

Abstract
Heterozygous loss-of-function mutations in SHANK2 are associated with autism spectrum disorder (ASD). We generated cortical neurons from induced pluripotent stem cells (iPSC) derived from neurotypic and ASD-affected donors. We developed Sparse coculture for Connectivity (SparCon) assays where SHANK2 and control neurons were differentially labeled and sparsely seeded together on a lawn of unlabeled control neurons. We observed increases in dendrite length, dendrite complexity, synapse number, and frequency of spontaneous excitatory postsynaptic currents. These findings were phenocopied in gene-edited homozygous SHANK2 knockout cells and rescued by gene correction of an ASD SHANK2 mutation. Dendrite length increases were exacerbated by IGF1, TG003, or BDNF, and suppressed by DHPG treatment. The transcriptome in isogenic SHANK2 neurons was perturbed in synapse, plasticity, and neuronal morphogenesis gene sets and ASD gene modules, and activity-dependent dendrite extension was impaired. Our findings provide evidence for hyperconnectivity and altered transcriptome in SHANK2 neurons derived from ASD subjects.
iPSC models of severe syndromic forms of ASD and engineered human embryonic stem cell (hESC) knockouts of ASD candidate genes provide evidence for reduced synaptic function of cortical neurons in vitro1-5. Despite these initial findings, extensive heterogeneity among neurons generated from different iPSC lines has come to be a widely acknowledged issue in the field6,7. This experimental variability hinders accurate modeling of altered connectivity of affected neurons relative to controls and may prevent discovery of all but the most extreme phenotypes. To overcome variability caused by neuronal density, Shcheglovitov and colleagues utilized equal co-culture of differentially labelled control and Phelan-McDermid Syndrome iPSC-derived neurons on a bed of rat cortical astrocytes6. Here, we adapted this approach to define changes in neuron morphology and electrophysiology by sparse seeding labelled control and mutant neurons on a lawn of unlabeled control neurons and mouse astrocytes. We applied these SparCon assays to iPSC-derived neurons from nonsyndromic ASD cases with mutations in the scaffolding protein SHANK2 (SH3- and multiple ankyrin repeats protein 2).
Generation of iPSCs from SHANK2 ASD families and by isogenic SHANK2 knockout or rescue
We generated iPSCs from two families with ASD-affected males with SHANK2 haploinsufficiency (Fig. 1a). One subject harbours a nonsense mutation (R841X) and the other a 66 kb deletion (DEL) that causes a frameshift and premature stop8. Whole genome sequencing revealed no additional variants of potential clinical significance beyond those already reported in the literature for SHANK2 DEL (CHRNA7 duplication and ARHGAP11B deletion)9. Multiple lines were isolated from both subjects, and control lines were generated from 3 unaffected parents and one unrelated individual (Fig. 1a and Supplementary Fig. 1). We generated an isogenic CRISPR/Cas9n-edited homozygous knockout (KO) from the unrelated CTRL1 (previously reported as 19-2) using a selection-free strategy based on sequential enrichment of targeted cells (Supplementary Fig. 2)10. Finally we CRISPR/Cas9n-edited R841X followed by Cre-excision of a floxed neomycin-resistance gene to isolate an isogenic corrected line (R841X-C). (Supplementary Fig 3). Whole genome sequencing confirmed correct targeting (Supplementary Table 1) and did not reveal any non-allelic alterations in close proximity to possible gRNA binding sites predicted by in silico means10. More distant off-target effects in the rest of the genome were not excluded by this analysis. STR profiling confirmed R841X-C was derived from R841X (Supplementary Table 2).
Fig. 1. Sparse co-culture for connectivity (SparCon) assays of iPSC-derived SHANK2 ASD neurons compare marked mutant and control neurons seeded on the consistent synaptogenic environment of a lawn of unlabeled control or mutant neurons.

(A) Induced pluripotent stem cells (iPSC) generated from multiple control and affected individuals are differentiated into neural precursor cells (NPCs). NPCs are differentiated in separate wells for 4 weeks and then differentially fluorescently labeled control and mutant cells are sparsely seeded onto a large unlabeled neuronal population (the lawn) and co-cultured with astrocytes. (B) Timeline of the experiment starting with seeding of NPCs. Measurements of mutant cells are normalized to control cells in the same well. (C) Sparse seeding allows simultaneous analyses of cell morphology and connectivity (total number of SYN1 puncta) of single neurons. Scale bars 100 μm. (D) To compare cell morphology, paired representative traces of control and SHANK2 ASD or engineered SHANK2 knockout neurons grown in the same well. (E) To compare synaptic function, sEPSCs are recorded from neurons grown in the same well. Confocal images and traces shown in (C) and (D) are representative of iPSC-derived neurons imaged in experiments depicted in Figure 2 (A-C). sEPSC traces shown in (E) are representative of patch-clamp recordings of iPSC-derived neurons described in Figure 3.
We used an embryoid-body based differentiation protocol to generate SOX1+ neural precursor cells (NPCs) that were expanded and frozen for use in multiple experiments11 (Supplementary Fig. 4a,b). The NPCs gave rise to neurons of mixed cortical layers (Supplementary Fig. 4c). SHANK2 protein level at 4 weeks was halved in neurons from R841X and DEL and absent in KO cells (Supplementary Fig. 5a) but restored in R841X-C (Supplementary Fig 3d). SHANK2 protein plateaued after 4 weeks of differentiation, demonstrating expression before and during the onset of synaptogenesis in vitro12,13 and developmental regulation in human neurons (Supplementary Fig. 5b). Thus the differentiation protocol produces SHANK2-expressing control and mutant cortical neurons suitable for connectivity studies.
Establishment of SparCon assays on SHANK2 and control neurons
We observed that directed differentiation produced neurons with variable cell densities in monocultures (Supplementary Fig. 6), and reasoned that the final cell type composition or maturity of monocultures may also differ over the prolonged experimental timecourse necessary for synaptic development in human neurons. The accompanying differences in neuronal synaptogenesis, which is modulated by extracellular cues, may increase experimental noise and obscure biological differences in connectivity between control and mutant neurons. In principle, maximal consistency would result if a controlled synaptogenic environment was used in every experiment. This is the foundation of the SparCon assay, in which a small number of differentially labeled pairs of SHANK2 mutant and control neurons are sparsely seeded together onto a lawn of unlabeled control neurons.
To set up SparCon assays (Fig. 1a), we transduced 3 week-old differentiating control and SHANK2 mutant neurons with lentiviruses expressing either red (mKO2 or mKate2), or green (GFP) fluorescent proteins driven by the CaMKIIα promoter, which further enriches for SHANK2-expressing neurons4 and has been shown by single cell RNASeq to mark well-differentiated excitatory glutamatergic neurons differentiated in vitro from iPSC14. Following a 1 week recovery, differentially labeled control and SHANK2 mutant neurons were sparsely seeded (at 2% of total neurons each) onto an unlabeled control lawn (96% of total neurons) on a layer of mouse astrocytes to promote synaptogenesis over 5 weeks (Fig. 1b). We established 14 experimental batches of neurons with multiple (3-12) cover slips from each batch of differentiated neurons. A single batch was used to test each SHANK2 genotype. Neurons from 3 lines each of R841X and DEL were cocultured with 2-5 control lines (including at least one parental control) in pairs using an experimental design in which the reporter used to mark each genotype was blinded. The KO was compared to its isogenic CTRL1 and one unrelated control line, and R841X was compared to the isogenic corrected R841X-C (Supplementary Tables 3-6). In addition to being cultured on the wild-type control lawns, the R841X isogenic pair was also evaluated on unlabeled R841X mutant lawns to control for cell non-autonomous effects of the lawns. After a total of 9 weeks of differentiation, marked neurons expressed cortical and excitatory markers4 (Supplementary Fig. 7a). These neurons were electrophysiologically active using physiological salt solutions in intracellular and extracellular environments and capable of generating action potentials (Supplementary Fig. 7b). SHANK2 mutant neurons did not differ from controls in intrinsic membrane properties (Supplementary Fig. 7c). Detailed SparCon methods, controls and validation of normalization of SHANK2 mutants to control cells in the same well are described (Online Methods and Supplementary Figs 8-11).
Because sparse seeding allows visualization of entire marked neurons by immunostaining, we directly measured connectivity as total synapse number per neuron by counting puncta of Synapsin-1 (SYN1), a universal marker of mammalian cortical synapses15 previously used to estimate synapse density in iPSC-derived neurons3,5, along fluorescently labeled neuronal dendrites (Fig. 1c). This was accompanied by simultaneous acquisition of morphological data (dendrite length and complexity) from the same cells (Fig. 1d) and spontaneous excitatory postysynaptic current (sEPSC) recordings (Fig. 1e), providing a heretofore unrealized level of detail on connectivity of human stem cell-derived neurons.
Increased synapse number and dendrite length in SHANK2 mutant human neurons
SYN1 puncta localized on the labeled neurons revealed a significant increase in total synapse number in all SHANK2 mutants after SparCon normalization (Fig. 2a), showing that lowered SHANK2 dosage causes an increase in synaptic connections. Using the same neurons, we also found increases in normalized dendrite length in all SHANK2 mutants (Fig. 2b). Consistent with a previous report on SHANK2 knockdown in rat neurons in vitro16, we found that normalized dendrite branching complexity, measured by Sholl analysis, was increased in all SHANK2 mutants (Fig. 2c). When R841X was compared to R841X-C seeded on an unlabeled R841X neuron lawn thereby mimicking the in vivo situation, similar-fold effects on synapse number, dendrite length and branching complexity were seen (Fig 2a-c), demonstrating that these effects were cell-autonomous and not due to the presence of control neurons in the lawn. The statistical summary of normalized and raw data comparisons is shown in Supplementary Table 7 and Supplementary Fig. 12a,b. Taken together, these findings indicate that lowered SHANK2 dosage increases the total synapse number resulting in neuronal hyperconnectivity, which is partly driven by increases in dendrite length and complexity.
Fig. 2. Synapse numbers, dendrite length and neuron complexity are enhanced in SHANK2 mutant neurons and the length phenotype is exacerbated by treatment with IGF-1 and BDNF.

(A) Total synapse number, normalized within-well by dividing synapse number of individual cells by the geometric mean of the synapse number of control cells in a given well. (B) Total dendrite length, normalized within-well by dividing dendrite length of individual cells by the geometric mean of dendrite length of control cells in a given well. (C) Sholl analysis, normalized within-well by subtracting the mean of control crossings from the number of crossings made by individual cells at a given radius. Radius step 10 μm. (D) IGF-1, BDNF and TG003 treatments increase dendrite length in R841X compared to isogenic R841X-C control cells. For (A-C) SHANK2 mutant cells are compared to co-cultured control cells from the same wells: SHANK2 KO (n=40) vs. CTRL (CTRL1, isogenic, n=46), SHANK2 R841X (n=89) vs. CTRL (CTRL1, CTRL2 (paternal), CTRL3 (maternal), n=96), SHANK2 DEL (n=74) vs. CTRL (CTRL1, CTRL2, CTRL3, CTRL4 (paternal), n=68). For control vs mutant lawn experiments in (A-C) SHANK2 R841X cells (n=22 for control lawn, n=40 for mutant lawn) are compared to isogenic R841X-C cells (n=27 for control lawn, n=33 for mutant lawn) in the same wells. Means + s.e.m. plotted. Total number of neurons = 535 from 5 WT lines from 4 CTRL individuals, 6 SHANK2 +/− lines from 2 ASD individuals,1 engineered SHANK2 −/− KO line, and 1 engineered isogenic SHANK2 correction line (R841X-C). For (D) SHANK2 R841X neurons (total n=595) were compared to isogenic R841X-C neurons (total n=651) in all treatment conditions. Total number of neurons = 1186. See Supplementary Tables 7 and 8 for details of all treatment comparisons and relevant statistics. Means +/− s.e.m. plotted,* P <0.05, ** P <0.01, *** P<0.005; Anderson-Darling k-samples test for (A) (B), and (D) and two-way ANOVA for (C).
