

Abstract
Aims
Increases in fat-free mass and fat mass have been associated with higher risk of atrial fibrillation (AF) in observational studies. It is not known whether these associations reflect independent causal processes. Our aim was to evaluate independent causal roles of fat-free mass and fat mass on AF.
Methods and results
We conducted a large observational study to estimate the associations between fat-free mass and fat mass on incident AF in the UK Biobank (N = 487 404, N events = 10 365). Genome-wide association analysis was performed to obtain genetic instruments for Mendelian randomization (MR). We evaluated the causal effects of fat-free mass and fat mass on AF with two-sample method by using genetic associations from AFGen consortium as outcome. Finally, we evaluated independent causal effects of fat-free mass and fat mass with multivariate MR. Both fat-free mass and fat mass had observational associations with incident AF [hazard ratio (HR) = 1.77, 95% confidence interval (CI) 1.72–1.83; HR = 1.40, 95% CI 1.37–1.43 per standard deviation increase in fat-free and fat mass, respectively]. The causal effects using the inverse-variance weighted method were 1.55 (95% CI 1.38–1.75) for fat-free mass and 1.30 (95% CI 1.17–1.45) for fat mass. Weighted median, Egger regression, and penalized methods showed similar estimates. The multivariate MR analysis suggested that the causal effects of fat-free and fat mass were independent of each other (causal risk ratios: 1.37, 95% CI 1.06–1.75; 1.28, 95% CI 1.03–1.58).
Conclusion
Genetically programmed increases in fat-free mass and fat mass independently cause an increased risk of AF.

Keywords: Fat-free mass, Fat mass, Bioimpedance, Genetics, Atrial fibrillation, Causal effect
See page 1283 for the editorial comment on this article (doi: 10.1093/eurheartj/ehz037)
Introduction
Atrial fibrillation (AF) is the most common arrhythmia affecting 2–3% of the population, and its prevalence is expected to increase globally due to the aging population.1,2 Major risk factors for AF are shared with other cardiovascular diseases; in particular, the growing obesity epidemic is expected to contribute to an increase in the prevalence of AF.1
Several epidemiological studies have shown that obesity increases the risk of AF, potentially through promoting inflammation, oxidative stress, autonomic dysfunction, cardiac fibrosis, insulin resistance, and hypertension.3–6 An alternative hypothesis suggests that due to the increased haemodynamic and musculoskeletal load, obesity leads to atrial hypertrophy and larger atrial mass, and thus, increased AF risk.6 However, increased body size could have these effects, independent of obesity. In fact, a recent study suggested fat-free mass (comprised of mostly muscle mass) to be the key anthropometric driver of elevated AF risk,7 questioning the independent role of obesity in AF development. Further, other studies have shown that the risk-increasing effect of fat-free mass for AF is stronger compared to body fat and other measures of obesity.4,5,8 However, due to high inter-correlations among different body composition traits, it is difficult to assess their causality in AF development by the use of observational data. Evaluating distinct causal effects of fat-free mass and fat mass would shed light on the AF pathophysiology, which would have direct clinical and public health implications.
Mendelian randomization (MR) is a method for evaluating causality of risk factors on disease by using genetic variants as proxies for risk factors. The causal role of obesity in AF has been supported by a MR study using body mass index (BMI) associated genetic variants as instrumental variable.9 However, BMI is not an ideal measure to address the role of different anthropometric aspects, as it does not distinguish between fat and muscle mass. In fact, two individuals with very different composition of fat-free mass and fat mass can have the same BMI.
Our aim was to evaluate the potential causal roles of fat-free mass and fat mass in AF development by use of well-powered genetic instruments from the UK Biobank in a multivariate MR framework. In particular, we had two questions: (i) does an increase in fat-free mass cause AF, independent of obesity; and (ii) does an increase in fat mass cause AF, independent of total fat-free mass?
Methods
Data sources and statistical analyses used in this study are illustrated in Figure 1. UK Biobank was used for our observational analysis, and for obtaining genetic instruments for MR analysis. The summary statistics data from AFGen consortium10 were used to obtain genetic associations for AF in MR analysis.
Figure 1.
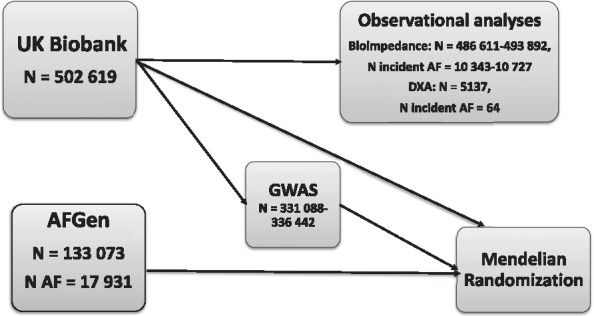
Flowchart of the data sources and statistical analyses.
