

Abstract
Ticarcillin–clavulanate covers a broad spectrum of pathogens that are common in premature infants. In infants <30 weeks gestational age, pharmacokinetic data to guide ticarcillin–clavulanate dosing are lacking. We enrolled 15 premature infants <30 weeks gestational age, determined pharmacokinetic parameters, and performed dosing simulations to determine optimal dosing for ticarcillin–clavulanate. The infants had a median (range) postnatal age (PNA) of 18 days (6–44 days) and gestational age of 25 weeks (23–28 weeks). Clearance was lower in infants with a PNA <14 days (0.050 L/kg/h [range 0.043–0.075]) compared with a PNA ≥14–45 days (0.078 L/kg/h [0.047–0.100]), consistent with maturation of renal function. Dosing simulations determined that ticarcillin 75 mg/kg q12h (PNA <14 days) or q8h (PNA ≥ 14–45 days) achieved the target exposure for organisms with a minimum inhibitory concentration ≤16 μ/mL in >90% of simulated infants. For highly resistant organisms (minimum inhibitory concentration 32 μg/mL), increased dosing frequency or extended infusion are necessary.
Keywords: clavulanate, infants, pharmacokinetics, premature, ticarcillin
What is already known about this subject
Infection is common in premature infants and results in substantial morbidity and mortality.
Ticarcillin–clavulanate covers the most common pathogens in premature infants, but appropriate dosing in premature infants <30 weeks gestational age is unknown.
What this study adds
This pharmacokinetic trial of ticarcillin–clavulanate enrolled 15 premature infants <30 weeks gestational age.
Clearance increased with postnatal age (PNA) and gestational age, reflecting maturation of renal function.
Ticarcillin 75 mg/kg q12h (PNA < 14 days) or q8h (PNA ≥ 14–45 days) achieved the target exposure for common organisms. For highly resistant organisms, increased dosing frequency or prolonged infusion are necessary.
1. INTRODUCTION
Seventy percent of late‐onset sepsis in the neonatal intensive care unit is due to Gram‐positive organisms.1 Coagulase‐negative Staphylococcus is the most commonly isolated pathogen,1, 2 and Staphylococcus aureus is the second most common.2, 3, 4, 5, 6, 7 Infants with these infections have prolonged hospitalizations and an increased risk of neurodevelopmental impairment and death.2, 8, 9, 10, 11, 12, 13, 14
Ticarcillin–clavulanate covers a broad spectrum of pathogens, including methicillin‐sensitive Staphylococcus aureus, coagulase‐negative Staphylococcus species, and many Gram‐negative bacteria.15 Ticarcillin–clavulanate is primarily eliminated unchanged via the kidneys, and clearance (CL) in infants will be impacted by maturation of renal function. Maturation‐related change in ticarcillin–clavulanate CL is supported by PK studies in premature and term infants where CL increased with postnatal age (PNA) and gestational age (GA).16, 17, 18, 19, 20, 21 Of note, only 3 infants in the above studies were < 30 weeks GA at birth.
The pharmacodynamic (PD) target for β‐lactam efficacy is the fraction of time the concentration of unbound drug remains above the minimum inhibitory concentration (fT > MIC). For β‐lactam drugs, bactericidal effects in adults are achieved with 50% fT > MIC.22, 23 However, a target of 75% fT > MIC has been proposed in premature infants due to their immunocompromised state.24, 25 The goal of this study was to characterize the PK of ticarcillin in premature infants <30 weeks GA and perform simulations to identify the optimal dose in this population.
2. METHODS
2.1. Participant population
Pharmacokinetic (PK) samples were obtained from a multicentre, prospective PK study of the antistaphylococcal agents clindamycin, rifampin and ticarcillin–clavulanate in infants (Clinicaltrials.gov #NCT01728363; IND #115,396). The ticarcillin–clavulanate cohort enrolled premature infants (PNA <91 days; GA <30 weeks) with normal renal function and suspected or confirmed infection. Complete inclusion/exclusion criteria can be found on ClinicalTrials.gov. The study protocol was reviewed and approved by the institutional review board of each participating institution.
2.2. Drug dosing and sample collection
Ticarcillin–clavulanate dosing was based on the ticarcillin component at 75 mg/kg infused over 30 minutes. Dosing frequency was determined based on PNA, with PNA <14 days administered every 12 hours and PNA ≥ 14–45 days administered every 8 hours. The local standard‐of‐care dose could be administered if the site principal investigator deemed it appropriate. Up to 7 optimally timed plasma PK samples were collected after dose 3, 4, 5 or 6.