To test pathways known to impact dendrite growth or rescue function in SHANK3-mutant human neurons4,17, we applied various compounds from weeks 5-9 in SparCon assays (batches 12-14; coculture details and statistical summary in Supplementary Tables 6 and 8). Specifically, we measured dendrite lengths of R841X and R841X-C after treatment with DMSO, IGF1, the mTOR inhibitor rapamycin, the CLK-2 inhibitor TG003, BDNF, and the MNK inhibitor eFT508. These results confirmed that DMSO-treated R841X dendrites were longer than isogenic controls. When treated R841X neurons were compared to the DMSO R841X neurons, dendrites were significantly longer with IGF1, TG003 and BDNF (Fig 2d) but unchanged with rapamycin and eFT508 (Supplementary Fig. 13). These findings demonstrate that increased dendrite length is caused by heterozygous SHANK2 mutation, and that these cells remain responsive to IGF1, TG003, and BDNF, which exacerbate the phenotype.
Increased sEPSC frequency in SHANK2 mutant human neurons
To test whether SHANK2 regulates excitatory synaptic function, we performed patch-clamp recordings of AMPA receptor-mediated sEPSCs (Fig. 3a and Supplementary Tables 4 and 6). There were small but statistically significant differences in normalized sEPSC amplitude in R841X and KO, but not DEL (Fig. 3b). R841X compared to R841X-C exhibited an increase in sEPSC amplitude on both the control lawn and mutant (R841X) lawn, providing further evidence that SHANK2 regulates the strength of synaptic connections in the R841X background under basal conditions (Fig. 3b). The statistical summary of normalized and raw data comparisons is shown in Supplementary Table 9 and Supplementary Fig 12c,d. In addition, when we increased sEPSC frequency with BDNF18, no significant difference was observed in R841X sEPSC amplitude (Supplementary Fig. 14a).
Fig. 3. Excitatory synaptic function is enhanced in SHANK2 mutant neurons on control and mutant lawns.

(A) Representative sEPSC traces from experiments described in (B) and (C) with an averaged sEPSC trace (inset) from single control, ASD, and engineered SHANK2 KO neurons. (B) sEPSC amplitude and (C) sEPSC frequency normalized within-well by dividing measurements of individual cells by the geometric mean of control cells in the same well. SHANK2 mutants are compared to co-cultured control neurons from the same wells: SHANK2 R841X (n=60, 20 each from line #2, #5 and #13) vs. CTRL (n=60, 30 each from line #2 of CTRL1 and line #39 of maternal CTRL3), SHANK2 DEL (n=60, 20 each from line #1, #2 and #4) vs. CTRL (n=60, 30 each from line #3 and #5 of paternal CTRL4), SHANK2 KO (n=60 from line #H6) vs. CTRL (n=60, 30 from line #2 of isogenic CTRL1 and 30 from line #4 of CTRL2). SHANK2 R841X +/− (n=50) and R841X-C +/+ (n=50) neurons on control and mutant lawns. Total number of neurons = 560 from 5 WT lines from 4 CTRL individuals, 6 SHANK2 +/− lines from 2 ASD individuals and 1 engineered SHANK2 −/− KO line. Mean +/− s.e.m. plotted, * P <0.05, ** P <0.01, *** P<0.005.; Anderson-Darling k-samples test. A detailed statistical summary including exact P-values can be found in Supplementary Table 9.
We found a striking increase in sEPSC frequency across all SHANK2 genotypes under basal conditions (Fig. 3c) and with BDNF treatment (Supplementary Fig. 14b). Moreover, R841X compared to R841X-C exhibited increased sEPSC frequency when grown on both control and mutant lawns (Fig. 3c). Finally, as deficiency of SHANK3 was previously shown to cause dysfunction of hyperpolarization-activated cyclic nucleotide-gated channel proteins (HCN proteins) forming Ih-channels3, we measured Ih-currents in SHANK2 mutant neurons. Acute application of the Ih-channel blocker ZD7288 resulted in no significant change between genotypes in the firing frequency of action potentials evoked by injection of current steps (Supplementary Table 6 and Supplementary Fig. 15). These data demonstrate that heterozygous loss-of-function in SHANK2 is solely responsible for the hyperconnectivity phenotype observed in neurons derived from subjects with ASD and that SHANK2 does not disturb Ih-channels. Given that a common population of neurons in the lawn in SparCon assays drives the input to both control and mutant neurons, increased sEPSC frequency is consistent with the increase we observed in synapse number in the neurons lacking SHANK2. Taken together, our data provide strong evidence that reduced dosage of SHANK2 increases synaptic connectivity.
Perturbed neurodevelopment gene sets and activity-dependent dendrite extension
To investigate molecular perturbations that occur in SHANK2 neurons, we performed RNASeq at timepoints relevant for the SparCon assay. R841X and R841X-C neuron monocultures at the 4 week timepoint and after 5 more weeks maturing on mouse astrocytes were subjected to magnetic activated cell sorting (MACS) to deplete contaminating human NPCs and astrocytes19. Duplicate RNA samples from each of two independent differentiations were sequenced. At 4 weeks, we found similar levels of cortical layers and neuronal subtype markers (Fig 4a, Supplementary Tables 10 and 11), suggesting that cortical cell identity was largely similar when SparCon cocultures are established. After the subsequent 5 weeks of maturation, increases in several cell identity markers and synapse genes were detected in 9 week R841X neurons. At 4 weeks 3029 genes were significantly altered, and 2809 genes were perturbed at 9 weeks (Fig 4b, Supplementary Table 11). However, only 724 genes were differentially expressed in common at these two timepoints, suggesting that SHANK2 mutant neurons diverge from controls during development. Consistent with this idea, continuous gene set enrichment analysis20 at 4 weeks showed that R841X cultures are associated with gene sets related to cell cycle, DNA replication, mitochondrial function, transcriptional and translation regulation, whereas R841X-C cultures are associated with greater expression in ion channel and synapse function genes (Fig 4b and Supplementary Table 12). At 9 weeks, following 5 weeks of culture with mouse astrocytes, there is a significant upregulation of gene sets associated with neuronal differentiation, neurite development, and synapse function in R841X neurons. Upregulation of gene sets associated with translation and mitochondrial function persists at 9 weeks in R841X neurons, whereas RNA processing changes are equivocal (Fig 4b). In contrast to changes at 4 weeks, there is a relative suppression of cell cycle-related genes in 9 week-old R841X neurons. The latter gene sets would be predicted to change neuronal density in monocultures contributing to the variability we previously observed (Supplementary Fig 6).
Fig. 4. Deeply perturbed transcriptome and defective activity-dependent dendrite extension in R841X neurons.
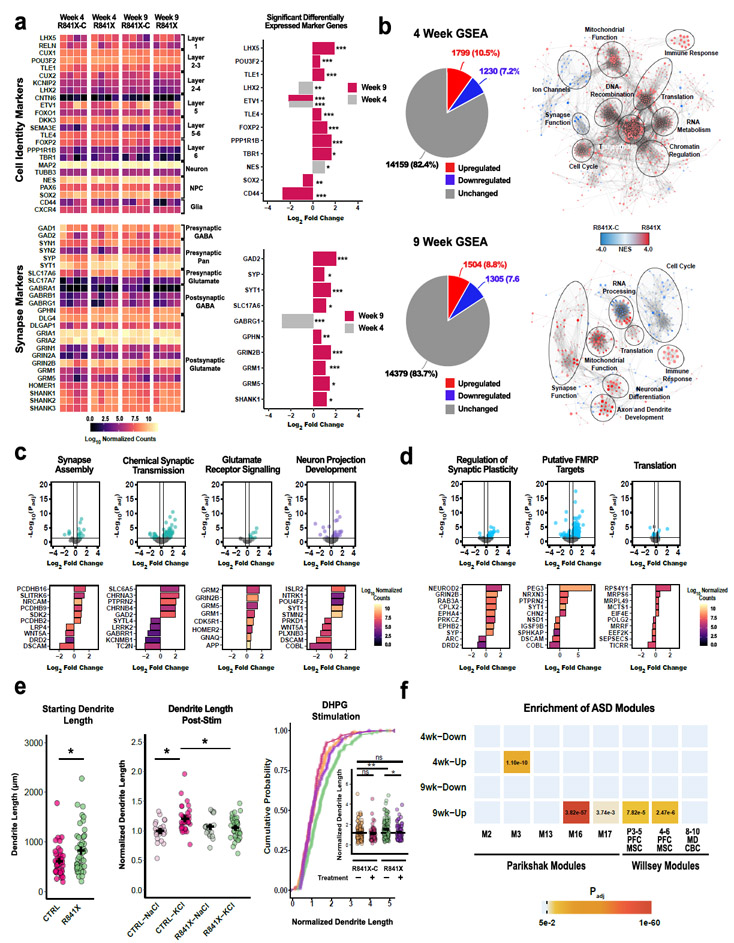
(A) Heat maps of cortical identity and synapse gene transcript levels and significant fold changes found by RNASeq of R841X and isogenic R841X-C controls. See Supplementary Table 10 for all marker gene expression fold change values and relevant statistics. (B) Number of differentially expressed genes in pie charts and enrichment of neurodevelopment, translation and cell cycle gene sets in R841X neurons. (C) Volcano plots and examples of differentially expressed genes in gene sets related to synapse function and neuron projection development. (D) Volcano plots and examples of differentially expressed genes in gene sets related to synaptic plasticity and translation. Highlighted points have absolute log2 Fold Change > 1.25 and Padj < 0.05. (E) Starting dendrite length is greater in R841X neurons, and impaired acute KCl activity-dependent dendrite extension (SHANK2 R841X n=47 from lines #2, 5, #13 vs. CTRL (n=44 from CTRL1 line #2 and CTRL3 #39), and rescue of R841X dendrite length by chronic DHPG treatment (R841X-C DMSO n=141, R841X-C + DHPG n=65, R841X DMSO n=120, R841X + DHPG n = 76). Mean +/− s.e.m. plotted, * P < 0.05, Anderson-Darling k-samples test. (F) Enrichment of M16 and M17 ASD gene modules in 9 week and the M3 ASD module in 4 week R841X neurons. For (A-D) and (F), n = 4 independent differentiations for both genotypes at each timepoint. * Padj < 0.05, ** Padj < 0.01, *** Padj < 0.005, Wald test with Benjamini-Hochberg correction for multiple testing.