UK Biobank
UK Biobank is a large, prospective cohort study aiming to improve human health, and the prevention, diagnosis, and treatment of chronic diseases.11 In 2006–10, over 500 000 individuals aged 40–69 years underwent a range of physical measurements, detailed assessments about health-related factors, and sampling of blood, urine, and saliva.
Fat-free mass and fat mass were assessed using bioelectrical impedance technique (N = 492 441 and N = 491 643, respectively, Tanita BC418MA). For sensitivity analysis, we utilized lean mass and fat mass from dual-energy X-ray absorptiometry (DXA, N = 5170). We used BMI [estimated as weight (kg) divided by height squared (m2), N = 499 514] as a measure of total body mass.
AF was defined as primary or secondary cause of hospitalization or death based on ICD-9 code 427.3 and ICD-10 code I48. The follow-up of hospitalizations ended on 31 March 2015 in England, 31 August 2014 in Scotland, and 28 February 2015 in Wales; whereas the death registry included all deaths that occurred before 31 January 2016 in England and Wales and 30 November 2015 in Scotland. Individuals were censored on these dates, time of AF, or time of death; whichever occurred first.
The genome-wide genetic data was available for 488 377 UK Biobank individuals. Genotyping has been conducted with the UK BiLEVE and UK Biobank Axiom arrays and imputed with IMPUTE2 by using both HRC and 1000 Genomes Phase 3 merged with the UK10K haplotype reference panels. Quality control (QC) of genetic markers consisted of tests for batch effects, plate effects, departures from Hardy–Weinberg equilibrium, sex effects, array effects, and discordance. Samples were checked for their missing rate and heterozygosity. Detailed description of the data QC and imputation has been provided by Bycroft et al.12 We used the March 2018 release of the imputed genetic marker data (including corrected imputations from the UK10K and 1000 Genomes Phase 3 reference panel). We restricted the sample to unrelated individuals with self-reported British descent and European ethnicity based on principal component analysis (N = 336 442 for BMI, N = 331 615 for fat-free, and N = 331 088 for fat mass). Further, we excluded genetic markers with minor allele count ≤30 and imputation quality <0.8.
The UK Biobank study was approved by the North West Multi-Centre Research Ethics Committee and all participants provided written informed consent to participate in the UK Biobank study. The study protocol is available in Supplementary material online.11
Statistical analyses
We conducted a large observational study in the UK Biobank to evaluate the associations of body composition measures and incident AF events. For observational analyses, we excluded prevalent AF events (N = 5684). All anthropometric measures were rank transformed to normality to allow for easier comparisons across measures. Associations between body composition measures and incident AF were analysed using Cox proportional hazards models in two sets of models: (i) adjusting for age, sex, and region of the UK Biobank assessment centre; (ii) further adjustment for additional covariates (physical activity, smoking, alcohol consumption, blood pressure, diabetes, and lipid medication) to account for possible confounding effects. Regression splines of Cox proportional hazards were used to assess non-linear associations. Proportional hazards assumption was inspected using Schoenfeld’s tests. Further, we performed some sensitivity analyses with DXA-derived measurements. We observed high correlation between bioimpedance-derived fat-free mass and DXA-derived lean mass (r = 0.96), as well as between bioimpedance-derived fat mass and DXA-derived fat mass (r = 0.86). Based on these observations, and to maximize power, we decided to perform our main analyses with the bioimpedance-derived measures, but also to perform some sensitivity analyses using DXA measurements.
To obtain genetic instruments for MR analysis providing comparable statistical power, we conducted genome-wide association studies (GWAS) for bioimpedance-derived fat-free mass and fat mass in the UK Biobank. The GWAS were conducted with a linear regression assuming additive models for association between genotype dosages and phenotypes using PLINK (version 2.0).13 Age, sex, genotype array, and 10 principal components were used as covariates. Independent variants (Supplementary material online, Tables S1 and S2) were determined using conditional analysis for a region around the lead single-nucleotide polymorphism (SNP). Effect sizes for association with AF were obtained from the AFGen consortium summary statistics10 (N AF cases = 17 931, N controls = 115 142, Supplementary material online, Tables S1 and S2).
We then performed two-sample MR for fat-free mass and fat mass individually with inverse-variance weighted (IVW) regression, as well as using several robust MR methods in sensitivity analyses (weighted median, Egger regression, and penalized methods). Egger regression was used to assess horizontal pleiotropy.