2.3. Analytical methods
Total ticarcillin concentrations in plasma were quantified using a validated HPLC/MS/MS assay. Accuracy and precision were within the Food and Drug Administration bioanalytical assay validation criteria (e.g. ±15%).26 The lower limit of quantification was 500 ng/mL (see supplementary materials for details).
2.4. Population pharmacokinetic analysis
Because ticarcillin–clavulanate is supplied in a 30:1 fixed ratio and dosing is based on ticarcillin component, PK analysis focused on ticarcillin. Ticarcillin plasma PK data were analysed with a nonlinear mixed effects modelling approach using NONMEM (version 7.2, Icon Solutions, Ellicott City, MD, USA). The first‐order conditional estimation method with interaction was used for all model runs. Data manipulation and visualization were performed using STATA 15 (College Station, TX, USA).
One‐ and two‐compartment PK models were explored with assumed linear PK.27 Between‐subject variability was assessed for PK model parameters using an exponential relationship. Both diagonal and block Omega matrices for covariance were explored. Proportional, additive and combined (additive plus proportional) residual error models were explored. Body weight (WT) was assumed to be a significant covariate for CL and volume of distribution (V) and was included in the base model. The relationship between WT and PK parameters was allometrically characterized using a fixed exponent (0.75 and 1) for CL and V parameters, respectively.
Other covariates were tested for model inclusion based on physiological relevance and by visual inspection of scatter and box plots of the individual deviations from the population‐typical value PK parameters (etas) against covariates. The covariates explored were postmenstrual age (PMA), PNA, serum creatinine (SCR), total bilirubin, serum albumin, haematocrit, race, ethnicity and sex. The relationship between age and CL was also explored using a sigmoidal maximum efficacy (Emax) maturation function as shown in equation 1. As a measure of age, PNA and PMA were explored.
| (1) |
where F age denotes the fraction of the adult CL value; TM 50 represents the value of age (days for PNA and weeks for PMA) when 50% adult CL is reached; and Hill (Hill coefficient) is a slope parameter for the sigmoidal maturation model. This relationship was first tested by estimating TM 50 and Hill and then by fixing TM 50 to 47.7 weeks and Hill to 3.4, values representing maturation of glomerular filtration in infants.28
With the exception of WT, other continuous covariates were normalized to the population median value. A forward inclusion (p < .05) and backward elimination (p < .01) approach was used to evaluate statistical significance.
2.5. Population PK model evaluation and validation
During the model‐building process, successful minimization, diagnostic plots, plausibility and precision of parameter estimates, as well as objective function value (OFV) and shrinkage values were used to assess model appropriateness. Parameter precision for the final model was evaluated using non‐parametric bootstrapping (1000 replicates) to generate the 95% confidence intervals for parameter estimates. A visual predictive check (VPC) was performed, whereby the final model was used to generate 1000 Monte‐Carlo simulation replicates per time point of ticarcillin exposure. The number of observed concentrations outside of the 90% prediction interval for each time point was quantified.
2.6. Dose–exposure evaluation
Multiple dosing regimens were evaluated using Monte‐Carlo simulations based on PK parameters and associated variability generated from the final ticarcillin PK model (Table S1). We evaluated the regimen used in the current trial (referred to as Pediatric Trials Network [PTN]); the PTN regimen with increased frequency (PTN‐increased frequency); the PTN regimen with extended infusion durations (PTN‐extended infusion); as well as regimens from common dosing guidelines (Harriet Lane, Neofax).29, 30
A population of 1000 virtual premature (GA <30 weeks) infants was generated using PK‐Sim® (version 5.3.2; Bayer Technology Services GmbH, Leverkusen, Germany) population builder.31 Each simulated infant was assigned an SCR value. SCR values (mg/dL) were determined using the “rnormal” function in STATA from the following distributions centred around the mean (standard deviation): PNA <3 days: SCR 1.1 (0.06); PNA 3 to < 15 days: SCR 0.7 (0.06); PNA 15 to < 22 days: SCR 0.6 (0.05); and PNA 22–45 days: SCR 0.4 (0.03).32 The demographics and distributions of SCR values were comparable between the study population and the simulated population.
