

SUMMARY
Metazoan cell death mechanisms are diverse and include numerous non-apoptotic programs. One program called entosis involves the invasion of live cells into their neighbors and is known to occur in cancers. Here, we identify a developmental function for entosis: to clear the male-specific linker cell in C. elegans. The linker cell leads migration to shape the gonad and is removed to facilitate fusion of the gonad to the cloaca. We find that the linker cell is cleared in a manner involving cell-cell adhesions and cell-autonomous control of uptake through linker cell actin. Linker cell entosis generates a lobe structure that is deposited at the site of gonad-to-cloaca fusion and is removed during mating. Inhibition of lobe scission inhibits linker cell death, demonstrating that the linker cell invades its host while alive. Our findings demonstrate a developmental function for entosis: to eliminate a migrating cell and facilitate gonad-to-cloaca fusion, which is required for fertility.
Graphical Abstract
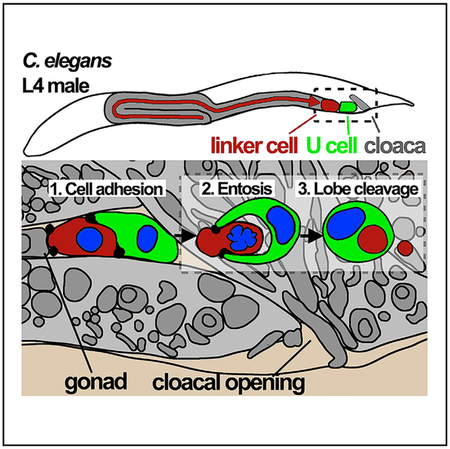
In Brief
Entosis is a cell death mechanism, previously observed in cancer cell populations, that involves the invasion of live cells into their neighbors. Lee et al. now show that entosis has a developmental function in C. elegans, clearing the linker cell during gonad formation.
INTRODUCTION
Programmed cell death mechanisms eliminate cells that have outlived their lifespan or are damaged or infected. Although it was once considered that most metazoan programmed cell deaths occur by apoptosis, numerous alternative mechanisms are now known (Galluzzi et al., 2012). For example, regulated forms of necrosis can be triggered by viruses or other pathogens to alert immune responses to infection (Vanden Berghe et al., 2014). While damage or infection can trigger numerous mechanisms, few have been shown to participate in normal development, suggesting stringent selection for mechanisms that are used to sculpt normal tissues.
One cell death that is critical for tissue development eliminates the male-specific linker cell in C. elegans (Abraham et al., 2007). The linker cell shapes and elongates the male gonad by leading a programmed migration to the cloaca, a critical event that generates the exit route for sperm. As the linker cell is positioned in between the gonad and cloaca after migration, it must undergo cell death and be removed in a manner that does not perturb, and is rather thought to facilitate, the joining of these two structures (Abraham et al., 2007). While genes acting within the linker cell to control its death have been identified (Blum et al., 2012; Kinet et al., 2016; Malin et al., 2016), here we examined the mechanism of linker cell clearance. We find evidence that the linker cell is removed by entosis, a cell-cell-adhesion-based mechanism originally discovered in cancers (Overholtzer et al., 2007).
RESULTS
Linker Cell Clearance Results in Separation of a Lobe Structure
To investigate linker cell clearance, we examined the temporal dynamics by time-lapse imaging in 3 dimensions (4D imaging) utilizing a C. elegans strain with linker cell GFP expression (lag-2 promoter::GFP) (Abraham et al., 2007). After completing migration, linker cells rounded and moved left or right of the midline and anterior, presumably due to engulfment by either the left or right U cell (U.lp or U.rp) (Abraham et al., 2007). We noted that as linker cells moved left or right, a subcellular piece extended from the cell body and detached, remaining at the midline (Figures 1A and S1A; Video S1). This separating lobe was 2.1 ± 0.74 μm in diameter and was detected in 65 out of 67 worms examined. To determine the relative timing of lobe separation and engulfment, worms were generated with expression of GFP in engulfing U cells (lin-48 promoter::GFP) and a marker of cortical actin in the linker cell, the calpoinin homology domain of the actin-binding protein Utrophin (UtrCH) (Morris et al., 1999) fused to mCherry (lag-2 promoter::mCherry::UtrCH). By 4D imaging, we found that a lobe formed from the linker cell and separated as it became engulfed, detaching from the back, opposite the direction of engulfment (Figure S1B).
Figure 1. Linker Cell Engulfment and Entotic Cell Death Involve Separation of a Lobe Structure.

(A) 4D imaging of linker cell engulfment shows the formation and separation of a lobe (arrowhead). Images are maximum projections, times are h:min. See Video S1.
(B) Entotic cells form lobes. Images show MCF-7 cells labeled with green and red Cell Tracker dyes imaged by 4D microscopy; times are h:min. Arrowhead indicates lobe that undergoes cleavage. See Video S3A.
(C) Lobe cleavage is a feature of entotic cell death. Top graph shows percent entotic MCF-7 cells imaged for 20 h that exhibit lobe cleavage (black bars) and one of three possible fates: remaining inside of hosts without dying (“no change”), escape from hosts, or cell death. Gray bars show the percentage of cells without lobe cleavage. For no change, n = 16; escape, n = 34; and cell death, n = 14; n represents the total number of cells imaged from more than three biological replicates. Bottom graph shows five representative lobe cleavages and entotic cell death events; relative times start at lobe cleavage (blue bars, arrow), and cell deaths are indicated by black bars. Scale bars, 10 μm.
(D)Graph shows cortical to cytoplasmic ratio of GFP::UtrCH (blue line, left y-axis) in a linker cell from the time of engulfment marked by lobe formation (arrowhead). Green line shows GFP intensity over time; black line (righty axis) shows distance of lobe separation from linker cell. Hatched box represents timing of linker cell death (arrow) determined by cortical actin ratio and GFP intensity (see Figure S1D for additional examples). Right images show linker cell quantified in graph. Top rows show maximum projections of GFP::UtrCH fluorescence; arrowhead indicates lobe. Bottom row shows the x-y confocal plane of GFP::UtrCH fluorescence from the same cell. Note cortical GFP::UtrCH fluorescence at 80 min redistributes to the cytoplasm by 90 min. See also Video S4B.
(E)Lysosome fusion to the linker cell vacuole precedes loss of GFP fluorescence. Images are x-y confocal planes of U cell lmp-1::mDsRed-labeled lysosomes (red) and linker cell GFP fluorescence. Time 0 is the time of lobe separation (L) from the linker cell (LC). Note that the linker cell vacuole becomes labeled with lmp-1::mDsRed fluorescence 20 min after lobe separation (bottom row, blue arrow), which is followed by loss of linker cell GFP. See also Video S5. Graph shows quantification of fluorescence intensities of the linker cell (black line) versus separated lobe (green line) over time after lysosome fusion (time 0). Data are means ± SD from three independent biological repeats. Scale bars, 10 μm.
