

Abstract
Mutations in genes encoding sarcomeric proteins are the most important causes of inherited cardiomyopathies, which are a major cause of mortality and morbidity worldwide. Although genetic screening procedures for early disease detection have been improved significantly, treatment to prevent or delay mutation-induced cardiac disease onset is lacking. Recent findings indicate that loss of protein quality control (PQC) is a central factor in the disease pathology leading to derailment of cellular protein homeostasis. Loss of PQC includes impairment of heat shock proteins, the ubiquitin-proteasome system, and autophagy. This may result in accumulation of misfolded and aggregation-prone mutant proteins, loss of sarcomeric and cytoskeletal proteins, and, ultimately, loss of cardiac function. PQC derailment can be a direct effect of the mutation-induced activation, a compensatory mechanism due to mutation-induced cellular dysfunction or a consequence of the simultaneous occurrence of the mutation and a secondary hit. In this review, we discuss recent mechanistic findings on the role of proteostasis derailment in inherited cardiomyopathies, with special focus on sarcomeric gene mutations and possible therapeutic applications.
Keywords: Cardiomyopathy, Protein quality control, Sarcomeric mutation, Heat shock proteins, Ubiquitin-proteasome system, Autophagy
Classification of cardiomyopathies
Cardiomyopathies (CM) constitute one of the most common causes of sudden cardiac death in young adults and represent major causes for cardiac transplantation [88]. Disease onset generally ranges between 20 and 50 years of age. CMs are defined by abnormal myocardial structure and function in the absence of any other diseases sufficient to cause these abnormalities [24]. These can be sub-classified based on their functional phenotype and their specific morphological changes. The most common types are hypertrophic CM (HCM), characterized by increased left ventricular (LV) wall thickness often occurring asymmetrically, and dilated CM (DCM), in which the presence of LV dilatation is accompanied by contractile dysfunction [24]. Besides HCM and DCM, there are less frequent forms, such as restrictive CM (RCM) and desmin-related cardiomyopathy [24]. All these cardiomyopathies can be familial and are typically inherited in an autosomal dominant manner. Mutations in genes encoding sarcomeric proteins are the most common cause of these types of CMs [3]. However, the genotype-phenotype relationship is far from clear. The variations in age of CM onset and disease phenotype suggest that additional factors play a role in CM pathogenesis.
Accumulating evidence indicates the presence of derailed proteostasis in CMs as well as its contribution to CM onset and progression. This derailment could either be caused directly by the mutation or indirectly due to a compensatory mechanism. In the former case, the mutant protein might be instable or improperly folded leading to direct activation of the protein quality control (PQC). In the latter case, the mutation does not interfere with protein folding or stability but causes functional impairment, which in turn leads to cellular stress and indirect activation of the PQC. Furthermore, the “secondary-hit” model may apply in CMs, in which a primary sarcomere mutation enhances vulnerability to secondary stressors, which increases cellular burden resulting in PQC derailment. This review summarizes the current knowledge about perturbations in the different components of the PQC in CMs that are caused by mutations in sarcomeric proteins.
Proteostasis network ensures cardiac health
The heart has a very limited regenerative capacity and therefore requires surveillance by a system that maintains protein homeostasis to ensure cardiac health [106]. The PQC system sustains proteostasis by refolding misfolded proteins or removing them if refolding is impossible. It is composed of heat shock proteins (HSPs), the ubiquitin-proteasome system (UPS), and autophagy. PQC is only then functional when all three components are operative and interact with each other. This means that derailment of one of the parts might impair the function of the others in a direct or indirect manner. In a physiological state, protein folding and refolding is ensured by HSPs and their regulators. Terminally misfolded and aggregation-prone proteins are cleared by the two degradation systems, i.e., the UPS and autophagy that work in collaboration with the HSPs (Fig. 1).
Fig. 1.

Collaboration of the protein quality control components. Stress leads to misfolding of proteins, which may result in abnormal interaction and subsequent aggregation. Small HSPs (white/gray rectangle) and HSPs with ATPase activity (blue moon shape with black rectangle) prevent aggregation formation by binding to the hydrophobic surfaces of misfolded proteins. They either refold the misfolded proteins to its native structure or initiate its polyubiquitination (Ub, orange hexagon). Misfolded proteins with polyubiquitin chains linked to lysine 48 (K48) are mainly degraded by the proteasome. Misfolded proteins carrying K63-linked polyubiquitin chains and aggregated proteins enter the autophagic pathway
First, the different parts of the PQC in normal physiology are described, before addressing their role in CMs.
Heat shock proteins
HSPs, originally identified as heat responsive proteins, are constitutively expressed in the cell to serve as molecular chaperones to ensure correct folding and assembly of proteins. HSPs are classified in two categories: the small HSPs with a low molecular weight (15–30 kDa) and the chaperones with a high molecular weight (> 30 kDa).
One functional group of HSPs are chaperonin complexes, which are ATP-dependent chaperones with a barrel-like structure that provide correct folding of nascent proteins after translation. Besides their folding function during protein translation, HSPs are also induced in response to cellular and environmental stressors to maintain a healthy cellular proteostasis by clearance of misfolded proteins [66, 111].
As reviewed by Garrido et al., small HSPs show an ATP-independent holdase activity. This means that they bind to misfolded proteins, keep them in a state competent for either refolding or degradation, and thereby prevent or attenuate their aggregation. Due to the association of small HSPs with the HSPs that have an ATP-dependent folding activity, the misfolded proteins can be refolded into their native and functional conformation [28]. The binding affinity of HSPs to the misfolded protein is dependent on the chaperone cofactors bound to the HSPs. Furthermore, this binding of chaperone cofactors determines the processing of the misfolded protein for either refolding or degradation. Chaperone cofactors involved in degradation pathways can switch off the refolding activity of HSPs by inhibiting their ATPase activity and assist the HSPs and the UPS or autophagy in the breakdown of misfolded proteins (Fig. 2) [13, 25]. The degradation of the thick filament protein myosin-binding protein C (MyBP-C), for instance, is mediated via the chaperone cofactor HSC70 playing a major role in regulating MyBP-C protein turnover [32]. These degradation pathways are addressed in the following sections.
Fig. 2.
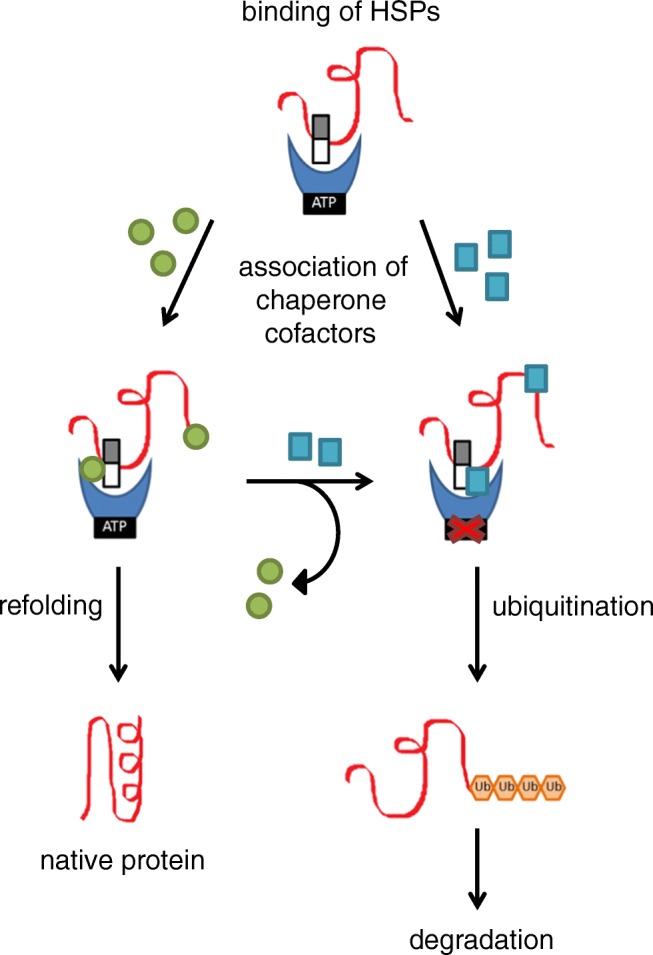
Chaperone cofactor binding determines the heat shock protein (HSP) function. Small HSPs (white/gray rectangle) and HSPs with ATPase activity (blue moon shape with black rectangle) bind to the misfolded protein to stabilize it. Dependent on the chaperone cofactors (green circles or turquoise squares), the misfolded protein gets either refolded or ubiquitinated for subsequent degradation. If refolding is impossible, the chaperone cofactors can be exchanged to promote degradation. In case of ubiquitination, the chaperone cofactors can switch off the HSP refolding activity by blocking the ATPase activity and, together with HSPs, assist in clearance of the misfolded protein via the degradation pathways
To maintain the structure and function of the highly dynamic cardiac sarcomeres, HSPs play an important role. The molecular chaperones GimC (Prefoldin), chaperonin TCP-1 Ring Complex (TRiC), αB-crystallin, and HSP27 ensure correct folding and assembly of proteins, maturation of actin and prevent aggregate formation [10, 14, 34, 36]. HSP27 is mostly found as high-molecular weight oligomers in the cytosol of unstressed cells [23]. Upon stress, HSP27 deoligomerizes and translocates to F-actin and thereby stabilizes the F-actin network [16]. To assemble the myosin thick filament, the chaperones UNC-45, HSP90, and HSP70 are required, whereas the actin filament is self-assembled [7, 8, 94]. Several members of the small HSPs family are expressed in the heart and associate with cytoskeletal proteins [33, 103]. These HSPBs stabilize cytoskeletal structures and improve coping with stress situations [33, 48, 49].
