

SUMMARY
The classic view of nucleosome organization at active promoters is that two well-positioned nucleosomes flank a nucleosome-depleted region (NDR). However, this view has been recently disputed by contradictory reports as to whether wider (≳150 bp) NDRs instead contain unstable Micrococcal Nuclease-sensitive (“fragile”) nucleosomal particles. To determine the composition of fragile particles we introduce CUT&RUN.ChIP, in which targeted nuclease cleavage and release is followed by chromatin immunoprecipitation. We find that fragile particles represent the occupancy of the RSC (remodeling the structure of chromatin) nucleosome remodeling complex and RSC-bound partially unwrapped nucleosomal intermediates. We also find that general regulatory factors (GRFs) bind to partially unwrapped nucleosomes at these promoters. We propose that RSC-binding and its action cause nucleosomes to unravel, facilitate subsequent binding of GRFs, and constitute a dynamic cycle of nucleosome deposition and clearance at the subset of wide Pol II promoter NDRs.
Keywords: fragile nucleosome, nucleosome-depleted region, CUT&RUN, general regulatory factor, nucleosome dynamics
GRAPHICAL ABSTRACT
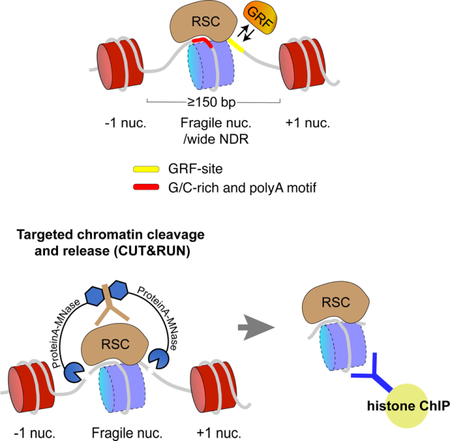
eTOC Blurb
Brahma et al. resolve the composition of MNase-sensitive particles at yeast promoters by showing that these are RSC and GRF-associated partially-unwrapped nucleosomal intermediates. For this, Brahma et al. combined a targeted chromatin cleavage and release approach with native ChIP, which would be widely applicable for genomic profiling of chromatin-factor co-occupancies.
INTRODUCTION
Nucleosomes are the fundamental repeating unit of chromatin and their precise positioning in the genome as well as their composition and modifications regulate accessibility of sites to DNA-templated machineries. Of particular importance to transcription regulation is the location, occupancy, histone subunit composition, and covalent modification states of nucleosomes near promoters of genes (Henikoff, 2016; Lai and Pugh, 2017; Struhl and Segal, 2013). Genome-wide analysis of nucleosome landscapes have provided a consensus view of nucleosome organization at promoters of active protein-coding genes in yeast which also holds for active genes in higher eukaryotes (Jiang and Pugh, 2009; Lieleg et al., 2015). According to this view, two well-positioned nucleosomes (designated as +1 and −1) flank a nucleosome-depleted region (NDR) of variable length. NDRs are sites for transcription pre-initiation complex (PIC) assembly and RNA polymerase II loading, and in yeast, transcription start sites (TSSs) are located about one helical turn inside of +1 nucleosomes. This organization has been attributed to DNA sequence features, binding of sequence-specific general regulatory factors (GRFs) at NDRs that create potential barriers to nucleosome formation, and ATP-dependent chromatin remodeling, which together create regularly phased nucleosome arrays around the NDRs. However, the individual or combined contribution of these factors in regulating the nucleosome landscape at promoters is not completely understood (Krietenstein et al., 2016; Struhl and Segal, 2013).
The major approach for determining chromatin accessibility and nucleosome occupancy involves digesting chromatin with micrococcal nuclease (MNase) (Lieleg et al., 2015). MNase is an endo-/exo-nuclease that digests DNA that is not protected by a nucleosome or DNA-binding protein, including linker regions between nucleosomes and NDRs. A genome-wide MNase sensitivity analysis in yeast suggested that many previously annotated NDRs are not nucleosome-free, challenging the classic view of promoter nucleosome organization (Kubik et al., 2015). In that study, NDRs in yeast were broadly divided into two classes based on their size, and wider (≳150 bp) NDRs associated with about a third of all protein-coding genes were found to be populated by particles referred to as “fragile” nucleosomes (FNs) owing to their nucleosome-like footprint and MNase sensitivity (Xi et al., 2011). Wider NDRs in yeast are generally associated with highly transcribed house-keeping genes (Kubik et al., 2015; Weiner et al., 2010). However, the nucleosome fragility concept has been fundamentally challenged by the failure to detect histones at promoters in genome-wide chromatin immunoprecipitation sequencing (ChIP-seq) and chromatin immunoprecipitation combined with lambda exonuclease digestion (ChIP-exo) experiments (Chereji et al., 2017; Rhee et al., 2014), and by chemical cleavage mapping experiments, which do not detect H4-DNA contacts at FN sites (Chereji et al., 2017; Henikoff et al., 2014). These observations instead suggest that nucleosome-size MNase-sensitive footprints at wider NDRs are due to non-histone protein complexes, likely to be transcription factors and/or chromatin remodelers (Chereji et al., 2017). Nevertheless, it is intriguing that nucleosome-size MNase-sensitive particles are detected particularly when the gap between +1 and −1 nucleosomes is ~150 bp or larger (Kubik et al., 2017).
To resolve this controversy, we hypothesize that if nucleosomes are present at wide NDRs, they are likely to be fragile due to destabilization by RSC, the only essential chromatin remodeler in yeast, which mobilizes nucleosomes to catalyze NDR formation (Lorch and Kornberg, 2017; Yague-Sanz et al., 2017). Several studies indicate direct involvement of RSC in generating NDRs (Badis et al., 2008; Hartley and Madhani, 2009; Krietenstein et al., 2016; Kubik et al., 2015; Kubik et al., 2018; Parnell et al., 2015; Yague-Sanz et al., 2017). RSC dynamically repositions nucleosomes, and by extensive interaction with both DNA and histones, destabilizes and restructures nucleosomes (Chaban et al., 2008). The altered structure of RSC-remodeled nucleosomes at wide NDRs due to unwrapping, and their low occupancy would reconcile the lack of H4-DNA contacts and the lack of detection by ChIP-seq, respectively. However, direct testing of this hypothesis requires a robust and sensitive method for profiling RSC-bound nucleosomes that are expected to be highly transient. Our lab has recently introduced “Cleavage Under Target & Release Using Nuclease” (CUT&RUN), a non-ChIP-based method that uses targeted in situ chromatin cleavage and release for profiling factors binding to chromatin genome-wide (Skene and Henikoff, 2017). Since CUT&RUN is performed on native unfixed chromatin, it avoids many problems with epitope masking, chromatin fragmentation and solubilization, and high background that are inherent to ChIP, while providing high sensitivity and base-pair resolution. CUT&RUN for RSC was more efficient with higher yield than was obtained using Native ChIP-seq, with similar profiles (Skene and Henikoff, 2017).
Here we demonstrate a widely applicable extension of CUT&RUN that allows direct interrogation of the histone content of RSC-associated chromatin genome-wide. In this approach, we use CUT&RUN to generate the input for histone-ChIP-seq, all under gentle native conditions (CUT&RUN.ChIP). We find that CUT&RUN.ChIP is highly efficient with negligible background, and is effective for mapping RSC-nucleosome complexes at promoters. We show that previously annotated fragile nucleosome sites are populated at a low level by subnucleosomal RSC-histone complexes with active histone-marks, resolving the controversy. CUT&RUN.ChIP also shows that the general regulatory factors Abf1 and Reb1 bind to partially unwrapped nucleosomes at wide NDRs. We propose that RSC binds to nucleosomes deposited at ≳150 bp-wide NDRs and mobilizes and destabilizes them, exposing the sites for GRF binding. Therefore, fragile nucleosomes are transient intermediates of RSC remodeling.
RESULTS
CUT&RUN.ChIP profiles histone co-occupancy on single nucleosomes
In CUT&RUN a factor- or histone-specific antibody is bound to chromatin in situ followed by binding to the antibody of a protein A-micrococcal nuclease (pA-MNase) fusion. This is followed by calcium-induced MNase cleavage of DNA on both sides of the bound factor, and cleavages begin within seconds. The reaction is performed at 0°C, which minimizes off-rates and diffusion of the complexes, preventing indirect and background cleavage events. Because there is no crosslinking, the factor-bound DNA that is cleaved on both sides of the bound particle is efficiently released into solution upon incubation at 37°C after inhibition of the MNase by chelating calcium (Figure 1A) (Skene and Henikoff, 2017). Limited digestion of chromatin only around target sites minimizes fragments from the bulk chromatin as compared to whole genome fragmentation in ChIP approaches, thereby greatly reducing sequencing depth and cell numbers required to extract signal from background noise (Skene et al., 2018; Skene and Henikoff, 2017). Paired-end sequencing of DNA extracted from chromatin released into solution allows efficient and precise mapping of factor-binding sites. Because there is little to no background cleavage for calibration of reads from different experiments or replicates, we add a fixed amount of heterologous spike-in DNA (Figure 1A) (Skene and Henikoff, 2017).