While engulfed linker cells underwent cell death and were degraded, by contrast, separated lobes retained their size and were long-lived (Video S2A; Figure 1E). Out of 10 lobes examined by long-term 4D imaging, using linker cell expression of a GFP-tagged version of UtrCH (lag2p::GFP::UtrCH), 9 persisted after engulfment until the end of the imaging experiment, from 1 to over 10 h (Video S2A). The remaining lobe disappeared from view more than 5 h after engulfment (not shown). Further analysis of confocal z series of U cell (lin48p::DsRed) and linker cell (lag2p::tagBFP2) markers revealed that separated lobes were not engulfed by either U cell (Video S2B). These observations are consistent with recent reports that linker cells split into two pieces (Abraham et al., 2007; Keil et al., 2017), reported to reflect engulfment by both left and right U cells (Kutscher et al., 2018), while our imaging reveals that separating lobes appear to be unengulfed.
Entosis Results in Lobe Separation
During entosis, specialized structures called uropods form on internalizing cells, oriented opposite the engulfing interface, similar to linker cell lobes (Purvanovetal., 2014). Entotic uropods promote cell internalization and are sites of actin accumulation (Purvanov et al., 2014). Whether these structures detach from internalizing cells is not known. We examined MCF-7 breast cancer cells that exhibit high rates of entosis (Overholtzer et al., 2007) and found that 55% (12 out of 22) of entotic structures had lobes extending from internalizing cells, and some indeed detached over time (Figure 1B; Video S3A). Entotic cells underwent cell death after lobe detachment, ceasing movement and decreasing in size (Florey et al., 2011; Overholtzer et al., 2007) (Figure S1C; Video S3B). 93% of entotic cells with lobe cleavage died, while 93–94% of cells that survived or escaped retained their lobes (Figure 1C), demonstrating that lobe detachment is a morphological feature of entotic cell death.
Linker Cells Undergo Death after Lobe Separation
To examine the relative timing of linker cell death and lobe separation, we took advantage of an observation that the mCherry::UtrCH actin reporter rapidly redistributed from the linker cell cortex to the cytoplasm upon cell death (Video S4A). We utilized a GFP-tagged version of this reporter in linker cells (lag-2 promoter::GFP::UtrCH) to examine of the timing of cytoplasmic redistribution and loss of GFP fluorescence (Abraham et al., 2007). By 4D imaging, we observed that cortical GFP::UtrCH fluorescence was maintained in linker cells, even after lobe formation that marks the timing of cell engulfment, and was relocalized to the cytoplasm ~30–40 min later (Figures 1D and S1D; Video S4B). The fluorescence intensity of GFP also diminished after lobe formation, reaching a plateau with similar timing as the relocation of GFP::UtrCH from the cortex (Figures 1D and S1D). We concluded based on these observations that linker cell death occurs 30–40 min after lobe formation. Consistent with this, the fusion of lysosomes from U cells around engulfed linker cells also occurred around this time, as shown by expression of a fluorescently tagged lysosome-associated membrane protein 1 protein in U cells (lin-48 promoter::lmp-1::DsRed) (Figure 1E; Video S5). Lysosome fusion occurred 27.6 ± 11.5 min after lobe formation (n = 5) and prior to the loss of linker cell GFP florescence (Figure 1E; Video S5).
Lobe Cleavage and Entotic Cell Death Are Inhibited by Expression of a PIP2 Reporter
Entotic cells are engulfed alive and can be rescued from death if entotic vacuole maturation is disrupted. Inhibition of the class III phosphoinositide-3 (PI-3) kinase Vps34, which controls phosphatidylinositol-3-phosphate (PI3P) formation on the entotic vacuole, reduces entotic cell death (Florey etal., 2011). On phagosomes, PI3P formation is preceded by loss of the plasmamembrane-associated phosphatidylinositol-4,5-bisphosphate (PIP2), which contributes to phagosome closure by promoting membrane scission (Sarantis et al., 2012), and on entotic vacuoles, we identified the same temporal pattern (data not shown). When host cells expressed a PIP2 reporter, a GFP-tagged version of the PH domain of phospholipase C delta (GFP-PLCδ-PH), entotic cells failed to die at a normal frequency (Figure 2A), suggesting that reporter expression might disrupt vacuole maturation. Internalized cells that failed to die inside of GFP-PLCδ-PH-expressing hosts also retained protruding lobes, consistent with a defect in scission, and the vacuoles harboring live cells maintained localization of the PIP2 reporter (Figure 2A).
Figure 2. Entotic and Linker Cell Death, and Lobe Scission, Are Disrupted by Expression of a PIP2 Sensor.

(A) Entotic death in MCF-7 cells is inhibited by GFP-PLCδ-PH expression. Graph shows the mean percentage of entotic cells from three independent biological repeats that underwent cell death over 24 h of time-lapse imaging. Error bars represent SEM. n = 35 total cells analyzed for GFP control, n = 36 for GFP-PLCδ-PH. Right images show representative entotic cell that did not undergo cell death and maintained a lobe (arrow) protruding from a host cell expressing GFP- PLCδ-PH (green). H2B-mCherry expression in cell nuclei is shown in red. **p = 0.04 (Student’s t test).
(B) Expression of PLCδ-PH::Venus in U cells inhibits linker cell cytoplasmic translocation of mCherry::UtrCH. Graph shows the cortical-to-cytoplasmic ratio of mCherry::UtrCH fluorescence (blue line, left y axis) in a linker cell from the time of engulfment marked by lobe formation. Red line shows the cortical actin ratio from control linker cell for comparison (from Figure 1D). Black line (right y axis) shows the distance of lobe separation from linker cell over time.
(C) High-level PLCδ-PH::Venus expression (>2-fold over background fluorescence intensity), but not low-level expression (<2-fold over background), in U cells inhibits linker cell death. Graph shows the percentage of linker cells that underwent cell death (determined by loss of cortical actin) within 45 min of lobe formation inside of U cells expressing GFP (n = 32) or low (n = 12) and high (n = 23) levels of PLCδ-PH::Venus. **p < 0.015; ***p < 0.001 (Fisher’s exact test).
(D) Representative images of a U cell expressing lin-48p::PLCδ-PH::Venus (green) with an engulfed mCherry::UtrCH-expressing linker cell 25 min (top) and 120 min (bottom) after lobe formation. Grayscale images show mCherry::UtrCH and PLCδ-PH::Venus fluorescence from the region indicated by the hatched box. Note that the linker cell maintains cortical actin as compared to the linker cell from a control experiment (right images, GFP-expressing U cell). See also Video S6.
(E) A surviving linker cell inside a PLCδ-PH::Venus-expressing U cell maintains an attached lobe. Images show linker cell mCherry::UtrCH fluorescence over the indicated time course. Note that the lobe (arrow, top images, maximum projections) stays attached, and cortical actin (bottom images, individual x-y plane images) is maintained. See also Video S7.
Scale bars, 10 μm.
