

Abstract
Aims
Pregnancy is associated with physiological changes that alter the pharmacokinetics (PK) of drugs. The aim of this study was to predict the PK of ziprasidone in pregnant women.
Methods
A full physiologically‐based pharmacokinetic (PBPK) model of ziprasidone was developed and validated for the non‐pregnant population (healthy adults, paediatrics, geriatrics), and this was extended to the pregnant state to assess the change in PK profile of ziprasidone throughout pregnancy.
Results
The PBPK model successfully predicted the ziprasidone disposition in healthy adult volunteers, wherein the predicted and observed AUC, C max and t max were within the fold‐difference of 0.94–1.09, 0.89–1.40 and 0.80–1.08, respectively. The paediatric and geriatric population, also showed predicted AUC, C max and t max within a two‐fold range of the observed values. The simulated exposure in pregnant women using a p‐PBPK model showed no significant difference when compared to non‐pregnant women.
Conclusions
The PBPK model predicted the impact of physiological changes during pregnancy on PK and exposure of ziprasidone, suggesting that dose adjustment is not necessary in this special population.
Keywords: PBPK, pregnancy, ziprasidone
What is already known about this subject
Evidence regarding the use of antipsychotics in pregnancy has been insufficient to provide adequate support for this practice and is a concern for clinicians and pregnant women alike.
Numerous physiological changes can significantly affect drug disposition during pregnancy and dose adjustments may be necessary.
PK studies of antipsychotics in pregnant women are sparse and alternative approaches that can predict changes in drug disposition are needed.
What this study adds
This study is the first pregnancy PBPK model aimed at predicting maternal drug concentrations of ziprasidone throughout pregnancy. The current example provides the opportunity for using the PBPK model to guide dose adjustment in pregnancy.
1. INTRODUCTION
Nearly one in five US adults lives with someone who has a mental illness (44.7 million in 2016),1 including depression and bipolar disorder. However, when the disorder involves distorted awareness and thinking, these illnesses are classified as psychiatric disorders. The prevalence of psychiatric disorders has increased over the years.2 Schizophrenia is one of these psychiatric disorders and it is classified as a chronic and severe mental illness which affects about 23 million people worldwide.3 The age of onset in women occurs during childbearing age between 25 and 35 years old, with a second peak occurring after menopause.4
The age range at the time of the first peak of the disease coincides with the child‐bearing potential in women. Pregnancy can induce physiological changes, including the increase in the size of the fetal‐placenta compartment, increase in renal filtration, body fluid volume and hepatic portal blood flow, changes in the expression and activity of drug metabolizing enzymes and drug transporters.5, 6 These changes affect absorption, distribution, metabolism and elimination of drugs, potentially resulting in a modification of the PK behaviour of drugs, including antipsychotics.7, 8, 9 Pregnancy is known to affect hepatic drug metabolism but the underlying mechanisms are still unknown. Clearances of drugs metabolized by CYP2D6 and 3A4 increased, whereas those drugs metabolized by CYP1A2 decreased during pregnancy as compared to non‐pregnant women.10 For example, plasma levels of midazolam and metoprolol decrease in pregnant women due to the effect of changes in hepatic CYP3A4 and 2D6, respectively.10, 11
The clinical and ethical discussions on drug prescription during pregnancy focus on the safety of the fetus. The maintenance of drug efficacy in pregnant mothers amid all physiological alterations is just as important. Running clinical trials in the pregnant population is a controversial topic for obvious ethical reasons. Consequently, there is a lack of exposure and outcome data12 supporting drug monitoring and dose adjustment of antipsychotics in the pregnant population.13
Physiologically based pharmacokinetic modelling (PBPK) has been used as a tool to understand the PK behaviour in special populations. A number of successful applications of PK prediction in special populations, such as paediatric, geriatric and pregnancy, have been reported.9, 14, 15, 16, 17, 18 Regulatory agencies, such as the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), recognize that clinicians need more information to make appropriate dosing decisions for their pregnant patients. In the absence of PK data from clinical pregnancy and lactation studies, alternative approaches, such as PBPK modelling and simulation, could provide additional dosing information.19 These agencies have adopted PBPK modelling and simulation to facilitate the review of Investigational New Drug and New Drug Application submissions to address a variety of clinical issues, including the assessment of the effect of intrinsic or extrinsic factors on drug disposition20, 21, 22 and to inform drug labelling to support dosing recommendations as well.23
Therefore, the pregnancy PBPK model that incorporates the pregnancy‐induced changes in various anatomical, physiological and biological parameters is a feasible alternative for empirical dosage selection when there are no clinical studies available to guide dose selection.24, 25 However, application of PBPK modelling in drug labels in treating pregnant women is limited. Applying PBPK to predict changes in PK profiles of antipsychotic medicine by stages of gestation was the focus of the current study.
Ziprasidone (ZIP) was selected for the PBPK simulation in pregnancy, given that the information of its exposure in pregnant women is lacking. This drug is classified as an atypical antipsychotic drug commonly used for the treatment of schizophrenia and related psychoses.26 ZIP is considered a relatively safe antipsychotic for the fetus when used during pregnancy. It is a category C drug, meaning that there is not enough research done to determine if the drug is safe for pregnancy. Safety data on ziprasidone remain scarce and insufficient for a quantitative safety evaluation.27 Prescription could only be made when the benefit outweighs the potential risk to the fetus.28, 29 Given that there is the possibility for this drug to be prescribed during pregnancy, the question remains whether efficacy could be altered during pregnancy due to changes in plasma drug levels.
Considering the paucity of data to ensure appropriate dosing of ZIP in pregnant women, the aim of the modelling is to evaluate whether physiological changes during the pregnancy could affect PK behaviour of ZIP.
2. METHODS
Simcyp Simulator® version 16 (Simcyp Ltd, Sheffield, UK) was used as a platform for PBPK simulation. The PBPK model was first developed by using the information on ZIP physicochemical properties (pKa, log P, molecular weight, etc.) and followed by verification of the predicted exposures with the observed data from healthy adult population after intravenous (i.v.) and oral administrations. All simulations of PK profiles after oral administrations were performed under fed conditions, as this is the recommended usage.30 After performance verification by comparing the observed PK parameters with the simulated data in healthy adult, paediatric and geriatric populations, the PK profiles at first, second and third trimesters of pregnancy were then simulated. Ten virtual trials were conducted in 10 subjects per trial (10 × 10) randomly selected by the simulator; the proportion of female and age were controlled to match the observed data.