Ubiquitin-proteasome system
In case of terminally misfolded proteins, that failed be refolded, HSPs and their chaperone cofactors recruit enzymes to mediate polyubiquitination of the target substrate and thereby mark them for the appropriate degradation pathway. Short-lived proteins are typically degraded by the UPS, whereas autophagy is mainly used for degrading long-lived proteins and entire organelles [17, 39].
The polyubiquitination of the target substrate requires the sequential action of three enzymes. The ubiquitin-activating enzyme (E1) activates ubiquitin, which is then transferred to a ubiquitin-conjugating enzyme (E2). In the last step, a ubiquitin ligase (E3) links ubiquitin from the E2 enzyme to a lysine residue of the target protein. There are only two E1 enzymes, several E2 enzymes, and many E3 ligases, each of which recognizes one or several specific protein motifs. Therefore, the substrate specificity is achieved by the selectivity of the different E3 ligases [26, 80]. Dependent on the combination of E2 enzyme and E3 ligase, polyubiquitin chains are linked to the preceding ubiquitin molecule either via lysine 48 (K48) or via lysine 63 (K63), which marks the protein for degradation. Therefore, the polyubiquitination process determines the degradation pathways: Proteins carrying K48-linked polyubiquitin chains are predominantly targeted to proteasomal degradation, and proteins carrying K63-linked polyubiquitin chains enter the autophagic pathways as discussed in the following paragraph [1].
K48-linked polyubiquitinated proteins are transferred to the proteasome, which is almost exclusively the 26S proteasome in eukaryotic cells. This protein complex consists of one 20S core- and two 19S regulatory subunits forming a barrel-like structure. The regulatory subunits have ubiquitin-binding sites to recognize polyubiquitinated proteins and unfold them using their ATPase activity. The unfolded proteins are transferred to the catalytic core and proteolytically cleaved [104].
Sarcomeric proteins have an average turnover rate of 5–10 days [12, 112]. Therefore, they rely on a proper functioning UPS to regulate their clearance. Once dissociated from the myofibrils, ubiquitin-conjugating enzymes mark the proteins for proteasomal degradation by adding K48-linked polyubiquitin chains [91, 112]. In cardiomyocytes, this step is mediated by the MuRF family of E3 ligases [59, 93].
Autophagy
Autophagy cleans up aggregates or proteins via lysosomal breakdown that cannot be refolded by chaperones or processed by the UPS [46]. During macroautophagy, herein referred to as autophagy, membrane-enclosed vesicles are formed containing the targeted cellular components. First, an isolation membrane is formed engulfing the cytoplasmic material. The membrane expands until the edges fuse to form the autophagosome [118]. Fusion of the autophagosome with a lysosome leads to an autolysosome, which breaks down the cargo [5]. During selective autophagy, proteins carrying a K63-linked polyubiquitin chain are degraded. In the case the proteasome is overwhelmed with proteins carrying a K48-linked ubiquitin chain, such as aggregated proteins, they can also be cleared via autophagy. Their polyubiquitin chain docks to the adaptor protein p62/SQSTM1, which enables the translocation into the autophagosome [45]. Acidic lysosomal hydrolases degrade the captured material together with the inner membrane, and the resulting macromolecules are recycled into ATP, amino acids and fatty acids.
Autophagy is found to be upregulated in response to starvation, growth factor withdrawal, or high bioenergetic demands [50, 56, 65, 86]. The ability to sequester and break down entire organelles, such as mitochondria, peroxisomes, endoplasmic reticulum, and intact intracellular microorganisms, makes autophagy a unique and essential process in the cell. Especially in post-mitotic cells like cardiomyocytes, basal activation of autophagy is important to maintain a balanced proteostasis by degradation of long-lived proteins, lipid droplets, and dysfunctional organelles [17].
Cardiomyocytes have a low basal autophagic activity under normal conditions. Upon stress, the formation of protein aggregates is facilitated and triggers activation of autophagy [96]. Furthermore, cardiac autophagy is initiated in response to energy stress during periods of nutrient deprivation or high metabolic demand [35].
Proteostasis derailment in inherited cardiomyopathies
The PQC is of great importance in many cardiac diseases caused by “wear and tear,” including cardiac amyloidosis, myocardial infarction, and atrial fibrillation [37, 60, 73, 115]. The activation of PQC in a variety of cardiac stress conditions can be considered as a positive compensatory response to maintain proteostasis. This might be especially true in the case of inherited CMs, where mutant protein expression is the disease-causing mechanism. Recent studies provide evidence for a causative role of the PQC in CM. On one hand, mutations in components of the PQC itself can cause CM. This has been described for the R120G mutation in CRYAB encoding for the chaperone αB-crystallin, causing desmin-related CM, and the P209L mutation in the chaperone cofactor BAG3, leading to juvenile DCM [87, 105]. Mutations in PQC components as causes of inherited CM are rare, but PQC impairment can also occur as a result of CM-causing sarcomeric mutations. In this case, mutant sarcomeric proteins may impair the function of the PQC through overload of its components including HSPs, UPS, and autophagy. This could lead to increased levels of mutant protein, exacerbating CM disease progression.
So far, the role of PQC has been investigated only in a limited number of studies on CM caused by sarcomeric gene mutations. In vitro information is available for HCM- and DCM-causing mutations in ACTC1. Furthermore, it has been studied in vivo with HCM-causing mutations in MYBPC3, MYH7, and MYOZ2; DCM-causing NEBL mutations; and RCM-causing TNNI3 mutations (Table 1). In the following sections, the interaction between CMs and derailments of the different parts of the PQC are described in detail.
Table 1.