Linker Cell Death Is Inhibited by PIP2 Reporter Expression in U Cells
Since GFP-PLCδ-PH expression in host cells reduced entotic cell death, we generated C. elegans expressing a fluorescently tagged PLCδ-PH in U cells (lin-48 promoter::PLCδ-PH::Venus) and examined the effect on linker cell death. While linker cells were engulfed inside of U cells expressing this reporter, linker cell death, measured by mCherry::UtrCH cytoplasmic relocalization, was delayed when U cells expressed high levels, but not low levels, of PLCδ-PH::Venus (Figures 2B and 2C; Video S6). High levels of reporter expression have been shown to disrupt various processes by sequestering PIP2, such as cell spreading (Szymańska et al., 2008) and myoblast fusion in Drosophila, while low levels are permissive (Bothe et al., 2014). 87% of linker cells inside of U cells with high PLCδ-PH::Venus expression failed to die by 45 min after engulfment and retained cortical localization of mCherry::UtrCH, whereas 58% inside of U cells with lower expression (<2-fold above background) and 78% inside of U cells expressing GFP underwent cell death by this time (Figure 2C). For cells that survived inside of high-PLCδ-PH::Venus-expressing U cells, nearly 50% appeared viable even after 5 h at the end of the time-lapse imaging, and the PIP2 reporter remained localized to the vacuole membrane (Figure 2D).
Lobe Cleavage Is Inhibited by PIP2 Reporter Expression in U Cells
We further examined surviving linker cells and found that lobes often remained connected to the linker cell body when U cells expressed high levels of the PIP2 reporter (Figure 2E; Video S7). Out of six lobes tracked by long-term 4D imaging, four stayed connected to the linker cell body for the duration of the experiment (from 2 to 5 h after engulfment) by extended, thin projections (Figure 2E; Video S7). Of the remaining two lobes, one eventually separated from the linker cell body after nearly 2 h and as the linker cell died, while the other appeared to break after becoming significantly extended, yet the linker cell stayed alive, as judged by cortical mCherry::UtrCH localization (Video S6). These data demonstrate that linker cell death and lobe scission can be disrupted by expression of the PLCδ-PH reporter within engulfing cells.
Nuclear Crenellation Occurs during Entosis
We sought to examine further potential similarities between linker cell clearance and entosis and considered if a reported morphological feature of linker cell death, crenellation, or invagination of the nucleus (Abraham et al., 2007), might occur during entotic cell death. By electron microscopy, 83% of internalized cells versus 21% of host cells exhibited nuclear invaginations resembling those reported in linker cells (Figure 3A) (Blum et al., 2012). Linker cell crenellation occurs prior to engulfment (Keil et al., 2017; Kutscher et al., 2018), and we also observed that crenellation occurred within entotic cells that were not completely engulfed, consistent with this morphological feature occurring prior to complete ingestion (Figure S2A).
Figure 3. Linker Cell Clearance and Entosis Involve Nuclear Crenellation, Cell Adhesions, and Cell-Autonomous Control through Actin.

(A) Entotic cells exhibit nuclear invagination. Image shows a scanning electron micrograph (serial block face) of an internalized entotic cell and host cell. Nuclear invaginations indicated with blue arrowheads. Graph shows the percentage of entotic cell (n = 6) or host cell (n = 14) nuclei with invaginated (black bars) or normal (gray bars) morphology. **p < 0.02 (Fisher’s exact test).
(B) DsRed-tagged HMP-2 expressed in the linker cell (lag-2p::hmp-2::DsRed) forms foci that colocalize with the ends of engulfing U cell arms(arrows). U cell GFP (lin-48p::GFP) is shown in green.
(C) β-catenin forms foci during entosis. Images show immunofluorescence staining of β-catenin (green) in MCF-7 cells undergoing entosis. DAPI-stained nuclei are shown in blue.
(D) A partially engulfed linker cell exhibits cell adhesions with a U cell. Transmission electron micrograph of a partially engulfed linker cell from a late L4-stage worm is shown (courtesy of WormAtlas). Green outline shows the shape of an engulfing U cell, and the blue hatched line indicates a linker cell. Boxed region shows lobe structure with cell-cell adhesions evident between the linker cell and U cell (arrows). The linker cell nucleus (pseudocolored in blue) appears crenellated.
(E) Linker cell actin is required for engulfment. Graph shows the percentage of linker cells expressing wild-type actin (act-2, left bar) or a temperature-sensitive (ts), dominant-negative mutant of actin (lag-2p::act-2(or621)::DsRed) (right bars) that are engulfed at the permissive (16°C) versus restrictive (26°C) temperature into U cells expressing GFP (lin-48p::GFP). *p < 0.05 (Fisher’s exact test). Images show representative engulfed linker cell at the permissive temperature (top) and a representative linker cell in contact with a U cell, but not engulfed at the restrictive temperature (middle and bottom). Dashed lines indicate linker cell (white) and U cell (blue) outlines. n indicates the total number of worms analyzed from more than three independent experiments. Scale bars, 10 μm.
(F)Linker cell death is inhibited by the him-4 mutation that disrupts migration. Graph shows the percent linker cells that undergo cell death (determined by loss of cortical actin and NLS-GFP) by 8 h after the onset of the L4-to-adult transition in control or him-4 mutant C. elegans with disrupted migration. n indicates the total number of worms analyzed from more than three independent experiments. *p < 0.05 (Fisher’s exact test). Images show a control linker cell at the onset of the L4-to -adult transition (time 0, blue arrow), with cortical actin and nuclear-localized GFP, and 2 h after the L4-to-adult transition (+2 hr), by which point it is engulfed and dead, as evidenced by loss of cortical actin and NLS-GFP.
(G)Images show a linker cell in him-4 mutant that migrates toward the head and is still alive with cortical actin and NLS-GFP 10 h after the onset of the L4-to-adult transition. Sperm are visible (black arrowhead) in the gonad that extends behind the linker cell. See also Video S8.
Scale bars, 10 μm.
Linker Cell Clearance Involves Cell-Cell Adhesions
Entosis occurs through an adherens-junctions-mediated mechanism (Overholtzer et al., 2007). To investigate if the linker cell forms adherens junctions with engulfing U cells, an exogenous DsRed-fused β-catenin protein, HMP-2, was expressed in linker cells (lag-2 promoter::hmp-2::DsRed) and examined by 4D imaging. HMP-2 formed foci localized to the tips of engulfing U cell arms, at the advancing engulfment interface (Figure 3B), in a pattern mimicking the localization of β-catenin during entosis (Figure 3C) (Overholtzer et al., 2007; Sun et al., 2014a). We further examined transmission electron micrographs of late-stage L4 males from Sulston and colleagues, accessed through WormAtlas (Altun et al., 2002-2006), and identified a late L4-stage linker cell that was nearly completely engulfed with the exception of a protruding lobe structure (Figures 3D and S2B). At the interface between the linker cell and U cell, we noted the presence of cell adhesions identified by characteristic electron density, which resemble junctions formed during entosis (Figures 3D and S2) (Overholtzer et al., 2007; Sun et al., 2014a).