Given that GRIN2B, GRM1 and GRM5 transcripts were elevated at 9 weeks in R841X (Fig 4a), we explored potential changes in genes and functional processes that are regulated by neuronal activity, development and synaptic plasticity. We observed no enrichment among genes previously shown to be activated in response to activity in iPSC-derived neurons21 (Supplementary Fig 16a). However, synapse assembly and chemical synaptic transmission gene sets were significantly enriched among upregulated DEGs in R841X neurons (Fig 4c). Consistent with this, the upregulation of glutamate receptor signaling genes in R841X cells suggested that hyperconnectivity may be in part supported by higher expression of glutamate receptors. Similarly, upregulation of genes involved in neuronal projection development in R841X neurons parallels our observation of increased dendrite length and complexity (Fig 4c). As a possible functional consequence of these alterations, there was a striking upregulation of genes associated with synaptic plasticity (Fig 4d). Consistent with this idea, a high proportion of Fragile-X mental retardation protein (FMRP) targets22 were increased in R841X neurons, although we found no obvious enrichment of translation-associated genes (Fig 4d).
We therefore tested how R841X neurons respond to activity. Live imaging experiments assaying KCl-induced dendrite extension revealed that GFP-marked dendrites were already longer at baseline in R841X compared to control neurons at week 6, but only control neurons extended further at 500 mins after activation (Fig 4e and Supplementary Fig 17). We also performed SparCon experiments testing chronic DHPG activation of mGluR, which regulates FMRP-dependent translation, from week 5-9 at a concentration that had no effect on R841X-C neurons. Dendrite length in R841X treated neurons was significantly reduced compared to R841X DMSO controls and equivalent to R841X-C (Fig 4e). These findings show that acute activity-dependent dendrite extension was compromised in R841X neurons in vitro. However, chronic DHPG exposure normalized dendrite length, demonstrating that the dendrite phenotype can be suppressed in R841X neurons during this developmental period, and may be related to increased expression of FMRP targets.
We next examined whether gene expression changes in R841X neurons could be related to ASD generally. When modules of coregulated genes during neurodevelopment defined by Parikshak et al.23 and Willsey et al.24 were examined in our RNASeq data (Fig 4f and Supplementary Fig 16b,c), we observed that several previously-described ASD associated modules associated with mid-fetal development M16, M1723 and localized to the prefrontal and primary motor-somatosensory cortex24 were significantly enriched among upregulated DEGs at 9 weeks. ASD module M3, which is associated with early fetal development, was enriched among upregulated DEGs at 4 weeks. Overall, these findings demonstrate that transcription is deeply perturbed in SHANK2 neurons and these changes are enriched in specific gene sets related to neurodevelopment and ASD.
DISCUSSION
Our findings show that human neurons with ASD-associated haploinsufficeint SHANK2 mutations make more functional excitatory connections relative to controls, and this phenotype is distinct from the lowered connectivity associated with mutations in SHANK3 or other ASD-associated genes2-4. The presence of the same hyperconnectivity phenotype in homozygous KO neurons and its reversion in gene corrected R841X-C neurons demonstrates that SHANK2 mutations cause this phenotype. The experiments we report simultaneously evaluated the total synapse number, dendrite length and complexity of SHANK2 neurons and demonstrated increases in these measures. We found small increases in sEPSC amplitude particularly in the 3 R841X lines compared to family and unrelated controls, and in the isogenic pair evaluations of R841X and KO neurons, but not in the 3 DEL lines. Large increases were observed on sEPSC frequency in all genotypes demonstrating that this measure of connectivity was most prominently and reproducibly affected. IGF1, TG003, and BDNF treatment exacerbated the dendrite length in both mutant R841X and corrected R841X-C neurons, suggesting that these modes of regulating global translation are not contributing to the altered morphology. Our results provide further evidence that SHANK2 functions in early neuronal development25 and serves an important role as a suppressor of dendrite branching26.
SparCon normalization overcame the challenge presented by variable cell densities in monocultured SHANK2 and control neurons that result from genetic background or batch effects7,27. We showed cell autonomous effects by assessing morphology and electrophysiology of R841X on control and mutant lawns. In future experiments, lawns could be engineered to contain specific excitatory subtypes by Ngn2 over-expression28 or to express optically- or chemically-gated modulators of activity that expand the range of possible testable phenotypes. Moreover, SparCon can be adapted to use other cell-type specific promoters such as hvGAT to selectively label inhibitory neurons29 or GDAP1L130 to restrict analyses to neurons of similar age and electrophysiological properties. As new approaches are developed to specify stem cell-derived neurons or mature them to later stages, our isogenic SHANK2 and control iPSC lines will be a valuable resource for examining the dynamics of connectivity and circuitry of human neurons during development and disease.
The phenotype of SHANK2 human neurons contrasts with variable findings reported for Shank2 and Shank3 mouse neurons. Two different Shank2 null mice with no evidence of Shank2 protein expression exhibited either no change or a decrease in miniature EPSC (mEPSC) frequency in the hippocampus, a site of relatively high Shank2 protein level31,32. One of those mice, Shank2 ex7−/− which is most similar to our engineered SHANK2 KO line, had enhanced LTP31 and decreased synapse maturation33. Consistent with this, RNA-Seq data in our cortical cultures showed a relative upregulation of GRIN2B in R841X neurons, a feature of immature synapses. Similarly, knockdowns of Shank2 in vitro in rodent neurons have typically led to 20-50% decreases in synapse density with a concomitant increase in dendrite complexity16,34. Only the shared increase in dendrite complexity was found in our human SHANK2 mutant neurons, possibly due to a species-specific difference in SHANK2 function, difference in the cell types assessed35, or in developmental timepoints at which the phenotypes were assayed. Concordant with the latter idea, AMPAR mEPSC hyperconnectivity was found in Shank3b null mice shortly after birth that disappeared at postnatal day 6036, suggesting the existence of a critical developmental window of hyperconnectivity to be found after SHANK mutation. Furthermore, the relatively high prevalence of nonsynonymous SHANK2 mutations in ASD, their association with other disorders like schizophrenia, as well the variable phenotypes reported for different Shank2 mouse mutants suggests a broader repertoire of possible functional alterations in SHANK2 that contribute to neuropsychiatric disorders37. In this context, our results contribute to defining the neuronal phenotype associated with reduced SHANK2 dosage, an expected consequence of deletions and non-sense mutations that show a near-complete penetrance of ASD in humans9,38,39.
SHANK2 is a post-synaptic density scaffolding protein that anchors a protein complex connecting NMDAR, AMPAR and mGluR receptors40, which participate in pathways that regulate activity-dependent transcription and translation41. Concordant with this, our RNA-Seq results show consistent increases in NMDA receptor subunit and mGluR transcripts, as well as enrichment of gene sets important for synapse plasticity and targets of FMRP, a well-known regulator of local activity-dependent translation at dendrites and synapses. Curiously, Fmr1 knockout mice demonstrate early hyperconnectivity in the medial prefrontal cortex42, similar to our findings in SHANK2 mutant human neurons. Impaired acute activity-dependent dendrite extension coupled with increased basal dendrite length in mutant neurons suggests that neurotypic levels of SHANK2 suppress excess dendritogenesis during development and enable acute activity-dependent extension. In contrast, at haploinsufficient SHANK2 levels, dendrite extension is no longer suppressed and acute activity-dependent growth regulation is lost. Further highlighting the critical role of activity-dependent translation in this phenotype, chronic agonism of mGluRs with DHPG in SparCon co-cultures differentially suppressed dendrite length in R841X neurons relative to controls. Here, return of dendrite length to wild-type levels suggests that mGluR activation of SHANK2 mutant neurons can rescue the dendrite phenotype. These findings, along with enrichment of gene sets regulating dendrite morphology in RNASeq, support the idea that SHANK2 haploinsufficiency disrupts the intricate interplay between synapse formation and dendritogenesis critical to appropriate establishment of neuronal connectivity43. It is possible that DHPG may act by enhancing synapse elimination44, which may concomitantly destabilize nascent neurites during development and result in shorter dendrite length.
Enrichment of ASD-associated modules among upregulated DEGs at 4 and 9 weeks suggests that the phenotypes we report here may be a general feature of ASD. The M3 module harbors cell cycle and chromatin/transcriptional regulators expressed in early fetal development and is upregulated relatively early at 4 weeks in SHANK2 mutant neurons. In contrast, upregulated DEGs at 9 weeks are enriched in modules M16 and M17 containing synaptic proteins and to the prefrontal and primary motor-somatosensory cortex modules during mid-fetal development. The abundance of risk genes for idiopathic ASD in these modules suggests that lowered SHANK2 dosage triggers an ASD-specific transcriptional program. Our findings contrast with the downregulation of these modules in human NPCs with knockdown of CHD845, another high-risk ASD gene, implying the existence of multiple distinct etiologies that manifest as ASD. Curiously, our finding of hyperconnectivity reflects observations of increased synapse density in postmortem ASD brain samples. Previously, this increase was linked to impaired mTOR-dependent autophagy, leading to a deficit in pruning and hyperconnectivity in teenage years46. Here, the alterations in SHANK2 neurons lead to a distinct, early hyperconnected phenotype. Unsuppressed elaboration of the dendritic tree in developing SHANK2 mutant neurons may increase the probability of establishing synaptic connections, leading to functional hyperconnectivity measured as strikingly increased sEPSC frequencies. It is possible that this mechanism applies broadly to ASD cases given the recent observation of increased abundance of SHANK2-AS long non-coding RNA and concurrently lowered SHANK2 protein levels in ASD samples without verified SHANK2 mutations47. As further ASD associated genes are shown to cause hyperconnectivity48, it will be important to determine how signalling pathways and common ASD enriched gene modules suppress or activate dendritic and synaptic regulation in excitatory neurons at both the transcriptional and translational levels. It will be crucial to assess the developmental specificity and reversibility of these perturbations to pinpoint druggable candidates that have therapeutic potential for neurodevelopmental disorders.
Methods
Methods in online version of the paper
Accession Codes
The RNA-Seq dataset can be accessed at GEO (GSE122550). The whole genome sequence dataset used for off-target analysis can be accessed at EGA (EGAS00001003436).
Online Methods
Generation of iPSCs
All pluripotent stem cell work was approved by the Canadian Institutes of Health Research Stem Cell Oversight Committee. With approval from the SickKids Research Ethics Board, we obtained dermal fibroblasts from skin-punch biopsies at the Hospital for Sick Children, after informed consent was obtained from all subjects in compliance with all relevant ethical regulations. Induced pluripotent stem cells were generated by either retroviral reprogramming (CTRL1, CTRL3, CTRL4, SHANK2 R841X) as described49 or Sendai virus reprogramming (CTRL2, SHANK2 DEL) using the Invitrogen CytoTune-iPS kit. During reprogramming and clonal expansion, iPSC colonies were grown on gelatin-coated plates on a layer of Mitomycin-C inactivated mouse embryonic fibroblasts extracted at embryonic day 12.5 (acquired from Hospital for Sick Children Embryonic Stem Cell Facility) in hESC media (Knockout DMEM supplemented with 15% Knockout serum replacement, 1x GlutaMAX, 1x penicillin/streptomycin, 1x non-essential amino acids, 0.5 mM 2-mercaptoethanol, and 10 ng / mL basic fibroblast growth factor (bFGF)). At this stage, the cells were passaged manually or with collagenase IV as described. Established iPSC lines were transitioned to plates coated with matrigel and grown in either mTeSR or StemMACS iPS Brew medium. From this point on, the cells were passaged either manually, using Dispase or ReleSR every 5-7 days.