2.1. A PBPK model for ZIP in non‐pregnant subjects
The physicochemical, biopharmaceutics and PK parameters of ziprasidone obtained from the literature or in silico prediction tool are summarized in Table 1.The oral absorption of ZIP was predicted using a first‐order absorption model. The effective permeability (Peff,man) was estimated using data obtained from an in vitro permeability study in Caco‐2 cells, which consisted of 36 different compound standards of high (eg propranolol) and low (eg cimetidine) permeability.32 The fraction absorbed (fa) was set to 0.90,33 the first‐order absorption rate constant (ka) was set to 0.32 h−1, fraction unbound in the gut (fugut) was set to 0.35 to match the gut availability (Fg) of 0.85,33 and the flow rate for overall delivery of drug to the gut (Qgut) was 9.01 L h−1, which was predicted by the system. The lag time was adjusted in the range of 2–3 h: for lower doses (5–20 mg) it was set to 2 h, and for higher doses (40–80 mg) it was set to 3 h. The rationale for this adjustment is that ZIP is poorly water‐soluble (free‐base solubility in pH 6.5 buffered media <0.1 mg/mL),40 classified as a class II (low solubility, high permeability) drug according to the biopharmaceuticals classification system (BCS). Its absorption is dependent on the drug dissolution.41 The lag time was attributed to delayed gastric emptying because of the presence of food at the time of drug administration, as shown in a food effect study.34
Table 1.
Parameter values used for ziprasidone PBPK model
| Parameters | Value | Reference/comments |
|---|---|---|
| MW (g Mol−1) | 412.94 | 31 |
| Log P | 3.60 | 31 |
| Compound type | Monoprotic base | 31 |
| pKa | 6.58 | 31 |
| Absorption | ||
| Model | First‐order | |
| Peff,man (10−4 cm s−1) | 1.66 | Predicted using Papp, Caco‐2 |
| Papp, Caco‐2 (10−6 cm s−1) | 12.30 | 32 |
| fa | 0.90 | 33 |
| k a (h−1) | 0.32 | Adjusted |
| Lag time (h) | 2–3 | 34 |
| fugut | 0.35 | Predicted |
| Qgut (L h−1) | 9.01 | Predicted |
| Distribution | ||
| Model | Full PBPK model | |
| Vss/F (L kg−1) | 1.03 | Predicted using the Rodgers and Rowland method35, 36 |
| fu | 0.01 | 37 |
| B:P | 0.64 | Predicted |
| k p Scalar | 0.63 | Fitted 35, 36 |
| Elimination | ||
| CLiv (L h−1) | 22.80 | 38 |
| % contribution 3A | 33.00 | 39 |
B:P, blood‐to‐plasma partition ratio; CLiv, intravenous clearance; fa, fraction absorbed from dosage form; fu, fraction of drug unbound in plasma; fugut, fraction unbound in the gut; ka, first‐order absorption rate constant; kp, tissue‐plasma partition coefficient; log P, log of the octanol–water partition coefficient for the neutral compound; MW, molecular weight; Papp, Caco‐2, apparent permeability coefficient using CACO‐2 cells; PBPK, physiologically‐based pharmacokinetic; Peff,man, effective permeability in man; pKa, acid dissociation constant; Qgut, gut blood flow; Vss/F, apparent volume of distribution at steady state.
Upon reaching the portal vein, the PK of ZIP was predicted by a full PBPK model. The apparent volume of distribution at steady state (Vss/F) and the tissue–plasma partition (k p) coefficients were predicted using the Rodger and Rowland method.35, 36 The k p scalar parameter was set to 0.63 to match the Vss/F of 1.03 ± 0.16 L kg−1.38 The fraction of unbound drug in plasma (fu) of 0.01 was applied.42 The in vivo clearance of 22.80 ± 14 L h−1 after i.v. administration was used.38 The percentage of contribution of CYP3A enzymes was set to 33% (± 30%), as CYP3A4 is responsible for one third of ziprasidone metabolism.39
For the PBPK model performance verification, the simulated data was compared with the observed in vivo PK profiles obtained after i.v. administration31 and oral administration37, 38, 43, 44 of the standard dosage in healthy adult volunteers. The observed data were digitized from the literature using the Plot Digitizer software. Additional qualification was based on the area under the plasma concentration–time curve (AUC), peak plasma concentration (C max) and time to maximum concentration (t max) values obtained by PK analyses after administration of different dosages and routes. Table 2 summarizes the input values and study design for each simulation, including age and the proportion of males and females evaluated, based on the information reported in the referenced clinical trials.
Table 2.