Overview of structural changes and adaptations in the protein quality control system related to cardiomyopathies
| Gene | Phenotype | Morphological abnormalities | Chaperones | UPS | Autophagy |
|---|---|---|---|---|---|
| ACTC1 | HCM | not reported | + (in vitro) [102] | not reported | not reported |
| DCM | not reported | + (in vitro) [102] | not reported | not reported | |
| RCM | not reported | not reported | not reported | not reported | |
| ACTN2 | HCM | cytoplasmic vacuolization, perinuclear halo, dysmorphic nuclei (human) [31] | not reported | not reported | not reported |
| DCM | not reported | not reported | not reported | not reported | |
| MYBPC3 | HCM | large irregular vacuoles (infant) [109] | αB-crystallin ↑ (mice) [116] | ↓ (mice) [85] | ↑ (human) [92] ↓ (mice) [85] |
| DCM | not reported | not reported | not reported | not reported | |
| MYH6 | HCM | not reported | not reported | not reported | not reported |
| DCM | not reported | not reported | not reported | not reported | |
| MYH7 | HCM | not reported | not reported | not reported | ↑ (human) [92] |
| DCM | not reported | not reported | not reported | not reported | |
| RCM | not reported | not reported | not reported | not reported | |
| MYL2 | HCM | not reported | not reported | not reported | not reported |
| MYL3 | HCM | not reported | not reported | not reported | not reported |
| RCM | ultrastructural defects (mice) [119] | not reported | not reported | not reported | |
| MYOZ2 | HCM | not reported | not reported | ↑ (mice) [41] | not reported |
| NEBL | HCM | myocyte vacuolization (human) [75] | not reported | not reported | not reported |
| DCM | enlarged and deformed mitochondria, lipid accumulation (mice) [78] | not reported | not reported | abnormal lysosomes (mice) [78] | |
| TNNC1 | HCM | not reported | not reported | not reported | not reported |
| DCM | no evidence of vacuolization (human) [43] | not reported | not reported | not reported | |
| RCM | degeneration of myocardial fibers (human) [76] | not reported | not reported | not reported | |
| TNNI3 | HCM | not reported | not reported | not reported | not reported |
| DCM | not reported | not reported | not reported | not reported | |
| RCM | irregularly shaped megamitochondria (human) [117] | not reported | ↓ proteasomal activity (mice) [20] | not reported | |
| TNNT2 | HCM | myocyte atrophy (mice) [97] | not reported | ↓ (mice) [30] | not reported |
| DCM | not reported | not reported | not reported | not reported | |
| RCM | abnormal mitochondria (human)* [74] | not reported | not reported | not reported | |
| TPM1 | HCM | nuclear gigantism [68] | not reported | not reported | not reported |
| DCM | accumulation of TPM1 (mice) [79] | not reported | not reported | not reported | |
| RCM | not reported | not reported | not reported | not reported |
Mixed genotypes are indicated with “*” and a “+” indicates a positive finding
Diverse abnormalities in heat shock protein function
HSP impairment or activation contribute to disease pathology in CMs. Desmin-related CM displays HSP impairment and is either caused by mutant desmin itself or mutant chaperone αB-crystallin. In a normal state, αB-crystallin binds to desmin and thereby prevents its aggregation [10]. Mutant desmin, however, impairs the interaction with αB-crystallin leading to desmin accumulation and cardiomyocyte dysfunction [54]. This suggests aberrant protein aggregation can cause CM. Correspondingly, the R120G mutation in CRYAB results in desmin-related-CM as well and also presents with aggregates containing desmin and mutant αB-crystallin [105]. Sanbe et al. showed that upregulation of HSPB8 due to geranylgeranylacetone treatment reduces the amount of mutant αB-crystallin-containing aggregates [83]. This implies that other HSPs can compensate for the loss of function to remove aggregates. Furthermore, in vitro experiments have shown that HCM- or DCM-causing mutations in ACTC1, encoding cardiac actin, can interfere with its folding by the TRiC chaperonin complex resulting in inefficient incorporation of actin into the myofilament and its subsequent aggregation [102]. Mutations in one specific subdomain of actin affect protein stability or polymerization, making actin more prone for degradation. Whereas mutations in other subdomains of actin cause alterations in protein-protein interactions [67]. A gene co-expression analysis of human controls and HCM samples identified the TRiC chaperonin complex as the most differential pathway, thereby further highlighting its importance in HCM [19].
By contrast, various studies on the role of PQC in CMs report on increased levels of HSPs due to PQC activation. However, it still remains unresolved whether the increased levels of HSPs are a direct effect of the mutant protein or a compensatory secondary effect due to increased cellular stress. Therefore, the direct interaction of mutant protein and HSPs needs to be studied. In mice with a truncating MYBPC3 mutation and an HCM phenotype, increased levels of αB-crystallin have been found [116]. In other CM mouse models, independent of a sarcomeric mutation, increased levels of HSP70 have been observed [62]. A study in patients with chronic heart failure due to DCM revealed a correlation of serum HSP60 levels with disease severity [69]. Since increased levels of HSP27 and HSP70 are associated with a protective effect in models for atrial fibrillation, by maintaining cardiomyocyte function, one can speculate that increased expression of these HSPs might be part of a compensatory protective mechanism in CM [15, 62].
In general, research findings indicate that HSP impairment is detrimental for cardiomyocyte function due to a higher risk of impaired protein folding and aggregate formation. By contrast, HSP activation in CM is considered as a beneficial effect and is most likely a compensatory mechanism of the cell.
Derailment of the ubiquitin-proteasome system
Derailed UPS function in CM affects the degradation of terminally misfolded proteins. MYBPC3 mutations often lead to expression of truncated protein, which is not incorporated into the sarcomere because the most C-terminal domain needed for incorporation is missing [64]. Truncated MyBP-C has not been detected in cardiac samples of HCM patients [101]. In addition, very low levels (< 4%) of truncated MyBP-C, which were not incorporated into the sarcomeres, were found in engineered heart tissue made of MYBPC3 knock-out mouse cardiomyocytes transfected with a truncating MYBPC3 mutation [110]. Therefore, it is likely that either the mutant mRNA is degraded via nonsense-mediated mRNA decay and/or the truncated MyBP-C forms a substrate for immediate degradation by the UPS or autophagy. Since MyBP-C is highly expressed in cardiomyocytes, high levels of truncated protein may lead to an increased UPS burden and competitive inhibition of the proteasome [84, 85]. In this case, the UPS is overwhelmed by the amount of truncated protein that needs to be degraded. In line with this hypothesis are analyses of myectomy samples from HCM patients with sarcomeric mutations, which show a decrease in proteolytic activity (Table 1) [77, 85]. Decreased processing through the UPS system is also indicated by the increase in overall levels of protein ubiquitination in HCM patients and animal models, which is already detectable at an early postnatal phase prior to any other symptom development [6, 30, 77, 85]. Consistent with the studies in MYBPC3-mutant samples, UPS perturbations have also been found in mouse heart tissue with TNNT2 mutations [30]. Patient samples with a sarcomeric mutation showed higher levels of polyubiquitination and decreased proteolytic activity compared to healthy controls [77].
In addition to overload of the UPS by mutant protein, increased oxidative stress can also impair the function of the proteasome. In this case, the proteasomal dysfunction would not be a direct effect of the mutant protein but a consequence of secondary cellular changes. In CM samples, an increase in oxidation of cytosolic protein content as well as the 19S proteasome, thereby decreasing the overall proteolytic function of the 26S proteasome subunit, has been identified [21, 30, 77].
In addition to the proteasome itself, the expression of ubiquitin ligases can be altered. In an HCM mouse model with mutant Mybpc3, the muscle specific E3 ligase Asb2β showed decreased mRNA levels compared to wild-type mice [99]. Since one of its targets is desmin, accumulation of desmin could contribute to the HCM phenotype, as observed for desmin-related CMs.
A large HCM patient cohort and matched healthy controls were screened for genetic variants in all three members of the MuRF family, since mutations in the gene encoding MuRF1 were reported to cause HCM [18]. In this study, a higher prevalence of rare variants of the cardiac-specific E3 ligases MuRF1 and MuRF2 was found in HCM patients [95]. These were associated with earlier disease onset and higher penetrance implying that disturbances of the UPS might act as a disease modifier contributing to HCM.
In contrast to HCM, in DCM, the reported UPS derailments could not yet be linked to sarcomeric mutations. A likely reason for this is that the DCM patient samples did not carry a sarcomeric mutation and/or the underlying disease cause was not known. Tissue analysis from explanted DCM hearts revealed increased expression of both E1 and/or E2 enzymes [47, 108]. Further evidence of increased ubiquitin-conjugating enzyme activity was detected in end-stage DCM. Here, increased levels of MuRF1 and MAFbx were associated with increased UPS degradation activity, which might be the cause of ventricle wall thinning as observed in end-stage DCM patients [9]. In line with the increased ubiquitin-conjugating enzyme levels, increased levels of polyubiquitinated proteins have been detected in DCM samples [11, 47, 72, 108]. This finding is further supported by a 2.3-fold reduced expression of the deubiquitinating enzyme isopeptidase-T in DCM patients [47]. Furthermore, increased proteolytic activity of the 26S proteasome as well as the 20S subunit peptidase activity has been found [9, 11, 72].