To further evaluate whether fat-free mass and fat mass have potential causal effects on AF independently of each other, we used a multivariable MR weighted regression-based method, in which the effects of multiple related risk factors can be estimated simultaneously.14,15 In this method, we extracted the effect sizes for SNPs associated (P ≤ 5 × 10−8) with fat-free mass, fat mass, and BMI from the summary statistics for all traits and AF. As the lead SNPs for body composition traits might be intercorrelated, we pruned the variants by linkage disequilibrium (LD) with PLINK (r2 = 0.05, clumping window = 500 kbp), by using the SNP with the smallest P-value for association with AF as the index variant in each locus. Then, we inspected relations between the SNP effects for body composition traits (Figure 2); because for multivariate MR to be useful, there needs to be some variants that have stronger effects with one trait than the other (i.e. the collinearity of genetic effects cannot be too high). This seemed to hold for fat-free mass and fat mass (r = 0.84) and fat-free mass and BMI (r = 0.67). Thus, we then conducted multivariate MR for two models; First, the SNP effects on AF (βAF) were modelled against the SNP effects for fat-free mass, adjusted for the effects for fat mass (βAF ∼ βFat-free mass + βFat mass) using a weighted linear regression model, where the weights were defined by inverse standard errors of βAF. Then, to further evaluate whether fat-free mass was causally related with AF independently of total body mass, we adjusted the SNP effects of fat-free mass for the SNP effects for BMI (βAF ∼ βFat-free mass + βBMI). Due to high correlations between the fat mass and BMI genetic effects (r = 0.96), the model βAF ∼ βFat mass + βBMI is likely to be invalid and thus was not further considered.
Figure 2.

Effect sizes for genetic instruments for body composition traits used in multivariate Mendelian randomization analysis.
Analyses were conducted with R (version 3.3.0) packages survival, TwosampleMR and MendelianRandomization.
Results
The demographic and clinical characteristics of study participants are shown in Table 1. Mean age at baseline was 56.5 years [standard deviation (SD) 8.1 years] and 54% of subjects were females. During follow-up (median 6.1 years; interquartile range 5.4–6.7 years; 3 005 107 person-years at risk), 10 852 incident AF cases occurred in participants free from the disease at baseline.
Table 1.
Baseline characteristics of the UK Biobank participants (N = 502 619)
| Gender | |
| Females | 273 455 (54) |
| Males | 229 164 (46) |
| Baseline age (years) | 56.5 (8.1) |
| Ethnicity | |
| White | 472 803 (94.1) |
| Black | 8064 (1.6) |
| Asian | 11 456 (2.3) |
| Mixed | 7518 (1.5) |
| Smoking status | |
| Never | 273 590 (54.4) |
| Previous | 173 091 (34.4) |
| Current | 52 986 (10.5) |
| Alcohol consumption | |
| Daily or almost daily | 101 787 (20.3) |
| Three or four times a week | 115 459 (23.0) |
| Once or twice a week | 129 317 (25.7) |
| One to three times a month | 55 870 (11.1) |
| Special occasions only | 58 025 (11.5) |
| Never | 40 658 (8.1) |
| Diabetes | 26 407 (5.3) |
| Blood pressure (mmHg) | |
| Systolic | 139.8 (19.7) |
| Diastolic | 82.3 (10.7) |
| Lipid medication | 82 366 (16.4) |
| Physical activity,a (MET-hours/week) | 43.8 (43.7) |
| Body mass index (kg/m2) | 27.4 (4.8) |
| Fat mass (kg) | 24.9 (9.6) |
| Fat-free mass (kg) | 53.2 (11.5) |
| Prevalent atrial fibrillation | 5684 (1.1) |
| Incident atrial fibrillation | 10 852 (2.2) |
Data are presented as mean (SD) or N (%).
Missing values of the variable were imputed with predictive mean matching.16
Observational analyses
Both fat-free mass and fat mass had strong associations with incident AF in observational analyses, but the effect was much stronger for fat-free mass than for fat mass [hazard ratio (HR) = 1.77, 95% confidence interval (CI) 1.72–1.83; HR = 1.40, 95% CI 1.37–1.43 per SD-increase in fat-free mass and fat mass, respectively, Take home figure, Supplementary material online, Table S3]. Adjusting for additional covariates slightly attenuated these associations (HR = 1.70, 95% CI 1.65–1.76; HR = 1.34, 95% CI 1.31–1.37 per SD-increase in fat-free mass and fat mass, respectively, Take home figure, Supplementary material online, Table S3). Although not a focus of the present study, cross-sectional associations of fat-free mass [odds ratio (OR) = 1.77, 95% CI 1.69–1.85] and fat mass (OR = 1.51, 95% CI 1.47–1.56) with prevalent AF were similar to the main results. In comparison, we estimated associations between some additional factors and incident AF (Supplementary material online, Table S4). For example, left ventricular ejection fraction showed strong association with incident AF (HR = 0.57, 95% CI 0.43–0.76). The point estimates for DXA-derived measures were stronger than corresponding bioimpedance-derived measures (HR = 1.99, 95% CI 1.33–2.93; HR = 1.50, 95% CI 1.16–1.94 per SD-increase in lean and fat mass, respectively). However, it should be noted that the number of incident AF events was low (N = 64) in these analyses.