Summaries of observed data of ziprasidone
| Posology | Observed data | Simulated data | |||||
|---|---|---|---|---|---|---|---|
| Scheme | Dose | n | Study reference | Population | Duration of the study (h) | Age | Proportion of female |
| i.v. single dose | 20 mg | 7 | 31 | Healthy adult volunteers | 36 | 20–50 | 0.50 |
| Oral single dose | 20 mg | 12 | 38 | Healthy adult volunteers | 36 | 20–45 | 0.00 |
| Oral single dose | 5 mg | 6 | 43 | Healthy adult volunteers | 72 | 20–29 | 0.00 |
| Oral single dose | 20 mg | 8 | 43 | Healthy adult volunteers | 72 | 20–45 | 0.00 |
| Oral single dose | 20 mg | 8 | 43 | Healthy adult volunteers | 72 | 18–34 | 0.00 |
| Oral single dose | 20 mg | 7 | 43 | Healthy adult volunteers | 72 | 22–40 | 0.00 |
| Oral single dose | 20 mg | 8 | 44 | Healthy adult volunteers | 72 | 19–31 | 0.00 |
| Oral single dose | 40 mg | 8 | 44 | Healthy adult volunteers | 72 | 19–31 | 0.00 |
| Oral single dose | 80 mg | 8 | 44 | Healthy adult volunteers | 72 | 19–31 | 0.00 |
| Oral single dose | 20 mg | 16 | 42 | Healthy adult volunteers | 12 | 18–44 | 0.5 |
| Oral twice daily dose, SS | 5 mg | 6 | 43 | Healthy adult volunteers | 72 | 20–29 | 0.00 |
| Oral twice daily dose, SS | 20 mg | 6 | 43 | Healthy adult volunteers | 72 | 20–45 | 0.00 |
| Oral twice daily dose, SS | 40 mg | 6 | 43 | Healthy adult volunteers | 72 | 18–34 | 0.00 |
| Oral twice daily dose, SS | 60 mg | 6 | 43 | Healthy adult volunteers | 72 | 22–40 | 0.00 |
| Oral twice daily dose, SS | 40 mg | 14 | 43 | Healthy adult volunteers | 72 | 20–42 | 0.75 |
| Oral twice daily dose, SS | 20 mg | 16 | 37 | Healthy adult volunteers | 180 | 18–44 | 0.5 |
| Oral single dose | 5 mg | 8 | 45 | Paediatric | 32 | 6–9 | 0.5 |
| Oral single dose | 10 mg | 7 | 45 | Paediatric | 32 | 9–15 | 0.5 |
| Oral single dose | 20 mg | 8 | 45 | Paediatric | 32 | 15–16 | 0.5 |
| Oral single dose | 20 mg | 16 | 37 | Geriatric | 12 | 65–76 | 0.5 |
| Oral twice daily dose, SS | 20 mg | 16 | 37 | Geriatric | 180 | 65–76 | 0.5 |
| Oral twice daily dose, SS | 40 mg | 3 | 46 | Non‐pregnant women = baseline | 72 | 20–45 | 1.00 |
| Oral twice daily dose, SS | 40 mg | 3 | 46 | Pregnant women—GA = 6 weeks | 72 | 20–45 | 1.00 |
| Oral twice daily dose, SS | 40 mg | 3 | 46 | Pregnant women—GA = 20 weeks | 72 | 20–45 | 1.00 |
| Oral twice daily dose, SS | 40 mg | 3 | 46 | Pregnant women—GA = 34 weeks | 72 | 20–45 | 1.00 |
GA, gestational age in week; i.v., intravenous; SS, steady‐state
Note: 10 virtual trials were conducted, 10 subjects per trial, randomly selected by the simulator such that the proportion of female and age were fitted to match the observed data.
The performance of simulations was assessed by the mean fold error (MFE) (Equation 1) for PK parameters (AUC, C max and t max) extracted from Simcyp®:
| (1) |
The model was accepted if all predicted PK parameters were within two‐fold of the corresponding observed values from the single and multiple ascending dose PK studies (MFE 0.5–2.0), as is commonly applied in assessing PBPK model performance.17, 47
Using the final model for healthy volunteers, the extrapolation to the paediatric and geriatric populations was accomplished using Simcyp® default parameters for paediatric48, 49 and geriatric50 populations. The PK profiles in these special populations were simulated and compared with the observed data for ZIP in paediatric45 and geriatric37 populations, using the same criteria previously described.
2.2. A PBPK model for ZIP in pregnant subjects
The PBPK model developed in healthy non‐pregnant subjects was used to create the ZIP PBPK model in pregnant women. Again, the Simcyp® default parameters for pregnancy were utilized to simulate the PK profile in this special population. The pregnancy tab was selected, then all drug parameters (eg physicochemical properties) were fixed and only parameters related to physiology (eg body weight, blood flow, glomerular filtration rate [GFR], etc.) were automatically modified by the software in order to mimic the physiology of pregnant women.
Various physiological modifications occurring during pregnancy were taken into account using the pregnancy module, including weight gain, plasma protein concentration, individual organ/tissue volumes, blood flow and GFRs.25 The PBPK model was extended by addition of an extra compartment for the feto‐placental unit to create a pregnancy physiologically‐based pharmacokinetic (p‐PBPK) model.24
Simulations of twice‐daily 40 mg ziprasidone were performed during the 6th, 20th and 34th weeks (ie first, second and third trimester, respectively), which are the same gestational ages as those reported previously.46 Non‐pregnant condition simulations, which were considered as baseline, were performed using the female healthy volunteer population.
2.3. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY,51 and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18.52
3. RESULTS
3.1. Model evaluation for the non‐pregnant subjects
The predicted and observed PK parameters of ziprasidone after single i.v. dose infused over an hour and after single and multiple oral ascending doses are summarized in Table 3. The simulated PK parameters in healthy adult volunteers were consistent with the observed values: AUC MFEs ranged from 0.94 to 1.09; C max MFEs ranged from 0.89 to 1.40; t max MFEs ranged from 0.80 to 1.08. The predicted AUC, C max and t max were all within a 50–200% boundary of the observed values (Figure 1). Figure 2 compares the model predictions against observed ZIP concentrations in healthy adult volunteers after a single dose of 20 mg i.v., and 20, 40 and 80 mg oral, showing that the overall PK profile was well‐described.
Table 3.