In contrast to HSPs, the answer to the question whether UPS activation or inhibition would be beneficial in HCM and DCM is not as straight forward. In an HCM phenotype, proteasome activation might improve the hypertrophic phenotype due to increased mutant protein degradation. However, in DCM, increased proteasome function might augment wall thinning, and therefore, DCM might benefit from proteasome inhibition.
Unresolved role of the autophagic response in cardiomyopathies
Autophagy is a crucial mechanism in CMs that only fulfills its cytoprotective mechanisms when it is in balance [50]. Moderate activation of autophagy has beneficial effects in CM patients by removing aggregates and supplying the cell with energy.
However, protein degradation due to excessive autophagy has been associated with different types of CM, including HCM, DCM, ischemic CM, and chemotherapy-induced CM [22, 44, 55, 58, 71, 89]. This could lead to loss of myofibrils, as observed in end-stage HCM and DCM patients [40, 70]. In a recent study, the expression of vacuolar protein sorting 34 (Vps34), an important autophagy regulator, was shown to be decreased in the myocardium of HCM patients and deletion of Vsp34 resulted in a HCM-like phenotype in mice. Furthermore, decreased expression of Vsp34 impaired the HSP-autophagy axis, as indicated by αB-crystallin-positive aggregates [44]. HCM patients with mutations in MYBPC3 or MYH7 revealed an upregulation of autophagic vacuoles and markers, indicated increased autophagic activity [92]. In a homozygous Mybpc3-mutant HCM mouse model, levels of autophagy markers were increased at the protein level implying autophagic activation. However, mRNA levels of these markers were not increased. This rather suggests an accumulation of autophagic proteins due to defective autophagic-lysosomal degradation instead of activation on transcriptional level [85]. In explanted hearts from DCM patients, the imbalance of high ubiquitination rate and insufficient degradation may contribute to autophagic cell death [47]. Vacuolization in CMs has been reported with mutations in ACTN2, MYBPC3, and NEBL [31, 75, 109]. This observation suggests that the accumulation of autophagic vacuoles implies cardiomyocyte stress. However, the interpretation of vacuole accumulation remains unclear, since it could reflect an increase in autophagic activity or an impairment of autophagosome-lysome fusion.
For a correct interpretation of the role of autophagy in CM, autophagic flux in combination with gene and protein expression data have to be studied in the future.
Environmental stressors influencing the proteostasis network
Besides the above mentioned effects of the sarcomeric gene mutations on the PQC, other environmental stressors, including physiological stress, genetic and epigenetic pathways, and inflammation, can also impair its function [82, 120]. In CM patients with a sarcomeric mutation, these stressors can act as second hit and thereby determine disease severity. Since most of CM patients become symptomatic only in a later stage of their life, the influence of drugs directed at the PQC system as treatment modality for co-morbidities and the age-related decline of the PQC are discussed below.
Several anti-cancer agents block the PQC to cause a lethal proteotoxicity in cancer cells. Anthracyclines, for example, directly impair its function by enhancing proteasomal degradation due to increased expression of E3 ligases and increased proteasome activity as well as inhibition of autophagy in cardiomyocytes [2, 27, 51, 63]. Furthermore, they disturb Ca2+ homeostasis, leading to endoplasmic reticulum stress, which derails protein folding [27]. In CM patients, dealing already with a sarcomeric mutation, treatment of another non-cardiac disease can trigger the onset of CM or worsen the clinical outcome. Cardiotoxic side effects of anthracyclines can lead to anthracycline-associated cardiomyopathy (AACM), which presents as LV dysfunction and DCM in adults and RCM in children [53, 61]. Treatment with a low dose of anti-cancer agents induced CM in cancer patients without a history of cardiac disease. Genetic screening of these patients revealed truncating titin variants, which are known as a genetic cause of DCM. These variants may increase the susceptibility for anti-cancer agents-induced CM [52]. In general, patients having a genetic predisposition for DCM are more prone to develop AACM after anthracycline treatment [100, 107]. It can be speculated that the impairment of the PQC due to the anti-cancer treatment is an additional burden to the cardiomyocyte. The clinical cardiac phenotype is caused by insufficient clearance of the mutant protein via UPS and/or autophagy. Therefore, these findings suggest that PQC impairment by anthracyclines can act as catalysts in the development of CM in patients with underlying sarcomeric gene mutation.
Aging represents another cellular stressor leading to toxic mutation effects because of the late disease onset and development of symptoms in inherited CMs. Clinical characteristics, such as wall thickness and diastolic function, worsen with increasing age [57]. This could be related to an age-associated decline in proteostatic function, which is supported by the presence of damaged macromolecules and mitochondria in aged cardiomyocytes [98]. Dysfunctional mitochondria generate high levels of reactive oxygen species, which promote proteotoxic stress and accelerate detrimental effects on the cardiomyocyte [90]. Also, the activity of the 26S proteasome is decreased during aging, which is possibly caused by oxidation of its components [21, 29, 30, 42]. The age-dependent decline in proteasome function increases the burden for the autophagic pathway. However, not only the proteasome, but also the autophagy-lysosomal system declines during aging [81]. As an example, mTOR, a negative regulator of autophagy, was upregulated during aging in a mouse study which indicates decreased autophagic activity [4]. As a result, the activity of the autophagic response might not be sufficient. However, similar to findings related to the UPS, autophagy was enhanced during aging in some animal models, suggesting an increased need for autophagy in aged cells [113]. Further research is warranted to investigate whether the age-related decline of the PQC is causative for CM onset and/or progression.
Future therapeutic implications
To improve the clinical outcome of CM patients, modulation of PQC components might serve as a novel therapeutic strategy. Figure 3 summarizes the three different ways and illustrates how a sarcomeric gene mutation can lead to PQC derailment. In case of a direct mutation effect on the PQC as well as in combination with secondary hits, targeting of the PQC would be most beneficial and the most direct way to prevent cardiomyocyte dysfunction. In case where the PQC derailment is a consequence of mutation-induced cellular disturbances, it is important to also target the cellular dysfunction to prevent further worsening of the PQC.
Fig. 3.

Effects of sarcomeric gene mutations on the protein quality control (PQC) system. Sarcomeric gene mutations can directly derail PQC function leading to cardiomyocyte dysfunction. PQC derailments in cardiomyopathies (CMs) can also be a compensatory mechanism to counteract cardiomyocyte dysfunction caused by the sarcomeric gene mutation. The secondary-hit hypothesis suggests that the PQC of cardiomyocytes carrying a sarcomeric gene mutation is more prone to derail in response to additional cellular stressors, thereby resulting in cardiomyocyte dysfunction
As extensively discussed in this review, PQC alterations in CMs are disease- and mutation-specific leading to either increased or reduced function in one or several of its components. Therefore, personalized treatment strategies are required to restore a balanced proteostasis. Potentially, all three PQC components can be therapeutically targeted with the appropriate compounds.
HSP expression can be induced by the drug geranylgeranylacetone. In animal models of desmin-related CM, the induction of HSP expression by geranylgeranylacetone resulted in a beneficial effect on heart function because desmin-aggregate formation was reduced [83]. This example suggests that activation of HSPs might also be beneficial in other types of inherited CMs, since HSPs play a crucial role in coping with the mutant protein.
The derailment of the UPS is context dependent: proteasomal function is decreased in HCM, RCM, and desmin-related CM and increased in DCM [9]. Decreased proteasomal function suggests that the misfolded proteins hamper the UPS by overwhelming it due to permanent degradation of misfolded proteins. As a consequence, the activity of the UPS is reduced. Therefore, UPS activation might be a beneficial therapeutic strategy in HCM and desmin-related CM [9]. In line with this, in HCM patients with a TNNT2 mutation increased proteasomal activity was correlated with a better clinical outcome [30]. In contrast, over-activation of the UPS indicates a direct response of the UPS to the misfolded proteins to ensure optimal clearance. However, excessive activation of the UPS transforms the initially beneficial effects into a detrimental maladaptation that possibly contributes to loss of myofibrils [9]. Nevertheless, complete proteasome inhibition itself triggered cardiac dysfunction and a CM-like phenotype in healthy pigs [38]. Therefore, it is important to achieve a moderate UPS response in DCM to prevent the detrimental effects of complete proteasome inhibition.