To verify genome integrity, G-band karyotyping was performed by the Centre for Applied Genomics (Hospital for Sick Children) and only iPSC lines without karyotypic abnormalities were used for analyses. To test expression of pluripotency associated factors OCT4, NANOG, TRA1-60, and SSEA4, immunocytochemistry was performed. To test whether cells were functionally pluripotent in vitro, iPSCs underwent spontaneous differentiation through an embryoid-body based step as described50, fixed, and stained for expression of endoderm (α-fetoprotein, AFP), mesoderm (smooth muscle actin, SMA), and ectoderm (β3-tubulin, TUBB3). Functional pluripotency in vivo was tested using teratoma assays. IPSC lines were grown in 10 cm matrigel-coated dishes, digested with collagenase IV and injected into the flank of 6 week old NOD/SCID female mice as cell clumps immersed as described50. Two months later or when obvious tumors appeared, the mice were sacrificed and the tumors were resected, stained with hematoxylin and eosin, and imaged. All animal work was approved by The Hospital for Sick Children Animal Care Committee and complies with the guidelines established by The Canadian Council of Animal Care.
Generation of SHANK2 knockout cells.
Two million iPSCs were nucleofected using the Amaxa Nucleofector II with program B-016 in Solution I with 5 μg CRISPR D10A nickase plasmid (Addgene, #44720), 2.5 μg each of two gRNA cloning plasmids (Addgene, #41824) containing paired gRNAs 1 and 2 targeting exon 16 of SHANK2, and 1 μl of 10 μM ssODN containing two 60 nucleotide-long arms of homology and SHANK2 KO sequence comprised of V5 tag, Mre1 restriction site and stop codons in all three reading frames. Following nucleofection, the cells were distributed in a 96-well plate. Following 2 weeks of growth, 1/3 of the cells were passed to a new 96-well plate and maintained, 1/3 were passed to a new 96-well plate and frozen in 90% KOSR - 10% DMSO mix with 10 µM ROCK inhibitor, and 1/3 had gDNA extracted as in Miyaoka et al.51 Digital droplet PCR was used to determine the proportion of SHANK2 tag alleles in each well with a Fam-conjugated probe for the SHANK2 WT sequence and Vic-conjugated probe for the SHANK2 KO (Vic-tag) sequence. The well with the highest proportion was expanded into a new 96-well- plate and the procedure was repeated until wells with 100% SHANK2 tag allele and 0% SHANK2 WT were found. The isolated wells were expanded and the resulting iPSC lines were characterized to assay pluripotency (in vitro via ICC for pluripotency markers, in vivo via teratoma assays) and karyotyping. Off-target effects were assessed using whole-genome sequencing performed by The Centre for Applied Genomics and analyzed as described below.
Gene correction in SHANK2 R841X IPSCs
The pPNT plasmid backbone with a floxed neomycin positive selection cassette and a HSV-TK negative selection cassette was used to generate the targeting plasmid for correction. First, genomic DNA from CTRL2 #4 (R841X paternal control) IPSCs was used to amplify a blunt-ended 2316 bp PCR fragment from the affected locus, which was subsequently subcloned into the pCR-Blunt II-TOPO vector using the Zero Blunt PCR Cloning kit (ThermoFisher). Bacterial clones bearing the plasmid with the wild-type sequence were isolated and their integrity verified using Sanger sequencing.
Following this, a Gibson cloning strategy was used to generate the targeting vector. First, the pPNT plasmid was digested with EcoRI and BamHI enzymes to cut out the floxed neomycin resistance cassette. Both the cassette and the digested plasmid were gel-purified. Then, two homology arms were amplified from the blunt-end cloned template fragment with overhangs complementary to the digested pPNT plasmid and to each other containing EcoRI and BamHI restriction enzyme sites in the intronic region for subcloning the floxed neo cassette. The amplified homology arms were combined with the digested pPNT plasmid in a Gibson cloning reaction to generate a plasmid with the negative selection marker HSV-TK and two homology arms separated by a region containing EcoRI and BamHI restriction enzyme sites. The floxed neomycin resistance cassette was subsequently cloned into that region, generating the final targeting plasmid.
To preserve the plasmid during CRISPR/Cas9n-mediated homology-directed repair, target specifically the mutant sequence, and increase recombination efficiency in IPSCs, we generated two pairs of gRNAs for the Cas9n nickase. The first pair targeted the junction between two homology arms such that the PAM site of one of the gRNAs would be contained on the other homology arm and thus prevent digestion of plasmid. The second pair was designed to be specific to the mutant sequence to spare the wild-type sequence-bearing plasmid and minimize cutting of the wild-type allele.
Six million IPSCs from the SHANK2 R841X line #5 were then nucleofected with 15 μg CRISPR D10A nickase plasmid (Addgene, #44720), 1.25 μg each of the four gRNA plasmids, and 20 μg of the targeting plasmid. Following nucleofection, the cells were plated on 10 cm dish and allowed to recover for 3d, after which the feeding media was supplemented with 25 μg / mL G418 every day. The surviving colonies were manually picked into a 96-well plate. Following expansion in the 96-well plate, 1/3 of the cells were passed to a new 96-well plate and maintained, 1/3 were passed to a new 96-well plate and frozen in 90% KOSR - 10% DMSO mix with 10 µM ROCK inhibitor, and 1/3 had gDNA extracted as in Miyaoka et al.51 Colonies were screened using PCR amplification and XhoI digestion of the targeted region. The mutation causing the premature termination codon in SHANK2 R841X destroys a naturally occurring XhoI restriction site. Because all mutant alleles will fail to digest with XhoI treatment, we picked only those clones whose PCR products were fully digested, signifying the presence of wild-type sequence at both alleles. These clones were then expanded, and 2 million cells were nucleofected with 10 μg Cre-IRES-Puro plasmid and plated in a 10 cm dish. The cells were subjected to 1 μg / mL puromycin on days 2 to 4 to select for transfected cells. The colonies were again picked into a 96-well-plate and expanded. Following expansion in the 96-well plate, 1/3 of the cells were passed to a new 96-well plate and maintained, 1/3 were passed to a new 96-well plate and frozen in 90% KOSR - 10% DMSO mix with 10 µM ROCK inhibitor, and 1/3 were passed to another 96-well plate where they were exposed to 25 μg / mL G418 for 1 week. Failure to survive under these conditions indicated a high likelihood of excision of the floxed neoymicin cassette. The corresponding IPSC colonies on the non-G418 treated plate were expanded, PCR-sequence verified for WT sequence, subjected to short-tandem repeat analysis to rule out contamination with other cell lines (Supplementary Table 2), and karyotyped. Then, western blots with protein lysates from the isolated clones were used to test whether SHANK2 protein levels returned to those seen in control IPSCs and 4wk old neurons.
Characterization of off-target effects resulting from CRISPR/Cas9n editing
Genomic positions with designed gRNA sequences matching the human genome (hg38) were downloaded from the CRISPR Design Tool at the Zhang Lab website (http://crispr.mit.edu/). Because our gRNA cloning protocol sets the first 5’ nucleotide as G which is dispensable for specificity, mismatches at that position were ignored. Likewise, mismatches at the PAM sequence were ignored. Sequences with up to 3 mismatches in the remaining fragments were then selected for analysis. Because hg38 genome may be missing parts of hg19 reference genome that may contain additional gRNA binding sites, the positions of those fragments were remapped to hg19, but did not yield any additional possible binding sites.
Whole genome sequencing was performed on both starting lines and the derived lines at The Centre for Applied Genomics. The reads were mapped using the bwa algorithm (0.7.12)52, duplicate reads were marked with Picard (1.33), and the genome was analyzed with GATK (3.4.6) 53.
To verify that the SHANK2 KO (Vic tag) sequence was inserted at the right location, 100 bp of SHANK2 KO sequence was reconstructed in each direction at each locus predicted by the CRISPR Design Tool. The only insertion was found at the on-target site, and reconstruction revealed it matched the SHANK2 KO (Vic tag) sequence (Supplementary Table 1). The sequence was verified by PCR and Sanger sequencing.
To search for off-target SHANK2 KO (Vic Tag) insertions at sites not predicted by CRISPR Design Tool, GATK (GenotypeConcordance) was used to identify de novo indel and SNP calls in SHANK2 KO genome with respect to the control genome. Because of the length of the SHANK2 KO sequence, it could have appeared as a series of GATK calls in close proximity to one another. Therefore, all calls occurring within a 200bp window were grouped, and GATK FastaAlternateReferenceMaker was used to reconstruct the SHANK2 KO sequence within that window to identify any potential insertion. We only found the SHANK2 KO (Vic Tag) insertion at the expected locus.
To determine if the SNPs/indels unique to the starting line can create additional gRNA binding sites and facilitate off-target mutagenesis, we reconstructed 100 bp of genome sequence of the starting line in both directions at each of the identified unique calls. The gRNA sequences 1 and 2 were then mapped to those sequences using bwa to identify potential gRNA binding sites. Both 1 and 2 gRNAs mapped only to regions containing the exact match of the sequence near the on-target location of SHANK2 exon 16, suggesting an absence of off-target effects caused by CRISPR/Cas9n editing.
To determine if any SNPs/indels may be attributable to CRISPR/Cas9n-editing, we first determined all possible binding positions for gRNAs, tolerating up to 5 mismatches. Next, we reconstructed 100 bp of sequence in each direction from the binding site in the derived lines. No new SNPs/indels relative to the starting lines were discovered.
The same process was repeated to search for off-target effects during CRISPR/Cas9n correction of R841X to R841X-C. In addition, sequence of the targeted locus confirmed the presence of loxP site and correction of the R841X-causing mutation.
Differentiation to Neural Precursor Cells (NPCs)
NPCs were generated using dual SMAD and Wnt inhibition with manual isolation of neuronal rosettes54-56. Briefly, iPSC colonies were digested for with collagenase IV (1 mg / mL) until they lifted off the plate. Subsequently, the colonies were grown as free-floating cellular aggregates for 4 days in EB media (20% knockout serum replacement, DMEM/F12, 1x NEAA, 1x penicillin/streptomycin, 20ng / mL bFGF), after which they were adhered to a matrigel coated plate (1/71) and grown for an additional 4 days in N2B27 media (DMEM/F12, N2 1x, B27 1x, 1x non-essential amino acids, 1x penicillin/streptomycin, 20ng / mL bFGF). For the first 8 days of differentiation, the cells were grown in the presence of SB431542 (10 μM) and dorsomorphin (2.5 μM). The cells were additionally treated with XAV939 (2 μM) during the first 4 or 8 days of differentiation, depending on the cell line, to facilitate more efficient differentiation. After 8 days, neural rosettes were lifted by a brief incubation (3-5 min) with Dispase and tapping, washed with PBS, and adhered to another matrigel-coated plate and fed with N2B27 media. Following that, secondary or tertiary rosettes were manually dissected to purify neural precursor cells (NPCs). NPCs were then expanded and frozen in 10% DMSO-90% N2B27 media. NPCs were characterized by immunocytochemistry for expression of NPC marker SOX1 and neural crest marker AP2 using the ThermoFisher Cellomics automated imaging and analysis platform. NPCs were seeded at a density of 15000 / well of a 96-well Greiner microclear plate. IMR-90 fibroblasts were used as a negative control and seeded at a density of 10000 cells/well. Using a customized TargetActivation algorithm, DAPI nuclei were detected and defined as SOX1 and AP2 positive if the average intensity of signal was greater than that observed in 99% of IMR-90 fibroblasts. Only NPC cultures with 60% SOX1+ cells and less than 5% AP2+ cells were used for subsequent differentiation into neurons and phenotyping.