To assess the number of infants who achieved the surrogate PD target of efficacy, concentration of unbound drug (fC) was calculated assuming the fraction unbound to be 55%.15 Unbound concentrations at 75% of the dosing interval at steady state (fC75ss) were calculated. The probability of fC75ss of ticarcillin exceeding different MIC levels was determined and plotted against MICs. Maximum plasma concentration (CMAX) was used as a surrogate for safety and values were comparable to adult CMAX values (218–333 μg/mL) after doses that showed favourable safety.33, 34, 35 Optimal ticarcillin dosing was selected when >90% of infants achieved therapeutic exposures.
3. RESULTS
3.1. Participant baseline characteristics
Fifteen participants from 2 centres were enrolled (Table 1) and contributed 65 PK samples. The median (range) ticarcillin dose was 75 mg/kg (70–81). Pharmacokinetics sampling began after 3 (2–5) doses. The number of samples collected per participant was 5 (3‐6). No ticarcillin concentrations were below the quantitation limit.
Table 1.
Clinical characteristics
| Variable | Median (range) or n (%) | ||
|---|---|---|---|
| Postnatal age < 14 d | Postnatal age 14–45 d | All | |
| n | 5 | 10 | 15 |
| Gestational age (weeks) | 25 (24–28) | 26 (23–28) | 25 (23–28) |
| Postnatal age (d) | 7 (6–10) | 23 (14–44) | 18 (6–44) |
| Postmenstrual age (weeks) | 26.0 (24.9–29.3) | 29.4 (26.0–31.7) | 29.3 (24.9–31.7) |
| Body weight (kg) | 0.68 (0.56–1.01) | 0.92 (0.59–1.22) | 0.83 (0.56–1.22) |
| Female | 2 (40) | 2 (20) | 4 (27) |
| Race | |||
| White | ‐ | 1 (10) | 1 (7) |
| Black or African American | 5 (100) | 9 (90) | 14 (93) |
| Ethnicity | |||
| Hispanic or Latino | ‐ | 1 (10) | 1 (7) |
| Not Hispanic or Latino | 5 (100) | 8 (80) | 13 (86) |
| Not reported | ‐ | 1 (10) | 1 (7) |
| Laboratory values | |||
| Serum creatinine (mg/dL) | 0.8 (0.4–1.3) | 0.4 (0.3–0.9) | 0.5 (0.3–1.3) |
| Serum albumin (g/dL) | 3.0 (3.0–3.5) | 2.9 (2.2–3.4) | 3.0 (2.2–3.5) |
3.2. Population pharmacokinetics model development
A 1‐compartment PK model characterized the ticarcillin data well (Figure S1, S2). Estimates of between‐subject variability on V (etaV) resulted in high residual standard error and shrinkage; etaV was consequently fixed at zero.
On univariable analysis, incorporation of PMA on CL resulted in the largest drop in the OFV (−17.19), followed closely by a sigmoidal maturation function on CL using PMA and fixing TM50 and the Hill coefficient at 47.7 weeks and 3.4, respectively (−17.045). Other covariates that resulted in significant drops in the OFV for ticarcillin were PNA on CL (−10.11) and SCR on CL (−8.562). In the ticarcillin multivariable analysis, SCR was tested with PNA‐ and PMA‐based models and was retained in both models in the backward elimination step. The PMA‐based maturation function with SCR on CL was chosen as the final model because it resulted in the largest drop in the OFV and was determined to be more generalizable due to its physiological relevance.
Final model equations and parameter estimates are presented in Table 2. Individual empirical Bayesian posthoc parameter estimates showed that CL was lower in infants with a PNA <14 days with median (range) 0.050 L/kg/h (0.043–0.075) compared with a PNA ≥14–45 days with 0.078 L/kg/h (0.047–0.100). Median CL for the overall population was 0.074 L/kg/h (0.043–0.100).
Table 2.
Ticarcillin population pharmacokinetic equations and parameter estimates
| Equations | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
|
| ||||||
| Parameter | Estimate | RSE (%) | 2.5th percentile | Bootstrapa median | 97.5th percentile | |
| Structural model | ||||||
| θCL (L/h) | 11.0 | 4.1 | 10.1 | 11.0 | 11.8 | |
| θV (L) | 32.3 | 4.5 | 29.1 | 32.3 | 35.0 | |
| θCL~SCR | −0.257 | 35.5 | −0.426 | −0.265 | −0.072 | |
| TM50 (weeks) | 47.7 FIX | ‐ | ‐ | ‐ | ‐ | |
| Hill | 3.4 FIX | ‐ | ‐ | ‐ | ‐ | |
| Between‐subject variability | ||||||
| CL (%CV) | 11.5 | 51.9 | 2.6 | 10.8 | 16.7 | |
| V (%CV) | 0 FIX | ‐ | ‐ | ‐ | ‐ | |
| Residual error | ||||||
| Proportional error (%) | 21.0 | 26.0 | 14.9 | 20.7 | 25.6 | |
CL, clearance; CV, coefficient of variation; Hill, Hill coefficient; RSE, relative standard error; SCR, serum creatinine; TM50, age (weeks) when 50% adult clearance is reached; V, volume of distribution; V70KG, volume of distribution (L) scaled to 70 kg; PNA, postnatal age.