Predicted and observed pharmacokinetic parameters of ziprasidone in different populations and doses. Results expressed as mean predicted (pred) and observed (obs) parameters
| Population | Study reference | Dose | AUC (ng ml −1 h −1 ) a | C max (ng ml −1 ) | t max (h) | |
|---|---|---|---|---|---|---|
| Healthy adult volunteers | 31 | 20 mg, i.v. single dose | Observed | 817.68 | 297.05 | 0.93 |
| Predicted | 891.99 | 415.33 | 1.00 | |||
| Pred/obs ratio | 1.09 | 1.40 | 1.08 | |||
| Single i.v. dose MFE | 1.09 | 1.40 | 1.08 | |||
| 38 | 20 mg, oral single dose | Observed | 514.00 | 64.00 | 8.20 | |
| Predicted | 349.74 | 43.21 | 5.74 | |||
| Pred/obs ratio | 0.68 | 0.68 | 0.70 | |||
| 43 | 5 mg, oral single dose | Observed | 86.70 | 12.20 | 5.00 | |
| Predicted | 88.85 | 11.03 | 3.73 | |||
| Pred/obs ratio | 1.02 | 0.90 | 0.75 | |||
| 20 mg, oral single dose | Observed | 226.30 | 26.60 | 4.80 | ||
| Predicted | 347.36 | 43.10 | 3.73 | |||
| Pred/obs ratio | 1.53 | 1.62 | 0.78 | |||
| 20 mg, oral single dose | Observed | 376.70 | 60.00 | 3.80 | ||
| Predicted | 359.42 | 44.58 | 3.75 | |||
| Pred/obs ratio | 0.95 | 0.74 | 0.99 | |||
| 20 mg, oral single dose | Observed | 308.40 | 34.30 | 4.00 | ||
| Predicted | 349.44 | 43.31 | 3.71 | |||
| Pred/obs ratio | 1.13 | 1.26 | 0.93 | |||
| 44 | 20 mg, oral single dose | Observed | 467.00 | 50.00 | 5.60 | |
| Predicted | 358.89 | 44.70 | 3.76 | |||
| Pred/obs ratio | 0.77 | 0.89 | 0.67 | |||
| 40 mg, oral single dose | Observed | 902.00 | 87.00 | 6.40 | ||
| Predicted | 710.82 | 88.09 | 4.76 | |||
| Pred/obs ratio | 0.79 | 1.01 | 0.74 | |||
| 80 mg, oral single dose | Observed | 1911.00 | 165.00 | 6.60 | ||
| Predicted | 1421.64 | 176.18 | 4.76 | |||
| Pred/obs ratio | 0.74 | 1.07 | 0.72 | |||
| 37 | 20 mg, single dose oral | Observed | 344.00 | 56.00 | 4.00 | |
| Predicted | 271.15 | 42.61 | 3.58 | |||
| Pred/obs ratio | 0.79 | 0.76 | 0.90 | |||
| Single oral dose MFE | 0.94 | 0.99 | 0.80 | |||
| 43 | 5 mg, b.i.d. | Observed | 109.80 | 14.80 | 5.20 | |
| Predicted | 88.88 | 12.95 | 3.53 | |||
| Pred/obs ratio | 0.81 | 0.88 | 0.68 | |||
| 20 mg, b.i.d. | Observed | 259.20 | 44.60 | 3.80 | ||
| Predicted | 347.58 | 50.66 | 3.53 | |||
| Pred/obs ratio | 1.34 | 1.14 | 0.93 | |||
| 40 mg, b.i.d. | Observed | 658.00 | 118.60 | 3.70 | ||
| Predicted | 719.34 | 104.65 | 3.55 | |||
| Pred/obs ratio | 1.09 | 0.88 | 0.96 | |||
| 60 mg, b.i.d. | Observed | 1027.90 | 139.40 | 4.70 | ||
| Predicted | 1048.85 | 152.93 | 3.51 | |||
| Pred/obs ratio | 1.02 | 1.10 | 0.75 | |||
| 44 | 40 mg, b.i.d. | Observed | 940.00 | 163.00 | 3.40 | |
| Predicted | 672.76 | 99.44 | 3.29 | |||
| Pred/obs ratio | 0.72 | 0.61 | 0.97 | |||
| 37 | 20 mg, b.i.d. | Observed | 465.00 | 69.00 | 4.00 | |
| Predicted | 344.65 | 50.49 | 3.38 | |||
| Pred/obs ratio | 0.74 | 0.73 | 0.85 | |||
| Oral dose b.i.d. (SS) MFE | 0.95 | 0.89 | 0.85 | |||
| Paediatric | 45 | 5 mg, oral single dose | Observed | 247.00 | 36.00 | 5.00 |
| Predicted | 259.59 | 37.90 | 3.39 | |||
| Pred/obs ratio | 1.05 | 1.05 | 0.68 | |||
| 10 mg, oral single dose | Observed | 338.00 | 45.00 | 5.10 | ||
| Predicted | 366.10 | 50.15 | 3.57 | |||
| Pred/obs ratio | 1.08 | 1.11 | 0.70 | |||
| 20 mg, oral single dose | Observed | 457.00 | 51.00 | 5.50 | ||
| Predicted | 581.07 | 73.84 | 3.72 | |||
| Pred/obs ratio | 1.27 | 1.44 | 0.67 | |||
| Single oral dose MFE—paediatric | 1.11 | 1.17 | 0.69 | |||
| Geriatric | 37 | 20 mg, oral single dose | Observed | 382.00 | 60.00 | 5.00 |
| Predicted | 269.46 | 42.54 | 3.50 | |||
| Pred/obs ratio | 0.71 | 0.71 | 0.70 | |||
| Single oral dose MFE—geriatric | 0.71 | 0.71 | 0.70 | |||
| 20 mg, b.i.d. | Observed | 560.00 | 85.00 | 4.00 | ||
| Predicted | 354.20 | 51.52 | 3.30 | |||
| Pred/obs ratio | 0.63 | 0.61 | 0.83 | |||
| Oral dose b.i.d. (SS) MFE—geriatric | 0.63 | 0.61 | 0.83 | |||
AUC, area under the plasma concentration–time curve; b.i.d., twice daily; C max, maximum concentration; i.v., intravenous; MFE, mean fold error; SS, steady‐state; t max, time to maximum concentration.
0–∞ for single dose studies and 0–12 h for multiples dose twice‐daily studies.
Note: Virtual trials were conducted in 100 (10 × 10) subjects randomly selected by simulator the proportion of female and age were fitted to match the observed data.
Figure 1.
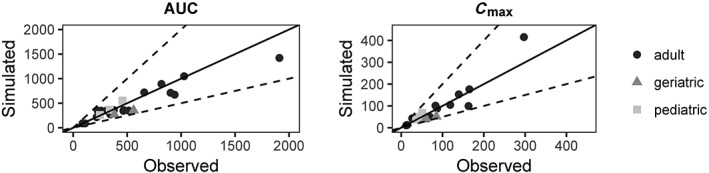
Comparison between simulated and observed PK parameters from several studies in the literature for non‐pregnant population. Solid lines represent line of unity, dashed lines represent two‐fold difference
Figure 2.