Neuronal Differentiation
NPCs were seeded at a density of roughly 1000 cells / mm2 (1000000 cells / well of a 6-well plate) on matrigel coated plastic plates (matrigel concentration 4x that used for iPSC, as determined by the dilution factor supplied by Corning with each matrigel batch). The cells were fed with Neuronal Differentiation (ND) media (Neurobasal, 1x N2, 1x B27, 1x glutamax, 1x penicillin/streptomicin, 1/500 laminin) supplemented with ascorbic acid (200 nM), BDNF (10 ng/μL), GDNF (10 ng/μL), cAMP (10 μM) every other day by changing all of the media in a given well. For days 10-14, the cultures were treated with 5 μM DAPT to attempt to curb excess NPC proliferation and promote differentiation (Fig. 1, Supplementary Fig. 3).
Sparse-seeding co-culture
To begin neuronal differentiation, NPCs were seeded at 1 million cells per well of a 6 well matrigel coated plate, as above, with one well per line per color. Nine to twelve wells of lawn NPCs (CTRL1 line #2 was used as the lawn; see Supplementary Tables 3-6) were seeded at the same time. For the first four weeks, the cells were fed as above. At day 21, neurons were infected with virus bearing different fluorescent colors (e.g. CaMKIIα-GFP for ASD and CaMKIIα-mKO2 or CaMKIIα-mKate24 for control cells; 10 µL virus / well from a standard preparation, see Virus Preparation). At this time, an appropriate number of vials of mouse astrocytes were thawed into a T-75 flask such that at least 1.25 million astrocytes would be present at day 26. At day 26, astrocytes at a minimum density of 250 cells / mm2 (~50000 per well of 24 well plate) were seeded on poly-L-ornithine and laminin coated coverslips (12 mm in diameter) or coated plastic wells. At day 27, neurons of the lawn were digested with Accutase for 15 minutes, carefully triturated using a Gilson P1000 pipetman a maximum of 15 times, washed with PBS, counted, and seeded at a density of at least 1000 cells/mm2 (200000 cells / well of a 24 well plate). Neuronal clumps were not removed at this stage to ensure there were enough cells of the lawn to seed. On day 28, infected neurons were digested with accutase for 15 minutes, carefully triturated using a Gilson P1000 pipetman a maximum of 15 times, strained with a 70 µm cell strainer to remove clumps, washed with PBS, and counted. After counting, each infected neuronal line was diluted to a concentration of 200000 cells / mL, and equal volumes of lines to be compared with each other were combined into desired comparisons and seeded onto the lawn. In order to account for error associated with efficiency of differentiation, infection, pipetting, and counting, three coverslips were seeded per comparison, with 2000, 4000, or 6000 cells (1%, 2%, or 3% of total neuron number) per line. Over the following week, the coverslips were monitored - those where there were at least 10 visible cells per line that were seeded sparsely enough so as to facilitate acquisition of images of full individual neurons were kept. Following re-seeding, the co-cultured neurons were fed with ND media supplemented with ascorbic acid (200 nM) or with ascorbic acid and BDNF (10 ng / mL) by replacing 1/2 the media every other day. See supplementary code file for the unnormalized and normalized connectivity data for every neuron for imaging (n=601) and electrophysiology experiments (n=680), respectively. These files will contain information regarding which lines were co-cultured together for each experiment (Supplementary Tables 3 and 4), as well as other relevant metadata.
Pharmacological Screen using SparCon
Neurons from R841X #5 and R841X-C lines were differentiated to 4 weeks and re-seeded for SparCon as above. For the following five weeks, they were grown in the presence of either DMSO or a pharmacological agent, such as IGF1 (20 ng /ul), BDNF (20 ng/ul), rapamycin (10 nM), TG003 (20 µM), eFT508 (10 nM), and DHPG (100 µM).
Validation of SparCon
See statistical analyses section.
Dendrite Extension Assay
NPCs from R841X #2, #5, #13 and CTRL1, CTRL2, and CTRL3 were differentiated as neuronal monocultures as described above in 24 well-plates. At 41 days of differentiation, they were transfected with an EF1α-EGFP expressing plasmid (500 ng plasmid and 2.5μl Lipofectamine 2000 in 50 μl Opti-MEM for 1 hr added to well), followed by washing twice with PBS with Ca2+ and Mg2+ and re-immersion in 500 μl of ND medium, as described in ‘Neuronal Differentiation’ section. On day 42, the plates were transferred to a Nikon Biostation CT at the Centre for Commercialization of Regenerative Medicine. The wells were imaged using the 20x fluorescence microscope of the Biostation CT, and locations of neurons were marked and stored in memory. The plates were then removed from the Biostation CT and 2 different salt solutions were added to different wells: 170 μL of 55 mM KCl (resulting in final concentration of 30 mM KCl) or 170 μL of 55 mM NaCl. The plates were then returned to the Biostation and each neuron was imaged at 25 min intervals for 24 hr. Dendrite length was characterized using the Simple Neurite Tracer ImageJ (3.1.3) plugin. Pilot experiments showed that maximal increase in length in control neurons occurred at approximately 500 min post-stimulation and this time-point was used for all future comparisons. The dendrite length at 500 min was normalized to the starting dendrite length for each neuron. The relative increase in dendrite length was compared across groups using a one-way ANOVA with post-hoc Tukey’s tests.
Virus preparation
Seven million HEK293T cells were plated in a T-75 flask, grown in 10% fetal bovine serum in DMEM. The next day, cells were transfected using Lipofectamine 2000 with plasmids for gag-pol (10 µg), rev (10 µg), VSV-G (5 µg), and the target construct (15 µg, CaMKII⍺-GFP or CaMKII⍺-mKate24 or CaMKII⍺-mKO2. Next day, the media was changed. The day after that, the media was spun down in a high-speed centrifuge at 30000g at 4°C for 2 hrs. The supernatant was discarded and 80 µl PBS was added to the pellet and left overnight at 4°C. The next day, the solution was triturated and distributed into 10 µl aliquots and frozen at −80°C. As noted, above, one full aliquot was used to infect neurons in one well of a 6-well plate.
Astrocyte Preparation
Astrocytes were prepared from postnatal day 1 CD-1 mice of unknown sex as described.57 Before use, astrocytes are checked for mycoplasma contamination. All animal work was approved by The Hospital for Sick Children Animal Care Committee and complies with the guidelines established by The Canadian Council of Animal Care.
Cloning
Guide RNAs were cloned into the gRNA cloning plasmid (Addgene #41824) by following option B in the instructions supplied with the plasmid (https://www.addgene.org/static/data/93/40/adf4a4fe-5e77-11e2-9c30-003048dd6500.pdf). Guide RNAs were designed using the CRISPR design tool on the Zhang Lab website (crispr.mit.edu).58 The CaMKII⍺-mKO2 plasmid is described in the a recent publication.11
Western blot
Protein was extracted from cells grown in 6-well plates by washing the cells 2 times with ice-cold PBS, followed by immersion in RIPA buffer (150 mM sodium chloride. 1.0% NP-40 or Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS (sodium dodecyl sulfate), 50 mM Tris, pH 8.0) with a complete Mini protease inhibitor. The cells in RIPA buffer were kept on ice for 10 mins and vortexed every 2-3 min. After 10 min, the samples were sonicated with on a Fisher Scientific sonic dismembrator F-60 and stored at −80°C. Proteins were quantified using the BioRad DC Assay kit. At least 20 µg of protein was loaded per sample per lane of a BOLT 4-12% gradient gel loaded into a BOLT mini gel tank. Proteins were transferred overnight onto nitrocellulose membrane at 60V. The membranes were blocked in either 5% BSA in TBS-T or 5% milk in PBS-T for 1 hr at room temperature. The membranes were then incubated with primary antibodies (1:1000 to 1:5000 dilution depending on antibody) overnight at 4°C. The following day, the membranes were washed 3 times for 5 minutes and incubated with HRP-conjugated secondary antibodies (1:5000) for 1 hr at room temperature. The membranes were then washed 3 times for 5 minutes. To visualize protein, either SuperSignal West Pico Chemiluminescent Substrate (used for loading controls) or SuperSignal West Femto Maximum Sensitivity Substrate (used for all other proteins) were applied to the membrane as evenly as possible. Shortly thereafter the membranes were exposed and imaged on a Biorad Gel Doc XR system. The resulting images were quantified using BioRad ImageLab 5.1.4) software.
Mycoplasma testing
All IPSC, NPC, IMR90 and HEK293T cell lines were shown to be mycoplasma-free every 2-3 months using a standard method,59 except Platinum Taq was used for the PCR reactions.
Immunocytochemistry
IPSCs and NPCs were fixed in 4% PFA in PBS for 7 minutes at room temperature. The cells were then washed 3 times for 5 min with PBS and permeabilized with 0.1% Triton-X in PBS for 10 minutes and blocked for 1 hr in 10% donkey or goat serum in PBS (blocking solution). After blocking, the cells were incubated with primary antibodies in blocking solution overnight at 4°C. The cells were then washed with PBS and incubated with secondary antibodies in blocking solution for 1 hr. The cells were then washed three times in PBS or incubated with DAPI (1:1000) in PBS and washed, if the experimental protocol required it.
Neurons were fixed by 1 min immersion in 37°C PKS (4% PFA in Krebs-Sucrose buffer, 50 mM KCl, 1.2 mM CaCl2, 1.3 mM MgCl2, 20 mm HEPES pH 7.4, 12 mM NaH2PO4, 400 mM sucrose, 145 mM NaCl, 10 mM glucose in water) to preserve fine structure60 followed by 10 min in ice cold methanol. PKS was added immediately after removal of culture medium, and pipetting was done slowly to prevent dislodging cells from coverslips. The neurons were then washed 3 times for 5 min with PBS and blocked with blocking solution for 1 hr at room temperature. After blocking, the cells were incubated with primary antibodies diluted in blocking solution overnight at 4°C. The cells were washed 3 times for 5 min with PBS and incubated with secondary antibodies at room temperature for 1 hr. If the goal of staining was to visualize synaptic puncta, all antibody solutions were mixed thoroughly and spun in a centrifuge at max speed for 3 min to remove antibody clumps. Coverslips were rinsed in sterile water, briefly dried with a Kimwipe and mounted on a frosted glass slide with 7-8 µL of Prolong Gold Anti-Fade mounting medium. The mounted coverslips were cured in the dark at room temperature for at least 24 hrs before imaging.