1000 bootstrap runs were performed.
3.3. Population pharmacokinetics model evaluation
Bootstrap analysis resulted in 96.2% of bootstrap datasets converging to >3 significant digits. The median of bootstrap fixed effects parameter estimates were within 3.1% of population estimates from the original dataset for all parameters (Table 2). The VPC revealed a good fit between the observed and predicted concentrations with 95.4% (62/65) of observed ticarcillin concentrations falling within the 90% prediction interval.
3.4. Dosing simulations
All of the simulated dosing regimens achieved an fC75ss for 90% of simulated infants against organisms with an MIC distribution of 0.5–16 μg/mL (Figure 1, Table S2). Only the PTN‐increased frequency regimen and the PTN‐extended infusion regimen with at least a 2 hour infusion achieved the target for highly resistant organisms (MIC 32 μ/mL). Simulated ticarcillin steady‐state CMAX in infants for all regimens were comparable to or lower than mean measured ticarcillin CMAX (218–333 μg/mL) in adults who tolerated a single 2–4g dose.33, 34, 35
Figure 1.
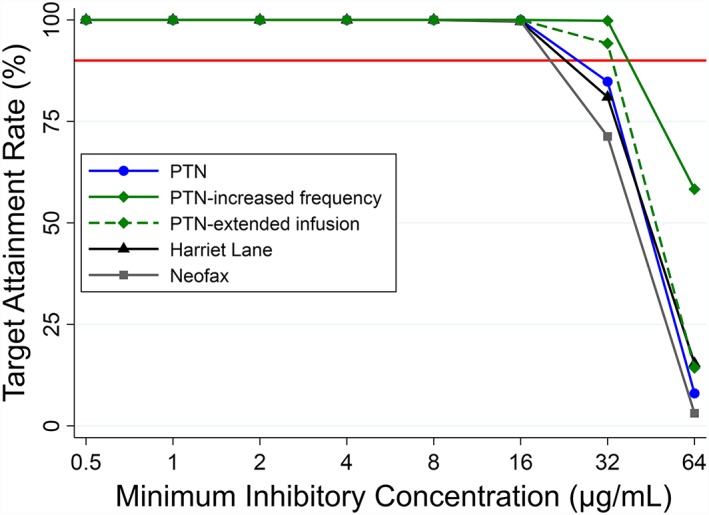
Target attainment rates for different ticarcillin dosing regimens for fraction of time the concentration of unbound drug remains above the minimum inhibitory concentration of 75%. Target attainment rate refers to the % of simulated infants who maintained an unbound ticarcillin concentration above the minimum inhibitory concentration for 75% of the dosing interval. The red horizontal line refers to a target attainment rate of 90%
4. DISCUSSION
The present study evaluated the population PK‐PD of ticarcillin in a cohort of 15 premature infants <30 weeks GA at birth with suspected serious infection. After accounting for weight, age was the most important covariate for CL. A PMA‐based sigmoidal maturation function resulted in a significant drop in OFV for ticarcillin CL. Because ticarcillin is renally eliminated, the PMA‐based maturation function used values for Hill and TM50 that characterize maturation of glomerular filtration rate.28, 36, 37, 38
Our estimate of CL in a standardized individual of 70 kg (11.0 L/h) was comparable to CL estimates reported in adults (6.7–9.2 L/h),33, 34, 35, 39, 40 showing that the maturation scaling worked well. Clearance estimates for the premature infants in the study population (0.074 L/kg/h) were higher than previously published CL estimates in premature infants with GA >30 weeks (0.047–0.067 L/kg/h)16, 17 and lower than full‐term infants and children (0.126–0.150 L/kg/h).20, 21 These differences are probably due to a combination of PNA (median PNA in study population and other premature infants were 8 and 18 days, respectively) and the maturation of renal function.41 The population V estimate of ticarcillin in the study population (0.48 L/kg) was higher than estimates of V in premature infants >30 weeks GA (0.26–0.34 L/kg)16, 17 and full‐term infants and children (0.22–0.23 L/kg).20, 21 The higher V in younger, more premature populations is probably multifactorial, including increased total body water41 and higher unbound fraction of drug42, 43 compared to adults.