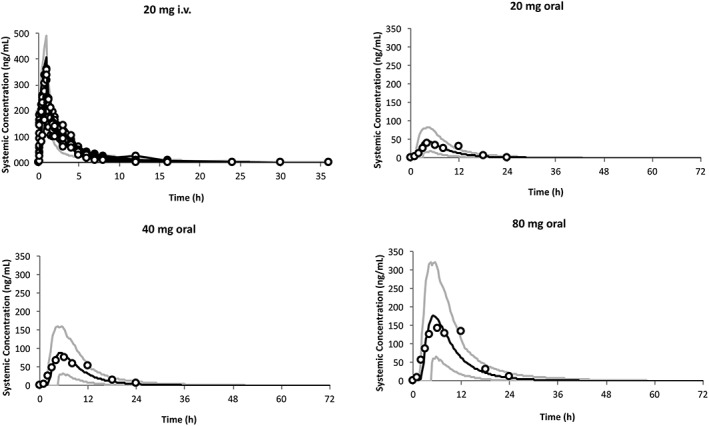
PK profiles in the non‐pregnant population. Simulation (mean predictions in black lines and 5th–95th percentiles of predictions in grey lines) of PK profiles for an i.v. administration of 20 mg infused over 1 hour and for oral administrations of 20, 40 and 80 mg of ziprasidone (under fed conditions). Simulations were compared with observed clinical data (circle) from 20 mg i.v. administration,31 20, 40 and 80 mg oral administration44
For the paediatric and geriatric populations, the predicted AUC, C max and t max were also within the 50–200% boundary of the observed values. In paediatrics, AUC MFE ranged from 1.05 to 1.27; C max MFE ranged from 1.05 to 1.44; t max MFE ranged from 0.67 to 0.70. In the geriatric population, AUC MFE ranged from 0.63 to 0.71; C max MFE ranged from 0.61 to 0.71; t max MFE ranged from 0.70 to 0.83) (Figure 1).
3.2. Evaluation of the predictive performance of the ZIP PBPK model in pregnant women
Simulations of PK profiles at steady state were performed during the first (6th week), second (20th week) and third (34th week) trimesters of pregnancy, and compared with that of baseline non‐pregnant women. Figure 3 shows the comparison between predicted profiles of ZIP for non‐pregnant and pregnant women at different periods of pregnancy.
Figure 3.
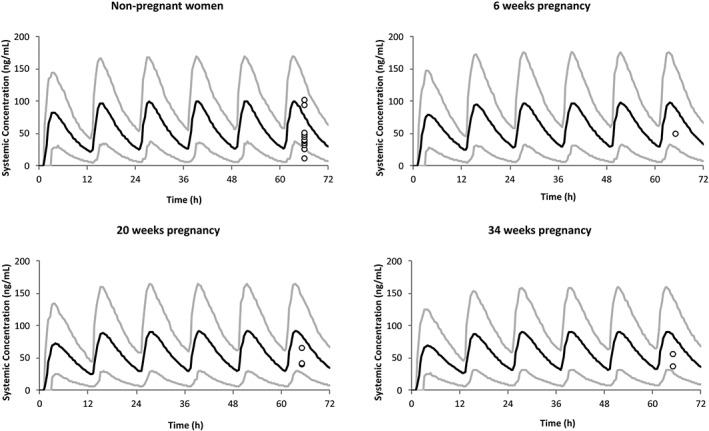
PK profiles in non‐pregnant and pregnant women. Simulation (mean predictions in black lines and 5th–95th percentiles of predictions in grey lines) of PK profiles for oral administration of 40 mg twice‐daily, in non‐pregnant situation and during the 6th, 20th and 34th weeks of pregnancy. Simulations were compared with the observed clinical data (circle) after 40 mg oral twice‐daily administration46
The reliability of the developed model was verified by comparison of the predicted PK profiles with the observed data for pregnant women at three stages of pregnancy (6th, 20th and 34th weeks). However, only one study of ZIP in pregnancy from three women was found in the literature.46 Simulation of changes in PK profile of ZIP throughout pregnancy weeks was also performed. Predicted PK parameters of ZIP after multiple oral doses (40 mg twice‐daily) during different periods of pregnancy are summarized in Table 4.
Table 4.
Predicted steady‐state pharmacokinetic parameters of ziprasidone during different periods of pregnancy (expressed as mean ± SD data)
| Parameters | Baselinea | 6 weeksa | 20 weeksa | 34 weeksa |
|---|---|---|---|---|
| Dose | 40 mg, b.i.d. | 40 mg, b.i.d. | 40 mg, b.i.d. | 40 mg, b.i.d. |
| fa | 0.81 ± 0.17 | 0.81 ± 0.17 | 0.81 ± 0.17 | 0.81 ± 0.17 |
| fu | 0.01007 ± 0.001 | 0.01038 ± 0.0010 | 0.01131 ± 0.0011 | 0.01285 ± 0.0012 |
| CLint (L h−1) | 3914.11 ± 1430.92 | 3690.45 ± 1503.84 | 3461.41 ± 1419.57 | 3115.07 ± 1264.15 |
| CL/F (L h−1)b | 69.94 ± 43.06 | 71.23 ± 51.71 | 74.41 ± 54.91 | 71.48 ± 49.36 |
| AUC0‐12h,SS (ng mL−1 h−1) | 655.39 ± 314.65 | 666.64 ± 349.45 | 641.00 ± 339.99 | 656.12 ± 332.99 |
| C max (ng mL−1) | 96.57 ± 43.24 | 92.11 ± 45.81 | 86.17 ± 43.19 | 85.99 ± 41.06 |
| C trough (ng mL−1) | 26.72 ± 16.12 | 29.74 ± 17.52 | 30.30 ± 17.61 | 32.36 ± 17.96 |
| t max (h) | 3.22 ± 0.70 | 3.32 ± 0.75 | 3.33 ± 0.76 | 3.34 ± 0.76 |
| Vss/F (L kg−1) | 1.15 ± 0.24 | 1.19 ± 0.22 | 1.29 ± 0.24 | 1.43 ± 0.26 |
AUC0‐12h,ss, area under the plasma concentration–time curve from 0–12 h at steady‐state; b.i.d., twice daily; CLint, intrinsic clearance; C max, maximum concentration; C trough, trough concentration; fa, fraction absorbed from dosage form; fu, fraction of drug unbound in plasma; Vss/F, apparent volume of distribution at steady‐state; t max, time to maximum concentration.
Baseline, non‐pregnant women; 6 weeks, first trimester pregnancy; 20 weeks, second trimester pregnancy; 34 weeks, third trimester pregnancy.
Clearance computed as F × Dose/AUC.
The simulations showed an increase in the fraction unbound of ZIP across the first (3%), second (12%) and third (28%) trimester compared with non‐pregnant women. The intrinsic clearance decreased by 6%, 11% and 20% in the first, second and third trimester of pregnancy, respectively, when compared with non‐pregnant women. The change in intrinsic clearance of ZIP was complemented by an increase in the unbound fraction that cancels out the effect on total clearance. Consequently, the total clearance increased by only 2%, 6% and 2% over the same periods. Vss/F increased by 12% and 24% in the second and third trimester of pregnancy, respectively, compared to non‐pregnant women. A change in Vss/F usually affects the half‐life of the drug but not the AUC. An increase in Vss/F results in a decrease in C max as the drug has to distribute over a larger volume. A corresponding decrease of 10% in C max from the second trimester of pregnancy was accompanied by an increase in trough concentration (C trough) of 13% and 21% in the second and third trimester of pregnancy, respectively. This suggests that the half‐life of ZIP is increased during pregnancy.