Image acquisition
For simultaneous morphometric and synaptic connectivity analysis, we acquired images of neurons at 400x magnification using the Nikon A1R laser scanning confocal microscope. First, we identified neurons by tracking axons to their origin. To take images of entire neurons, we acquired 4×4 tiled z-stacks at 0.75 µm spacing spanning 4.5 µm total. The pinhole was widened to 2.6 Airy units, which increased speed of image acquisition by allowing to increase Z-spacing and lower pixel dwell time without sacrificing Z-resolution to the extent that would compromise co-localization of synapsin I puncta to neurites. Each tile was 512×512 pixels, with 10% overlap, bringing the total field of view that was acquired to ~760 by 760 µm (0.4 µm / pixel). Image acquisition was done in a semi-automated manner, with manual picking of individual neurons and batch acquisition. Individual channels were acquired in sequential manner to prevent bleed-through of fluorophores. We aimed to acquire at least 7 neurons per color per coverslip, acquired in separate batches. Three channels would be acquired per batch for Synapsin I, GFP or mKO2. Image acquisition was done in a blinded manner. While most neurons would be acquired successfully with the above setup, we omitted those neurons with neurites that either extended in x,y, or z direction beyond the 760 × 760 × 4.5 µm space, encountered regions of high cell density which resulted in blurriness, or overlapped significantly with other fluorescent nearby.
To image iPSCs, embryoid bodies, as well as acquire phase-contrast images of live neurons, a Leica DMI4000B microscope with a DFC 340 FX camera was used. To acquire images of fluorescent neurons co-stained with cortical and synaptic markers, Zeiss Axiovert 200M epifluorescence microscope with a Hamamatsu C9100-13 EM CCD camera was used. To acquire images of NPCs and unlabeled neurons (Supplementary Fig. 3), the high-throughput imaging platform ThermoFisher Cellomics VTi was used. To quantify images of NPCs, the Cellomics Target Activation algorithm was used, with thresholds defined using the IMR-90 cells as a negative control.
Synapse and morphology analysis
All analyses were completed in Fiji (ImageJ 2.0.0-rc-65/1.51w). ND2 files from the Nikon NIS Elements software (4.3) were imported using the Bio-Formats plugin with channels pseudocolored such that the fluorescent protein and synapse channels were always colored in the same way from image to image to prevent the experimenter from inadvertently deciphering the genotype of the cells. The analyses were completed on maximum intensity projections of z-stacks. For morphological analysis, the longest neurite was assumed to be an axon and not traced. Dendrites were traced and morphology was analyzed using the Simple Neurite Tracer (version 3.1.3 plugin to obtain measurements for total dendrite length and Sholl analysis with 10 µm radius). Total synapse number was quantified by counting the number of Synapsin I puncta overlapping with signal from the fluorescent protein using the Cell Counter plugin (2010/12/07 version). Images in which portions of the neurons could not be unambiguously distinguished, either due to blurriness or to excessive density of nearby cells were not analyzed. All image analysis was completed in a blinded manner.
Statistics
No sample-size calculations were performed for experimental batches 1 to 5 (see Supplementary Table 5 for detailed information). Sample size was determined to be adequate based on the magnitude and consistency of measurable differences between groups. Monte Carlo simulation to estimate sample sizes required to achieve a power of 0.8 were then calculated using data from batches 1 to 5 and used to guide subsequent experiments (see below). The data met the assumptions of the statistical test used, and normality was formally tested. The fluorescent reporters were randomized in the SparCon assays but randomization was not used in other assays. Blinding was used in all SparCon assays, neuron morphology and electrophysiology experiments. No animals were excluded from the analysis. Cell culture experiments that failed were excluded based on neuronal cultures that did not survive or lifted off the culture plate/coverslip by the specified experimental timepoint, or were judged to be inadequate by lab personnel. Inadequate cultures may have had excessive cell death, excessive contamination with unwanted cell types, or inappropriate cell density (too low to allow for robust establishment of connectivity or too high to permit adequate imaging or patch-clamp).
Statistical validation and development of SparCon
SparCon within-well normalization overcomes variability and increases statistical power
For development of the method, we aimed to characterize the variability in connectivity across batches and wells. Because we could not derive an estimate for this variability in advance, we characterized 479 neurons by imaging for total synapse number and dendrite length from batches 1 to 3 and 390 neurons for sEPSC frequency and amplitude from batches 4 and 5 and across 39 and 48 wells, respectively. Probability distributions were fitted to the data using the fitdistrplus (1.0-11) R package. The distribution with the lowest Akaike information criterion score was considered best fit.
To examine synaptic connectivity, we first quantified absolute numbers of synapses per neuron to explore the range of variation between batches and between wells within a batch (Supplementary Fig. 8a). Variation was immediately evident with total synapse number per cell in batch 2 being lower and batch 3 being higher than batch 1 (Supplementary Fig. 8a). To minimize sources of error and facilitate comparisons across wells and batches, the SparCon setup allows relative calculations of total synapse numbers by within-well normalization. Because absolute synapse number and dendrite length in control neurons both fit lognormal distributions (Supplementary Fig. 8b), we normalized measurements of each neuron in a given well to the geometric mean of control neuron values in the same well. Following normalization, data across batches still fit the lognormal distribution for control neurons, but was largely devoid of systematic error (Supplementary Fig. 8c-e). The same held true for sEPSC frequency and amplitude of control neurons (Supplementary Fig. 9). We also performed colour swapping experiments that compared the same line to itself in different colors (Supplementary Tables 3 and 4). We did not detect any reporter gene bias as normalized synapse number and dendrite length were not dependent on which fluorescent protein was expressed (Supplementary Fig. 10a), and the same held true for normalized sEPSC frequency and amplitude (Supplementary Fig. 10b). Taken together, we show that normalization of SparCon data addresses confounding sample variability, and that the reporter genes have no effect on neuronal connectivity.
Since the data fit lognormal distributions, we hypothesized that nonparametric statistical tests applied to within-well normalized data may be more suitable for subsequent calculations. We performed Monte Carlo simulations to determine which statistical test was the most sensitive and whether relative within-well normalization increased statistical power at α=0.05. These simulations revealed that the Anderson-Darling k-samples (AD) test is more sensitive than the Kolmogorov-Smirnov and more sensitive or equal to Student’s t-test for all measures (Supplementary Fig. 11a-b). Critically, while within-well normalization significantly improved sensitivity for all measures, the effect was most pronounced on connectivity measures, such as synapse number and sEPSC frequency. For unnormalized synapse number, 45 neurons per group are necessary to detect a 1.5-fold difference for a power > 0.8 at α = 0.05, but only 20 are required after within-well normalization (Supplementary Fig. 11c). At this sample size of 20, skipping the normalization step requires a 1.9 fold-difference in order to achieve a power > 0.8 (Supplementary Fig. 11d). Similarly, for sEPSC frequency, within-well normalization reduces the required sample size to detect a 2-fold difference from 55 neurons to 35. At this sample size of 35, a 2.4 fold-difference is necessary to achieve a power > 0.8 if the data are unnormalized. While similar trends hold true for dendrite length and sEPSC amplitude, the impact is most pronounced on synapse number and sEPSC frequency, suggesting that these two measures of connectivity are particularly susceptible to experimental variation arising from heterogeneity of iPSC-derived neuronal cultures (Supplementary Fig. 11). We therefore used within-well normalization with the Anderson-Darling k-samples test to evaluate SHANK2 connectivity phenotypes.
Sparse-seeding co-culture
To control for well-to-well variability, measurements of all cells in a given well were normalized to the control cells in that well. For synapse number, dendrite length, sEPSC frequency and amplitude, normalization was done by dividing the values of all cells by the geometric mean of the controls. The standard deviation differed between control and mutant groups both before and after normalization (Supplementary Tables 7 and 9). Anderson-Darling k-samples test was used to compare pooled samples that were co-cultured together. For Sholl analysis, normalization was done by subtracting the mean number of crossings of the controls from that of a given cell in the same well at a particular radius. The change in crossings at a particular radius was then compared. Two-way ANOVA was used to compare Sholl analysis curves between conditions. P-values of less than 0.05 were considered statistically significant. All analyses were performed with R. R Code, unnormalized and normalized data are provided as supplementary files.
Pharmacological screen using SparCon
Results were normalized to the R841X-C in DMSO condition. Anderson-Darling k samples tests were used to compare groups.
Other experiments
When two groups were compared, two-tailed Student’s t-test was used. When multiple groups were compared, a one-way ANOVA was used with a post-hoc Tukey’s test. P-values of less than 0.05 were considered statistically significant. Where suitable, analyses were completed in either R or GraphPad Prism (6.0c).
Power Simulations
To determine which statistical tests were most sensitive and whether normalization improved sensitivity of those tests, we performed power simulations across a range of sample sizes with fold change held constant and vice versa. Conditions reaching a power of 0.8 with lower n were considered more sensitive. The following procedure was repeated for each of the four generated datasets (unnormalized synapse number and dendrite length, within-well normalized synapse number and dendrite length, unnormalized sEPSC frequency and amplitude, normalized sEPSC frequency and amplitude; see Supplementary File R-code.zip). To determine the influence of sample size to detect a specified fold-change, control cells from a given dataset grown in ND + AA were sampled with replacement at sample sizes ranging from 5 to 180 in steps of 5. The values of sampled control cells were then multiplied by a fold-change factor to obtain hypothetical values for mutant cells (1.5-fold for synapse number, 1.25-fold for dendrite length, 2-fold for sEPSC frequency, 1.25-fold for sEPSC amplitude). Statistical tests (Anderson-Darling, Kolmogorov-Smirnov, Student’s t-test) were then used to compare the samples. To determine the influence of different fold-changes on power when the sample size was held constant, control cells from a given dataset grown in ND + AA were sampled with replacement at a specified sample size (20 neurons for synapse number, 30 for dendrite length, 35 for sEPSC frequency, 20 for sEPSC amplitude) for fold changes ranging from 1 to 2.5 in 0.1 increments. The procedure was repeated 1000 times at each n, and the proportion of tests reaching significance (i.e. power) at α=0.05 was recorded. The smallest sample size or fold change which achieved a power > 0.8 is reported as the minimum required sample size or fold change.
RNA Sequencing and analysis
Growth protocol and RNA extraction
NPCs from R841X #5 and R841X-C lines were differentiated into neuronal monocultures as described above in 6×10 cm dishes per line per batch. At 4 weeks of age, one-half of the cultures were dissociated with Accutase, and subjected to negative selection using magnetic activated cell sorting (MACS) as described previously to deplete NPCs and glia19. Mouse astrocytes were added to the other half, which was grown until 9 weeks of age, at which point it was also processed with MACS. The flow-through (neuronal fraction) was harvested, and total RNA was extracted using a standard Trizol-based protocol. The quality of RNA was checked by BioAnalyzer.