The altered autophagic flux in CMs can be caused on the one hand directly by the misfolded protein itself or on the other hand indirectly by compensating for the impaired functionality of the UPS. To optimize the degradation response, the autophagic activity needs to be pharmacologically titrated into its proteostasis promoting range [114].
Conclusion
The PQC is crucial for cardiac health and requires the collaboration of all its components to be functional. Key modulators of the PQC are disease- and mutation-specifically altered and derailed in CM. Pharmacological targeting of PQC components represents a novel therapeutic strategy to treat CMs. Since most of the described findings are retrieved from single CM patients or experimental animal models, systematic studies in larger CM patient populations are warranted to untie the knot of disease- and mutation-specific derailments of the PQC.
Acknowledgements
We acknowledge support from the Netherlands Cardiovascular Research Initiative—an initiative with support of the Dutch Heart Foundation, CVON: The Netherlands CardioVascular Research Committee, CVON2014-40 DOSIS. Furthermore, this research has been supported by LSH-TKI (40-43100-98-008), the Dutch Heart Foundation (2013T096, 2013T144), and the Netherlands Organisation for Scientific Research (NWO), as part of their joint strategic research programme: “Earlier recognition of cardiovascular diseases” (AFFIP: 14728). Van der Velden is supported by a VICI grant from the Netherlands Organization for Scientific Research (NWO-ZonMW; 91818602).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
This article is part of the special issue on Sarcomeric Mutations in Pflügers Archiv – European Journal of Physiology
Larissa M. Dorsch and Maike Schuldt contributed equally to this work.
Contributor Information
Larissa M. Dorsch, Phone: +31-204448110, Email: l.dorsch@vumc.nl
Maike Schuldt, Phone: +31-204448110, Email: m.schuldt@vumc.nl.
References
- 1.Amm I, Sommer T, Wolf DH. Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim Biophys Acta. 2014;1843:182–196. doi: 10.1016/j.bbamcr.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 2.Arai M, Yoguchi A, Takizawa T, Yokoyama T, Kanda T, Kurabayashi M, Nagai R. Mechanism of doxorubicin-induced inhibition of sarcoplasmic reticulum Ca(2+)-ATPase gene transcription. Circ Res. 2000;86:8–14. doi: 10.1161/01.res.86.1.8. [DOI] [PubMed] [Google Scholar]
- 3.Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, Marwick T, Pinney S, Bellazzi R, Favalli V, Kramer C, Roberts R, Zoghbi WA, Bonow R, Tavazzi L, Fuster V, Narula J. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the World Heart Federation. J Am Coll Cardiol. 2013;62:2046–2072. doi: 10.1016/j.jacc.2013.08.1644. [DOI] [PubMed] [Google Scholar]
- 4.Baar EL, Carbajal KA, Ong IM, Lamming DW. Sex- and tissue-specific changes in mTOR signaling with age in C57BL/6J mice. Aging Cell. 2016;15:155–166. doi: 10.1111/acel.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baba M, Takeshige K, Baba N, et al. Ultrastructural analysis of the autophagic process in yeast: detection of autophagosomes and their characterization. J Cell Biol. 1994;124:903–913. doi: 10.1083/jcb.124.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bahrudin U, Morisaki H, Morisaki T, Ninomiya H, Higaki K, Nanba E, Igawa O, Takashima S, Mizuta E, Miake J, Yamamoto Y, Shirayoshi Y, Kitakaze M, Carrier L, Hisatome I. Ubiquitin-proteasome system impairment caused by a missense cardiac myosin-binding protein C mutation and associated with cardiac dysfunction in hypertrophic cardiomyopathy. J Mol Biol. 2008;384:896–907. doi: 10.1016/j.jmb.2008.09.070. [DOI] [PubMed] [Google Scholar]
- 7.Barral JM, Epstein HF. Protein machines and self assembly in muscle organization. Bioessays. 1999;21:813–823. doi: 10.1002/(SICI)1521-1878(199910)21:10<813::AID-BIES3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 8.Barral JM, Hutagalung AH, Brinker A, et al. Role of the myosin assembly protein UNC-45 as a molecular chaperone for myosin. Science. 2002;295:669–671. doi: 10.1126/science.1066648. [DOI] [PubMed] [Google Scholar]
- 9.Baumgarten A, Bang C, Tschirner A, Engelmann A, Adams V, von Haehling S, Doehner W, Pregla R, Anker MS, Blecharz K, Meyer R, Hetzer R, Anker SD, Thum T, Springer J. TWIST1 regulates the activity of ubiquitin proteasome system via the miR-199/214 cluster in human end-stage dilated cardiomyopathy. Int J Cardiol. 2013;168:1447–1452. doi: 10.1016/j.ijcard.2012.12.094. [DOI] [PubMed] [Google Scholar]
- 10.Bennardini F, Wrzosek A, Chiesi M. Alpha B-crystallin in cardiac tissue. Association with actin and desmin filaments. Circ Res. 1992;71:288–294. doi: 10.1161/01.res.71.2.288. [DOI] [PubMed] [Google Scholar]
- 11.Birks EJ, Latif N, Enesa K, Folkvang T, Luong LA, Sarathchandra P, Khan M, Ovaa H, Terracciano CM, Barton PJR, Yacoub MH, Evans PC. Elevated p53 expression is associated with dysregulation of the ubiquitin-proteasome system in dilated cardiomyopathy. Cardiovasc Res. 2008;79:472–480. doi: 10.1093/cvr/cvn083. [DOI] [PubMed] [Google Scholar]
- 12.Boateng SY, Goldspink PH. Assembly and maintenance of the sarcomere night and day. Cardiovasc Res. 2008;77:667–675. doi: 10.1093/cvr/cvm048. [DOI] [PubMed] [Google Scholar]
- 13.Bozaykut P, Ozer NK, Karademir B. Regulation of protein turnover by heat shock proteins. Free Radic Biol Med. 2014;77:195–209. doi: 10.1016/j.freeradbiomed.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 14.Brown DD, Christine KS, Showell C, Conlon FL. Small heat shock protein Hsp27 is required for proper heart tube formation. Genesis. 2007;45:667–678. doi: 10.1002/dvg.20340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brundel BJ, Shiroshita-Takeshita A, Qi X, et al. Induction of heat shock response protects the heart against atrial fibrillation. Circ Res. 2006;99:1394–1402. doi: 10.1161/01.RES.0000252323.83137.fe. [DOI] [PubMed] [Google Scholar]
- 16.Bryantsev AL, Loktionova SA, Ilyinskaya OP, Tararak EM, Kampinga HH, Kabakov AE. Distribution, phosphorylation, and activities of Hsp25 in heat-stressed H9c2 myoblasts: a functional link to cytoprotection. Cell Stress Chaperones. 2002;7:146–155. doi: 10.1379/1466-1268(2002)007<0146:dpaaoh>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cecconi F, Levine B. The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell. 2008;15:344–357. doi: 10.1016/j.devcel.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen SN, Czernuszewicz G, Tan Y, Lombardi R, Jin J, Willerson JT, Marian AJ. Human molecular genetic and functional studies identify TRIM63, encoding Muscle RING Finger Protein 1, as a novel gene for human hypertrophic cardiomyopathy. Circ Res. 2012;111:907–919. doi: 10.1161/CIRCRESAHA.112.270207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen XM, Feng MJ, Shen CJ, He B, du XF, Yu YB, Liu J, Chu HM. A novel approach to select differential pathways associated with hypertrophic cardiomyopathy based on gene coexpression analysis. Mol Med Rep. 2017;16:773–777. doi: 10.3892/mmr.2017.6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui Z, Venkatraman G, Hwang SM, et al. Effect of the troponin I restrictive cardiomyopathy mutation R145W on protein expression in murine hearts. Biophys J. 2013;104:312a. [Google Scholar]
- 21.Day SM, Divald A, Wang P, Davis F, Bartolone S, Jones R, Powell SR. Impaired assembly and post-translational regulation of 26S proteasome in human end-stage heart failure. Circ Heart Fail. 2013;6:544–549. doi: 10.1161/CIRCHEARTFAILURE.112.000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Decker RS, Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am J Pathol. 1980;98:425–444. [PMC free article] [PubMed] [Google Scholar]