Linker Cells Control Clearance in an Actin-Dependent Manner
Internalizing entotic cells contribute actively to their ingestion through a mechanism requiring actin and myosin and regulated by Rho-GTPase (Overholtzer et al., 2007; Sun et al., 2014b). To examine if the linker cell participates actively in engulfment, we expressed a temperature-sensitive, dominant-negative allele of actin (lag-2 promoter::act-2(or621)::DsRed) in the linker cell and investigated the effects on uptake into U cells. As shown in Figures 3E and S3, while the majority of linker cells were engulfed and killed at the permissive temperature (16°C), switching to the nonpermissive temperature (26°C) when linker cells were in contact with U cells, or as they approached the completion of migration, inhibited engulfment. By contrast, linker cells expressing a wild-type act-2 construct (lag-2 promoter::act-2::DsRed) underwent efficient engulfment at 26°C (Figures 3E and S3). These data demonstrate a linker cell-autonomous role of actin in controlling uptake into U cells.
Disrupting Linker Cell Migration Inhibits Cell Death
To examine if linker cell death and engulfment require U cell contact, we determined the fate of linker cells that migrate to the head region in him-4 mutant worms, where disrupted matrix deposition alters linker cell migration (Vogel and Hedgecock, 2001). Linker cells in wild-type and him-4 mutant (e1267) male worms were engineered to express the F-actin reporter (lag2p::mCherry::UtrCH) and a nuclear-localized GFP (lag2p::4xNLS-GFP) recently utilized to indicate cell viability in C. elegans (Johnsen and Horvitz, 2016). 26 of 27 linker cells examined in him-4 mutant worms exhibited disrupted migration, either remaining in the midbody region (n = 12) or migrating toward the head (n = 14), while one linker cell migrated to the tail. As shown in Figure 3F, when linker cells migrated to the tail in wild-type worms, they underwent engulfment and efficient cell death marked by loss of cortical actin and nuclear GFP. By contrast, linker cells with disrupted migration in him-4 mutant worms were inhibited for cell death, as 88.5% of linker cells maintained cortical actin and also nuclear localization of NLS-GFP for at least 8 h post-onset of the L4-to-adult transition (marked by tail morphology), compared to only 14% of linker cells in wild-type worms (Figures 3F and 3G; Video S8). The one linker cell that migrated to the tail in a him-4 mutant worm underwent cell death (not shown). These data demonstrate that linker cell death is delayed when proper migration is disrupted.
Linker Cell Lobes Persist at the Site of Gonad-to-Cloaca Fusion and Are Cleared during Mating
To examine the fate of separated linker cell lobes, we imaged their positioning in worms expressing a marker of cell adhesions, the apical junction molecule 1 (ajm-1) protein fused to GFP (ajm-1p::ajm-1::GFP), which marks cell adhesions in the cloaca and between the linker cell and vas deferens cells (Figures 4A and S4) (Kato et al., 2014). The linker cell lobe was oriented between the gonad and cloaca as the linker cell body was engulfed (Figures 4A and S4). After linker cell clearance, the lobe persisted in the same position as the gonad and cloaca moved closer together (Figure 4A). We also noted that linker cell lobes were observable in adult worms, positioned near the gonad and directly posterior to sperm (Figure 4B). As linker cell lobes persisted into adulthood, we considered that they may remain until mating and be cleared during sperm exit. To examine this, we mated males expressing lag-2p::GFP with hermaphrodites and examined the effect on the presence of lobe structures. Whereas more than half of adult male worms that did not mate maintained lobes, all of the worms that mated lost their lobes (Figure 4C), demonstrating that lobes formed by linker cell entosis are long-lived and that their clearance is facilitated by mating.
Figure 4. Persistence of the Linker Cell Lobe and Entosis Model.

(A) The linker cell lobe remains at the site of gonad-to-cloaca fusion after cleavage. Images from 4D imaging show the linker cell (LC) and lobe (L) in red (DsRed) and the cell adhesion marker apical junction molecule 1 (AJM-1) in green (ajm-1p::ajm-1::GFP) that marks cell junctions. The gonad is outlined in by the dashed blue region and the cloaca by the dashed purple region, and the hypothetical positioning of the engulfing U cell (left) is shown by the dashed orange region. Bottom images show regions indicated by hashed white boxes in the top images. The lobe (left) lies between the gonad and cloaca and remains at this site even after the linker cell body is cleared and the gonad and cloaca appear to undergo fusion (right images). Times are indicated as h:min; scale bar, 10 μm.
(B) The linker cell lobe is observable in adult male worms posterior to sperm. Green fluorescence shows lag-2p::GFP expression; the lobe and sperm are indicated in the boxed region (right). Scale bar, 5 μm.
(C) Mating facilitates lobe removal. Graph shows the percentage of adult male worms with lag-2p::GFP-labeled lobes after mating with hermaphrodites (left bar, n = 12) or growth in the absence of hermaphrodites (right bar, n = 13). *p < 0.02 (Fisher’s exact test).
DISCUSSION
Here, we investigated the mechanism of clearance for the linker cell in C. elegans and identified numerous similarities with entosis. Both involve the establishment of cell-cell adhesions, are promoted by an invasion-like mechanism requiring actin, lead to the separation of a lobe from internalizing cells, result in the ingestion of viable cells whose death can be delayed, and are associated with nuclear crenellation. While definitive experiments to probe the requirement of cell-cell adhesion molecules and Rho-GTPase for linker cell engulfment await further studies, based on these criteria, we propose that the linker cell is cleared by entosis. While entosis occurs in human cancers (Overholtzer et al., 2007), our data and a previous report linking entosis to embryo implantation (Li et al., 2015) implicate this process in regulating key events required for fertility. Entosis was also recently implicated in controlling the turnover of developing spermatids by Sertoli cells, suggesting yet further links between fertility and this process (Ahmed et al., 2017).
The control over linker cell clearance by entosis raises the question of why this program would be selected for this cell. One possibility is that an adhesion-based mechanism ensures that the linker cell completes migration and establishes adhesion between the gonad and cloaca prior to being eliminated. Another possibility is that the migratory activity of the linker cell might prime it to undergo this mechanism once reaching its target, as entosis involves invasive activity (Overholtzer et al., 2007; Sun et al., 2014a, 2014b). A further possibility is that the lobe could have a specialized function. Similar lobes were recently shown to form and shed from primordial germ cells (PGCs), which may remodel the cytoplasm to facilitate PGC maturation (Abdu et al., 2016). Lobes are also reported to be cleaved from leukocytes undergoing transendothelial migration and are left behind in blood vessels, where they could conceivably promote junctional resealing between endothelial cells (Hyun et al., 2012). Our data suggest that linker cell lobes could participate in the fusion of the gonad and cloaca. Lobes could provide a positional or timing cue that signifies linker cell clearance or perhaps participate more directly in fusion. We find that lobe clearance is facilitated by mating, suggesting that lobes could also serve as a barrier to protect the developing gonad from the external environment until adulthood. Central to this model is our observation that the linker cell lobe persists for long periods of time while the cell body is rapidly degraded (Video S2A), which differs from a recent model of competitive phagocytosis involving rapid lobe clearance (Kutscher et al., 2018). Whether technical differences could underlie these different observations awaits further study.