Figure 1: CUT&RUN.ChIP maps histone co-occupancies in single nucleosomes.

(A) Schematic diagram of the CUT&RUN.ChIP strategy. Yeast nuclei are successively treated with factor (or tag)-specific antibody (here anti-FLAG) and Protein A-MNase that diffuse into the nucleus. Ca++ induces MNase cleavage, and particles cleaved on both sides are released and diffuse out of the nucleus. FLAG-antibody is competed off from the released particles and antibody for a second epitope is added for ChIP followed by Protein-A beads to isolate immunoprecipitated particles. DNA extracted from the CUT&RUN supernatant and ChIP is used to prepare libraries for paired-end sequencing. Heterologous spike-in DNA can be added either before or after ChIP. (B) Spike-normalized (using the second spike-in DNA) CUT&RUN.ChIP profiles (121–200 bp) of histone modifications in H2A.Z and H2B nucleosomes (blue tracks). In each set (H2A.Z and H2B), the CUT&RUN supernatant was used as input for the subsequent ChIP-seq. IgG is a non-specific negative control for ChIP. The “Nucleosomes” track represents nucleosomal reads derived from H3Q85C chemical cleavage mapping of single-nucleosome dyads (Chereji et al., 2018). Crosslinking ChIP-seq of H2A.Z and the specified histone modifications (green tracks) are shown for comparison (Weiner et al., 2015). Tracks in each set of CUT&RUN.ChIP are scaled to the same intensity (y-axes). The positions of the annotated yeast open reading frames are shown at the bottom with the gene names. (C) Enrichment of spike-in normalized densities of 121–200 bp (nucleosomal) fragments from H2A.Z CUT&RUN and histone ChIP-seq using H2A.Z CUT&RUN as input, over the ±1-kb region spanning the dyads of 5542 +1 nucleosomes identified by H3Q85C cleavage mapping (Chereji et al., 2018). (D) Heat maps of 121–200 bp (nucleosomal) fragments from H2A.Z CUT&RUN.ChIP spanning +1 nucleosome centers ±1 kb, and sorted by NDR width. Heat maps are total count-normalized (total number of reads for each sample and size class are indicated at the bottom of each heat map), and shown in log2 scale, with log2 center = 5 and contrast = 3. Dotted lines separate wide NDRs (≥150-bp) from the rest. See also Figure S1.
As a proof of principle for CUT&RUN.ChIP, we applied CUT&RUN to FLAG-tagged yeast histones H2A.Z (Htz1-FLAG) and H2B (Htb1-FLAG). We then tested the CUT&RUN supernatants as input for ChIP-seq of a variety of histone modifications (Figure 1A, B). To do this, the pA-MNase-bound first antibody was competed off the particles in the supernatant using a FLAG peptide before adding the second antibody for ChIP (Figure 1A). DNA was purified from the immunoprecipitated material and sequenced. H2A.Z is a variant of histone H2A that is incorporated into chromatin in a DNA replication-independent pathway. H2A.Z is enriched at promoters of both active and silent genes, and has been implicated in both transcriptional activation and repression (Weber and Henikoff, 2014). When compared to H2A.Z ChIP-seq with crosslinked and MNase-digested chromatin (Weiner et al., 2015), H2A.Z CUT&RUN profiles appear much cleaner with negligible background (Figure 1B first blue track). Furthermore, ChIP-seq of H2A.Z CUT&RUN supernatant using an H2A.Z antibody shows that only the nucleosomes immediately flanking TSSs are recovered, not the shoulders seen in the input (Figure 1B, compare first and third blue track, and Figure 1C, D), which are present because CUT&RUN also releases adjacent nucleosomes (Skene and Henikoff, 2017). Therefore, H2A.Z is confined to nucleosomes immediately flanking the NDR, and H2A.Z signals seen farther downstream in ChIP-seq experiments are attributable to background. The clean release and specific recovery of H2A.Z nucleosomes is also evident genome-wide as shown by average plots and heat maps aligned over +1 nucleosome dyads (Figure 1C, D). Strong correlations between CUT&RUN.ChIP experiments for H2A.Z-H2B nucleosomes in which we switched the order (H2A.Z CUT&RUN followed by H2B ChIP, and H2B CUT&RUN followed by H2A.Z ChIP), reveal reproducibility of the approach (Figures 1D and S1A, B).
We also performed ChIP-seq on H2A.Z CUT&RUN supernatant using antibodies to histone modifications (Figure 1B-D). We find that promoter-proximal H2A.Z nucleosomes are enriched in trimethylated H3K4 (H3K4me3) and to some extent in H4 with a tetra-acetylated N-terminal tail (H4ac), but the input material is depleted for H3K36me3, which characterizes transcribed gene bodies (Carrozza et al., 2005; Joshi and Struhl, 2005). In contrast, H3K36me3 is highly enriched in gene-body nucleosomes obtained from histone H2B CUT&RUN (Figure 1B). These results are consistent with previous studies mapping these modifications individually using ChIP-seq (Weiner et al., 2015). More importantly, because CUT&RUN.ChIP interrogates two chromatin epitopes, we can directly identify H2A.Z nucleosomes that contain H3K4me3 or H4 acetylation, which is not possible with separate ChIP experiments for the individual epitopes. Consistent with the representative nucleosome landscape shown, H2A.Z nucleosomes show strong correlations with H3K4me3 and H4ac, but not with H3K36me3 (Figures 1D and S1C-E). Recovery of specific peaks with ChIP, lack of signal in the IgG negative controls, and the absence of H2A.Z-H3K36me3 reveal the specificity of CUT&RUN.ChIP. Both CUT&RUN and CUT&RUN.ChIP provide much higher signal-to-noise than standard ChIP-seq as evident from our data (Figures 1 and S1).
RSC occupies NDRs and +1 and −1 nucleosomes with active marks
We next applied CUT&RUN to analyze the composition of the suspected fragile nucleosomes thought to occupy some NDRs. If nucleosomes are present at wide NDRs, they may be destabilized and ‘fragile’ due toremodeling by RSC. ChIP of proteins that transiently interact with and mobilize nucleosomes, such as ATP-dependent nucleosome remodeling complexes including RSC, is inefficient (Gelbart et al., 2005; Yen et al., 2012). However, using approaches that do not involve crosslinking, such as Native ChIP-seq (ORGANIC) and CUT&RUN, our lab has been successful in profiling remodeler complexes (Kasinathan et al., 2014; Ramachandran et al., 2015; Skene and Henikoff, 2017; Zentner et al., 2013).
CUT&RUN by tethering pA-MNase to FLAG-tagged Sth1, the catalytic subunit of RSC, shows high RSC occupancy mostly around the NDRs and 5′-end of genes (Figure 2A), consistent with RSC being involved in sliding promoter nucleosomes (Badis et al., 2008; Krietenstein et al., 2016; Yen et al., 2012). Additionally, RSC is strongly enriched at other non-protein-coding regions of the genome, examples being TLC1 (coding for a telomerase associated RNA) and a H/ACA small nucleolar RNA (Figure 2A). RSC CUT&RUN and Native-ChIP show similar profiles as seen in the snapshot from chromosome II as well as in genome-wide heat maps aligned over +1 nucleosome dyads, confirming that the CUT&RUN peaks observed are direct sites of RSC action (Figure 2A-C). To map nucleosomal and subnucleosomal particles separately, we grouped Native-ChIP and CUT&RUN reads into 121–200 bp (nucleosomal) and ≤120 bp (subnucleosomal or short) size classes. Although canonical nucleosomes contain 147 bp of DNA, MNase has the propensity to nibble away at some extra base pairs of DNA at the edges of the nucleosome exposed by nucleosome breathing or internal fluctuations. Therefore, DNA that is slightly shorter than 147 bp could still be obtained from a canonical nucleosome. We set the cutoff for subnuclesomal particles at 120 bp because that is approximately the size of DNA protected from MNase in nucleosomes that are partially unwrapped equally from the edges but contain all the histone octamer subunits (Ramachandran et al., 2017b). Therefore, particles smaller than 120 bp represent RSC bound by itself to DNA or to nucleosome-like particles that have substantially lost histone-DNA contacts, or hexasomes or tetrasomes that have lost either one or both H2A-H2B dimers. The two size classes reveal that RSC mostly maps to +1 and −1 nucleosomes, with short RSC-chromatin fragments at the NDRs (Figure 2A-D).