- 23.Ehrnsperger M, Lilie H, Gaestel M, et al. The dynamics of Hsp25 quaternary structure. Structure and function of different oligomeric species. J Biol Chem. 1999;274:14867–14874. doi: 10.1074/jbc.274.21.14867. [DOI] [PubMed] [Google Scholar]
- 24.Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–276. doi: 10.1093/eurheartj/ehm342. [DOI] [PubMed] [Google Scholar]
- 25.Esser C, Alberti S, Hohfeld J. Cooperation of molecular chaperones with the ubiquitin/proteasome system. Biochim Biophys Acta. 2004;1695:171–188. doi: 10.1016/j.bbamcr.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 26.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu HY, Sanada S, Matsuzaki T, Liao Y, Okuda K, Yamato M, Tsuchida S, Araki R, Asano Y, Asanuma H, Asakura M, French BA, Sakata Y, Kitakaze M, Minamino T. Chemical endoplasmic reticulum chaperone alleviates doxorubicin-induced cardiac dysfunction. Circ Res. 2016;118:798–809. doi: 10.1161/CIRCRESAHA.115.307604. [DOI] [PubMed] [Google Scholar]
- 28.Garrido C, Paul C, Seigneuric R, Kampinga HH. The small heat shock proteins family: the long forgotten chaperones. Int J Biochem Cell Biol. 2012;44:1588–1592. doi: 10.1016/j.biocel.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 29.Gilda JE, Gomes AV. Proteasome dysfunction in cardiomyopathies. J Physiol. 2017;595:4051–4071. doi: 10.1113/JP273607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilda JE, Lai X, Witzmann FA, Gomes AV. Delineation of molecular pathways involved in cardiomyopathies caused by troponin T mutations. Mol Cell Proteomics. 2016;15:1962–1981. doi: 10.1074/mcp.M115.057380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Girolami F, Iascone M, Tomberli B, Bardi S, Benelli M, Marseglia G, Pescucci C, Pezzoli L, Sana ME, Basso C, Marziliano N, Merlini PA, Fornaro A, Cecchi F, Torricelli F, Olivotto I. Novel alpha-actinin 2 variant associated with familial hypertrophic cardiomyopathy and juvenile atrial arrhythmias: a massively parallel sequencing study. Circ Cardiovasc Genet. 2014;7:741–750. doi: 10.1161/CIRCGENETICS.113.000486. [DOI] [PubMed] [Google Scholar]
- 32.Glazier AA, Hafeez N, Mellacheruvu D, Basrur V, Nesvizhskii AI, Lee LM, Shao H, Tang V, Yob JM, Gestwicki JE, Helms AS, Day SM (2018) HSC70 is a chaperone for wild-type and mutant cardiac myosin binding protein C. JCI Insight 3. 10.1172/jci.insight.99319 [DOI] [PMC free article] [PubMed]
- 33.Golenhofen N, Perng MD, Quinlan RA, et al. Comparison of the small heat shock proteins alphaB-crystallin, MKBP, HSP25, HSP20, and cvHSP in heart and skeletal muscle. Histochem Cell Biol. 2004;122:415–425. doi: 10.1007/s00418-004-0711-z. [DOI] [PubMed] [Google Scholar]
- 34.Grantham J, Ruddock LW, Roobol A, Carden MJ. Eukaryotic chaperonin containing T-complex polypeptide 1 interacts with filamentous actin and reduces the initial rate of actin polymerization in vitro. Cell Stress Chaperones. 2002;7:235–242. doi: 10.1379/1466-1268(2002)007<0235:ecctcp>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gustafsson AB, Gottlieb RA. Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol. 2008;44:654–661. doi: 10.1016/j.yjmcc.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hansen WJ, Cowan NJ, Welch WJ. Prefoldin-nascent chain complexes in the folding of cytoskeletal proteins. J Cell Biol. 1999;145:265–277. doi: 10.1083/jcb.145.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henning RH, Brundel B. Proteostasis in cardiac health and disease. Nat Rev Cardiol. 2017;14:637–653. doi: 10.1038/nrcardio.2017.89. [DOI] [PubMed] [Google Scholar]
- 38.Herrmann J, Wohlert C, Saguner AM, Flores A, Nesbitt LL, Chade A, Lerman LO, Lerman A. Primary proteasome inhibition results in cardiac dysfunction. Eur J Heart Fail. 2013;15:614–623. doi: 10.1093/eurjhf/hft034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 40.Hoorntje ET, Bollen IA, Barge-Schaapveld DQ et al. (2017) Lamin A/C-related cardiac disease: late onset with a variable and mild phenotype in a large cohort of patients with the Lamin A/C p.(Arg331Gln) founder mutation. Circ Cardiovasc Genet 10. 10.1161/CIRCGENETICS.116.001631 [DOI] [PubMed]
- 41.Ivandic BT, Mastitsky SE, Schonsiegel F, et al. Whole-genome analysis of gene expression associates the ubiquitin-proteasome system with the cardiomyopathy phenotype in disease-sensitized congenic mouse strains. Cardiovasc Res. 2012;94:87–95. doi: 10.1093/cvr/cvs080. [DOI] [PubMed] [Google Scholar]
- 42.Jana NR. Protein homeostasis and aging: role of ubiquitin protein ligases. Neurochem Int. 2012;60:443–447. doi: 10.1016/j.neuint.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 43.Kaski JP, Burch M, Elliott PM. Mutations in the cardiac Troponin C gene are a cause of idiopathic dilated cardiomyopathy in childhood. Cardiol Young. 2007;17:675–677. doi: 10.1017/S1047951107001291. [DOI] [PubMed] [Google Scholar]
- 44.Kimura H, Eguchi S, Sasaki J, et al. Vps34 regulates myofibril proteostasis to prevent hypertrophic cardiomyopathy. JCI Insight. 2017;2:e89462. doi: 10.1172/jci.insight.89462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirkin V, Mcewan DG, Novak I, et al. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 46.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 47.Kostin S, Pool L, Elsasser A, et al. Myocytes die by multiple mechanisms in failing human hearts. Circ Res. 2003;92:715–724. doi: 10.1161/01.RES.0000067471.95890.5C. [DOI] [PubMed] [Google Scholar]
- 48.Landry J, Huot J. Modulation of actin dynamics during stress and physiological stimulation by a signaling pathway involving p38 MAP kinase and heat-shock protein 27. Biochem Cell Biol. 1995;73:703–707. doi: 10.1139/o95-078. [DOI] [PubMed] [Google Scholar]
- 49.Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol Cell Biol. 1995;15:505–516. doi: 10.1128/mcb.15.1.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ling YH, Priebe W, Perez-Soler R. Apoptosis induced by anthracycline antibiotics in P388 parent and multidrug-resistant cells. Cancer Res. 1993;53:1845–1852. [PubMed] [Google Scholar]
- 52.Linschoten M, Teske AJ, Baas AF, Vink A, Dooijes D, Baars HF, Asselbergs FW. Truncating titin (TTN) variants in chemotherapy-induced cardiomyopathy. J Card Fail. 2017;23:476–479. doi: 10.1016/j.cardfail.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 53.Lipshultz SE, Lipsitz SR, Sallan SE, Dalton VM, Mone SM, Gelber RD, Colan SD. Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J Clin Oncol. 2005;23:2629–2636. doi: 10.1200/JCO.2005.12.121. [DOI] [PubMed] [Google Scholar]
- 54.Liu J, Tang M, Mestril R, Wang X. Aberrant protein aggregation is essential for a mutant desmin to impair the proteolytic function of the ubiquitin-proteasome system in cardiomyocytes. J Mol Cell Cardiol. 2006;40:451–454. doi: 10.1016/j.yjmcc.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 55.Lu L, Wu W, Yan J, Li X, Yu H, Yu X. Adriamycin-induced autophagic cardiomyocyte death plays a pathogenic role in a rat model of heart failure. Int J Cardiol. 2009;134:82–90. doi: 10.1016/j.ijcard.2008.01.043. [DOI] [PubMed] [Google Scholar]
- 56.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 57.Luo HC, Pozios I, Vakrou S, Sorensen L, Abraham RM, Abraham T. Age-related changes in familial hypertrophic cardiomyopathy phenotype in transgenic mice and humans. J Huazhong Univ Sci Technolog Med Sci. 2014;34:634–639. doi: 10.1007/s11596-014-1329-6. [DOI] [PubMed] [Google Scholar]