Library Preparation, read mapping and downstream analysis
Libraries prepared from the isolated RNA were sequenced on Illumina HiSeq 2500 with paired-end reads (125 bp each) to a depth of approximately 30 million reads per sample. Reads were aligned to the human genome (Ensembl release 84 for GRCh38) using HISAT2 (version 2.1.0) 61, with default parameters for the paired-end reads. Aligned reads overlapping protein coding genes were counted by featureCounts (subread package version 1.5.3) 62 with default parameters, using GENCODE basic gene annotation from a release 27 for GRCh38. Expression levels of genes in terms of transcript per million (TPM) was separately estimated by Sailfish (version 0.10.0) 63, using an index prepared from a transcriptome of protein coding genes (Ensembl release 86 for GRCh38). Differential gene expression analysis was carried out in DESeq2 (1.20.0)64 using pairwise comparisons between R841X and R841X-C at each timepoint. Differentially expressed genes were defined as those with adjusted P-value < 0.05. To gain further insight into perturbed cell function, continuous gene set enrichment analysis20 was performed by first ranking differentially expressed genes by −log10(adjusted P-value) * sign(log2FoldChange). Gene sets smaller than 15 or bigger 500 genes were excluded from the analysis, and 1000 permutations were performed. Gene sets from multiple databases excluding those inferred from electronic annotation were used for enrichment, as compiled by the Bader Lab at http://download.baderlab.org/EM_Genesets in August 2018 (Human_GOBP_AllPathways_no_GO_iea_August_01_2018_symbol.gmt). The table of negative and positive associations at each developmental time point (4wk and 9wk) were used to generate an enrichment map using the Cytoscape65 plugin Enrichment Map (2.1.0) 66 with q-value cutoff of 0.01. The resulting network was first automatically annotated using the AutoAnnotate (1.2) 67 plugin to aid in discovery of shared function among closely clustered gene sets. The labels for closely clustered gene sets were then edited for readability and comprehension and reproduced using Adobe Illustrator CS6. Only the largest interconnected network is shown in the main figure. The full unannotated network is shown in supplementary figures. The RNA-Seq dataset can be accessed at GEO (GSE122550).
Electrophysiology
Electrophysiological recordings
Conventional patch-clamp recordings were made at room temperature (22°C), as described previously68, in human iPSC-derived neurons after 9-11 weeks in culture and experimenter was blinded to genotype in electrophysiological recordings and data analysis. An Axopatch 1-D amplifier (Molecular Devices, USA) and a DigiData 1200 series interface (Molecular Devices, USA) were used for patch-clamp recordings. Electrical signals were sampled at 10 kHz and filtered at 2 kHz. Recordings were acquired online using Clampex 9.2 software and analyzed off-line using Clampfit 9.2 software (Axon Instruments). Recording pipettes with resistance of about 5 to 8 MΩ were pulled on a P-87 pipette puller (Sutter Instrument Co., USA) using capillary glass (World Precision Instruments, Inc., USA). The pipettes, for the recordings on intrinsic membrane properties, were filled with a solution that contained (in mM): 144 K+-gluconate, 10 KCl, 2 EGTA, 10 HEPES, and 2 Mg-ATP, and pH was adjusted to 7.2 with KOH. Extracellular recording solutions were composed of (in mM): 140 NaCl, 1 MgCl2, 5.4 KCl, 10 glucose, 15 HEPES, and 2 CaCl2, and pH was adjusted to 7.35 with NaOH. Under current-clamp condition, a liquid junction potential of 16 mV with K+-gluconate based internal solutions was subtracted from the measured membrane potential values. Action potentials from the membrane potentials of approximate −75 mV were evoked by the injection of a series of current steps from −5 pA to +50 pA (to +30 pA in studying the effect of ZD7288; in 5 pA increments) for 1 s. ZD7288 at 100 µM was applied to examine its effect on firing frequency of action potentials evoked in both control and SHANK2 mutant iPSC-derived neurons. Under voltage-clamp condition, membrane potentials were held at −70 mV and voltage-gated currents were elicited by depolarizing the membrane potentials to a series of potentials.
sEPSCs were recorded in voltage-clamp condition with the holding membrane potentials of −60 mV. Extracellular recording solutions were composed of (in mM): 140 NaCl, 15 HEPES, 5.4 KCl, 25 glucose, 0.003 glycine, 0.001 strychnine, 0.01 bicuculline, 1 MgCl2, and 1.3 CaCl2, and pH was adjusted to 7.35 with NaOH. Recording pipettes were filled with a solution that contained (in mM): 137 CsF, 1.5 CsCl, 10 BAPTA, 10 HEPES, 5 QX-314, and 4 Mg-ATP, and pH was adjusted to 7.2 with CsOH. Detection and analysis of sEPSCs were performed by Mini Analysis Program (version 5.0) in a blinded manner (Synaptosoft Inc, NJ, USA).
Reporting Summary
The Nature Research Reporting Summary appended to this article provides further information on research design, methods and reagents used.
Code Availability
The RNASeq R scripts used to generate the figures in the manuscript are available in the Supplementary Software Zip file and at https://github.com/kzaslavsky/SparCon. The ‘GENERIC_SPARCON_ANALYSIS’ folder contains scripts for users to analyze their own co-culture data. It performs within-well normalization, plotting, and statistical analysis. Sample data (excerpted from current study) is provided as outlined under Data Availability.
Scripts in Supplementary Software Zip File
Running the scripts will generate plots and reproduce statistical analyses used in the paper.
In the ‘SparCon’ folder, the SpaCon_Analysis_v1.1.R file will perform within-well normalization and output plots used for the main figures of the manuscript. The Sholl_Analysis.R file will perform within-well normalization and output the Sholl analysis plots. The Power_Sims.R file will perform the power simulations used in the paper. The Distributions.R file will produce plots describing the distributions of the data. The SparCon_instrinsic_ephys.R file will produce plots and analyze data on intrinsic membrane properties. The SparCon_fn.R and Power_Sims_Fn.R contain functions used by above scripts.
In the ‘Dendrite extension’ folder, the dend_extension_exp.R file will perform analyses and generate plots for the dendrite extension experiment.
In the ‘MiniScreen’ folder, the Miniscreen_plots_script.R file will perform analyses and generate plots for the SparCon screen with IGF1, BDNF, TG003, eFT508, Rapamycin, and DHPG.
In the ‘RNASeq’ folder, the Deseq2_SHANK2.R file will perform differential gene expression analysis, output differential expression tables, and prepare ranked files and normalized counts files for gene set enrichment analysis. Activity_and_ASD_Modules.R will analyze abundance of transcripts known to be involved in activity-dependent transcription in iPSC-derived neurons, as well as check for enrichment of DEGs among previously defined co-expression modules known to be enriched for ASD risk genes. Heatmap_SHANK2.R will generate heatmaps of transcript levels of manually curated neuronal, NPC, and glial markers across all samples. Volcano_plots.R will generate volcano plots and bar graphs in Fig 4.
The SparCon_fn.R, RNASeq_FN.R, GOfnKZ.R, and Power_Sims_Fn.R files contain functions used throughout the scripts.
In the ‘GENERIC_SPARCON_ANALYSIS’ folder, the Generic_SparCon.R file can be used to analyze custom data. Sample datasets are provided. Generic_SparConFN.R contains normalization and plotting functions.
R packages used include: data.table (1.11.2), tidyr (0.8.0), purrr (0.2.4), dplyr (0.7.4), stringr (1.3.1), ggplot2 (2.2.1), ggthemes (3.5.0), RColorBrewer (1.1–2), scales (0.5.0), psych (1.8.4), kSamples (1.2–7), fitdistrplus (1.0–11), DESeq2 (1.20.0), biomaRt (2.36.1), forcats (0.3.0), viridis (0.5.1), gridExtra (2.3), gtable (0.2.0)
Data Availability
The whole genome sequence dataset used for off-target analysis can be accessed at EGA (EGAS00001003436). These raw data are associated with Fig. 1a, Supplementary Fig. 1-3, and Supplementary Tables 1-2. SparCon and dendrite extension datasets used to generate the figures in the manuscript are provided in the Supplementary Software Zip file and on GitHub (https://github.com/kzaslavsky/SparCon). These raw data are associated with Fig.1-4, Supplementary Fig. 7-15, Supplementary Tables 3-9. The RNA-Seq dataset can be accessed at GEO (GSE122550). These raw datasets are associated with Fig. 4a-d,f, Supplementary Fig.16, and Supplementary Tables 10-12.
SparCon Datasets in Supplementary Software Zip file
The SparCon folder contains four connectivity datasets: imaging (SC_ICC_DB.txt and SC_ICC_DB_norm.txt, Synapse Number and Dendrite Length for n=601 neurons, unnormalized and normalized) and electrophysiology (SC_ephys_db.txt and SC_ephys_db_norm.txt, sEPSC Frequency and Amplitude for n=680 neurons, unnormalized and normalized). Additionally, there are datasets for Sholl analysis (shollDB_unnorm.txt and shollDB_norm.txt, n = 601), a dataset describing the intrinsic membrane properties (intrinsic_ephys_db.txt, n = 148), a ZD7288 stimulation dataset (ZD7288_exp_final.txt, n = 96).
The ‘Dendrite Extension’ folder contains baseline and normalized extension data for the dendrite extension experiment (‘Dend_extend_baseline.txt’ and Dend_extend_normalize.txt’, n = 91).
The ‘MiniScreen’ folder contains several datasets for the SparCon screen with IGF1, BDNF, TG003, eFT508, Rapamycin, and DHPG (n = 1246). Please see Zip file readme file for detailed descriptions of datasets.
The ‘RNASeq’ folder contains data and scripts for analysis of the RNA-Seq experiment. The simple_coding_counts_4wk.txt and simple_coding_counts_9wk.txt contain counts of aligned transcripts suitable for analysis with DESeq2. Pruunslid_Bic_Stim_7wk.txt is a dataset of activity-dependent genes in 7 week old iPSC-derived neurons stimulated with bicuculline from Pruunslid et al. ASD_Modules.txt contains ASD-associated modules from weighted gene network correlation analyses by Parikshak et al. and Willsey et al. FMRP.csv contains FMRP targets from Darnell et al. Neuron_markers.txt contains a list of manually curated markers of neuron layers, neuronal subtypes, as well as NPC and glial markers. geneset_list.csv contains gene set and gene ontology accession numbers for generating volcano plots.
Materials
Please refer to Supplementary Table 13 for a listing of all materials used.
Supplementary Material
Acknowledgements
This study was funded by grants from the National Institutes of Health (award R33MH087908 to J.E. and S.W.S.), and the Ontario Brain Institute (J.E. and S.W.S.), the Canadian Institutes of Health Research (EPS-129129 to J.E.; MOP-102649 and MOP-133423 to J.E. and M.W.S.) and the Simons Foundation/SFARI (#514918 to J.E.). We thank the MSSNG Open Science project for sharing data. Fellowship and studentship support: CIHR Canada Vanier Graduate Scholarship (K.Z.), MD/PhD Studentships at the University of Toronto and McLaughlin Centre (K.Z.), CIHR Banting Fellowship (E.D.), Ontario Stem Cell Initiative Fellowship (P.J.R.), Ontario Ministry of Research & Innovation Fellowship (P.J.R.), International Rett Syndrome Foundation Fellowship (D.C.R.), S.W.S. is the GlaxoSmithKline-CIHR Endowed Chair in Genome Sciences at The Hospital for Sick Children. M.W.S. is the Northbridge Chair in Paediatric Research at the Hospital for Sick Children. We thank Ryan Yuen for comments regarding whole genome sequencing analysis of the two children in the study and Janice Hicks for her technical help. We thank Drs. Wendy Roberts, Rosanna Weksberg, Brian Chung and Melissa Carter for obtaining skin biopsies. We also thank the participants and their family members for their contributions to this study. We thank the Centre for Commercialization of Regenerative Medicine for in-kind access to equipment and project resources.