Figure 2: CUT&RUN.ChIP maps and characterizes RSC-bound nucleosomes.

(A) Spike-normalized (using the second spike-in DNA) RSC CUT&RUN.ChIP profiles (blue) compared with RSC Native-ChIP (green) (Ramachandran et al., 2015). Reads of 121- to 200-bp were analyzed for the RSC CUT&RUN.ChIP profiles, where fragment sizes analyzed for the other tracks are indicated. NET-seq shows the relative number of reads at each nucleotide position corresponding to the 3′ ends of nascent RNA transcripts mapped to the yeast genome (Churchman and Weissman, 2011). Plus and minus denote reads mapped to the Watson and Crick strands, respectively. Yeast open reading frames are shown at the bottom in black, and a non-protein-coding RNA is shown in grey. (B, C) Heat maps of RSC Native-ChIP (B) and CUT&RUN (C) over 2-kb regions spanning the centers of the +1 nucleosomes, sorted by NDR width, and separated into 121–200 bp (nucleosomal) and ≤120 bp (subnucleosomal or short) size classes. Dotted lines separate wide NDRs (≥150-bp) from narrow NDRs. Heat maps are total count-normalized and shown in log2 scale, with log center = 1 and contrast = 2 (B, Native-ChIP) and log center = 5 and contrast = 4 (C, CUT&RUN). (D) Enrichment of spike-in normalized densities of ≤120 bp (subnucleosomal) and 121–200 bp (nucleosomal) fragments from RSC CUT&RUN.ChIP over the ±1-kb region spanning the midpoints of all annotated NDRs (Chereji et al., 2018). (E) Enrichment of spike-in normalized densities of 121–200 bp (nucleosomal) fragments from histone ChIP-seq of RSC CUT&RUN, over the ±1-kb region spanning the midpoints of all annotated NDRs. See also Figures S2 and S3.
Using RSC CUT&RUN as input for ChIP we find that the main nucleosomal peaks from RSC CUT&RUN contain H2A.Z, H3K4me3, and H4ac, demonstrating that these histone variant and modification features are present in RSC-bound nucleosomes (Figure 2A). This is not surprising given the enrichment of RSC as well as these modifications in H2A.Z nucleosomes at the 5′-end of genes (Figure 1), and we observe strong RSC occupancy of +1 and −1 nucleosomes containing H2A.Z, H3K4me3, and H4ac in genome-wide averages (Figure 2E). Consistent with RSC-nucleosomes containing the H4ac mark, histone acetylation mediated by SAGA and NuA4 histone acetyltransferases is reported to activate RSC in vitro, which aids in the passage of RNA Polymerase II through a nucleosome (Carey et al., 2006). Absence of signal in IgG (negative control) and low intensities of RSC-H3K36me3 around promoters reflect the specificity of RSC CUT&RUN.ChIP (Figure 2A, E).
RSC asymmetrically protects the nucleosome DNA-entry site in vivo
We next applied fragment midpoint-vs-length plotting (V-plotting) to RSC CUT&RUN data to precisely annotate fragment-size information and further characterize binding features of RSC. In a V-plot, pixels corresponding to the centers of each DNA fragment are placed on a 2D map where the x and y-axes represent the map position and fragment lengths, respectively (Henikoff et al., 2011). Pixel densities corresponding to regions protected from MNase digestion assume a V-shape, where the vertex of the “V” represents limit digestion, and cleavages precisely on both sides of the region represent a factor-binding site. As RSC is found mostly around gene promoters, we applied this procedure to generate plots centered on the midpoint of all 5542 NDRs annotated from H3Q85C single-nucleosome chemical cleavage mapping in yeast (Chereji et al., 2018). We observe pixel clusters corresponding to phased +1 and −1 nucleosomes, as well as to protection from MNase cleavage at the NDRs, with minimum protection of ~50 bp (Figure S2A). Interestingly, the +1 and –1 nucleosome pixel clusters each show a whisker protruding up and into the NDR region. This pattern is also seen with RSC CUT&RUN followed by ChIP for histones (e.g., H2A.Z as shown in Figure S2B), suggesting that this represents a general feature of RSC-bound nucleosomes. A whisker protruding from a nucleosome would be generated by fragments spanning the nucleosome as well as parts of the flanking DNA in the direction of the whisker (Figure S2A, illustration at the bottom). We attribute this feature to RSC engulfing a nucleosome and asymmetrically binding to and protecting DNA, or sterically blocking the access of pA-MNase, preferably at the NDR-proximal edge of +1 and –1 nucleosomes. Asymmetric binding of RSC to nucleosomes is consistent with RSC remodeling producing asymmetrically unwrapped nucleosomal intermediates (Ramachandran et al., 2015). We observe that the asymmetric protection is nuclease-sensitive, as the whisker is diminished upon longer incubation with pA-MNase, as seen with a CUT&RUN time course (Figure S2D-F) (Skene and Henikoff, 2017). However, whiskers protruding from nucleosomes are not observed with H2A.Z CUT&RUN followed by H2A.Z ChIP (compare Figures S2B and C), implying that asymmetric protection is a unique feature of nucleosomes flanking the NDRs when RSC is bound. pA-MNase tethered to H2A.Z on a well-positioned nucleosome cleaves DNA on either side of the ~150 bp nucleosome core particle generating a tighter cluster of pixels. By comparing the number of fragments whose centers overlap with the 147 bp core of +1 or −1 nucleosomes plus part of the NDR (nucleosome plus whisker), versus fragments that correspond only to the core nucleosomes, we estimate that 25% ± 6% of RSC-bound nucleosomes are asymmetrically protected at the NDR-proximal side (near the DNA-entry site).
RSC is associated with partially unwrapped nucleosomes at wide NDRs
In order to determine the composition of fragile particles, we next examined RSC-bound particles at the set of previously annotated MNase-sensitive fragile nucleosome sites (Kubik et al., 2015). We find that 1902 out of the 1953 previously annotated fragile nucleosome sites are within NDRs with ≥150 bp between the stable +1 and −1 nucleosomes (i.e., these are ≥150 bp-wide NDRs). Both Native-ChIP and CUT&RUN show that RSC occupancy of subnucleosomal particles at NDRs correlate well with NDR width as seen in the heat maps aligned over +1 nucleosome dyads and ordered by NDR width (Figure 2B, C), as well as genome-wide average plots (Figure S3A-D). Since Native-ChIP for a factor only profiles sites that are direct targets, it confirms that the NDR space between nucleosomes is directly bound by RSC. By V-plot analysis of RSC CUT&RUN fragments, wide NDRs show distinct features as compared to all other (narrow) NDRs (compare Figures 3A and 3B). At wide NDRs over the previously annotated fragile nucleosome sites, we observe a strong and precise footprint from particles that protect a minimum of ~100 bp of DNA from pA-MNase digestion, as seen by sharp diagonals forming a V-shape with a vertex at ~100 bp on the y-axis (Figure 3A). In contrast, at narrow NDRs, limit digestion by pA-MNase results in fragments that are much shorter in length (~50 bp), resembling the limit digestion average at all 5542 NDRs (Figure 3B, compare with Figure S2A). Phased +1 and −1 nucleosomes are seen flanking the NDRs with, on average, ~300 bp center-to-center spacing at promoters with wide NDRs, which is wider than the average for nucleosomes flanking all other NDRs (~220 bp), as expected (Kubik et al., 2015). V-plot analysis of RSC CUT&RUN thus shows that the two sets of promoters are characterized by distinct chromatin landscapes, with different sizes and occupancies of chromatin particles at wide versus narrow NDRs.
Figure 3: RSC occupies wide NDRs and is associated with fragile nucleosomes.