- 58.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 59.Mcelhinny AS, Perry CN, Witt CC, et al. Muscle-specific RING finger-2 (MURF-2) is important for microtubule, intermediate filament and sarcomeric M-line maintenance in striated muscle development. J Cell Sci. 2004;117:3175–3188. doi: 10.1242/jcs.01158. [DOI] [PubMed] [Google Scholar]
- 60.Meijering RA, Zhang D, Hoogstra-Berends F, et al. Loss of proteostatic control as a substrate for atrial fibrillation: a novel target for upstream therapy by heat shock proteins. Front Physiol. 2012;3:36. doi: 10.3389/fphys.2012.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meinardi MT, Van Der Graaf WT, Van Veldhuisen DJ, et al. Detection of anthracycline-induced cardiotoxicity. Cancer Treat Rev. 1999;25:237–247. doi: 10.1053/ctrv.1999.0128. [DOI] [PubMed] [Google Scholar]
- 62.Min TJ, Jo WM, Shin SY, Lim HE. The protective effect of heat shock protein 70 (Hsp70) in atrial fibrillation in various cardiomyopathy conditions. Heart Vessel. 2015;30:379–385. doi: 10.1007/s00380-014-0521-8. [DOI] [PubMed] [Google Scholar]
- 63.Minotti G, Licata S, Saponiero A, Menna P, Calafiore AM, di Giammarco G, Liberi G, Animati F, Cipollone A, Manzini S, Maggi CA. Anthracycline metabolism and toxicity in human myocardium: comparisons between doxorubicin, epirubicin, and a novel disaccharide analogue with a reduced level of formation and [4Fe-4S] reactivity of its secondary alcohol metabolite. Chem Res Toxicol. 2000;13:1336–1341. doi: 10.1021/tx000143z. [DOI] [PubMed] [Google Scholar]
- 64.Miyamoto CA, Fischman DA, Reinach FC. The interface between MyBP-C and myosin: site-directed mutagenesis of the CX myosin-binding domain of MyBP-C. J Muscle Res Cell Motil. 1999;20:703–715. doi: 10.1023/a:1005513312939. [DOI] [PubMed] [Google Scholar]
- 65.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mundia MM, Demers RW, Chow ML, Perieteanu AA, Dawson JF. Subdomain location of mutations in cardiac actin correlate with type of functional change. PLoS One. 2012;7:e36821. doi: 10.1371/journal.pone.0036821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muthuchamy M, Pieples K, Rethinasamy P, Hoit B, Grupp IL, Boivin GP, Wolska B, Evans C, Solaro RJ, Wieczorek DF. Mouse model of a familial hypertrophic cardiomyopathy mutation in alpha-tropomyosin manifests cardiac dysfunction. Circ Res. 1999;85:47–56. doi: 10.1161/01.res.85.1.47. [DOI] [PubMed] [Google Scholar]
- 69.Niizeki T, Takeishi Y, Watanabe T, Nitobe J, Miyashita T, Miyamoto T, Kitahara T, Suzuki S, Sasaki T, Bilim O, Ishino M, Kubota I. Relation of serum heat shock protein 60 level to severity and prognosis in chronic heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 2008;102:606–610. doi: 10.1016/j.amjcard.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 70.Nijenkamp L, Bollen IE, Van Velzen HG, et al. Sex differences at the time of myectomy in hypertrophic cardiomyopathy. Circ Heart Fail. 2018;11:e004133. doi: 10.1161/CIRCHEARTFAILURE.117.004133. [DOI] [PubMed] [Google Scholar]
- 71.Nowis D, Maczewski M, Mackiewicz U, et al. Cardiotoxicity of the anticancer therapeutic agent bortezomib. Am J Pathol. 2010;176:2658–2668. doi: 10.2353/ajpath.2010.090690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Otsuka K, Terasaki F, Shimomura H, Tsukada B, Horii T, Isomura T, Suma H, Shibayama Y, Kitaura Y. Enhanced expression of the ubiquitin-proteasome system in the myocardium from patients with dilated cardiomyopathy referred for left ventriculoplasty: an immunohistochemical study with special reference to oxidative stress. Heart Vessel. 2010;25:474–484. doi: 10.1007/s00380-010-0006-3. [DOI] [PubMed] [Google Scholar]
- 73.Patterson C, Ike C, Willis PWT, et al. The bitter end: the ubiquitin-proteasome system and cardiac dysfunction. Circulation. 2007;115:1456–1463. doi: 10.1161/CIRCULATIONAHA.106.649863. [DOI] [PubMed] [Google Scholar]
- 74.Peddy SB, Vricella LA, Crosson JE, et al. Infantile restrictive cardiomyopathy resulting from a mutation in the cardiac troponin T gene. Pediatrics. 2006;117:1830–1833. doi: 10.1542/peds.2005-2301. [DOI] [PubMed] [Google Scholar]
- 75.Perrot A, Tomasov P, Villard E, et al. Mutations in NEBL encoding the cardiac Z-disk protein nebulette are associated with various cardiomyopathies. Arch Med Sci. 2016;12:263–278. doi: 10.5114/aoms.2016.59250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ploski R, Rydzanicz M, Ksiazczyk TM, Franaszczyk M, Pollak A, Kosinska J, Michalak E, Stawinski P, Ziolkowska L, Bilinska ZT, Werner B. Evidence for troponin C (TNNC1) as a gene for autosomal recessive restrictive cardiomyopathy with fatal outcome in infancy. Am J Med Genet A. 2016;170:3241–3248. doi: 10.1002/ajmg.a.37860. [DOI] [PubMed] [Google Scholar]
- 77.Predmore JM, Wang P, Davis F, Bartolone S, Westfall MV, Dyke DB, Pagani F, Powell SR, Day SM. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation. 2010;121:997–1004. doi: 10.1161/CIRCULATIONAHA.109.904557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Purevjav E, Varela J, Morgado M, Kearney DL, Li H, Taylor MD, Arimura T, Moncman CL, McKenna W, Murphy RT, Labeit S, Vatta M, Bowles NE, Kimura A, Boriek AM, Towbin JA. Nebulette mutations are associated with dilated cardiomyopathy and endocardial fibroelastosis. J Am Coll Cardiol. 2010;56:1493–1502. doi: 10.1016/j.jacc.2010.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rajan S, Ahmed RP, Jagatheesan G, et al. Dilated cardiomyopathy mutant tropomyosin mice develop cardiac dysfunction with significantly decreased fractional shortening and myofilament calcium sensitivity. Circ Res. 2007;101:205–214. doi: 10.1161/CIRCRESAHA.107.148379. [DOI] [PubMed] [Google Scholar]
- 80.Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol. 2008;9:679–690. doi: 10.1038/nrm2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 82.Sadoul K, Boyault C, Pabion M, Khochbin S. Regulation of protein turnover by acetyltransferases and deacetylases. Biochimie. 2008;90:306–312. doi: 10.1016/j.biochi.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 83.Sanbe A, Daicho T, Mizutani R, Endo T, Miyauchi N, Yamauchi J, Tanonaka K, Glabe C, Tanoue A. Protective effect of geranylgeranylacetone via enhancement of HSPB8 induction in desmin-related cardiomyopathy. PLoS One. 2009;4:e5351. doi: 10.1371/journal.pone.0005351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sarikas A, Carrier L, Schenke C, et al. Impairment of the ubiquitin-proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc Res. 2005;66:33–44. doi: 10.1016/j.cardiores.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 85.Schlossarek S, Englmann DR, Sultan KR, Sauer M, Eschenhagen T, Carrier L. Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res Cardiol. 2012;107:235. doi: 10.1007/s00395-011-0235-3. [DOI] [PubMed] [Google Scholar]
- 86.Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004;7:167–178. doi: 10.1016/j.devcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 87.Selcen D, Muntoni F, Burton BK, et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol. 2009;65:83–89. doi: 10.1002/ana.21553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Semsarian C, Ingles J, Wilde AA. Sudden cardiac death in the young: the molecular autopsy and a practical approach to surviving relatives. Eur Heart J. 2015;36:1290–1296. doi: 10.1093/eurheartj/ehv063. [DOI] [PubMed] [Google Scholar]