4. DISCUSSION
The p‐PBPK model that incorporates the pregnancy‐induced changes in various anatomical, physiological and biological parameters is a feasible alternative for empirical dosage selection when there are no clinical studies available to guide dose selection.24, 25 In this study, the p‐PBPK modelling was applied to predict PK changes throughout pregnancy. The model predicted the impact of these changes on systemic exposure of ZIP during pregnancy. The pregnancy model utilized for ZIP incorporated time‐varying physiological change in a virtual female population.24, 25, 53 A number of studies have provided independent verification and application of a pregnancy module to study drugs in the pregnant population,16, 17, 18 which helped to increase confidence in the predictive performance of the respective models.19
Despite the fact that strong physiological changes associated with pregnancy can alter drug disposition,7, 11, 54 the simulated exposure in pregnant women showed no significant difference for ZIP when compared to non‐pregnant women. A number of hypotheses were postulated. The reduction in the levels of some plasma proteins (albumin and α‐1‐acid glycoprotein) by 20–30% in the third trimester of pregnancy affects the protein‐binding capacity of ZIP.8 This decrease in plasma protein levels is relevant for ZIP, which is highly protein bound.37 The lower plasma protein can result in higher levels of unbound fraction of ZIP in the blood during pregnancy, which becomes available for drug metabolism.47 Intrinsic clearance decreased by 20% in the third trimester of pregnancy when compared with non‐pregnant women, while the total clearance increased by a modest 2%, 6% and 2% in the first, second and third trimester of pregnancy.
Renal clearance of ZIP is not considered relevant in the change in total drug clearance, as less than 5% of the intact ZIP is eliminated by urinary excretion.39, 55 Metabolism could potentially be a more important contributing factor, as pregnancy is associated with an increased CYP3A4 activity. The importance of the change in CYP3A4 metabolism on ZIP disposition can be derived from drug–drug interaction studies. Some studies examined the effect of potent CYP3A4 inhibitors (ie ketoconazole),56 nonspecific inhibitors of cytochrome P450 (ie cimetidine) and antacids on ZIP pharmacokinetics and showed that these agents are unlikely to alter ZIP pharmacokinetics significantly.42 Another study had shown that induction of CYP3A4 with carbamazepine led to a modest reduction (<36%) in steady‐state exposure of ZIP.57 It can be inferred from this observation that changes in CYP3A4 activity during pregnancy will not impact ZIP disposition to an extent greater than two‐fold difference in its exposure.
The p‐PBPK model used in this study describes drug disposition in the maternal organs. However, the fetoplacental unit did not incorporate a detailed physiological organ system within the model structure. Nonetheless, the fetoplacental compartment was semi‐mechanistic in that its volume and the blood flow from the maternal circulation developed over the course of pregnancy. The fetoplacental compartment was assumed to be homogeneous without subdivision into fetus, placenta, membranes, amniotic fluid and the umbilical cord; this assumption limited any extrapolation to interpret drug concentrations within the fetoplacental unit. The partition coefficient for this fetoplacental compartment was assumed to be identical to that for the brain compartment, as the barriers for these tissues share some similar physiological and biochemical functions.35 Considering these assumptions, the increase in Vss/F is related to the increase in the size of the fetoplacental compartment, as expected, throughout the pregnancy. Another important assumption in this model was that the fetus does not contribute to the clearance of ZIP in the pregnant woman.
Constant care needs to be taken when considering the risks and benefits of medications for mothers and their unborn children; this prompted us to develop a rational analysis of an antipsychotic drug ziprasidone in pregnant women.58 The results of the current study indicate that the plasma drug level is within a desirable therapeutic range; consequently, pregnancy per se does not seem to change plasma ZIP exposure. The model result suggests that dose adjustment is not recommended for mothers taking ZIP during their pregnancy in order to prevent possible cases of overdosing, and to avoid side effects such as hypertonicity and dystonic reactions.
COMPETING INTERESTS
There are no competing interests to declare.
CONTRIBUTORS
C.B. and F.S.M. developed and analyzed the PBPK model. S.K.B.S. provided the pharmacokinetic and physiological discussion of the model and A.D. was the principal investigator who provided advice and concept to the overall research.
ACKNOWLEDGEMENTS
This research was made possible by the support of Certara through its Simcyp Simulator Academic License Program. The research was supported by Fundação Araucária Process n° 029/2017 – PROT. 48027.
Biesdorf C, Martins FS, Sy SKB, Diniz A. Physiologically‐based pharmacokinetics of ziprasidone in pregnant women. Br J Clin Pharmacol. 2019;85:914–923. 10.1111/bcp.13872
REFERENCES
- 1. National Institute of Mental Health . Any mental illness (AMI) among adults . (n.d.). http://www.nimh.nih.gov/health/statistics/prevalence/any‐mental‐illness‐ami‐among‐adults.shtml. Accessed June 29, 2018.
- 2. Moher D, Liberati A, Tetzlaff J, Altman DG, for the PRISMA Group . Preferred reporting items for systematic reviews and meta‐analyses: The PRISMA statement. PLoS Med. 2009;6(7):e1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. World Health Organization . Mental disorders. http://www.who.int/mediacentre/factsheets/fs396/en/. Accessed June 29, 2018.