Our findings implicate U cell contact in supporting entosis and linker cell death, as linker cells in him-4 mutant worms exhibited significantly increased survival. While these data are generally consistent with previous findings that have also shown reduced linker cell death in this context (Abraham et al., 2007), the extent of the defect that we observe by our time-lapse imaging approach is much greater. Phagocytosis of improperly migrated linker cells is also reported to occur at low frequency in the him-4 background, which has suggested a specialized phagocytic function of U cells (Kutscher et al., 2018). Our findings implicate U cells in mediating clearance through entosis, a process where host cell functions are not well described. Whether the engulfment machinery recently identified in U cells, including RAB-35, could play a specialized role to control entosis awaits further studies (Kutscher et al., 2018).
While we have investigated the mechanism of linker cell clearance, several previous studies have identified genes required for linker cell death. Linker cell death occurs independently of genes required for apoptosis (Abraham et al., 2007) and involves three parallel pathways identified by genetic studies (egl-20/wnt and lin-44/wnt, lin-29, and sek-1/mapkk upstream of pqn-41) that impinge on the induction of heat shock factor 1 (HSF-1) activity to induce the expression of let-70 and other components of the ubiquitin proteasome system that are required cell autonomously for linker cell death (Blum et al., 2012; Kinet et al., 2016; Malin et al., 2016). Importantly, loss of function of these key signaling pathways blocks linker cell death as well as engulfment. Our findings raise the possibility that these pathways participate in a linker cell-autonomous role in uptake into U.lp or U.rp and that U cells could play an active role in cell death execution. While we show that the execution of linker cell death can be delayed even after engulfment, our data do not rule out that linker cells could initiate cell death prior to engulfment, and entosis could enhance death execution. Entotic cells and linker cells exhibit nuclear crenellation prior to the completion of cell internalization (Figures 3D and S2) (Kutscher et al., 2018), and unengulfed linker cells in sek-1 and pqn-41 mutant backgrounds exhibit organelle swelling that is considered a hallmark of linker cell death (Blum et al., 2012). Phagocytosis can promote apoptotic cell death in C. elegans (Hoeppner et al., 2001; Johnsen and Horvitz, 2016; Reddien et al., 2001). It is conceivable that entosis could provide a similar function for linker cell death.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Overholtzer (overhom1@mskcc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All strains were maintained on Escherichia coli OP50 seeded nematode growth medium (NGM) plates. Temperature sensitive strains were maintained at 16°C. Transgenic lines were generated by injecting plasmid DNA directly into hermaphrodite gonad. To generate him-4 mutant males with wimEx83, him-8 mutant males with wimEx83 were mated with him-4 mutant hermaphrodites, and offspring were screened morphologically for him-4 phenotype (Vogel and Hedgecock, 2001).
METHOD DETAILS
Cell culture and constructs
MCF-7 cells (Lombardi Cancer Center, Georgetown University, Washington, D.C.) were cultured in DMEM (11965-092; Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (F2442; Sigma-Aldrich) and penicillin/streptomycin (30-002-CI; Mediatech). Cells expressing the H2B–mCherry nuclear marker were prepared by transducing cells with retroviruses made with the pBabe–H2B–mCherry construct, as described (Florey et al., 2011). Cells were transfected with expression constructs by nucleofection (Cell Line Nucleofector Kit V, VCA-1003; Lonza, Basel, Switzerland) according to manufacturer’s protocol and assayed 24 hours post transfection. The following expression constructs were used: pQCXIP–GFP and GFP–C1–PLCδ-PH (a gift from Tobias Meyer (Addgene plasmid # 21179; http://addgene.org/21179; RRID:Addgene_21179) (Stauffer et al., 1998)). To observe tail formation and cleavage during entosis, MCF-7 cells were labeled with 10 μm Cell Tracker dyes (green or red, C7025 and C34552, respectively; Life Technologies, Grand Island, NY) for 20 minutes at 37°C, then plated at a 1:1 ratio at a total cell density of 250,000 cells in 35 mm glass-bottom dishes overnight in media containing 10 μm Y-27632 (#1254, Tocris Bioscience) to block entosis. The next day cells were washed three times with PBS, fresh media was added, and analyzed by confocal microscopy.
Cell fate analysis by time-lapse microscopy
MCF-7 cells expressing either GFP or GFP–PLCδ-PH were mixed 1:1 with MCF-7 H2B-mCherry cells (250,000 cells total), plated on 35 mm glass-bottom dishes (P06G-1.5-20-F; MatTek, Ashland, MA), and allowed to adhere overnight. Entotic structures consisting of GFP or PLCδ-PH-expressing cells on the outside and H2B–mCherry-expressing cells on the inside were imaged for 24 hours and the cell fate recorded. Inner cell death was determined by DIC morphology and the diffusion of mCherry from the nucleus to the inner cell cytoplasm. Fluorescence and differential interference contrast (DIC) images were acquired every 15 minutes for 24 hours using a Nikon Ti-E inverted microscope attached to a CoolSNAP charge-coupled device camera (Photometrics, Tucson, AZ) and NIS Elements software (Nikon, Melville, NY).
Cloning of C. elegans expression vectors
Expression plasmids for transgenic worm lines were constructed using the pFx_Plag-2::venusT vector, which was a kind gift from Shohei Mitani (Gengyo-Ando et al., 2006). The lag-2 promoter was used for linker cell transgenic expression. Plasmids with different fluorescent proteins were generated by replacing the sequence encoding venusT with sequences encoding either EGFP or DsRedexpress1. For U cell expression, the lag-2 promoter sequence was replaced with the sequence encoding the lin-48 promoter amplified from pPK699, which was a kind gift from Patricia Kuwabara (Soloviev etal., 2011). Using BamHI and NotI site in the expression plasmid, the promoter region was cloned by amplification of the promoter region by polymerase chain reaction followed by In-Fusion cloning according to manufacturer’s protocol (638909; Takara Bio USA, Inc., Mountain View, CA). For translational fusion, chromosomal DNA of C. elegans was amplified and cloned at NotI site by In-Fusion cloning (638910; Takara Bio USA, Inc., Mountain View, CA) or ligation with T4 DNA ligase according to manufacturer’s protocol (M0202; New England BioLabs, Inc., Ipswich, MA). Sequences encoding the pleckstrin homology (PH) domain of human Phospholipase Cδ1 (PLCδ) and the calponin homology (CH) domain of human Utrophin (Utr) were cloned into the expression plasmid. GFP-C1-PLCδ-PH was a gift from Tobias Meyer (Addgene plasmid # 21179; http://addgene.org/21179 ; RRID:Addgene_21179) (Stauffer et al., 1998). GFP-UtrCH and mCherry-UtrCH were gifts from William Bement (Addgene plasmid # 26737 ; http://addgene.org/26737 ; RRID:Addgene_26737 and #26740 ;http://addgene.org/26740 ; RRID:Addgene_26740) (Burkel et al., 2007). Sequences encoding lmp-1, act-2, and hmp-2 were amplified from chromosomal DNA of the N2 strain and cloned into the expression plasmid. A semi-dominant temperature-sensitive allele of act-2 was amplified from chromosomal DNA of the act-2(or621) strain and cloned into the expression plasmid.