- 89.Shimomura H, Terasaki F, Hayashi T, Kitaura Y, Isomura T, Suma H. Autophagic degeneration as a possible mechanism of myocardial cell death in dilated cardiomyopathy. Jpn Circ J. 2001;65:965–968. doi: 10.1253/jcj.65.965. [DOI] [PubMed] [Google Scholar]
- 90.Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008;4:176–184. doi: 10.4161/auto.5269. [DOI] [PubMed] [Google Scholar]
- 91.Solomon V, Goldberg AL. Importance of the ATP-ubiquitin-proteasome pathway in the degradation of soluble and myofibrillar proteins in rabbit muscle extracts. J Biol Chem. 1996;271:26690–26697. doi: 10.1074/jbc.271.43.26690. [DOI] [PubMed] [Google Scholar]
- 92.Song L, Su M, Wang S, Zou Y, Wang X, Wang Y, Cui H, Zhao P, Hui R, Wang J. MiR-451 is decreased in hypertrophic cardiomyopathy and regulates autophagy by targeting TSC1. J Cell Mol Med. 2014;18:2266–2274. doi: 10.1111/jcmm.12380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Spencer JA, Eliazer S, Ilaria RL, Jr, Richardson JA, Olson EN. Regulation of microtubule dynamics and myogenic differentiation by MURF, a striated muscle RING-finger protein. J Cell Biol. 2000;150:771–784. doi: 10.1083/jcb.150.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Srikakulam R, Winkelmann DA. Chaperone-mediated folding and assembly of myosin in striated muscle. J Cell Sci. 2004;117:641–652. doi: 10.1242/jcs.00899. [DOI] [PubMed] [Google Scholar]
- 95.Su M, Wang J, Kang L, Wang Y, Zou Y, Feng X, Wang D, Ahmad F, Zhou X, Hui R, Song L. Rare variants in genes encoding MuRF1 and MuRF2 are modifiers of hypertrophic cardiomyopathy. Int J Mol Sci. 2014;15:9302–9313. doi: 10.3390/ijms15069302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tannous P, Zhu H, Nemchenko A, Berry JM, Johnstone JL, Shelton JM, Miller FJ, Rothermel BA, Hill JA. Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation. 2008;117:3070–3078. doi: 10.1161/CIRCULATIONAHA.107.763870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tardiff JC, Factor SM, Tompkins BD, Hewett TE, Palmer BM, Moore RL, Schwartz S, Robbins J, Leinwand LA. A truncated cardiac troponin T molecule in transgenic mice suggests multiple cellular mechanisms for familial hypertrophic cardiomyopathy. J Clin Invest. 1998;101:2800–2811. doi: 10.1172/JCI2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Terman A, Dalen H, Eaton JW, Neuzil J, Brunk UT. Mitochondrial recycling and aging of cardiac myocytes: the role of autophagocytosis. Exp Gerontol. 2003;38:863–876. doi: 10.1016/s0531-5565(03)00114-1. [DOI] [PubMed] [Google Scholar]
- 99.Thottakara T, Friedrich FW, Reischmann S, Braumann S, Schlossarek S, Krämer E, Juhr D, Schlüter H, van der Velden J, Münch J, Patten M, Eschenhagen T, Moog-Lutz C, Carrier L. The E3 ubiquitin ligase Asb2beta is downregulated in a mouse model of hypertrophic cardiomyopathy and targets desmin for proteasomal degradation. J Mol Cell Cardiol. 2015;87:214–224. doi: 10.1016/j.yjmcc.2015.08.020. [DOI] [PubMed] [Google Scholar]
- 100.Van Den Berg MP, Van Spaendonck-Zwarts KY, Van Veldhuisen DJ, et al. Familial dilated cardiomyopathy: another risk factor for anthracycline-induced cardiotoxicity? Eur J Heart Fail. 2010;12:1297–1299. doi: 10.1093/eurjhf/hfq175. [DOI] [PubMed] [Google Scholar]
- 101.Van Dijk SJ, Dooijes D, Dos Remedios C, et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- 102.Vang S, Corydon TJ, Borglum AD, et al. Actin mutations in hypertrophic and dilated cardiomyopathy cause inefficient protein folding and perturbed filament formation. FEBS J. 2005;272:2037–2049. doi: 10.1111/j.1742-4658.2005.04630.x. [DOI] [PubMed] [Google Scholar]
- 103.Vos MJ, Kanon B, Kampinga HH. HSPB7 is a SC35 speckle resident small heat shock protein. Biochim Biophys Acta. 2009;1793:1343–1353. doi: 10.1016/j.bbamcr.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 104.Wang J, Maldonado MA. The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases. Cell Mol Immunol. 2006;3:255–261. [PubMed] [Google Scholar]
- 105.Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, Hewett T, Robbins J. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- 106.Wang X, Su H, Ranek MJ. Protein quality control and degradation in cardiomyocytes. J Mol Cell Cardiol. 2008;45:11–27. doi: 10.1016/j.yjmcc.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wasielewski M, Van Spaendonck-Zwarts KY, Westerink ND, et al. Potential genetic predisposition for anthracycline-associated cardiomyopathy in families with dilated cardiomyopathy. Open Heart. 2014;1:e000116. doi: 10.1136/openhrt-2014-000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Weekes J, Morrison K, Mullen A, Wait R, Barton P, Dunn MJ. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics. 2003;3:208–216. doi: 10.1002/pmic.200390029. [DOI] [PubMed] [Google Scholar]
- 109.Wessels MW, Herkert JC, Frohn-Mulder IM, Dalinghaus M, van den Wijngaard A, de Krijger RR, Michels M, de Coo IFM, Hoedemaekers YM, Dooijes D. Compound heterozygous or homozygous truncating MYBPC3 mutations cause lethal cardiomyopathy with features of noncompaction and septal defects. Eur J Hum Genet. 2015;23:922–928. doi: 10.1038/ejhg.2014.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wijnker PJ, Friedrich FW, Dutsch A, et al. Comparison of the effects of a truncating and a missense MYBPC3 mutation on contractile parameters of engineered heart tissue. J Mol Cell Cardiol. 2016;97:82–92. doi: 10.1016/j.yjmcc.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 111.Willis MS, Patterson C. Hold me tight: role of the heat shock protein family of chaperones in cardiac disease. Circulation. 2010;122:1740–1751. doi: 10.1161/CIRCULATIONAHA.110.942250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Willis MS, Schisler JC, Portbury AL, et al. Build it up-tear it down: protein quality control in the cardiac sarcomere. Cardiovasc Res. 2009;81:439–448. doi: 10.1093/cvr/cvn289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wohlgemuth SE, Julian D, Akin DE, Fried J, Toscano K, Leeuwenburgh C, Dunn WA., Jr Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res. 2007;10:281–292. doi: 10.1089/rej.2006.0535. [DOI] [PubMed] [Google Scholar]
- 114.Xie M, Morales CR, Lavandero S, Hill JA. Tuning flux: autophagy as a target of heart disease therapy. Curr Opin Cardiol. 2011;26:216–222. doi: 10.1097/HCO.0b013e328345980a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, Yang G, Matsui Y, Sadoshima J, Vatner SF. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005;102:13807–13812. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yang Q, Osinska H, Klevitsky R, Robbins J. Phenotypic deficits in mice expressing a myosin binding protein C lacking the titin and myosin binding domains. J Mol Cell Cardiol. 2001;33:1649–1658. doi: 10.1006/jmcc.2001.1417. [DOI] [PubMed] [Google Scholar]
- 117.Yang SW, Hitz MP, Andelfinger G. Ventricular septal defect and restrictive cardiomyopathy in a paediatric TNNI3 mutation carrier. Cardiol Young. 2010;20:574–576. doi: 10.1017/S1047951110000715. [DOI] [PubMed] [Google Scholar]
- 118.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl 2):1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yuan CC, Kazmierczak K, Liang J, Kanashiro-Takeuchi R, Irving TC, Gomes AV, Wang Y, Burghardt TP, Szczesna-Cordary D. Hypercontractile mutant of ventricular myosin essential light chain leads to disruption of sarcomeric structure and function and results in restrictive cardiomyopathy in mice. Cardiovasc Res. 2017;113:1124–1136. doi: 10.1093/cvr/cvx060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]