor area (mm2) was measured with calipers as tumor width x tumor height, over the indicated time periods.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software. Normality of data was assessed using the D’Agostino & Pearson omnibus normality test in Prism, all data presented passed the normality assumption unless otherwise noted. Unless specifically noted, all data are representative of > 3 independent experiments. Error bars represent mean ± S.E.M. calculated using Prism unless otherwise noted. Specific statistical tests used were paired and unpaired parametric T tests, or unpaired nonparametric Mann-Whitney U test (if data failed normal assumption) and all p values < 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We thank L. Lanier, J. Roose and L. Fong for advice and generous sharing of materials and M. Spasic and N. Khurana for support with response data. This work was supported by NIH grant R01CA197363 awarded to M. Krummel. Acquisition and processing of human melanoma samples in cohort A described in this study was funded in part by contributions from AbbVie, Amgen, and Bristol-Myers Squibb as members of the Immunoprofiler Consortium. Further support came from NIH grant 5P30CA082103 awarded to the UCSF Hellen Diller Family Comprehensive Cancer Center. M. Broz was supported by the Genentech Predoctoral Research Fellowship, the Margaret A. Cunningham Immune Mechanisms in Cancer Research Fellowship Award, and the Achievement Reward for College Scientists Scholarship. K. Barry is supported by a postdoctoral fellowship from the Cancer Research Institute and Fibrolamellar Cancer Foundation.
Footnotes
Data availability
All RNA sequencing reads were uploaded to Gene Expression Omnibus (GEO) with accession number GSE113126. All other relevant data are available from the corresponding author on request.
Competing Interests
The authors declare no competing financial interests.
References
- 1.Topalian SL, Drake CG & Pardoll DM Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 27, 451–461 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rizvi NA et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (80-. ). 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broz ML et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 26, 638–652 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salmon H et al. Expansion and Activation of CD103+ Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 44, 924–938 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanchez-Paulete AR et al. Cancer immunotherapy with immunomodulatory anti-CD137 and ant-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov. 6, 71–79 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hildner K et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spranger S, Dai D, Horton B & Gajewski TF Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 31, 711–723.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lavin Y et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 169, 750–765.e17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spranger S, Bao R & Gajewski TF Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Bogunovic D et al. Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc. Natl. Acad. Sci. U. S. A. 106, 20429–20434 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.TCGA. Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei SC et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell (2017). doi: 10.1016/j.cell.2017.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu K & Nussenzweig MC Origin and development of dendritic cells. Immunol Rev 234, 45–54 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Engelhardt JJ et al. Marginating Dendritic Cells of the Tumor Microenvironment Cross-Present Tumor Antigens and Stably Engage Tumor-Specific T Cells. Cancer Cell 21, 402–417 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Böttcher JP et al. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 172, 1022–1037.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Du X et al. Genomic profiles for human peripheral blood T cells, B cells, natural killer cells, monocytes, and polymorphonuclear cells: Comparisons to ischemic stroke, migraine, and Tourette syndrome. Genomics 87, 693–703 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Bezman NA et al. Molecular definition of the identity and activation of natural killer cells. Nat. Immunol. 13, 1000–1009 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loo K et al. Partially exhausted tumor-infiltrating lymphocytes predict response to combination immunotherapy. JCI Insight 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Philip M et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krieg C et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med. (2018). doi: 10.1038/nm.4466 [DOI] [PubMed] [Google Scholar]
- 21.Zitvogel L Dendritic and natural killer cells cooperate in the control/switch of innate immunity. J. Exp. Med. 195, F9–14 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernandez NC et al. Dendritic cells directly trigger NK cell functions: Cross-talk relevant in innate anti-tumor immune responses in vivo. Nat. Med. 5, 405–411 (1999). [DOI] [PubMed] [Google Scholar]
- 23.Solanilla A et al. Expression of Flt3-ligand by the endothelial cell. Leukemia 14, 153–62 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Miloud T, Fiegler N, Suffner J, Hämmerling GJ & Garbi N Organ-specific cellular requirements for in vivo dendritic cell generation. J. Immunol. 188, 1125–35 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Goding SR, Yu S, Bailey LM, Lotze MT & Basse PH Adoptive transfer of natural killer cells promotes the anti-tumor efficacy of T cells. Clin. Immunol. 1–11 (2016). doi: 10.1016/j.clim.2016.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blake SJ et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. 6, 446–459 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Dougall WC, Kurtulus S, Smyth MJ & Anderson AC TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunological Reviews 276, 112–120 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Cheng PF, Dummer R ;, Levesque MP, Dummer R & Levesque MP Data mining the cancer genome atlas in the era of precision cancer medicine. Swiss Med Wkly 145, (2015). [DOI] [PubMed] [Google Scholar]
Methods only References
- 29.Benjamini Y & Hochberg Y Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society B 57, 289–300 (1995). [Google Scholar]
- 30.Wu C, Jin X, Tsueng G, Afrasiabi C & Su AI BioGPS: Building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res. 44, D313–D316 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruffell B et al. Leukocyte composition of human breast cancer. Proc. Natl. Acad. Sci. U. S. A. 109, 2796–801 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKenna HJ et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood 95, 3489–3497 (2000). [PubMed] [Google Scholar]
- 36.Cao X et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor γ chain. Immunity 2, 223–238 (1995). [DOI] [PubMed] [Google Scholar]
- 37.Gazit R et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat. Immunol. 7, 517–523 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Fidler IJ Biological Behavior of Malignant Melanoma Cells Correlated to Their Survival in Vivo. Cancer Res. 35, 218–224 (1975). [PubMed] [Google Scholar]
- 39.Graf LH, Kaplan P & Silagi S Efficient DNA-mediated transfer of selectable genes and unselected sequences into differentiated and undifferentiated mouse melanoma clones. Somat. Cell Mol. Genet. 10, 139–151 (1984). [DOI] [PubMed] [Google Scholar]
- 40.Corbett TH, Griswold DP, Roberts BJ, Peckham JC & Schabel FM Tumor Induction Relationships in Development of Transplantable Cancers of the Colon in Mice for Chemotherapy Assays, with a Note on Carcinogen Structure. Cancer Res. 35, 2434–2439 (1975). [PubMed] [Google Scholar]
- 41.Pinkard H, Stuurman N, Corbin K, Vale R & Krummel MF Micro-Magellan: open-source, sample-adaptive, acquisition software for optical microscopy. Nat. Methods 13, 807–809 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.