Worm microinjection
To obtain germ-line transformation of C. elegans expression plasmids with a selection marker were injected in the cytoplasm of the syncytial gonad. Microinjection was done generally by the method described in WormBook (Evans 2006). DNA solution was prepared by mixing the expression plasmid (10-20 μg/ml) and pRF4 (rol-6(su1006)) (100 μg/ml). Final concentration of DNA mixture was adjusted to 200 μg/ml with pBluescript. The DNA solution was loaded to a needle made from a borosilicate glass capillary (Kwik-Fil™ (1B100F-4), World Precision Instruments, Inc., Sarasota, FL) using microloader tip (Eppendorf AG, Germany). The needle was made using Sutter micropippette Puller (P-2000, Sutter Instrument, Novato, CA). DNA solution was injected to 20-30 well-fed gravid hermaphrodites using Axio Observer.A1 inverted microscope (Carl Zeiss Microscopy GmbH, Germany) equipped with three-axis hanging joystick oil hydraulic micromanipulator (MMO-202ND, Narishige, Japan), and microINJECTOR™ (Tritech Research, Inc., Los Angeles, CA).
Worm imaging
Male worms at mid L4 stage were picked from NGM plates and used for live imaging. Worms were transferred to an agarose pad placed on a 24×60 mm No. 1.5 cover glass (12-545M; Fisherbrand, Pittsburgh, PA) and anesthetized instantly by dropping 1 μL of 0.06% (−)-tetramisole hydrochloride (L9756; Sigma-Aldrich, St. Louis, MO) dissolved in water onto the agarose pad. The agarose pad was prepared by placing 20 μL of 3% molten agarose between 2 cover glasses separated by a 0.31 mm thick plastic spacer. After mounting worms, a 20×60 mm 0.25 mm-thick Press to seal silicone sheet (70338-29; Electron Microscopy Sciences, Hatfield, PA) with a 12.7 mm circular hole was placed on the cover glass. The worm mounting was finished by covering the same cover glass on top of the agarose pad. Confocal microscopy was performed with the Ultraview Vox spinning-disk confocal system (Perkin Elmer, Waltham, MA) equipped with a Yokogawa CSU-X1 spinning-disk head and an electron-multiplying charge-coupled device camera (Hamamatsu C9100-13) coupled to a Nikon Ti-E microscope equipped with a CFI Plan Apo VC 60 × oil objective. Z stacks (0.5 or 1 μm steps) were acquired with a Piezo z stack drive controlled by a nano drive (Mad City Lab, Madison, WI). All image analyses were performed using Volocity software (Perkin Elmer, Waltham, MA) or ImageJ(Schneider et al., 2012). Whole cell GFP intensities were quantified from maximum projections. Cortical to cytoplasmic ratios for GFP::UtrCH or mCherry::UtrCH were quantified on single midplane confocal images using regions drawn around the cell cortex or cytoplasm; ratios or GFP intensities were normalized to time 0. Incubation temperatures during confocal imaging were controlled by a CherryTemp microfluidic temperature control system (Cherry Biotech, France). The PDMS microfluidic device of the system was mounted on top of the cover glass. For imaging of temperature-sensitive allele of act-2, the dual channel temperature controller was set to 16°C and 26°C as permissive and non-permissive temperatures, respectively. For the timing of L4 to adult transition, time 0 was indicated by the first separation of the cuticle from the tail and observation of tail ray structures by DIC microscopy.
Worm mating
Late L4 stage male worms (8 to 15 worms), which have retracted tail-tip, expressing lag-2promoter::GFP were mixed with equal number of hermaphrodites for 10 hours at room temperature to allow for the completion of adult development and mating, or cultured alone for 10 hours. Males were then collected and imaged by confocal microscopy for the presence of GFP-labeled lobe structures.
Electron microscopy
For serial block face scanning electron microscopy (SBF-SEM), cells were prepared and mounted as described (Russell et al., 2017). SBF-SEM data was collected using a 3View2XP (Gatan, Pleasanton, CA) attached to a Sigma VP SEM (Zeiss), following our published protocol (Russell et al., 2017). Transmission electron microscopy was performed as described (Sun et al., 2014a). Images were obtained using a JEOL 1200 EX transmission electron microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification and statistical parameters are indicated in the legends of each figure, including error bars (SEM), n numbers, and p values. For statistical tests we applied either Fisher’s exact test or Student’s t test, as indicated. p values of < 0.05 were considered significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cell Tracker green | Life Technologies | Cat# C7025 |
| Cell Tracker red | Life Technologies | Cat# C34552 |
| Y-27632 | Tocris Bioscience | Cat# 1254 |
| (−)-tetramisole hydrochloride | Sigma-Aldrich | L9756 |
| Critical Commercial Assays | ||
| Cell Line Nucleofector Kit V | Lonza | Cat# VCA-1003 |
| In-Fusion cloning kit | Takara Bio | Cat# 638910 |
| Experimental Models: Cell Lines | ||
| MCF-7 | ATCC | HTB-22 |
| Experimental Models: Organisms/Strains | ||
| C. elegans: N2 | Caenorhabditis Genetics Center | N2 |
| C. elegans: him-4(e1267) X | Caenorhabditis Genetics Center | CB1267 |
| C. elegans: him-8 (e1489) IV | Caenorhabditis Genetics Center | CB1489 |
| C. elegans: act-2 (or621)V | Caenorhabditis Genetics Center | EU1295 |
| C. elegans: qIs56 V [Plag-2::GFP + unc-119(+)] | Caenorhabditis Genetics Center | JK2868 |
| C. elegans: syIs50 [Pcdh-3::GFP + dpy-20(+)] | Caenorhabditis Genetics Center | PS3352 |