(A, B) V-plots of RSC CUT&RUN spanning ±400 bp from annotated fragile nucleosome (FN) centers (Kubik et al., 2015) at ≳150 bp-wide NDRs (A) or the midpoints of narrow (all other) NDRs (B). Arrows show the positions corresponding to nucleosomal fragment midpoints, a subnucleosomal particle at a wide NDR (A), and a small particle over a narrow NDR (B) map. Horizontal dotted lines show the length in base pairs of limit CUT&RUN digestion around the central protected region in both plots. The average center-to-center distances between the +1 and −1 nucleosomes in both sets are shown at the bottom. For detailed interpretation of V-plots see Figure S3A. (C) Heat maps of RSC CUT&RUN.ChIP spanning +1 nucleosome centers ±1 kb, sorted by NDR width, and separated into 121–200 bp (nucleosomal) and ≤120 bp (subnucleosomal) size classes. Heat maps are total count-normalized (total number of reads for each sample and size class are indicated at the bottom of each heat map), and shown in log2 scale, with log center = 5 and contrast = 4. Dotted lines separate wide NDRs (≥150-bp) from narrow NDRs. (D, E) Enrichment of spike-in normalized densities of ≤120 bp fragments from RSC CUT&RUN.ChIP over the ±1-kb region spanning the NDRs, plotted separately for ≳150 bp-wide NDRs containing FN sites (D) and narrow (all other) NDRs (E). See also Figure S3.
We tested whether the ~100 bp particles at the wide NDRs contain histones by RSC CUT&RUN.ChIP with anti-histone antibodies. When aligned over +1 nucleosome dyads and ordered by NDR width, RSC CUT&RUN.ChIP heat maps reveal that a fraction of the particles occupying wide NDRs contain histones (Figure 3C). Separating reads by nucleosomal (121–200 bp) and shorter (≤120 bp) lengths reveals that histone-containing particles within the NDR are shorter than full-length nucleosomes, suggesting that these are subnucleosomal intermediates of RSC-nucleosome remodeling. As expected from previous work (Ramachandran et al., 2015), we observe that they contain H2A.Z, H4ac and H3K4me3. Histone occupancies within the NDRs are considerably higher at wide NDRs, and gradually diminish as the NDR shortens, revealing the MNase-sensitivity of NDRs (Kubik et al., 2015). Consistent with the heat maps, average plots of the distribution of ≤120 bp fragments at wide NDRs show three peaks of enrichment for H2B, H4ac, and H3K4me3 compared to a negative control, where one peak falls in the NDR between subnucleosomal fragments from the flanking +1 and −1 nucleosomes, coinciding with the centers of previously annotated fragile nucleosome sites (Figure 3D). Unlike the wide NDRs, narrow NDRs do not contain a defined site where we would expect RSC-associated histones. We therefore aligned the RSC CUT&RUN.ChIP data to the center of the narrow NDRs for the genome-wide average plots and V-plots. In contrast to the enrichment of histones over fragile nucleosome sites at wide NDRs relative to the flanking nucleosome positions, no enrichment of histones is seen between the −1 and +1 nucleosomes at the narrow NDRs (Figure 3E). V-plots of RSC CUT&RUN.ChIP reveal that limit digestion around RSC-associated subnucleosomes at wide NDRs is ~100 bp, in agreement with other analyses (Figure S3E, F), and the absence of a signal or a V-shape over the narrow NDRs again shows that RSC is not associated with histones at these genomic regions (Figure S3G). Instead, the ~50 bp protection by RSC at narrow NDRs (Figure 3B) suggest that RSC likely binds to DNA by itself at these sites. Composite plots of ≤120 bp fragments from RSC Native-ChIP (Figure S3B), RSC CUT&RUN (Figure S3A), and RSC CUT&RUN.ChIP for histones (Figure 3D) show sharp peaks at the center of fragile nucleosome sites within wide NDRs, which implies that the ChIPed histones at wide NDRs are directly bound by RSC. We observe that histone occupancies of wide NDRs as shown by the ChIPs are low compared to flanking nucleosomes and also compared to RSC occupancy as seen with the input (RSC CUT&RUN), indicating that only a small fraction of these sites in the population of yeast cells are occupied by nucleosomes at any time (compare the ≤120 bp profile in Figure S3A with Figure 3D). The low occupancy of RSC-associated histones may also reflect the unstable nature of these subnucleosomal particles, such that they are not immunoprecipitated efficiently. We estimate that on average 14% ± 6% of RSC-bound particles contain histones. Although we find H2A.Z and H2B at wide NDRs with RSC CUT&RUN.ChIP, heat maps of histone-ChIP with H2A.Z or H2B CUT&RUN as input do not show similar enrichment of ≤120 bp particles (Figure S1F, G). Possibly, only a fraction of +1 and −1 nucleosomes are RSC-bound, and therefore, the intensities of histones at NDRs relative to the flanking +1 and −1 nucleosomes is much higher in the material that is released upon CUT&RUN with RSC than CUT&RUN with H2A.Z or H2B.
Our data show that RSC and RSC-associated subnucleosomes occupy wide NDRs and recapitulate the MNase-sensitive footprint observed by Kubik et al. (Kubik et al., 2015). These fragile nucleosomes have less DNA wrapped around them compared to full-length nucleosomes. We envision that RSC directly engulfs nucleosomes deposited at wide NDRs, and RSC-mediated sliding and destabilization of these nucleosome causes their MNase-sensitive fragility. Consistent with fragile nucleosomes having disrupted histone-DNA interactions, single-nucleosome H3Q85C chemical cleavage mapping (Chereji et al., 2018) shows low fragment densities at both narrow and wide NDRs (Figure S3H, left panel). The smaller length of DNA (~100 bp) associated with RSC-bound histones at wide NDRs is also consistent with these particles being hexasomal intermediates that have lost one of the two H2A-H2B dimers from the full complement of histones in a canonical nucleosome. RSC remodeling of nucleosomes in the presence of the histone chaperone NAP1 has been shown to generate hexasomes in vitro (Kuryan et al., 2012). Our data therefore suggest a role of RSC remodeling at wide NDRs in the switch between two dynamic states – partially unwrapped subnucleosomes and nucleosome-depleted chromatin.
GRFs bind at sites within partially unwrapped nucleosomes at wide NDRs
In addition to chromatin remodelers, GRFs have been implicated in regulating the promoter landscape and as trans-acting factors involved in promoter nucleosome disruption (Badis et al., 2008; Ganapathi et al., 2011; Hartley and Madhani, 2009). The GRFs Abf1 (ARS binding factor 1) and Reb1 (rDNA enhancer binding protein) are seen to be highly enriched at promoters (Kasinathan et al., 2014; Rhee and Pugh, 2011), and their binding sites frequently overlap with sites for fragile nucleosomes (Kubik et al., 2015). We performed CUT&RUN.ChIP for Abf1 (Abf1-FLAG) and Reb1 (Reb1-FLAG) to investigate whether these GRFs bind to nucleosomes or to nucleosome-free DNA in NDRs. Heat maps of both Abf1 and Reb1 CUT&RUN.ChIP aligned over their previously annotated binding sites that are within NDRs and ordered by NDR width show enrichment of ≤120 bp fragments overlapping the factor-binding sites, specifically at wide NDRs (Figure 4A, B). Similar to RSC CUT&RUN.ChIP, we observe enrichment of H2A.Z, H4ac, and H3K4me3 over Abf1 and Reb1 sites at the wide NDRs. Consistent with the heat maps, genome-wide average plots show enrichment of Abf1 and Reb1-associated subnucleosomal histone-containing particles at wide NDRs but not at narrow NDRs (Figure 4C-F). In contrast, nucleosome-size fragments (121–200 bp) over Abf1 and Reb1 sites within NDRs are very few or absent (Figure S4A-D). Nevertheless, we observe 121- to 200-bp fragments corresponding to the +1 and −1 nucleosomes flanking the NDRs with Abf1 and Reb1 CUT&RUN (Figures S4A-D), which we attribute to transcription factors releasing the flanking nucleosomes in CUT&RUN due to flexibility of the MNase tether (Figure S4E), as previously shown (Skene and Henikoff, 2017). Our data suggest that GRFs bind to fragile nucleosomes when their binding sites are exposed, as the nucleosomes are partially unwrapped upon RSC remodeling. In agreement with Abf1 and Reb1 binding to RSC-remodeled nucleosomes with altered histone-DNA interactions, H3Q85C chemical cleavage mapping data aligned over Abf1 and Reb1 sites at NDRs show low fragment densities over the sites (Figure S3H, middle and right). We estimate that 5% ± 2% of Abf1 and 27% ± 9% of Reb1 bound at wide NDRs is likely associated with histones (compare Figures 4C, D and S4F). V-plots of Abf1 and Reb1 CUT&RUN.ChIP aligned over the respective GRF-binding sites within wide and narrow NDRs also show that histone occupancies of NDRs are low compared to the flanking +1 and −1 nucleosomes (Figure S5). We conclude that the GRF-subnucleosome at wide NDRs is a transient site-exposed state.
Figure 4: GRFs associate with subnucleosomes at wide NDRs.