Based on the population PK model and simulations, the PTN, Harriet Lane29 and Neofax,30 dosing strategies met the overall goals for exposure against organisms commonly found in clinical practice with an MIC distribution of 0.5–16 μg/mL. However, these dosing regimens were suboptimal for highly resistant organisms (MIC >16 μg/mL). To treat highly resistant organisms, we recommend a different agent or increasing the dosing frequency of the PTN regimen. Our simulations assumed the fraction of unbound ticarcillin to be 55%.15 The unbound fraction may have been higher in our population because infants have lower concentrations of albumin and higher concentrations of endogenous substances (e.g., bilirubin) that can displace drugs from albumin‐binding sites.42, 43 Simulated ticarcillin steady‐state CMAX concentrations in infants were comparable to mean measured ticarcillin CMAX in adults who tolerated a single 2–4g dose (218–333 μg/mL)33, 34, 35 and infants who tolerated a single dose of 83–100 mg/kg (159–447 μg/mL)17], suggesting a comparable safety profile.
In conclusion, for infants <30 weeks GA, we recommend a dose of 75 mg/kg infused over 30 minutes and dosed every 12 and 8 hours for PNA <14 and PNA 14–45 days, respectively.
COMPETING INTERESTS
This work was funded under National Institute of Child Health and Human Development contract HHSN275201000003I for the Pediatric Trials Network (PI Daniel K. Benjamin, Jr).
K.M.W. receives support from the Pediatric Critical Care and Trauma Scientist Development Program (5K12HD047349) and the National Institute of Child Health and Human Development (1K23HD075891). C.P.H. receives salary support for research from the National Institute of Child Health and Human Development (1K23HD090239) and the U.S. government for his work in paediatric and neonatal clinical pharmacology (Government Contract HHSN267200700051C, PI: Benjamin, under the Best Pharmaceuticals for Children Act). S.J.B. receives salary and research support from the National Institute of General Medical Sciences and the National Institute of Child Health and Human Development (2T32GM086330‐06). D.K.B. Jr. receives support from the National Institutes of Health (award 2K24HD058735‐10), National Institute of Child Health and Human Development (HHSN275201000003I), National Institute of Allergy and Infectious Diseases (HHSN272201500006I), ECHO Program (1U2COD023375‐02), and the National Center for Advancing Translational Sciences (1U24TR001608‐03); he also receives research support from Cempra Pharmaceuticals (subaward to HHSO100201300009C) and industry for neonatal and pediatric drug development, www.dcri.duke.edu/research/coi.jsp. P.B.S. was supported by National Institute of Child Health and Human Development contract HHSN275201000003I for the Pediatric Trials Network. M.L. receives support from the US government for his work in paediatric and neonatal clinical pharmacology: National Heart, Lung, and Blood Institute (R34 HL124038); Food and Drug Administration Office of Orphan Products Development (R01 FD005101); National Institute of Child Health and Human Development Government Contract (HHSN267200700051C) under the Best Pharmaceuticals for Children Act; National Institute of Child Health and Human Development (K23HD068497); and also from United Therapeutics for drug development. M.C.‐W. receives support for research from the NIH (1R01‐HD076676‐01A1), the National Center for Advancing Translational Sciences of the NIH (UL1TR001117), the National Institute of Allergy and Infectious Diseases (NIAID) (HHSN272201500006I and HHSN272201300017I), NICHD (HHSN275201000003I), the Biomedical Advanced Research and Development Authority (BARDA) (HHSO100201300009C), the nonprofit organization Thrasher Research Fund (www.thrasherresearch.org), and industry for drug development in adults and children (www.dcri.duke.edu/research/coi.jsp). The other authors have no competing interests to declare.
CONTRIBUTORS
K.M.W., C.P.H., D.K.B. Jr., R.A., M.L., P.B.S., and M.C.W. all contributed to the design of the study, acquisition of data, and analysis and interpretation of results. S.J.B. contributed to the analysis and interpretation of results. G.M. and C.M.C. contributed to acquisition of data and interpretation of results. B.H. contributed to the design of the study and acquisition of data. All authors contributed to drafting and/or revising the manuscript and approved the final published version.