| C. elegans: him-5(e1490) V; syIs78 [ajm-1::GFP + unc-119(+)] | Caenorhabditis Genetics Center | PS4657 |
| C. elegans: him-8 (e1489) IV; syIs50 [Pcdh-3::GFP + dpy-20(+)] | This paper | him-8- syIs50 |
| C. elegans: him-8 (e1489) IV; wimEx3 [Plag-2::hmp-2::DsRed + Plin-48::EGFP + pRF4(rol-6(su1006))] | This paper | him-8- wimEx3 |
| C. elegans: him-8 (e1489) IV; wimEx5 [Plag-2::GFP-UtrCH + pRF4(rol-6(su1006))] | This paper | him-8- wimEx5 |
| C. elegans: him-8 (e1489) IV; wimEx8 [Plin-48::lmp-1::DsRed + pRF4(rol-6(su1006))] | This paper | him-8- wimEx8 |
| C. elegans: him-8 (e1489) IV; wimEx30 [Plag-2::mCherry-UtrCH + Plin-48::PH::Venus + pRF4(rol-6(su1006))] | This paper | him-8- wimEx30 |
| C. elegans: him-8 (e1489) IV; wimEx52 [Plag-2::act-2::DsRed + Plin-48::EGFP + pRF4(rol-6(su1006))] | This paper | him-8- wimEx52 |
| C. elegans: him-8 (e1489) IV; syIs50 [Pcdh-3::GFP + dpy-20(+)], wimEx59 [Plag-2::act-2 (or621)::DsRed + pRF4(rol-6(su1006))] | This paper | him-8- syIs50- wimEx59 |
| C. elegans: him-8 (e1489) IV; wimEx63 [Plag-2::mCherry-UtrCH + Plin-48::EGFP + pRF4(rol-6(su1006))] | This paper | him-8- wimEx63 |
| C. elegans: him-8 (e1489) IV; wimEx74 [Plag-2::DsRed + pRF4(rol-6(su1006))] | This paper | him-8- wimEx74 |
| C. elegans: him-8 (e1489) IV; wimEx77 [Plag-2::tagBFP2 + Plin-48::DsRed + pRF4(rol-6(su1006))] | This paper | him-8- wimEx77 |
| C. elegans: him-8 (e1489) IV; wimEx83 [Plag-2::mCherry-UtrCH + Plag-2::NLS-GFP+ pRF4(rol-6(su1006))] | This paper | him-8- wimEx83 |
| C. elegans: him-4(e1267) X; wimEx83 [Plag-2::mCherry-UtrCH + Plag-2::NLS-GFP+ pRF4(rol-6(su1006))] | This paper | him-4- wimEx83 |
| C. elegans: him-5(e1490) V; syIs78 [ajm-1::GFP + unc-119(+)], wimEx77 [Plag-2::tagBFP2 + Plin-48::DsRed + pRF4(rol-6(su1006))] | This paper | syIs78- wimEx77 |
| Oligonucleotides | ||
| lin-48 promoter Forward primer for infusion: GGTTCCGCGTGGATCCCCTGCATTTTTTTCAGAGTCTATAATATCCGT | eurofins | N/A |
| lin-48 promoter Reverse primer for infusion: GCTCACCATGCGGCCGCCTGAAATTGAGCAGAGCTGAAAATTTTTGT | eurofins | N/A |
| lmp-1 forward primer: AAGCGGCCGCATGTTGAAATCGTTTGTCAT | eurofins | N/A |
| lmp-1 reverse primer: TTGCGGCCGCCGACGCTGGCATATCCTTGTC | eurofins | N/A |
| act-2 forward primer: AAATGATCGGCGGCCGCATGTGTGACGACGATGTTGCCGCTCTCGTA | eurofins | N/A |
| act-2 reverse primer: GCTCACCATGCGGCCGCCGAAGCATTTGCGATGAACAATTGAT | eurofins | N/A |
| hmp-2 forward primer for infusion: AAATGATCGGCGGCCGCATGCGATTATTCTCATATTTGGACG | eurofins | N/A |
| hmp-2 reverse primer for infusion: GCTCACCATGCGGCCGCCCAAATCGGTATCGTACCAATTGTGA | eurofins | N/A |
| Recombinant DNA | ||
| pBabe–H2B–mCherry | Joan Brugge Lab | N/A |
| pQCXIP-GFP | Xuejun Jiang Lab | N/A |
| GFP–C1–PLCdelta-PH | Stauffer et al., 1998 | Addgene Plasmid #21179 |
| pFx_Plag-2::venusT | Shohei Mitani Lab | N/A |
| pPK699 | Patricia Kuwabara Lab | N/A |
| GFP-UtrCH | Burkel et al., 2007 | Addgene plasmid #26737 |
| mCherry–UtrCH | Burkel et al., 2007 | Addgene plasmid #26740 |
| pRF4 (rol-6(su1006)) | Cole Haynes Lab | N/A |
| pBluescript | Cole Haynes Lab | N/A |
| Plin-48::EGFP | This paper | Plasmid #113 |
| Plag-2::hmp-2::DsRed | This paper | Plasmid #43 |
| Plag-2::GFP-UtrCH | This paper | Plasmid #35 |
| Plin-48::lmp-1::DsRed | This paper | Plasmid #42 |
| Plag-2::mCherry-UtrCH | This paper | Plasmid #36 |
| Plin-48::PH::Venus | This paper | Plasmid #51 |
| Plag-2::act-2::DsRed | This paper | Plasmid #68 |
| Plag-2::act-2 (or621)::DsRed | This paper | Plasmid #70 |
| Plag-2::DsRed | This paper | Plasmid #23 |
| Plag-2::tagBFP2 | This paper | Plasmid #123 |
| Plin-48::DsRed | This paper | Plasmid #114 |
| Plag-2::NLS-GFP | This paper | Plasmid #127 |
| Software and Algorithms | ||
| NIS Elements software | Nikon | N/A |
| Volocity image analysis software | Perkin Elmer | N/A |
| ImageJ | Schneider et al., 2012 | N/A |
| Other | ||
| 35 mm glass-bottom dishes | MatTek | Cat# P06G-1.5-20-F |
| Kwik-Fil borosilicate glass capillary | World Precision Instruments, Inc. | Cat# 1B100F-4 |
| 24×60 mm No. 1.5 cover glass | Fisherbrand | 12-545M |
| 0.25 mm-thick clear silicone sheet | Electron Microscopy Sciences | Cat# 70338-29 |
Highlights.
Linker cell engulfment and entosis generate subcellular lobe structures
Linker cell actin is required for clearance
Lobe cleavage is required for linker cell death
Entosis controls linker cell clearance
ACKNOWLEDGMENTS
We thank members of the Overholtzer lab for helpful discussions. This work was supported by grants from the NIH (RO1AG047182 to C.M.H.); the National Cancer Institute (RO1CA154649 to M.O.); the Benjamin Friedman Research Fund (to M.O.); Cancer Research UK (C47718/A16337 to O.F.); the MRC, BBSRC, and EPSRC under grant award MR/K01580X/1 (to L.M.C.); and the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC001999), the UK Medical Research Council (FC001999), and the Wellcome Trust (FC001999). The transmission electron microscopy (TEM) image from animal JSG shown in Figures 3D and S2B was provided from the Worm-Image website (http://www.wormimage.org), an archive maintained by David Hall at Albert Einstein College of Medicine. The JSG images were transferred to Dr. Hall through the generous efforts of Jonathan Hodgkin and John White from their MRC/LMB laboratory in Cambridge, England. Support for Worm-Image comes from NIH grant OD 010943 to D.H. Some C. elegans strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found with this article online at https://doi.org/10.1016/j.celrep.2019.02.073.