- 4. McGrath JJ, Saha S, Al‐Hamzawi AO, et al. Age of onset and lifetime projected risk of psychotic experiences: Cross‐national data from the World Mental Health Survey. Schizophr Bull. 2016;42(4):933‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carlin A, Alfirevic Z. Physiological changes of pregnancy and monitoring. Best Pract Res Clin Obstet Gynaecol. 2008;22(5):801‐823. [DOI] [PubMed] [Google Scholar]
- 6. Isoherranen N, Thummel KE. Drug metabolism and transport during pregnancy: How does drug disposition change during pregnancy and what are the mechanisms that cause such changes? Drug Metab Dispos. 2013;41(2):256‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Anderson GD. Pregnancy‐induced changes in pharmacokinetics: A mechanistic‐based approach. Clin Pharmacokinet. 2005;44(10):989‐1008. [DOI] [PubMed] [Google Scholar]
- 8. Ke AB, Rostami‐Hodjegan A, Zhao P, Unadkat JD. Pharmacometrics in pregnancy: An unmet need. Annu Rev Pharmacol Toxicol. 2014;54(1):53‐69. [DOI] [PubMed] [Google Scholar]
- 9. Ke AB, Nallani SC, Zhao P, Rostami‐Hodjegan A, Unadkat JD. Expansion of a PBPK model to predict disposition in pregnant women of drugs cleared via multiple CYP enzymes, including CYP2B6, CYP2C9 and CYP2C19. Br J Clin Pharmacol. 2014;77(3):554‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jeong H. Altered drug metabolism during pregnancy: Hormonal regulation of drug‐metabolizing enzymes. Expert Opin Drug Metab Toxicol. 2010;6(6):689‐699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pariente G, Leibson T, Carls A, Adams‐Webber T, Ito S, Koren G. Pregnancy‐associated changes in pharmacokinetics: A systematic review. PLoS Med. 2016;13(11):e1002160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abel KM. Fetal antipsychotic exposure in a changing landscape: Seeing the future. Br J Psychiatry. 2013;202(5):321‐323. [DOI] [PubMed] [Google Scholar]
- 13. Freeman MP. Pregnancy and psychiatric disorders: Inherent risks and treatment decisions. J Clin Psychiatry. 2013;74(4):373‐374. [DOI] [PubMed] [Google Scholar]
- 14. Sy SK, Innes S, Derendorf H, Cotton MF, Rosenkranz B. Estimation of intracellular concentration of stavudine triphosphate in HIV‐infected children given a reduced dose of 0.5 milligrams per kilogram twice daily. Antimicrob Agents Chemother. 2014;58(2):1084‐1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sy SK, Malmberg R, Matsushima A, et al. Effect of reducing the paediatric stavudine dose by half: A physiologically‐based pharmacokinetic model. Int J Antimicrob Agents. 2015;45(4):413‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. De Sousa Mendes M, Hirt D, Urien S, et al. Physiologically‐based pharmacokinetic modeling of renally excreted antiretroviral drugs in pregnant women. Br J Clin Pharmacol. 2015;80(5):1031‐1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Colbers A, Greupink R, Litjens C, Burger D, Russel FG. Physiologically based modelling of darunavir/ritonavir pharmacokinetics during pregnancy. Clin Pharmacokinet. 2016;55(3):381‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jogiraju VK, Avvari S, Gollen R, Taft DR. Application of physiologically based pharmacokinetic modeling to predict drug disposition in pregnant populations. Biopharm Drug Dispos. 2017;38(7):426‐438. [DOI] [PubMed] [Google Scholar]
- 19. Ke AB, Greupink R, Abduljalil K. Drug dosing in pregnant women: Challenges and opportunities in using physiologically based pharmacokinetic modeling and simulations. CPT Pharmacometrics Syst Pharmacol. 2018;7(2):103‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luzon E, Blake K, Cole S, Nordmark A, Versantvoort C, Berglund EG. Physiologically based pharmacokinetic modeling in regulatory decision‐making at the European Medicines Agency. Clin Pharmacol Ther. 2016;102(1):98‐105. [DOI] [PubMed] [Google Scholar]
- 21. Wagner C, Pan Y, Hsu V, et al. Predicting the effect of cytochrome P450 inhibitors on substrate drugs: Analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin Pharmacokinet. 2015;54(1):117‐127. [DOI] [PubMed] [Google Scholar]
- 22. Wagner C, Pan Y, Hsu V, Sinha V, Zhao P. Predicting the effect of CYP3A inducers on the pharmacokinetics of substrate drugs using physiologically based pharmacokinetic (PBPK) modeling: An analysis of PBPK submissions to the US FDA. Clin Pharmacokinet. 2016;55(4):475‐483. [DOI] [PubMed] [Google Scholar]
- 23. Jamei M. Recent advances in development and application of physiologically‐based pharmacokinetic (PBPK) models: A transition from academic curiosity to regulatory acceptance. Curr Pharmacol Rep. 2016;2(3):161‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gaohua L, Abduljalil K, Jamei M, Johnson TN, Rostami‐Hodjegan A. A pregnancy physiologically based pharmacokinetic (p‐PBPK) model for disposition of drugs metabolized by CYP1A2, CYP2D6 and CYP3A4. Br J Clin Pharmacol. 2012;74(5):873‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abduljalil K, Furness P, Johnson TN, Rostami‐Hodjegan A, Soltani H. Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: A database for parameters required in physiologically based pharmacokinetic modelling. Clin Pharmacokinet. 2012;51(6):365‐396. [DOI] [PubMed] [Google Scholar]
- 26. Vogel F, Gansmuller R, Leiblein T, et al. The use of ziprasidone in clinical practice: Analysis of pharmacokinetic and pharmacodynamic aspects from data of a drug monitoring survey. Eur Psychiatry. 2009;24(3):143‐148. [DOI] [PubMed] [Google Scholar]
- 27. Damkier P, Videbech P. The safety of second‐generation antipsychotics during pregnancy: A clinically focused review. CNS Drugs. 2018;32(4):351‐366. [DOI] [PubMed] [Google Scholar]
- 28. Cohen LS, Viguera AC, McInerney KA, et al. Reproductive safety of second‐generation antipsychotics: Current data from the Massachusetts General Hospital National Pregnancy Registry for Atypical Antipsychotics. Am J Psychiatry. 2016;173(3):263‐270. [DOI] [PubMed] [Google Scholar]