Take home figure.
Observational associations and causal effects of body composition measures on atrial fibrillation. All effects are in SD-units. Observational, Model 1: Cox regression model adjusted for age, sex, and region. Observational, Model 2: Cox regression model adjusted for age, sex, region, physical activity, smoking, alcohol consumption, blood pressure, diabetes, and lipid medication. Multivariate causal: the causal effects of fat-free and fat mass adjusted for each other. CI, confidence interval; IVW, inverse-variance weighted.
Due to differences in body composition in males and females, we also estimated these associations by sex. The association of fat mass with incident AF was higher in females (HR = 1.49, 95% CI 1.44–1.54) than in males (HR = 1.35, 95% CI 1.31–1.39, respectively, Pinteraction = 1.2 × 10−5). The effects of fat-free mass were similar for both genders (HR = 1.75, 95% CI 1.65–1.85 for females; HR = 1.77, 95% CI 1.71–1.84 for males; Pinteraction = 0.26). The associations of both measures with AF were non-linear (all Pnonlinearity <0.003, Figure 3). In particular, the associations were J-shaped for fat mass and AF in females; and fat-free mass and AF in males (Supplementary material online, Figure S1).
Figure 3.
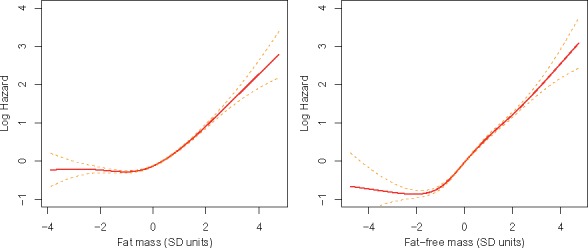
Relations of body composition and atrial fibrillation events. Lines are based on a regression spline of Cox proportional hazards.
Mendelian randomization
Our univariate MR analyses suggested causal effects of fat-free mass and fat mass on AF in the same direction as in the observational analysis (causal risk ratio = 1.55, 95% CI 1.38–1.75; causal risk ratio = 1.30, 95% CI 1.17–1.45 per SD-increase in fat-free mass and fat mass, respectively, Take home figure, Supplementary material online, Table S3). Penalized IVW regression, MR-Egger, and median-based regression showed consistent results with the regular IVW (Supplementary material online, Figures S2 and S3), and no directional horizontal pleiotropy was detected (PEgger intercept >0.14). At an alpha level 0.05, the statistical power was >99% to detect the effects of fat-free mass and fat mass on AF at the observed effect sizes of 1.77 and 1.40 per SD-increase, respectively (Supplementary material online, Table S3). We had >80% power to detect causal effects down to 1.08 and 1.12 per SD-increase for fat-free mass and fat mass on AF, respectively.
The multivariate MR showed that the potential causal effects of fat-free mass and fat mass on AF were independent of each other (Take home figure, Supplementary material online, Table S3). However, when adjusted for fat mass, the effect of fat-free mass was substantially lower compared to that of univariate MR [causal risk ratio = 1.37, 95% CI 1.06–1.75 (as compared to 1.55 in the univariate MR), Take home figure, Supplementary material online, Table S3]. In contrast, the effect for fat mass did not change substantially when adjusted for fat-free mass [causal risk ratio = 1.28, 95% CI 1.03–1.58 (as compared to 1.30 in the univariate MR), Take home figure, Supplementary material online, Table S3]. When keeping BMI constant, the effect of fat-free mass on AF was 1.50 (95% CI 1.27–1.77). For sensitivity analysis, we restricted instruments to those SNPs that had specific associations for fat mass or fat-free mass (Pfat mass < Pfat-free mass × 10 and vice versa). This resulted in 26 SNPs for fat mass, and 388 SNPs for fat-free mass. The point estimate for fat-free mass was identical to that of univariate MR (causal risk ratio = 1.55, 95% CI 1.16–2.07), but larger for fat mass (causal risk ratio = 1.44, 95% CI 1.08–1.91).