REFERENCES
- Abdu Y, Maniscalco C, Heddleston JM, Chew TL, and Nance J (2016). Developmentally programmed germ cell remodelling by endodermal cell cannibalism. Nat. Cell Biol 18, 1302–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham MC, Lu Y, and Shaham S (2007). A morphologically conserved nonapoptotic program promotes linker cell death in Caenorhabditis elegans. Dev. Cell 12, 73–86. [DOI] [PubMed] [Google Scholar]
- Ahmed N, Yang P, Huang Y, Chen H, Liu T, Wang L, Nabi F, Liu Y, and Chen Q (2017). Entosis acts as a novel way within sertoli cells to eliminate spermatozoa in seminiferous tubule. Front. Physiol 8, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altun ZF, Herndon LA, Wolkow CA, Crocker C, Lints R, and Hall DH (2002-2006). WormAtlas. http://www.wormatlas.org. [Google Scholar]
- Blum ES, Abraham MC, Yoshimura S, Lu Y, and Shaham S (2012). Control of nonapoptotic developmental cell death in Caenorhabditis elegans by a polyglutamine-repeat protein. Science 335, 970–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothe I, Deng S, and Baylies M (2014). PI(4,5)P2 regulates myoblast fusion through Arp2/3 regulator localization at the fusion site. Development 141, 2289–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkel BM, von Dassow G, and Bement WM (2007). Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil. Cytoskeleton 64, 822–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans TC (2006). Transformation and microinjection In WormBook, The C. elegans Research Community (WormBook). [Google Scholar]
- Florey O, Kim SE, Sandoval CP, Haynes CM, and Overholtzer M (2011). Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol 13, 1335–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagos-klonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al. (2012). Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gengyo-Ando K, Yoshina S, Inoue H, and Mitani S (2006). An efficient transgenic system by TA cloning vectors and RNAi for C. elegans. Biochem. Biophys. Res. Commun 349, 1345–1350. [DOI] [PubMed] [Google Scholar]
- Hoeppner DJ, Hengartner MO, and Schnabel R (2001). Engulfment genes cooperate with ced-3 to promote cell death in Caenorhabditis elegans. Nature 412, 202–206. [DOI] [PubMed] [Google Scholar]
- Hyun YM, Sumagin R, Sarangi PP, Lomakina E, Overstreet MG, Baker CM, Fowell DJ, Waugh RE, Sarelius IH, and Kim M (2012). Uropod elongation is a common final step in leukocyte extravasation through inflamed vessels. J. Exp. Med 209, 1349–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsen HL, and Horvitz HR (2016). Both the apoptotic suicide pathway and phagocytosis are required for a programmed cell death in Caenorhabditis elegans. BMC Biol. 14, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Chou TF, Yu CZ, DeModena J, and Sternberg PW (2014). LINKIN, a new transmembrane protein necessary for cell adhesion. eLife 3, e04449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil W, Kutscher LM, Shaham S, and Siggia ED (2017). Long-term high-resolution imaging of developing C. elegans larvae with microfluidics. Dev. Cell 40, 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinet MJ, Malin JA, Abraham MC, Blum ES, Silverman MR, Lu Y, and Shaham S (2016). HSF-1 activates the ubiquitin proteasome system to promote non-apoptotic developmental cell death in C. elegans. eLife 5, e12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutscher LM, Keil W, and Shaham S (2018). RAB-35 and ARF-6 GTPases mediate engulfment and clearance following linker cell-type death. Dev. Cell 47, 222–238.e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Sun X, and Dey SK (2015). Entosis allows timely elimination of the luminal epithelial barrier for embryo implantation. Cell Rep. 11, 358–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin JA, Kinet MJ, Abraham MC, Blum ES, and Shaham S (2016). Transcriptional control of non-apoptotic developmental cell death in C. elegans. Cell Death Differ. 23, 1985–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GE, Nguyen TM, Nguyen TN, Pereboev A, Kendrick-Jones J, and Winder SJ (1999). Disruption of the utrophin-actin interaction by monoclonal antibodies and prediction of an actin-binding surface of utrophin. Biochem. J 337, 119–123. [PMC free article] [PubMed] [Google Scholar]
- Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW, Cibas ES, and Brugge JS (2007). A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 131, 966–979. [DOI] [PubMed] [Google Scholar]
- Purvanov V, Holst M, Khan J, Baarlink C, and Grosse R (2014). G-protein-coupled receptor signaling and polarized actin dynamics drive cell-in-cell invasion. eLife 3, e02786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddien PW, Cameron S, and Horvitz HR (2001). Phagocytosis promotes programmed cell death in C. elegans. Nature 412, 198–202. [DOI] [PubMed] [Google Scholar]
- Russell MR, Lerner TR, Burden JJ, Nkwe DO, Pelchen-Matthews A, Domart MC, Durgan J, Weston A, Jones ML, Peddie CJ, et al. (2017). 3D correlative light and electron microscopy of cultured cells using serial blockface scanning electron microscopy. J. Cell Sci 130, 278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarantis H, Balkin DM, De Camilli P, Isberg RR, Brumell JH, and Grinstein S (2012). Yersinia entry into host cells requires Rab5-dependent dephosphorylation of PI(4,5)P2 and membrane scission. Cell Host Microbe 11, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soloviev A, Gallagher J, Marnef A, and Kuwabara PE (2011). C. elegans patched-3 is an essential gene implicated in osmoregulation and requiring an intact permease transporter domain. Dev. Biol 351, 242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer TP, Ahn S, and Meyer T (1998). Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr. Biol 8, 343–346. [DOI] [PubMed] [Google Scholar]
- Sun Q, Cibas ES, Huang H, Hodgson L, and Overholtzer M (2014a). Induction of entosis by epithelial cadherin expression. Cell Res. 24,1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Luo T, Ren Y, Florey O, Shirasawa S, Sasazuki T, Robinson DN, and Overholtzer M (2014b). Competition between human cells by entosis. Cell Res. 24, 1299–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymańska E, Sobota A, Czuryto E, and Kwiatkowska K (2008). Expression of PI(4,5)P2-binding proteins lowers the PI(4,5)P2level and inhibits FcgammaRIIA-mediated cell spreading and phagocytosis. Eur. J. Immunol 38, 260–272. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, and Vandenabeele P (2014). Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol 15, 135–147. [DOI] [PubMed] [Google Scholar]
- Vogel BE, and Hedgecock EM (2001). Hemicentin, a conserved extracellular member of the immunoglobulin superfamily, organizes epithelial and other cell attachments into oriented line-shaped junctions. Development 128, 883–894. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.