(A, B) Heat maps of ≤120 bp (subnucleosomal) fragments from Abf1 (A) and Reb1 (B) CUT&RUN.ChIP spanning ±1 kb of the respective GRF binding sites. Heat maps are total count-normalized and shown in log2 scale, with log center = 5 and contrast = 5. Dotted lines separate wide NDRs (≥150-bp) from the rest. Abf1 and Reb1 binding site locations were previously published (Skene and Henikoff, 2017). (C-F) Enrichment of spike-in normalized densities of ≤120 bp fragments from Abf1 (C, E) and Reb1 (D, F) CUT&RUN.ChIP over the ±1-kb region spanning the NDRs, plotted separately for ≳150 bp-wide NDRs containing fragile nucleosome (FN) sites (C, D) and narrow (all other) NDRs (E, F). See also Figures S4 and S5.
DISCUSSION
We have shown that CUT&RUN.ChIP is a simple and efficient approach for mapping chromatin complexes genome-wide under native conditions with high specificity, negligible background and wide dynamic range. Our mapping of H3 and H4 histone modifications in individual H2A.Z and H2B nucleosomes, and in RSC and GRF-associated nucleosomes, demonstrates that CUT&RUN.ChIP is applicable to a variety of chromatin complexes. In standard ChIP-based methods, because of crosslinking and sonication to fragment chromatin, it has been difficult to address if epitopes are on the same particle. This is evident in contradictory reports estimating the fraction of bivalent nucleosomes containing both H3K27me3 and H3K4me3 in ESCs using sequential-ChIP versus single-molecule imaging (Bernstein et al., 2006; Shema et al., 2016). Moreover, sequential-ChIP suffers from low recovery and inefficiency (Thakur and Henikoff, 2016; Weiner et al., 2016), limiting its genome-wide application. The high efficiency and the base-pair resolution of CUT&RUN that we (Skene et al., 2018) and others (Liu et al., 2018) have demonstrated in higher eukaryotic cells indicate that CUT&RUN.ChIP can replace sequential ChIP for any genome-wide chromatin-based co-occupancy study.
Using CUT&RUN.ChIP we characterized the proteins that occupy wide NDRs containing MNase-sensitive nucleosome-like particles between the strongly positioned +1 and −1 nucleosomes. We showed that these wide NDRs are occupied by RSC, which often engulfs a partially unwrapped nucleosome containing H2A.Z and active histone modifications. Given the role of RSC in promoter nucleosome clearance (Badis et al., 2008; Hartley and Madhani, 2009; Krietenstein et al., 2016; Kubik et al., 2015; Kubik et al., 2018; Parnell et al., 2015; Yague-Sanz et al., 2017), these particles are likely transient intermediates in RSC-remodeling, accounting for their MNase-sensitivity. A large fraction of RSC bound at wide NDRs is depleted of histones, suggesting that RSC-associated subnucleosomes are unstable and that RSC transiently binds to free DNA. The striking similarity between RSC and GRF-associated subnucleosomal particles at fragile nucleosome sites within wide NDRs, and the lack of GRF-associated full-length nucleosomes, lead us to suggest that RSC remodeling of nucleosomes at wide NDRs exposes sites for GRF binding.
Wide NDRs provide more binding sites for regulatory factors, likely attributable to genes with wide NDRs being highly transcribed, but at the same time they provide enough space for a nucleosome to form. Regulatory regions are inherently limited in size because they must be nucleosome-free to allow factor binding, therefore, NDRs require a mechanism to remove nucleosomes. The dynamic process at wide NDRs suggested by our study is in principle similar to that observed earlier at a few well-studied inducible systems such as the yeast PHO5 promoter and GAL1/10 upstream activating sequence (UASg). At the PHO5 promoter, transcriptional activation is associated with activator-driven opening of the promoter due to nucleosome unfolding and removal that involves RSC action (Boeger et al., 2003; Fascher et al., 1990; Musladin et al., 2014). A dynamic MNase-sensitive nucleosome-like footprint over UASg was one of the first observed indications of nucleosome fragility (Henikoff et al., 2011; Xi et al., 2011), and it was proposed that RSC engulfment and partial unwinding of a H2A.Z-containing nucleosome at UASg makes the Gal4 activator binding site more accessible (Boeger et al., 2003; Floer et al., 2010). Similarly, we propose a general model for RSC-mediated promoter nucleosome clearance at wide NDRs (Figure 5), that is consistent with the roles of RSC and GRFs in establishing the promoter architecture (Krietenstein et al., 2016; Kubik et al., 2015; Kubik et al., 2018). RSC engulfs and displaces +1 and −1 nucleosomes away from each other (Hartley and Madhani, 2009), and in association with other remodelers, positions them to generate nucleosome depleted regions (NDRs) upstream of TSSs (Krietenstein et al., 2016). When the gap between the +1 and −1 nucleosome is large (≳150 bp) a nucleosome is likely to be deposited. But RSC may be directed to wide NDRs by G/C-rich and poly-A sequence motifs enriched at these regions, which are binding sites for the RSC subunits Rsc3 and Rsc30 (Badis et al., 2008; Kubik et al., 2015), and have been implicated in RSC orientation (Krietenstein et al., 2016). RSC would then engulf, mobilize or slide and partially unwrap nucleosomes deposited at wide NDRs, creating subnucleosomal intermediates. Nucleosome displacement and partial unwrapping also may expose sites for GRF binding. In vitro experiments show that the dwell-times for transcription factors that are bound within a nucleosome are orders of magnitude shorter than when bound to exposed-DNA (Luo et al., 2014). GRFs bound to partially unwrapped nucleosomes may also rapidly dissociate, but a binding site that is more accessible due to nucleosome unwrapping would ensure rapid re-association, creating transient GRF-bound subnucleosomal intermediates. Eventually, subnucleosomal intermediates are cleared, likely by the combined action of RSC and GRFs, making room for RNA Polymerase II loading for transcription. It has been shown for transcription factors in yeast including GRFs that affinity and copy number of their binding motifs correlate closely with nucleosome depletion (Yan et al., 2018). Interestingly, the same study also described Rsc3, the DNA sequence-specific subunit of RSC in the same capacity of nucleosome depleting factor, but with weaker nucleosome depletion activity as compared to the GRFs. The presence of high-affinity binding motifs for GRFs within NDRs and their high abundance in the cell suggest that GRFs and nucleosomes compete directly for binding at NDRs. Our data therefore suggest that transient site exposure due to RSC remodeling would shift the equilibrium in favor of GRF binding. When the factors disengage, a nucleosome can reform at wide NDRs, resuming the cycle.
Figure 5: A dynamic model for RSC-mediated promoter nucleosome clearance and generation of fragile nucleosomes.

RSC, perhaps oriented by poly-A and G/C-rich sequence motifs, engulfs a nucleosome deposited at a ≳150 bp-wide NDR (step 1), and mobilizes and partially unwraps the nucleosome in an ATP-dependent manner (step 2). GRF binding is facilitated at sites exposed due to nucleosome displacement/unwrapping (step 3). RSC is still associated with the remodeled nucleosome at the two intermediate stages flanking step 3, but for simplicity RSC is not shown. RSC (and GRFs) eventually disrupt the nucleosome (step leading to stable GRF binding at sites on the exposed DNA. RSC may be also transiently bound to free DNA at the exposed NDRs, but not necessarily along with a GRF at the same time. When GRFs (and RSC) turn over (step 5), a nucleosome can reform at the NDR, and the cycle resumes.
This function of RSC in promoter nucleosome clearance is likely distinct from its role in positioning +1 and −1 nucleosomes, which was also suggested based on the contribution of RSC in genome-wide reconstitution of promoter nucleosome organization in a purified system (Krietenstein et al., 2016). Alternatively, a model wherein RSC, engulfing a nucleosome, transits back and forth between the +1 and −1 nucleosome positions (like a pendulum) is plausible. In this model, RSC-associated nucleosomes would spend more time at +1 and −1 positions, while the RSC-associated particles at the NDR would represent a transient intermediate, with the partially unwrapped state favored due to GRF binding. The RSC paralog switch/sucrose non-fermentable (SWI/SNF) remodeling complex has in fact been shown in vitro to be able to slide nucleosomes past a bound transcription factor, likely evicting it (Li et al., 2015).