As our observational analyses suggested non-linearity and larger effect sizes for fat mass in females, we applied two additional MR analyses using individual-level data from the UK Biobank: sex-stratified two-stage least-squares17 and non-linear MR.18,19 In these analyses, we included all (prevalent and incident) AF events in the UK Biobank (N events = 15 990 among unrelated, European descent individuals with genetic data passing quality control). The results are presented in Supplementary material online, Table S5. Consistent with the observational results, we indeed observed larger effects of fat mass on AF in females than in males (causal risk ratio = 1.30, 95% CI 1.22–1.39 in females; causal risk ratio = 1.21, 95% CI 1.15–1.29 in males). The effects of fat-free mass on AF were similar in both genders.
Non-linear MR using piecewise linear method18 suggested that the overall effects of fat-free mass and fat mass on AF were non-linear, but gender-specific analyses showed that the non-linear effects were significant in females, but not in males.
Discussion
Principal findings
We conducted a large observational and MR analysis assessing the effects of fat-free mass and fat mass in development of AF. In line with previous observational studies,4,5,7 the observational association of fat-free mass was stronger than the association of fat mass. Both observational and MR analyses suggested that the magnitude of the association of fat-free mass with AF was equal in both sexes, whereas fat mass showed stronger associations in women than in men. Our MR analyses suggested that both fat-free mass and fat mass are causally related with AF. Our multivariate MR analysis showed that these effects are independent of each other.
Our results allow us to draw the following conclusions. First, our MR analyses suggest that the association between fat-free mass and AF, which has been reported in several recent observational studies,4,5,7,8 is causal by nature. The mechanisms explaining this association are not well established, but atrial hypertrophy is likely to have a central role.6 However, our MR analysis showed clearly lower effect sizes for fat-free mass compared with those obtained using observational data. This indicates that observational estimates for associations of fat-free mass with AF are likely to be confounded—a notion also supported by the attenuation of point estimates upon adjustment for potential confounders—and highlights inherent limitations of observational methods. Observational analyses are prone to several biases, such as confounding, reverse causality, and multicollinearity. Instead, MR utilizes genetic variants as proxies for risk factors, which are, in under some assumptions, free from these biases. Second, in contrast to recent observational analyses questioning the independent role of fat mass (reflecting adiposity) in AF development,6,7 our multivariate MR analysis show that fat mass per se also predisposes to AF, as the causal effect of fat mass on AF was independent and not mediated through the effect on fat-free mass.
Clinical implications
Our analyses suggest that larger body mass, whether composed of fat-free mass or fat mass, promotes development of AF. Obesity might increase the risk of AF through several potential mediating mechanisms, including increasing the risk of clinical risk factors, such as hypertension, diabetes, and the metabolic syndrome. Obesity can also directly or indirectly have deleterious effects on inflammation, oxidative stress, autonomic dysfunction, cardiac fibrosis, and left ventricular and atrial enlargement.6 Thus, aggressively targeting obesity with multi-modality therapeutic intervention is likely to be an effective way to prevent AF and resultant comorbidities such as stroke.
In contrast, even if our results suggest that fat-free mass is also causally related with AF, strategies aiming to decrease fat-free mass (which is mostly composed of muscle mass) might have unfavourable health effects, as they would involve reduced physical activity and fitness. Limiting strength training may have deleterious effects on other cardiometabolic conditions, such as diabetes and atherosclerotic cardiovascular disease, particularly for secondary prevention.20 Furthermore, higher muscular strength (measured as relative grip strength, thus accounting for body mass) and increased overall cardiorespiratory fitness both decrease the risk for AF.21 That said, it is important to recognize the potential risks related to activities aiming extensive gains in muscle mass or consumption of bodybuilding supplements. Indeed, there are case reports of AF presumably caused by supplements aimed to increase muscle bulk and strength.22–25 More research is needed to identify biological mechanisms underlying the associations between fat-free mass and AF, and determine whether protective and harmful gains in fat-free mass can be distinguished, especially as our analyses indicate that the risk-increasing might be non-linear.