Footnotes
Competing Interests
The authors declare no competing interests.
Co-first
References
- 1.Djuric U et al. MECP2e1 isoform mutation affects the form and function of neurons derived from Rett syndrome patient iPS cells. Neurobiol. Dis. 76, 37–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pak C et al. Human Neuropsychiatric Disease Modeling using Conditional Deletion Reveals Synaptic Transmission Defects Caused by Heterozygous Mutations in NRXN1. Cell Stem Cell 17, 316–328 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yi F et al. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352, aaf2669–aaf2669 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shcheglovitov A et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503, 267–271 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Griesi-Oliveira K et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Molecular Psychiatry 20, 1350–1365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brennand KJ et al. Creating Patient-Specific Neural Cells for the In Vitro Study of Brain Disorders. Stem Cell Reports 5, 933–945 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandoe J & Eggan K Opportunities and challenges of pluripotent stem cell neurodegenerative disease models. Nature Neuroscience 16, 780–789 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Berkel S et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nature Genetics 42, 489–491 (2010). [DOI] [PubMed] [Google Scholar]
- 9.C Yuen RK et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci 20, 602–611 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deneault E et al. Complete Disruption of Autism-Susceptibility Genes by Gene Editing Predominantly Reduces Functional Connectivity of Isogenic Human Neurons. Stem Cell Reports 11, 1211–1225 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodrigues DC et al. MECP2 Is Post-transcriptionally Regulated during Human Neurodevelopment by Combinatorial Action of RNA-Binding Proteins and miRNAs. Cell Rep 17, 720–734 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Johnson MA, Weick JP, Pearce RA & Zhang S-C Functional neural development from human embryonic stem cells: accelerated synaptic activity via astrocyte coculture. J. Neurosci 27, 3069–3077 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta K, Hardingham GE & Chandran S NMDA receptor-dependent glutamate excitotoxicity in human embryonic stem cell-derived neurons. Neuroscience Letters 543, 95–100 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nehme R et al. Combining NGN2 Programming with Developmental Patterning Generates Human Excitatory Neurons with NMDAR-Mediated Synaptic Transmission. Cell Rep 23, 2509–2523 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Micheva KD, Busse B, Weiler NC, O’Rourke N & Smith SJ Single-synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron 68, 639–653 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berkel S et al. Inherited and de novo SHANK2 variants associated with autism spectrum disorder impair neuronal morphogenesis and physiology. Human Molecular Genetics 21, 344–357 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bidinosti M et al. CLK2 inhibition ameliorates autistic features associated with SHANK3 deficiency. Science 351, 1199–1203 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Tyler WJ & Pozzo-Miller LD BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J. Neurosci 21, 4249–4258 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Djuric U et al. Spatiotemporal Proteomic Profiling of Human Cerebral Development. Mol. Cell Proteomics 16, 1548–1562 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pruunsild P, Bengtson CP & Bading H Networks of Cultured iPSC-Derived Neurons Reveal the Human Synaptic Activity-Regulated Adaptive Gene Program. Cell Rep 18, 122–135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Darnell JC et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parikshak NN et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willsey AJ et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du Y, Weed SA, Xiong WC, Marshall TD & Parsons JT Identification of a novel cortactin SH3 domain-binding protein and its localization to growth cones of cultured neurons. Molecular and Cellular Biology 18, 5838–5851 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vessey JP & Karra D More than just synaptic building blocks: scaffolding proteins of the post-synaptic density regulate dendritic patterning. Journal of Neurochemistry 102, 324–332 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Kirwan P et al. Development and function of human cerebral cortex neural networks from pluripotent stem cells in vitro. Development 142, 3178–3187 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeRosa BA et al. hVGAT-mCherry: A novel molecular tool for analysis of GABAergic neurons derived from human pluripotent stem cells. Mol. Cell. Neurosci 68, 244–257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bardy C et al. Predicting the functional states of human iPSC-derived neurons with single-cell RNA-seq and electrophysiology. Molecular Psychiatry 21, 1573–1588 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmeisser MJ et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486, 256–260 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Won H et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Wegener S et al. Defective Synapse Maturation and Enhanced Synaptic Plasticity in Shank2 Δex7−/− Mice. eNeuro 5, ENEURO.0398–17.2018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grabrucker AM et al. Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. The EMBO Journal 30, 569–581 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Modi ME et al. Hyperactivity and Hypermotivation Associated With Increased Striatal mGluR1 Signaling in a Shank2 Rat Model of Autism. Frontiers in Molecular Neuroscience 11, 107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peixoto RT, Wang W, Croney DM, Kozorovitskiy Y & Sabatini BL Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B(−/−) mice. Nature Neuroscience 19, 716–724 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eltokhi A, Rappold G & Sprengel R Distinct Phenotypes of Shank2 Mouse Models Reflect Neuropsychiatric Spectrum Disorders of Human Patients With SHANK2 Variants. Frontiers in Molecular Neuroscience 11, 147 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leblond CS et al. Genetic and Functional Analyses of SHANK2 Mutations Suggest a Multiple Hit Model of Autism Spectrum Disorders. PLoS Genet 8, e1002521 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leblond CS et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: a gradient of severity in cognitive impairments. PLoS Genet 10, e1004580 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sheng M & Kim E The Shank family of scaffold proteins. Journal of Cell Science 113, 1851–1856 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Santini E & Klann E Reciprocal signaling between translational control pathways and synaptic proteins in autism spectrum disorders. Sci Signal 7, re10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Testa-Silva G et al. Hyperconnectivity and slow synapses during early development of medial prefrontal cortex in a mouse model for mental retardation and autism. Cereb. Cortex 22, 1333–1342 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cline H & Haas K The regulation of dendritic arbor development and plasticity by glutamatergic synaptic input: a review of the synaptotrophic hypothesis. The Journal of Physiology 586, 1509–1517 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hasegawa S, Sakuragi S, Tominaga-Yoshino K & Ogura A Dendritic spine dynamics leading to spine elimination after repeated inductions of LTD. Sci Rep 5, 7707 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sugathan A et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proceedings of the National Academy of Sciences 111, E4468–77 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang G et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo T et al. Effect of the autism-associated lncRNA Shank2-AS on architecture and growth of neurons. J. Cell. Biochem. 57, 19 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Deneault E et al. CNTN5−/+ or EHMT2−/+ iPSC-Derived Neurons from Individuals with Autism Develop Hyperactive Neuronal Networks. bioRxiv 368928 (2018). doi: 10.1101/368928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hotta A et al. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nature Methods 6, 370–376 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Cheung AYL et al. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Human Molecular Genetics 20, 2103–2115 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miyaoka Y et al. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nature Methods 11, 291–293 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H & Durbin R Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKenna A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chambers SM et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature Biotechnology 27, 275–280 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maroof AM et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 12, 559–572 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brennand KJ et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 473, 221–225 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim HJ & Magrané J Isolation and culture of neurons and astrocytes from the mouse brain cortex. Methods Mol. Biol. 793, 63–75 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Hsu PD, Scott DA, Weinstein JA & Ran FA DNA targeting specificity of RNA-guided Cas9 nucleases. Nature (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Otto E, Zalewski C, Kaloss M & Del Giudice RA Quantitative detection of cell culture Mycoplasmas by a one step polymerase chain reaction method. Methods in Cell Science 18, 261–268 (1996). [Google Scholar]
- 60.Kwiatkowski AV et al. Ena/VASP Is Required for neuritogenesis in the developing cortex. Neuron 56, 441–455 (2007). [DOI] [PubMed] [Google Scholar]
- 61.Kim D, Langmead B & Salzberg SL HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 63.Patro R, Mount SM & Kingsford C Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nature Biotechnology 32, 462–464 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cline MS et al. Integration of biological networks and gene expression data using Cytoscape. Nature Protocols 2, 2366–2382 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Merico D, Isserlin R, Stueker O, Emili A & Bader GD Enrichment Map: A Network-Based Method for Gene-Set Enrichment Visualization and Interpretation. PLoS ONE 5, e13984 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kucera M, Isserlin R, Arkhangorodsky A & Bader GD AutoAnnotate: A Cytoscape app for summarizing networks with semantic annotations. F1000Res 5, 1717 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang W-B et al. Fyn Kinase regulates GluN2B subunit-dominant NMDA receptors in human induced pluripotent stem cell-derived neurons. Sci Rep 6, 23837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The whole genome sequence dataset used for off-target analysis can be accessed at EGA (EGAS00001003436). These raw data are associated with Fig. 1a, Supplementary Fig. 1-3, and Supplementary Tables 1-2. SparCon and dendrite extension datasets used to generate the figures in the manuscript are provided in the Supplementary Software Zip file and on GitHub (https://github.com/kzaslavsky/SparCon). These raw data are associated with Fig.1-4, Supplementary Fig. 7-15, Supplementary Tables 3-9. The RNA-Seq dataset can be accessed at GEO (GSE122550). These raw datasets are associated with Fig. 4a-d,f, Supplementary Fig.16, and Supplementary Tables 10-12.
SparCon Datasets in Supplementary Software Zip file
The SparCon folder contains four connectivity datasets: imaging (SC_ICC_DB.txt and SC_ICC_DB_norm.txt, Synapse Number and Dendrite Length for n=601 neurons, unnormalized and normalized) and electrophysiology (SC_ephys_db.txt and SC_ephys_db_norm.txt, sEPSC Frequency and Amplitude for n=680 neurons, unnormalized and normalized). Additionally, there are datasets for Sholl analysis (shollDB_unnorm.txt and shollDB_norm.txt, n = 601), a dataset describing the intrinsic membrane properties (intrinsic_ephys_db.txt, n = 148), a ZD7288 stimulation dataset (ZD7288_exp_final.txt, n = 96).
The ‘Dendrite Extension’ folder contains baseline and normalized extension data for the dendrite extension experiment (‘Dend_extend_baseline.txt’ and Dend_extend_normalize.txt’, n = 91).
The ‘MiniScreen’ folder contains several datasets for the SparCon screen with IGF1, BDNF, TG003, eFT508, Rapamycin, and DHPG (n = 1246). Please see Zip file readme file for detailed descriptions of datasets.
The ‘RNASeq’ folder contains data and scripts for analysis of the RNA-Seq experiment. The simple_coding_counts_4wk.txt and simple_coding_counts_9wk.txt contain counts of aligned transcripts suitable for analysis with DESeq2. Pruunslid_Bic_Stim_7wk.txt is a dataset of activity-dependent genes in 7 week old iPSC-derived neurons stimulated with bicuculline from Pruunslid et al. ASD_Modules.txt contains ASD-associated modules from weighted gene network correlation analyses by Parikshak et al. and Willsey et al. FMRP.csv contains FMRP targets from Darnell et al. Neuron_markers.txt contains a list of manually curated markers of neuron layers, neuronal subtypes, as well as NPC and glial markers. geneset_list.csv contains gene set and gene ontology accession numbers for generating volcano plots.