Supporting information
Table S1. Dosing regimens evaluated
Table S2. Simulated exposures for different dosing regimens
Figure S1. Concentration vs time after last dose for ticarcillin
Figure S2. Ticarcillin model diagnostic plots. Observed vs population (A) and individual (B) predictions for the final ticarcillin model. Conditional weighted residuals (CWRES) vs population predictions (A) and time after last dose (B) for the final ticarcillin model. The solid black lines represent lines of identity. The dashed blue lines represent smooth lines by loess fit.
Figure S3. Visual predictive check for ticarcillin final model.
ACKNOWLEDGEMENTS
The assay measuring ticarcillin–clavulanic acid concentrations was performed at OpAns Laboratory (Durham, NC, USA) by Amanda Merrill (Analyst), Christine Grosse (Responsible Scientist), and Kenneth Lewis (Chief Executive Officer).
The PTN Publications Committee: Gary Furda, Duke Clinical Research Institute, Durham, NC; Danny Benjamin, Duke Clinical Research Institute, Durham, NC; Edmund Capparelli, University of California San Diego, San Diego, CA; Gregory L. Kearns, Arkansas Children's Hospital Research Institute, Little Rock, AR; Ian M. Paul, Penn State College of Medicine, Hershey, PA; Christoph Hornik, Duke Clinical Research Institute, Durham, NC; Kelly Wade, Children's Hospital of Philadelphia, Philadelphia, PA.
The Eunice Kennedy Shriver National Institute of Child Health and Human Development: David Siegel and Anne Zajicek
The Emmes Corporation (Data Coordinating Center): Ravinder Anand and Gina Simone
Pediatric Trials Network Ticarcillin‐Clavulanic Acid Study Team, Principal Investigators (PIs), and Study Coordinators (SCs): Kings County Hospital: Gratias Mundakel (PI), Subhatra Limbu (SC)
Duke: Michal Cotten (PI), Joanne Finkle (SC), Rebecca Jones (SC), Cynthia Ross (SC)
Watt KM, Hornik CP, Balevic SJ, et al. Pharmacokinetics of ticarcillin–clavulanate in premature infants. Br J Clin Pharmacol. 2019;85:1021–1027. 10.1111/bcp.13882
PI statement: The authors confirm that the Principal Investigator for this paper is P. Brian Smith and that he has direct clinical responsibility for patients.
REFERENCES
- 1. Stoll BJ, Hansen N. Infections in VLBW infants: studies from the NICHD Neonatal Research Network. Semin Perinatol. 2003;27(4):293‐301. [DOI] [PubMed] [Google Scholar]
- 2. Stoll BJ, Hansen N, Fanaroff AA, et al. Late‐onset sepsis in very low birth weight neonates: the experience of the NICHD Neonatal Research Network. Pediatrics. 2002;110(2):285‐291. [DOI] [PubMed] [Google Scholar]
- 3. Denniston S, Riordan FA. Staphylococcus aureus bacteraemia in children and neonates: a 10 year retrospective review. J Infect. 2006;53(6):387‐393. [DOI] [PubMed] [Google Scholar]
- 4. Jeong IS, Jeong JS, Choi EO. Nosocomial infection in a newborn intensive care unit (NICU), South Korea. BMC Infect Dis. 2006;6(1):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sohn AH, Garrett DO, Sinkowitz‐Cochran RL, et al. Prevalence of nosocomial infections in neonatal intensive care unit patients: Results from the first national point‐prevalence survey. J Pediatr. 2001;139(6):821‐827. [DOI] [PubMed] [Google Scholar]
- 6. Usukura Y, Igarashi T. Examination of severe, hospital acquired infections affecting extremely low birthweight (ELBW) infants. Pediatr Int. 2003;45(2):230‐232. [DOI] [PubMed] [Google Scholar]
- 7. Drews MB, Ludwig AC, Leititis JU, Daschner FD. Low birth weight and nosocomial infection of neonates in a neonatal intensive care unit. J Hosp Infect. 1995;30(1):65‐72. [DOI] [PubMed] [Google Scholar]
- 8. Payne NR, Carpenter JH, Badger GJ, Horbar JD, Rogowski J. Marginal increase in cost and excess length of stay associated with nosocomial bloodstream infections in surviving very low birth weight infants. Pediatrics. 2004;114(2):348‐355. [DOI] [PubMed] [Google Scholar]