In conclusion, reductions in fat mass and fat-free mass are likely to affect different biological pathways, and both of these mechanisms seem to lower AF risk independently. Thus, weight loss is likely to be a key factor in preventing AF, regardless of the person’s current body composition. This observation is also in line with the 2016 European Society of Cardiology (ESC) Guidelines for the management of AF developed in collaboration with European Association for Cardio-Thoracic Surgery (EACTS), where weight loss should be considered to reduce AF burden and symptoms in obese patients with AF.26
Strengths and limitations
The main strength of this study is its large study sample, which allowed us to perform comprehensive time-to-event analysis for incident AF and well-powered GWAS to obtain genetic instruments for MR analyses. Indeed, we identified hundreds of genetic variants for body composition traits, and even though many of them might not be valid genetic instruments for MR analysis, we applied established sensitivity analyses17 to minimize the effects of pleiotropic SNPs. These include weighted median method, which allows 50% of the genetic instruments to be invalid and Egger regression that adjusts for directional pleiotropy. Further, we applied multivariate MR,14,15 which is useful for evaluating independent causal effects of multiple related risk factors, as it accounts for pleiotropic effects within the model. One main advantage of this method is that it does not require exclusions of SNP associated with several of the investigated exposures, which might be subjective and lead to a genetic instrument that is no longer biologically meaningful.27 However, it is important to acknowledge that MR analysis has several other potential limitations,17 and while using multiple genetic instruments improves the power of MR, there is always some risk of pleiotropy despite extensive sensitivity analyses. Thus, the results from MR study should always be considered as another layer of evidence,28 strengthening or challenging the findings from observational studies rather than proving causality.
There are also limitations in our study. First, as DXA measurements were available only a small subset of individuals (N = 3695 available for GWAS), we conducted our main analysis with bioimpedance-derived fat-free mass and fat mass, which is not ideal as they might be influenced by factors, such as hydration. However, the correlations between bioimpedance measures and the DXA-derived ‘gold standard’ measures were high, and we conducted some sensitivity analyses with DXA-measures. Second, based on our observational analyses, the association of fat mass with AF was different in females and males. To assess causal effects by gender, we conducted individual-level MR analysis by gender in the UK Biobank, which might bias causal estimates slightly towards the confounded observational association,17 as the genetic instruments were obtained by using the same dataset. Unfortunately, we could not assess the causal effects by gender using the two-sample MR method, because the outcome data for MR analysis was from AFGen Consortium, which did not report gender-specific associations. Third, the AF prevalence and incidence rates are not generalizable to general population due to ‘healthy volunteer bias’ among the UK Biobank study participants. Moreover, the definition of AF was based on hospital and death registries, which presumably leads to under diagnosis of AF, especially of milder cases. However, while the AF prevalence as well as the strength of risk factor associations may be underestimated, the presence of associations between body composition traits and AF are still likely be generalizable to the broader population.29 Fourth, we could not adjust our observational models for low-density lipoprotein (LDL) cholesterol or other lipid fractions, as there were not yet available in the UK Biobank. However, we adjusted our models for lipid-lowering medication as a proxy for dyslipidaemia. Also, the MR analyses are by design unaffected by potential confounding by lipid levels (or other confounders). Finally, the majority of participants in the UK Biobank were of European ancestry, and the GWAS was conducted using only Europeans. Hence, the generalizability of the results to other ethnicities is unknown.
In conclusion, increases in fat-free mass and fat mass are associated with higher risk for AF, and these relationships are likely to be causal by nature. Treating and preventing obesity is likely to be the most effective way to reduce the AF risk caused by large body mass. To fully understand the causal processes, more research is needed to evaluate the mediating mechanisms between body composition traits and AF.
Supplementary Material
Acknowledgements
This research has been conducted using the UK Biobank Resource under Application Number 13721. Data on atrial fibrillation was contributed by the AFGen Consortium (https://www.afgen.org/).
Funding
This work was supported by National Institutes of Health [1R01HL135313-01, 1R01DK106236-01A1, and R01DK107437], Knut and Alice Wallenberg Foundation [2013.0126], Finnish Cultural Foundation, Finnish Foundation for Cardiovascular Research, Orion Research Foundation, Emil Aaltonen Foundation, Doris Duke Charitable Foundation, and the Stanford Diabetes Research centre award [P30DK116074].
Conflict of interest: E.I. is a scientific advisor for Precision Wellness and Olink Proteomics for work unrelated to the present project. E.T. is employee of Nightingale Health Ltd, a company offering NMR-based metabolic profiling. Other authors report no relationships with industry.