Although standard ChIP-seq or chemical mapping cannot detect histone occupancy of wide NDRs in budding yeast, histone H3 and H2B ChIP-seq with Drosophila embryo and mouse ES cells show the presence of MNase-sensitive subnucleosomal histone-containing particles upstream of TSSs (Chereji et al., 2016; Ishii et al., 2015). We attribute this difference to RSC-mediated clearance of nucleosomes at budding yeast promoters soon after they are deposited, and the absence of RSC function in metazoans. Although RSC orthologs are found in metazoans, they may not be as competent as RSC in promoter nucleosome clearance, and unlike RSC, the metazoan BRG1-associated factor (BAF) complex has not been reported to have any direct promoter DNA-binding property. Consistent with RSC-mediated dynamics at NDRs, promoter regions in budding yeast display a high rate of replication-independent histone turnover (Dion et al., 2007). Evidence in support of yeast-specific RSC function in promoter nucleosome clearance also comes from studies that assayed nucleosome re-establishment around promoters after the passage of replication fork in budding yeast and Drosophila (Fennessy and Owen-Hughes, 2016; Ramachandran et al., 2017a; Ramachandran and Henikoff, 2016; Vasseur et al., 2016). Whereas nucleosomes transiently fill up NDRs post-replication in Drosophila, NDRs in budding yeast remain depleted of nucleosomes behind the replication fork. RSC in budding yeast therefore may be specially adapted for promoter nucleosome clearance that results in a highly dynamic nucleosome landscape at a subset of gene promoters.
STAR METHODS
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Steven Henikoff (steveh@fredhutch.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast cultures
Saccharomyces cerevisiae strains used in this study are listed in Key Resources Table.
Yeast cultures were grown in YPD at 30oC.
METHOD DETAILS
Preparation of yeast nuclei for CUT&RUN
Yeast nuclei were prepared as described (Orsi et al., 2015). Briefly, yeast strains were grown in 500 mL YPD to O.D.600 ~0.7 (~5 × 109 cells), harvested, and yeast spheroplasts were prepared using Zymolase. Yeast nuclei were extracted using Ficoll, divided into 10 aliquots (each containing nuclei isolated from ~5 × 108 cells), snap-frozen and held at −80°C, then thawed on ice before use.
CUT&RUN.ChIP
CUT&RUN was performed following the published protocol (Skene and Henikoff, 2017), with some variations. Bio-Mag Plus Concanavalin A (lectin) coated beads were equilibrated with binding buffer (20 mM HEPES-KOH pH7.9, 10 mM KCl, and 1 mM each CaCl2 and MnCl2). The beads (~200 μl) were rapidly mixed with a thawed yeast nuclei aliquot and held at room temperature (RT) ≥5 min, placed on a magnet stand to clear (<1 min), and decanted on a magnet stand. The beads were then washed once with Dig-Wash buffer (20 mM Na-HEPES pH 7.5, 150 mM NaCl, 0.5 mM Spermidine, 0.05% Digitonin, supplemented with EDTA-free protease inhibitor cocktail tablet), supplemented with 2 mM EDTA, using the magnet stand to decant. From this step, samples were kept cold (on ice) as much as possible. The beads were incubated 2 hr to overnight at 4°C with mouse anti-FLAG antibody (1:2 00 final dilution) in cold Dig-Wash + EDTA, decanted, washed twice in cold Dig-Wash buffer (no EDTA from this point), then incubated 1 hr at 4°C with rabbit anti-mouse I gG antibody (1:100) in Dig-Wash buffer. The beads were washed twice with Dig-Wash buffer, then incubated 1 hr at 4°C with pA-MN (360 μg/ml, 1:400) in Dig-Wash buffer. The beads were washed three times in Dig-Wash buffer, brought up in 1.2 ml Dig-Wash buffer, and equilibrated to 0°C. CaCl2 was quickly mixed to a final concentration of 2 mM, incubated on ice for 5 min, and reactions were stopped with 1.2 ml of 2XSTOP (150 mM NaCl, 20 mM EDTA, 4 mM EGTA, 50 μg/ml RNase A, and 40 μg/ml glycogen, containing 5–50 pg/ml heterologous mostly mononucleosome-sized DNA fragments extracted from formaldehyde-crosslinked MNase-treated Drosophila chromatin as the first spike-in). After incubating at 37°C for 20 min, FLAG peptide was added to a final concentration of 13 μg/ml, mixed, and the beads were centrifuged 5 min at 13,000 rpm at 4°C. The supernatant was removed on a magnet stand and divided into 7–8 aliquots for ChIP, and one aliquot was saved (stored at 4°C) as the input. To the ChIP samples respective antibodies (for IgG, H2B, H2A.Z, H4ac, H3K4me3, and H3K36me3, see Key Resource Table) were added (1:100), and incubated at 4°C ove rnight. Protein A Dynabeads for rabbit polyclonal antibodies (or Protein G for the mouse monoclonal H3K36me3 antibody) were equilibrated in Wash buffer (same as Dig-Wash, but without digitonin) supplemented with 0.05% Tween-20, and 20 µl of beads were added to each ChIP sample (except the input), incubated at 4°C for 15 min, and washed once with Wash buffer+Tween-20. The ChIP samples were brought up with DNA-extraction buffer (150 mM NaCl 10 mM EDTA, 2 mM EGTA, 0.1% SDS, 0.2 mg/ml Proteinase K, and 40 µg/ml glycogen, containing 5–50 pg/ml heterologous mostly mononucleosome-sized DNA fragments extracted from MNase-treated Schizosaccharomyces pombe chromatin as a second spike-in). SDS (0.1%), Proteinase K (0.2 mg/ml) and S. pombe spike-in DNA (5–50 pg/ml) were also added to the input samples such that their concentrations match with the ChIP samples. Samples were incubated at 50°C for 20 min, then extracted at room temperature once with buffered phenol-chloroform-isoamyl alcohol (25:24:1, Sigma Catalog # P2069), transferred to a phase-lock tube (Qiagen, Hilden, Germany, Catalog #129046), re-extracted with one volume CHCl3, transferred to a fresh tube containing 2 μL of 2 mg/ml glycogen, precipitated by addition of 2–2.5 vol ethanol overnight at −20°C, and centrifuged 10 min at 13,000 rpm at 4°C. The pe llets were rinsed with 100% ethanol, air-dried and dissolved in 25 μL 0.1 × TE (=1 mM Tris pH 8, 0.1 mM EDTA). Note: Drosophila and S. pombe spike-in DNA can be interchangeably used as the first or second spike-in. CUT&RUN.ChIP for Sth1-FLAG (RSC) and Htz1-FLAG (H2A.Z) were repeated three times each with two independent preparations of yeast nuclei from separate cultures grown at different times. Abf1-FLAG and Reb1-FLAG CUT&RUN.ChIP experiments were repeated two times each with yeast nuclei prepared separately for each replicate. Sequencing datasets corresponding to two replicates of each experiment is deposited at GEO under accession GEO: GSE116853.
Library preparation and sequencing
Libraries were prepared for Illumina sequencing with Tru-Seq adapters as described (Skene et al., 2018; Skene and Henikoff, 2017). Briefly, libraries were constructed without size-selection, following the KAPA DNA polymerase library preparation kit protocol (https://www.kapabiosystems.com/product-applications/products/next-generation-sequencing-2/dna-library-preparation/kapa-hyper-prep-kits/), optimized to favor exponential amplification of the desired CUT&RUN fragments over linear amplification of large DNA fragments (Skene and Henikoff, 2017). Un-ligated adapters were removed using AMPure XP beads. Libraries were sequenced for 25 cycles in paired-end mode on the Illumina HiSeq 2500 platform at the Fred Hutchinson Cancer Research Center Genomics Shared Resource.
Data processing
Paired-end Saccharomyces cerevisiae reads were mapped to the sacCer3/V64 genome using Bowtie2 version 2.2.5 with options: --local --verysensitive-local --no-unal --no-mixed --no-discordant --phred33 -I 10 -X 700. Spike-in fragments were mapped to repeat-masked FlyBase r6.06 (Drosophila) and PomBase version 171101 (S. pombe) genomes, also using the --no-overlap --no-dovetail options to avoid cross-mapping of the experimental genome to that of the spike-in DNA.
QUANTIFICATION AND STATISTICAL ANALYSIS
Continuous-valued data tracks (bedGraphs) were generated using genomecov in bedtools v2.27.0 by calibrating with spike-in DNA reads mapped to repeat-masked reference genomes (Skene and Henikoff, 2017), and displayed as genomic tracks shown in Figures 1B and 2A using Integrated Genome Browser (IGB). Scatterplots were generated using custom scripts by triangular smoothing of total count-normalized 121–200 bp (nucleosomal) reads with 11 bp windows and displayed using gnuplot v3.13.0. Pearson correlation coefficients (r) were calculated using a custom script. Heatmaps were generated using Java Tree view v1.1.6r2 in “Auto style” with total count-normalized read depth per base pair (-d option in bedtools genomecov). V-plots were generated using custom scripts by plotting the length of each mapped paired-end fragment as a function of the distance from the fragment midpoint to the center of the site for each annotated feature. Heatmaps were generated using gnuplot v3.13.0 by quantifying the number of reads at each relative distance and length coordinate, normalized by the number of mapped spike-in reads in the sample. The maximum intensity of the heatmaps in Figures S2A-C were independently scaled to achieve the greatest contrast.