- 9. Fanaroff AA, Korones SB, Wright LL, et al. Incidence, presenting features, risk factors and significance of late onset septicemia in very low birth weight infants. The National Institute of Child Health and Human Development Neonatal Research Network. Pediatr Infect Dis J. 1998;17(7):593‐598. [DOI] [PubMed] [Google Scholar]
- 10. Deulofeut R, Critz A, Adams‐Chapman I, Sola A. Avoiding hyperoxia in infants < or = 1250 g is associated with improved short‐ and long‐term outcomes. J Perinatol. 2006;26(11):700‐705. [DOI] [PubMed] [Google Scholar]
- 11. Remington J, Klein J, Wilson C, Baker C. Infectious Diseases of the Fetus and Newborn Infant. 6th ed. Philadelphia, PA: Elsevier Saunders; 2006. [Google Scholar]
- 12. Healy CM, Hulten KG, Palazzi DL, Campbell JR, Baker CJ. Emergence of new strains of methicillin‐resistant Staphylococcus aureus in a neonatal intensive care unit. Clin Infect Dis. 2004;39(10):1460‐1466. [DOI] [PubMed] [Google Scholar]
- 13. Healy CM, Palazzi DL, Edwards MS, Campbell JR, Baker CJ. Features of invasive staphylococcal disease in neonates. Pediatrics. 2004;114(4):953‐961. [DOI] [PubMed] [Google Scholar]
- 14. Karlowicz MG, Buescher ES, Surka AE. Fulminant late‐onset sepsis in a neonatal intensive care unit, 1988‐1997, and the impact of avoiding empiric vancomycin therapy. Pediatrics. 2000;106(6):1387‐1390. [DOI] [PubMed] [Google Scholar]
- 15. FDA . TIMENTIN (ticarcillin disodium and clavulanate potassium) for Injection. In: U.S. Deptartment of Health and Human Services. GlaxoSmithKline; 2014.
- 16. Burstein AH, Wyble LE, Gal P, et al. Ticarcillin‐clavulanic acid pharmacokinetics in preterm neonates with presumed sepsis. Antimicrob Agents Chemother. 1994;38(9):2024‐2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fricke G, Doerck M, Hafner D, Horton R, Kresken M. The pharmacokinetics of ticarcillin/clavulanate acid in neonates. J Antimicrob Chemother. 1989;24(Suppl B):111‐120. [DOI] [PubMed] [Google Scholar]
- 18. Nelson JD. Neonatal ticarcillin dosage. Pediatrics. 1979;64:549‐550. [PubMed] [Google Scholar]
- 19. Nelson JD, Kusmiesz H, Shelton S, Woodman E. Clinical pharmacology and efficacy of ticarcillin in infants and children. Pediatrics. 1978;61:858‐863. [PubMed] [Google Scholar]
- 20. Reed MD, Yamashita TS, Blumer JL. Pharmacokinetic‐based ticarcillin/clavulanic acid dose recommendations for infants and children. J Clin Pharmacol. 1995;35(7):658‐665. [DOI] [PubMed] [Google Scholar]
- 21. Zobell JT, Stockmann C, Young DC, et al. Population pharmacokinetic and pharmacodynamic modeling of high‐dose intermittent ticarcillin‐clavulanate administration in pediatric cystic fibrosis patients. Clin Ther. 2011;33(11):1844‐1850. [DOI] [PubMed] [Google Scholar]
- 22. Drusano GL. Antimicrobial pharmacodynamics: critical interactions of 'bug and drug'. Nat Rev Microbiol. 2004;2(4):289‐300. [DOI] [PubMed] [Google Scholar]
- 23. Lodise TP, Lomaestro BM, Drusano GL. Application of antimicrobial pharmacodynamic concepts into clinical practice: focus on beta‐lactam antibiotics: insights from the Society of Infectious Diseases Pharmacists. Pharmacotherapy. 2006;26(9):1320‐1332. [DOI] [PubMed] [Google Scholar]
- 24. Le J, Bradley JS. Optimizing antibiotic drug therapy in pediatrics: current state and future needs. J Clin Pharmacol. 2018;58(Suppl 10):S108‐S122. [DOI] [PubMed] [Google Scholar]
- 25. Cohen‐Wolkowiez M, Watt KW, Zhou C, et al. Developmental pharmacokinetics of piperacillin and tazobactam using plasma and dried blood spots from infants. Antimicrob Agents Chemother. 2014;58(5):2856‐2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. FDA . Guidance for industry bioanalytical method validation. Rockville, MD: U.S. Dept. of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research; 2013.