References
- 1. Rahman F, Kwan GF, Benjamin EJ.. Global epidemiology of atrial fibrillation. Nat Rev Cardiol 2014;11:639–654. [DOI] [PubMed] [Google Scholar]
- 2. Ball J, Carrington MJ, McMurray JJ, Stewart S.. Atrial fibrillation: profile and burden of an evolving epidemic in the 21st century. Int J Cardiol 2013;167:1807–1824. [DOI] [PubMed] [Google Scholar]
- 3. Aune D, Sen A, Schlesinger S, Norat T, Janszky I, Romundstad P, Tonstad S, Riboli E, Vatten LJ.. Body mass index, abdominal fatness, fat mass and the risk of atrial fibrillation: a systematic review and dose-response meta-analysis of prospective studies. Eur J Epidemiol 2017;32:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Frost L, Benjamin EJ, Fenger-Grøn M, Pedersen A, Tjønneland A, Overvad K.. Body fat, body fat distribution, lean body mass and atrial fibrillation and flutter. A Danish cohort study. Obesity 2014;22:1546–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Karas MG, Yee LM, Biggs ML, Djousse L, Mukamal KJ, Ix JH, Zieman SJ, Siscovick DS, Gottdiener JS, Rosenberg MA, Kronmal RA, Heckbert SR, Kizer JR.. Measures of body size and composition and risk of incident atrial fibrillation in older people: the cardiovascular health study. Am J Epidemiol 2016;183:998–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nattel S. Atrial fibrillation and body composition: is it fat or lean that ultimately determines the risk? J Am Coll Cardiol 2017;69:2498–2501. [DOI] [PubMed] [Google Scholar]
- 7. Fenger-Gron M, Overvad K, Tjonneland A, Frost L.. Lean body mass is the predominant anthropometric risk factor for atrial fibrillation. J Am Coll Cardiol 2017;69:2488–2497. [DOI] [PubMed] [Google Scholar]
- 8. Azarbal F, Stefanick ML, Assimes TL, Manson JE, Bea JW, Li W, Hlatky MA, Larson JC, LeBlanc ES, Albert CM, Nassir R, Martin LW, Perez MV.. Lean body mass and risk of incident atrial fibrillation in post-menopausal women. Eur Heart J 2016;37:1606–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chatterjee NA, Giulianini F, Geelhoed B, Lunetta KL, Misialek JR, Niemeijer MN, Rienstra M, Rose LM, Smith AV, Arking DE, Ellinor PT, Heeringa J, Lin H, Lubitz SA, Soliman EZ, Verweij N, Alonso A, Benjamin EJ, Gudnason V, Stricker BH, Van Der Harst P, Chasman DI, Albert CM.. Genetic obesity and the risk of atrial fibrillation: causal estimates from Mendelian randomization. Circulation 2017;135:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Christophersen IE, Rienstra M, Roselli C, Yin X, Geelhoed B, Barnard J, Lin H, Arking DE, Smith AV, Albert CM, Chaffin M, Tucker NR, Li M, Klarin D, Bihlmeyer NA, Low S-K, Weeke PE, Müller-Nurasyid M, Smith JG, Brody JA, Niemeijer MN, Dörr M, Trompet S, Huffman J, Gustafsson S, Schurmann C, Kleber ME, Lyytikäinen L-P, Seppälä I, Malik R, Horimoto ARVR, Perez M, Sinisalo J, Aeschbacher S, Thériault S, Yao J, Radmanesh F, Weiss S, Teumer A, Choi SH, Weng L-C, Clauss S, Deo R, Rader DJ, Shah SH, Sun A, Hopewell JC, Debette S, Chauhan G, Yang Q, Worrall BB, Paré G, Kamatani Y, Hagemeijer YP, Verweij N, Siland JE, Kubo M, Smith JD, Van Wagoner DR, Bis JC, Perz S, Psaty BM, Ridker PM, Magnani JW, Harris TB, Launer LJ, Shoemaker MB, Padmanabhan S, Haessler J, Bartz TM, Waldenberger M, Lichtner P, Arendt M, Krieger JE, Kähönen M, Risch L, Mansur AJ, Peters A, Smith BH, Lind L, Scott SA, Lu Y, Bottinger EB, Hernesniemi J, Lindgren CM, Wong JA, Huang J, Eskola M, Morris AP, Ford I, Reiner AP, Delgado G, Chen LY, Chen Y-DI, Sandhu RK, Li M, Boerwinkle E, Eisele L, Lannfelt L, Rost N, Anderson CD, Taylor KD, Campbell A, Magnusson PK, Porteous D, Hocking LJ, Vlachopoulou E, Pedersen NL, Nikus K, Orho-Melander M, Hamsten A, Heeringa J, Denny JC, Kriebel J, Darbar D, Newton-Cheh C, Shaffer C, Macfarlane PW, Heilmann-Heimbach S, Almgren P, Huang PL, Sotoodehnia N, Soliman EZ, Uitterlinden AG, Hofman A, Franco OH, Völker U, Jöckel K-H, Sinner MF, Lin HJ, Guo X, Dichgans M, Ingelsson E, Kooperberg C, Melander O, Loos RJF, Laurikka J, Conen D, Rosand J, van der Harst P, Lokki M-L, Kathiresan S, Pereira A, Jukema JW, Hayward C, Rotter JI, März W, Lehtimäki T, Stricker BH, Chung MK, Felix SB, Gudnason V, Alonso A, Roden DM, Kääb S, Chasman DI, Heckbert SR, Benjamin EJ, Tanaka T, Lunetta KL, Lubitz SA, Ellinor PT; AFGen Consortium. Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat Genet 2017;49:946–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.UK Biobank. ukbiobank.ac.uk. https://www.ukbiobank.ac.uk/ (10 January 2019).