The percentage of RSC-associated +1 and −1 nucleosomes that are asymmetrically protected from MNase-cleavage at the NDR-flanking edge (whiskers in Figure S2A, B), was calculated the following way using the bedtools intersect tool: A = number of fragments whose centers overlap with the 147 bp core of +1 or −1 nucleosomes plus part of the NDR (nucleosome plus whisker). B = number of fragments whose centers overlap only with the core nucleosome. % of asymmetrically protected nucleosomes =
To calculate the fractions of RSC and GRF-associated subnucleosomal histone-containing particles, spike-in normalized numbers of ≤120 bp fragments that overlap with the centers of previously annotated fragile nucleosome sites (Kubik et al., 2015) in the ChIP samples (A) and the input (CUT&RUN supernatant) samples (B) were calculated separately using the bedtools intersect tool. The percentage of histone-containing particles in each ChIP was determined as (A/B) × 100. The averages from all four histone ChIP experiments from two biological replicates are reported as the percent of RSC or GRF bound at wide NDRs that are associated with histones.
DATA AND SOFTWARE AVAILABILITY
Sequencing data from this study have been deposited in GEO at the National Center for Biotechnology under the accession number GSE116853. The datasets deposited correspond to two replicates of each experiment.
Supplementary Material
Table 1: KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal ANTI-FLAG M2 antibody | Sigma-Aldrich | Cat# F1804 |
| Rabbit Anti-Mouse IgG H&L | abcam | Cat# ab46540 |
| Guinea Pig Anti-Rabbit IgG (Heavy & Light Chain) | Antibodies-Online | Cat# ABIN101961 |
| Anti-Histone H2A.Z antibody, Rabbit polyclonal | abcam | Cat# ab4626 |
| Rabbit polyclonal Histone H2B antibody | Active Motif | Cat# 39237 |
| Rabbit polyclonal Anti-acetyl-Histone H4 antibody | Millipore Sigma | Cat# Upstate 06–598 |
| Rabbit polyclonal Histone H3K4me3 antibody | Active Motif | Cat# 39159/60 |
| Mouse monoclonal Histone H3K36me3 antibody | Active Motif | Cat# 61021 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Protein A-MNase fusion protein | Henikoff Lab (in-house) | N/A |
| BioMag Plus Concanavalin A beads | Bangs Laboratories | Cat# BP531 |
| Spermidine Trihydrochloride | Sigma-Aldrich | Cat# S2501–5G |
| Digitonin | EMD-Millipore | Cat# 300410 |
| Roche complete protease inhibitor tablet | Sigma-Aldrich | Cat# 11836170001 |
| Proteinase K | Thermo Fisher Scientific | Cat# 25530049 |
| RNase A | Thermo Fisher Scientific | Cat# EN0531 |
| Glycogen | Sigma-Aldrich | Cat# 10901393001 |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat# 10004D |
| Dynabeads Protein A | Thermo Fisher Scientific | Cat# 10002D |
| Drosophila mononucleosomal DNA | Henikoff Lab (in-house) | N/A |
| S. pombe mononucleosomal DNA | Henikoff Lab (in-house) | N/A |
| Ampure XP beads | Beckman Coulter | Cat# A29153 |
| Deposited Data | ||
| All sequencing datasets have been uploaded in GEO under accession GEO: GSE116853 | This paper | GEO: GSE116853 |
| H3Q85C chemical cleavage mapping | (Chereji et al., 2018) | GEO: GSE97290 |
| RSC CUT&RUN | (Skene and Henikoff,2017) | GEO: GSE84474 |
| H2A.Z, H3K4me3 and H3K36me3 ChIP-seq | (Weiner et al., 2015) | GEO: GSE61888 |
| NET-seq | (Churchman and Weissman, 2011) | GEO: GSE25107 |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae: W1588–4C sth1–3FLAG-kanMX4 | Laboratory of Toshi Tsukyama | YTT3249 |
| S. cerevisiae: W1588–4C htz1–2L-3FLAG-kanMX4 | Laboratory of Toshi Tsukyama | YTT1724 |
| S. cerevisiae: htb1–3FLAG | Laboratory of Sue Biggins | SBY4554 |
| S. cerevisiae: W1588–4C abf1–3FLAG- kanMX | Henikoff Lab | (Kasinathan et al., 2014) |
| S. cerevisiae: W1588–4C reb1–3FLAG- kanMX | Henikoff Lab | (Kasinathan et al., 2014) |
| Software and Algorithms | ||
| bedtools | http://bedtools.readthedocs.io/en/latest/index.html | v2.27.0 |
| Bowtie2 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | v2.2.5 |
| Integrated genome browser | https://wiki.transvar.org/display/igbman/Home | v9.0.1 |
| gnuplot | http://gnuplot.info/ | v3.13.0 |
| Java Tree View | http://jtreeview.sourceforge.net/ | v1.1.6r2 |
| Custom shell scripts | This paper | N/A |
| Custom perl scripts | This paper | N/A |
Highlights.
CUT&RUN.ChIP maps chromatin-factor co-occupancies with (sub)nucleosomal histones
RSC and GRF-associated partially-unwrapped nucleosomes occupy ≳150 bp-wide NDRs
Fragile nucleosomes at wide NDRs are partially-unwrapped RSC-remodeling intermediates
Acknowledgements
We thank Jorja Henikoff and Srinivas Ramachandran for assistance with bioinformatics analysis, Christine Codomo for preparing Illumina sequencing libraries, Kami Ahmad for feedback, the Fred Hutch Genomics Shared Resource for sequencing, and the Howard Hughes Medical Institute and the NIH 4D Nucleome Program (TCPA A093) for funding.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
The authors declare no competing interests.