- 27. Jelliffe RW, Gomis P, Tahani B, Ruskin J, Sattler FR. A population pharmacokinetic model of trimethoprim in patients with pneumocystis pneumonia, made with parametric and nonparametric methods. Ther Drug Monit. 1997;19(4):450‐459. [DOI] [PubMed] [Google Scholar]
- 28. Rhodin MM, Anderson BJ, Peters AM, et al. Human renal function maturation: a quantitative description using weight and postmenstrual age. Pediatr Nephrol. 2009;24(1):67‐76. [DOI] [PubMed] [Google Scholar]
- 29. Harriet Lane Service (Johns Hopkins Hospital) , Flerlage J, Engorn B, Johns Hopkins Hospital . Children's Medical and Surgical Center. The Harriet Lane handbook: a manual for pediatric house officers.
- 30. NeoFax/Pediatrics . Truven Health Analytics IAM, 2017 through MICROMEDEX.
- 31. Claassen K, Thelen K, Coboeken K, et al. Development of a physiologically‐based pharmacokinetic model for preterm neonates: evaluation with in vivo data. Curr Pharm Des. 2015;21(39):5688‐5698. [DOI] [PubMed] [Google Scholar]
- 32. Martin RJ, Fanaroff AA, Walsh MC. Fanaroff and Martin's Neonatal‐Perinatal Medicine: Diseases of the Fetus and Infant. 9th ed. St. Louis, MO: Elsevier Health Science; 2010. [Google Scholar]
- 33. Bennett S, Wise R, Weston D, Dent J. Pharmacokinetics and tissue penetration of ticarcillin combined with clavulanic acid. Antimicrob Agents Chemother. 1983;23(6):831‐834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bodey GP, Yeo E, Ho DH, Rolston K, LeBlanc B. Clinical pharmacology of timentin (ticarcillin and clavulanic acid). Clin Pharmacol Ther. 1985;38(2):134‐139. [DOI] [PubMed] [Google Scholar]
- 35. Libke RD, Clarke JT, Ralph ED, Luthy RP, Kirby WM. Ticarcillin vs carbenicillin: clinical pharmacokinetics. Clin Pharmacol Ther. 1975;17(4):441‐446. [DOI] [PubMed] [Google Scholar]
- 36. Germovsek E, Kent A, Metsvaht T, et al. Development and evaluation of a gentamicin pharmacokinetic model that facilitates opportunistic gentamicin therapeutic drug monitoring in neonates and infants. Antimicrob Agents Chemother. 2016;60(8):4869‐4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Germovsek E, Lutsar I, Kipper K, et al. Plasma and CSF pharmacokinetics of meropenem in neonates and young infants: results from the NeoMero studies. J Antimicrob Chemother. 2018;73(7):1908‐1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Standing JF, Nika A, Tsagris V, et al. Oseltamivir pharmacokinetics and clinical experience in neonates and infants during an outbreak of H1N1 influenza A virus infection in a neonatal intensive care unit. Antimicrob Agents Chemother. 2012;56(7):3833‐3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guglielmo BJ, Flaherty JF, Batman R, Barriere SL, Gambertoglio JG. Comparative pharmacokinetics of low‐ and high‐dose ticarcillin. Antimicrob Agents Chemother. 1986;30(3):359‐360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoffken G, Tetzel H, Koeppe P, Lode H. The pharmacokinetics of ticarcillin, clavulanic acid and their combination. J Antimicrob Chemother. 1986;17(Suppl C):47‐55. [DOI] [PubMed] [Google Scholar]
- 41. Kearns GL, Abdel‐Rahman SM, Alander SW, et al. Developmental pharmacology‐‐drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349(12):1157‐1167. [DOI] [PubMed] [Google Scholar]
- 42. Ehrnebo M, Agurell S, Jalling B, Boreus LO. Age differences in drug binding by plasma proteins: studies on human foetuses, neonates and adults. Eur J Clin Pharmacol. 1971;3(4):189‐193. [DOI] [PubMed] [Google Scholar]
- 43. Rane A, Lunde PK, Jalling B, Yaffe SJ, Sjoqvist F. Plasma protein binding of diphenylhydantoin in normal and hyperbilirubinemic infants. J Pediatr. 1971;78(5):877‐882. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Dosing regimens evaluated
Table S2. Simulated exposures for different dosing regimens
Figure S1. Concentration vs time after last dose for ticarcillin
Figure S2. Ticarcillin model diagnostic plots. Observed vs population (A) and individual (B) predictions for the final ticarcillin model. Conditional weighted residuals (CWRES) vs population predictions (A) and time after last dose (B) for the final ticarcillin model. The solid black lines represent lines of identity. The dashed blue lines represent smooth lines by loess fit.
Figure S3. Visual predictive check for ticarcillin final model.