- 12. Bycroft C, Freeman C, Petkova D, Band G, Elliot LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O'Connell J, Cortes A, Welsh S, Young A, Effingham M, McVean G, Leslie S, Allen N, Donnelly P, Marchini J. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018;562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ.. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015;4:7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burgess S, Thompson SG.. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol 2015;181:251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burgess S, Dudbridge F, Thompson SG.. Re: ‘Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects’. Am J Epidemiol 2015;181:290–291. [DOI] [PubMed] [Google Scholar]
- 16. Marshall A, Altman DG, Holder RL, Royston P.. Combining estimates of interest in prognostic modelling studies after multiple imputation: current practice and guidelines. BMC Med Res Methodol 2009;9:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haycock PC, Burgess S, Wade KH, Bowden J, Relton C, Davey Smith G.. Best (but oft-forgotten) practices: the design, analysis, and interpretation of Mendelian randomization studies. Am J Clin Nutr 2016;103:965–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Staley JR, Burgess S.. Semiparametric methods for estimation of a nonlinear exposure-outcome relationship using instrumental variables with application to Mendelian randomization. Genet Epidemiol 2017;41:341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burgess S, Davies NM, Thompson SG; EPIC-InterAct Consortium. Instrumental variable analysis with a nonlinear exposure-outcome relationship. Epidemiology 2014;25:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wannamethee SG, Shaper AG, Whincup PH, Lennon L, Papacosta O, Sattar N.. The obesity paradox in men with coronary heart disease and heart failure: the role of muscle mass and leptin. Int J Cardiol 2014;171:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tikkanen E, Gustafsson S, Ingelsson E, Associations of fitness, physical activity, strength, and genetic risk with cardiovascular disease: longitudinal analyses in the UK Biobank Study. Circulation 2018;137:2583–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kammer RT. Lone atrial fibrillation associated with creatine monohydrate supplementation. Pharmacotherapy 2005;25:762–764. [DOI] [PubMed] [Google Scholar]
- 23. Sullivan ML, Martinez CM, Gallagher EJ.. Atrial fibrillation and anabolic steroids. J Emerg Med 1999;17:851–857. [DOI] [PubMed] [Google Scholar]
- 24. Liyanage CR, Kodali V. Bulk muscles, loose cables. BMJ Case Rep2014. pii:bcr2014204424. [DOI] [PMC free article] [PubMed]
- 25. Manoharan G, Campbell NP, O'Brien CJ.. Syncopal episodes in a young amateur body builder. Br J Sports Med 2002;36:67–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kirchhof P, Benussi S, Kotecha D, Ahlsson A, Atar D, Casadei B, Castella M, Diener H-C, Heidbuchel H, Hendriks J, Hindricks G, Manolis AS, Oldgren J, Popescu BA, Schotten U, Van Putte B, Vardas P, Agewall S, Camm J, Baron Esquivias G, Budts W, Carerj S, Casselman F, Coca A, De Caterina R, Deftereos S, Dobrev D, Ferro JM, Filippatos G, Fitzsimons D, Gorenek B, Guenoun M, Hohnloser SH, Kolh P, Lip GYH, Manolis A, McMurray J, Ponikowski P, Rosenhek R, Ruschitzka F, Savelieva I, Sharma S, Suwalski P, Tamargo JL, Taylor CJ, Van Gelder IC, Voors AA, Windecker S, Zamorano JL, Zeppenfeld K; ESC Scientific Document Group. 2016 ESC guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J 2016;37:2893–2962. [DOI] [PubMed] [Google Scholar]
- 27. Holmes MV, Ala-Korpela M, Smith GD.. Mendelian randomization in cardiometabolic disease: challenges in evaluating causality. Nat Rev Cardiol 2017;14:577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. O'Donnell CJ. Mendelian randomization evidence for cardiovascular precision medicine. JAMA Cardiol 2018;3:627–628. [DOI] [PubMed] [Google Scholar]
- 29. Fry A, Littlejohns TJ, Sudlow C, Doherty N, Adamska L, Sprosen T, Collins R, Allen NE.. Comparison of sociodemographic and health-related characteristics of UK Biobank participants with those of the general population. Am J Epidemiol 2017;186:1026–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.