References
- Badis G, Chan ET, van Bakel H, Pena-Castillo L, Tillo D, Tsui K, Carlson CD, Gossett AJ, Hasinoff MJ, Warren CL, et al. (2008). A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Molecular cell 32, 878–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326. [DOI] [PubMed] [Google Scholar]
- Boeger H, Griesenbeck J, Strattan JS, and Kornberg RD (2003). Nucleosomes unfold completely at a transcriptionally active promoter. Molecular cell 11, 1587–1598. [DOI] [PubMed] [Google Scholar]
- Carey M, Li B, and Workman JL (2006). RSC exploits histone acetylation to abrogate the nucleosomal block to RNA polymerase II elongation. Molecular cell 24, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, et al. (2005). Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123, 581–592. [DOI] [PubMed] [Google Scholar]
- Chaban Y, Ezeokonkwo C, Chung WH, Zhang F, Kornberg RD, Maier-Davis B, Lorch Y, and Asturias FJ (2008). Structure of a RSC-nucleosome complex and insights into chromatin remodeling. Nature structural & molecular biology 15, 1272–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chereji RV, Kan TW, Grudniewska MK, Romashchenko AV, Berezikov E, Zhimulev IF, Guryev V, Morozov AV, and Moshkin YM (2016). Genome-wide profiling of nucleosome sensitivity and chromatin accessibility in Drosophila melanogaster. Nucleic acids research 44, 1036–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chereji RV, Ocampo J, and Clark DJ (2017). MNase-Sensitive Complexes in Yeast: Nucleosomes and Non-histone Barriers. Molecular cell 65, 565–577 e563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chereji RV, Ramachandran S, Bryson TD, and Henikoff S (2018). Precise genome-wide mapping of single nucleosomes and linkers in vivo. Genome Biol 19, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, and Weissman JS (2011). Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion MF, Kaplan T, Kim M, Buratowski S, Friedman N, and Rando OJ (2007). Dynamics of replication-independent histone turnover in budding yeast. Science 315, 1405–1408. [DOI] [PubMed] [Google Scholar]
- Fascher KD, Schmitz J, and Horz W (1990). Role of trans-activating proteins in the generation of active chromatin at the PHO5 promoter in S. cerevisiae. EMBO J 9, 2523–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fennessy RT, and Owen-Hughes T (2016). Establishment of a promoter-based chromatin architecture on recently replicated DNA can accommodate variable inter-nucleosome spacing. Nucleic acids research 44, 7189–7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floer M, Wang X, Prabhu V, Berrozpe G, Narayan S, Spagna D, Alvarez D, Kendall J, Krasnitz A, Stepansky A, et al. (2010). A RSC/nucleosome complex determines chromatin architecture and facilitates activator binding. Cell 141, 407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathi M, Palumbo MJ, Ansari SA, He Q, Tsui K, Nislow C, and Morse RH (2011). Extensive role of the general regulatory factors, Abf1 and Rap1, in determining genome-wide chromatin structure in budding yeast. Nucleic acids research 39, 2032–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelbart ME, Bachman N, Delrow J, Boeke JD, and Tsukiyama T (2005). Genome-wide identification of Isw2 chromatin-remodeling targets by localization of a catalytically inactive mutant. Genes & development 19, 942–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley PD, and Madhani HD (2009). Mechanisms that specify promoter nucleosome location and identity. Cell 137, 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff JG, Belsky JA, Krassovsky K, MacAlpine DM, and Henikoff S (2011). Epigenome characterization at single base-pair resolution. Proceedings of the National Academy of Sciences of the United States of America 108, 18318–18323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S (2016). Mechanisms of Nucleosome Dynamics In Vivo. Cold Spring Harb Perspect Med 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Ramachandran S, Krassovsky K, Bryson TD, Codomo CA, Brogaard K, Widom J, Wang JP, and Henikoff JG (2014). The budding yeast Centromere DNA Element II wraps a stable Cse4 hemisome in either orientation in vivo. eLife 3, e01861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii H, Kadonaga JT, and Ren B (2015). MPE-seq, a new method for the genome-wide analysis of chromatin structure. Proceedings of the National Academy of Sciences of the United States of America 112, E3457–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, and Pugh BF (2009). Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 10, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AA, and Struhl K (2005). Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Molecular cell 20, 971–978. [DOI] [PubMed] [Google Scholar]
- Kasinathan S, Orsi GA, Zentner GE, Ahmad K, and Henikoff S (2014). High-resolution mapping of transcription factor binding sites on native chromatin. Nat Methods 11, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krietenstein N, Wal M, Watanabe S, Park B, Peterson CL, Pugh BF, and Korber P (2016). Genomic Nucleosome Organization Reconstituted with Pure Proteins. Cell 167, 709–721 e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubik S, Bruzzone MJ, Albert B, and Shore D (2017). A Reply to “MNase-Sensitive Complexes in Yeast: Nucleosomes and Non-histone Barriers,” by Chereji et al. Molecular cell 65, 578–580. [DOI] [PubMed] [Google Scholar]
- Kubik S, Bruzzone MJ, Jacquet P, Falcone JL, Rougemont J, and Shore D (2015). Nucleosome Stability Distinguishes Two Different Promoter Types at All Protein-Coding Genes in Yeast. Molecular cell 60, 422–434. [DOI] [PubMed] [Google Scholar]
- Kubik S, O’Duibhir E, de Jonge WJ, Mattarocci S, Albert B, Falcone JL, Bruzzone MJ, Holstege FCP, and Shore D (2018). Sequence-Directed Action of RSC Remodeler and General Regulatory Factors Modulates +1 Nucleosome Position to Facilitate Transcription. Molecular cell 71, 89–102 e105. [DOI] [PubMed] [Google Scholar]
- Kuryan BG, Kim J, Tran NN, Lombardo SR, Venkatesh S, Workman JL, and Carey M (2012). Histone density is maintained during transcription mediated by the chromatin remodeler RSC and histone chaperone NAP1 in vitro. Proceedings of the National Academy of Sciences of the United States of America 109, 1931–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai WKM, and Pugh BF (2017). Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nature reviews. Molecular cell biology 18, 548–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Hada A, Sen P, Olufemi L, Hall MA, Smith BY, Forth S, McKnight JN, Patel A, Bowman GD, et al. (2015). Dynamic regulation of transcription factors by nucleosome remodeling. eLife 4, e06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieleg C, Krietenstein N, Walker M, and Korber P (2015). Nucleosome positioning in yeasts: methods, maps, and mechanisms. Chromosoma 124, 131–151. [DOI] [PubMed] [Google Scholar]
- Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias-Trevino C, Rogers JM, Kurita R, et al. (2018). Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 173, 430–442 e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorch Y, and Kornberg RD (2017). Chromatin-remodeling for transcription. Q Rev Biophys 50, e5. [DOI] [PubMed] [Google Scholar]
- Luo Y, North JA, Rose SD, and Poirier MG (2014). Nucleosomes accelerate transcription factor dissociation. Nucleic acids research 42, 3017–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musladin S, Krietenstein N, Korber P, and Barbaric S (2014). The RSC chromatin remodeling complex has a crucial role in the complete remodeler set for yeast PHO5 promoter opening. Nucleic acids research 42, 4270–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi GA, Kasinathan S, Zentner GE, Henikoff S, and Ahmad K (2015). Mapping regulatory factors by immunoprecipitation from native chromatin. Curr Protoc Mol Biol 110, 21 31 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnell TJ, Schlichter A, Wilson BG, and Cairns BR (2015). The chromatin remodelers RSC and ISW1 display functional and chromatin-based promoter antagonism. eLife 4, e06073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Ahmad K, and Henikoff S (2017a). Capitalizing on disaster: Establishing chromatin specificity behind the replication fork. BioEssays : news and reviews in molecular, cellular and developmental biology 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Ahmad K, and Henikoff S (2017b). Transcription and Remodeling Produce Asymmetrically Unwrapped Nucleosomal Intermediates. Molecular cell 68, 1038–1053 e1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, and Henikoff S (2016). Transcriptional Regulators Compete with Nucleosomes Post-replication. Cell 165, 580–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Zentner GE, and Henikoff S (2015). Asymmetric nucleosomes flank promoters in the budding yeast genome. Genome Res 25, 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Bataille AR, Zhang L, and Pugh BF (2014). Subnucleosomal structures and nucleosome asymmetry across a genome. Cell 159, 1377–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, and Pugh BF (2011). Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147, 1408–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shema E, Jones D, Shoresh N, Donohue L, Ram O, and Bernstein BE (2016). Single-molecule decoding of combinatorially modified nucleosomes. Science 352, 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff JG, and Henikoff S (2018). Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nature protocols 13, 1006–1019. [DOI] [PubMed] [Google Scholar]
- Skene PJ, and Henikoff S (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6, e21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K, and Segal E (2013). Determinants of nucleosome positioning. Nature structural & molecular biology 20, 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur J, and Henikoff S (2016). CENPT bridges adjacent CENPA nucleosomes on young human alpha-satellite dimers. Genome Res 26, 1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasseur P, Tonazzini S, Ziane R, Camasses A, Rando OJ, and Radman-Livaja M (2016). Dynamics of Nucleosome Positioning Maturation following Genomic Replication. Cell Rep 16, 2651–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber CM, and Henikoff S (2014). Histone variants: dynamic punctuation in transcription. Genes & development 28, 672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner A, Hsieh TH, Appleboim A, Chen HV, Rahat A, Amit I, Rando OJ, and Friedman N (2015). High-resolution chromatin dynamics during a yeast stress response. Molecular cell 58, 371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner A, Hughes A, Yassour M, Rando OJ, and Friedman N (2010). High-resolution nucleosome mapping reveals transcription-dependent promoter packaging. Genome Res 20, 90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner A, Lara-Astiaso D, Krupalnik V, Gafni O, David E, Winter DR, Hanna JH, and Amit I (2016). Co-ChIP enables genome-wide mapping of histone mark co-occurrence at single-molecule resolution. Nat Biotechnol 34, 953–961. [DOI] [PubMed] [Google Scholar]
- Xi Y, Yao J, Chen R, Li W, and He X (2011). Nucleosome fragility reveals novel functional states of chromatin and poises genes for activation. Genome Res 21, 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yague-Sanz C, Vazquez E, Sanchez M, Antequera F, and Hermand D (2017). A conserved role of the RSC chromatin remodeler in the establishment of nucleosome-depleted regions. Curr Genet 63, 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Chen H, and Bai L (2018). Systematic Study of Nucleosome-Displacing Factors in Budding Yeast. Molecular cell 71, 294–305 e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Vinayachandran V, Batta K, Koerber RT, and Pugh BF (2012). Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell 149, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Tsukiyama T, and Henikoff S (2013). ISWI and CHD chromatin remodelers bind promoters but act in gene bodies. PLoS genetics 9, e1003317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data from this study have been deposited in GEO at the National Center for Biotechnology under the accession number GSE116853. The datasets deposited correspond to two replicates of each experiment.