- 29. Huybrechts KF, Hernandez‐Diaz S, Patorno E, et al. Antipsychotic use in pregnancy and the risk for congenital malformations. JAMA Psychiat. 2016;73(9):938‐946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Geodon (ziprasidone) prescribing information. New York: Pfizer; 2005. [Google Scholar]
- 31. Sutton SC, Nause R, Gandelman K. The impact of gastric pH, volume, and emptying on the food effect of ziprasidone oral absorption. AAPS J. 2017;19(4):1084‐1090. [DOI] [PubMed] [Google Scholar]
- 32. Yee S. In vitro permeability across Caco‐2 cells (colonic) can predict in vivo (small intestinal) absorption in man—Fact or myth. Pharm Res. 1997;14(6):763‐766. [DOI] [PubMed] [Google Scholar]
- 33. Nishimuta H, Nakagawa T, Nomura N, Yabuki M. Significance of reductive metabolism in human intestine and quantitative prediction of intestinal first‐pass metabolism by cytosolic reductive enzymes. Drug Metab Dispos. 2013;41(5):1104‐1111. [DOI] [PubMed] [Google Scholar]
- 34. Hamelin BA, Allard S, Laplante L, et al. The effect of timing of a standard meal on the pharmacokinetics and pharmacodynamics of the novel atypical antipsychotic agent ziprasidone. Pharmacotherapy. 1998;18(1):9‐15. [PubMed] [Google Scholar]
- 35. Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95(6):1238‐1257. [DOI] [PubMed] [Google Scholar]
- 36. Rodgers T, Rowland M. Mechanistic approaches to volume of distribution predictions: Understanding the processes. Pharm Res. 2007;24(5):918‐933. [DOI] [PubMed] [Google Scholar]
- 37. Wilner KD, Tensfeldt TG, Baris B, et al. Single‐ and multiple‐dose pharmacokinetics of ziprasidone in healthy young and elderly volunteers. Br J Clin Pharmacol. 2000;49(Suppl 1):15S‐20S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miceli JJ, Wilner KD, Swan SK, Tensfeldt TG. Pharmacokinetics, safety, and tolerability of intramuscular ziprasidone in healthy volunteers. J Clin Pharmacol. 2005;45(6):620‐630. [DOI] [PubMed] [Google Scholar]
- 39. Beedham C, Miceli JJ, Obach RS. Ziprasidone metabolism, aldehyde oxidase, and clinical implications. J Clin Psychopharmacol. 2003;23(3):229‐232. [DOI] [PubMed] [Google Scholar]
- 40. Thombre AG, Herbig SM, Alderman JA. Improved ziprasidone formulations with enhanced bioavailability in the fasted state and a reduced food effect. Pharm Res. 2011;28(12):3159‐3170. [DOI] [PubMed] [Google Scholar]
- 41. Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413‐420. [DOI] [PubMed] [Google Scholar]
- 42. Wilner KD, Hansen RA, Folger CJ, Geoffroy P. The pharmacokinetics of ziprasidone in healthy volunteers treated with cimetidine or antacid. Br J Clin Pharmacol. 2000;49(Suppl 1):57S‐60S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miceli JJ, Wilner KD, Hansen RA, Johnson AC, Apseloff G, Gerber N. Single‐ and multiple‐dose pharmacokinetics of ziprasidone under non‐fasting conditions in healthy male volunteers. Br J Clin Pharmacol. 2000;49(Suppl 1):5S‐13S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miceli JJ, Glue P, Alderman J, Wilner K. The effect of food on the absorption of oral ziprasidone. Psychopharmacol Bull. 2007;40(3):58‐68. [PubMed] [Google Scholar]
- 45. Sallee FR, Miceli JJ, Tensfeldt T, Robarge L, Wilner K, Patel NC. Single‐dose pharmacokinetics and safety of ziprasidone in children and adolescents. J Am Acad Child Adolesc Psychiatry. 2006;45(6):720‐728. [DOI] [PubMed] [Google Scholar]
- 46. Westin AA, Brekke M, Molden E, Skogvoll E, Castberg I, Spigset O. Treatment with antipsychotics in pregnancy: Changes in drug disposition. Clin Pharmacol Ther. 2018;103(3):477‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Polasek TM, Tucker GT, Sorich MJ, et al. Prediction of olanzapine exposure in individual patients using physiologically based pharmacokinetic modelling and simulation. Br J Clin Pharmacol. 2018;84(3):462‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Abduljalil K, Jamei M, Rostami‐Hodjegan A, Johnson TN. Changes in individual drug‐independent system parameters during virtual paediatric pharmacokinetic trials: Introducing time‐varying physiology into a paediatric PBPK model. AAPS J. 2014;16(3):568‐576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Johnson TN, Rostami‐Hodjegan A. Resurgence in the use of physiologically based pharmacokinetic models in pediatric clinical pharmacology: Parallel shift in incorporating the knowledge of biological elements and increased applicability to drug development and clinical practice. Paediatr Anaesth. 2011;21(3):291‐301. [DOI] [PubMed] [Google Scholar]
- 50. Polasek TM, Patel F, Jensen BP, Sorich MJ, Wiese MD, Doogue MP. Predicted metabolic drug clearance with increasing adult age. Br J Clin Pharmacol. 2013;75(4):1019‐1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new Guide to IMMUNOPHARMACOLOGY. Nucl Acids Res. 2017;46:D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol. 2017;174(Suppl 1):S272‐S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Corley RA, Mast TJ, Carney EW, Rogers JM, Daston GP. Evaluation of physiologically based models of pregnancy and lactation for their application in children's health risk assessments. Crit Rev Toxicol. 2003;33(2):137‐211. [DOI] [PubMed] [Google Scholar]
- 54. Tasnif Y, Morado J, Hebert MF. Pregnancy‐related pharmacokinetic changes. Clin Pharmacol Ther. 2016;100(1):53‐62. [DOI] [PubMed] [Google Scholar]
- 55. Stahl SM, Shayegan DK. The psychopharmacology of ziprasidone: Receptor‐binding properties and real‐world psychiatric practice. J Clin Psychiatry. 2003;64(Suppl 19):6‐12. [PubMed] [Google Scholar]
- 56. Miceli JJ, Smith M, Robarge L, Morse T, Laurent A. The effects of ketoconazole on ziprasidone pharmacokinetics—A placebo‐controlled crossover study in healthy volunteers. Br J Clin Pharmacol. 2000;49(Suppl 1):71S‐76S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Miceli JJ, Anziano RJ, Robarge L, Hansen RA, Laurent A. The effect of carbamazepine on the steady‐state pharmacokinetics of ziprasidone in healthy volunteers. Br J Clin Pharmacol. 2000;49(Suppl 1):65S‐70S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ruzic K, Dadic‐Hero E, Knez R, Medved P, Petric D. Pregnancy and atypical antipsychotics. Psychiatr Danub. 2009;21(3):368‐370. [PubMed] [Google Scholar]