

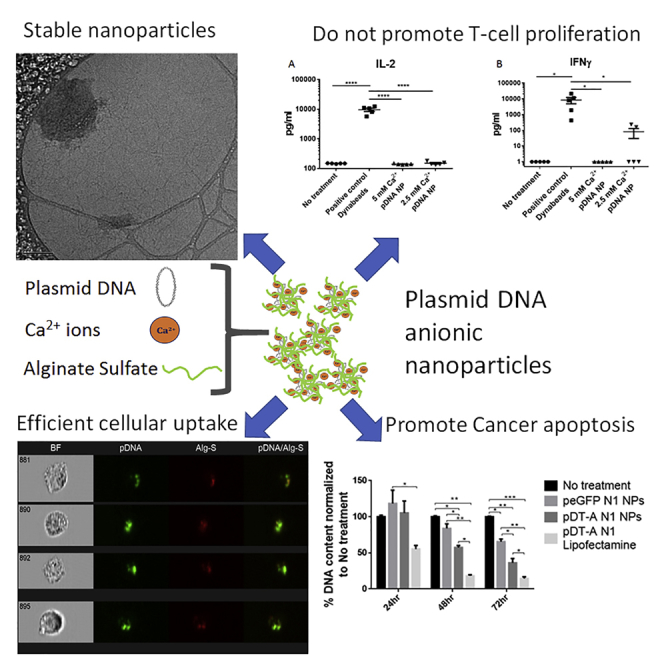
Abstract
Successful gene therapy requires the development of suitable carriers for the selective and efficient delivery of genes to specific target cells, with minimal toxicity. In this work, we present a non-viral vector for gene delivery composed of biocompatible materials, CaCl2, plasmid DNA and the semi-synthetic anionic biopolymer alginate sulfate (AlgS), which spontaneously co-assembled to form nanoparticles (NPs). The NPs were characterized with a slightly anionic surface charge (Zeta potential [ζ] = −14 mV), an average size of 270 nm, and their suspension was stable for several days with no aggregation. X-ray photoelectron spectroscopy (XPS) validated their ternary composition, and it elucidated the molecular interactions among Ca2+, the plasmid DNA, and the AlgS. Efficient cellular uptake (>80%), associated with potent GFP gene expression (22%–35%), was observed across multiple cell types: primary rat neonatal cardiac fibroblasts, human breast cancer cell line, and human hepatocellular carcinoma cells. The uptake mechanism of the NPs was studied using imaging flow cytometry and shown to be via active, clathrin-mediated endocytosis, as chemical inhibition of this pathway significantly reduced EGFP expression. The NPs were cytocompatible and did not activate the T lymphocytes in human peripheral blood mononuclear cells. Proof of concept for the efficacy of these NPs as a carrier in cancer gene therapy was demonstrated for Diphtheria Toxin Fragment A (DT-A), resulting in abrogation of protein synthesis and cell death in the human breast cancer cell line. Collectively, our results show that the developed AlgS-Ca2+-plasmid DNA (pDNA) NPs may be used as an effective non-viral carrier for pDNA.
Keywords: polymeric nanoparticles, plasmid DNA, gene therapy, calcium, alginate sulfate
Graphical Abstract
Introduction
Gene therapy using exogenous DNA has great potential for the treatment of a large variety of diseases, whether acquired or inherent.1, 2, 3 To be therapeutically active, the exogenous DNA must enter the target cell nucleus while surpassing several hurdles, including fast elimination and/or degradation rates, inadequate cellular entry, and trafficking to the nucleus.3, 4 To overcome these hurdles, DNA delivery is usually mediated by viral and non-viral vectors that protect the DNA cargo in the circulation and facilitate its entry into the cell. Over the years, several non-viral nanocarriers have been developed using biocompatible polymers or lipids, mostly positively charged.5, 6, 7, 8, 9 Recently, many of the positively charged carriers have shown toxicity resulting from their strong positive charge,10, 11 and the trend has been changed toward developing carriers with a slightly anionic or zwitterionic surface charge.
Recent work from our lab presented a new type of slightly anionic nanoparticles (NPs), self-assembled by the complexation of small interfering RNA (siRNA) with semi-synthetic polysaccharides, i.e., hyaluronan sulfate (HAS) or alginate sulfate (AlgS), mediated by calcium ion bridges.12, 13, 14, 15 The ternary complex was simply formed by mixing all components, under aqueous conditions, yielding NPs with an average size of 130–150 nm and a surface charge of ∼−10 mV.13, 15 The NPs were shown to be stable over time even in the presence of serum proteins, were cytocompatible, and silenced targeted genes very efficiently in different cell lines.12 Mechanistic studies revealed that the Ca2+ component is a major promoter of cell internalization and endosomal escape of the NPs14 and that these processes were Ca2+ concentration dependent. Further, the NPs were shown to enter the cell through multiple endocytosis pathways, dynamin, clathrin, and caveolin dependent, most likely due to the multi-faceted nature of Ca2+ and the participation of this ion in many cell processes.
Based on the potential advantages of these NPs as intracellular carriers, the aim of this study has been to investigate the prospect of the electrostatic-driven self-assembled NPs as a transfecting strategy for plasmid DNA (pDNA). The delivery of pDNA provides additional challenges, such as its larger size compared to the siRNA (typically 20–30 bp for siRNA compared to several thousand base pairs for a plasmid) and its site of action, which is the cell nucleus while the siRNA is active in the cytoplasm. Herein, we describe the complexation process of AlgS with pDNA, mediated by calcium ion bridges to form slightly anionic AlgS-Ca2+-pDNA NPs, and we analyze the physico-chemical features of the resultant NPs (size, surface charge, and ternary composition), the encapsulation efficiency of pDNA, their stability over time, cytocompatibility, and cellular uptake mechanism. Furthermore, we analyze systematically the in vitro influence of AlgS-Ca2+-pDNA NPs on peripheral blood mononuclear cells (PBMCs) from healthy individuals, revealing their effect on T cell activation and cytokine production. Ultimately, the protein expression induced by the developed platform for model and therapeutic pDNA, across multiple cell types, was evaluated.
Results
Physico-chemical Characterization of the AlgS-Ca2+-pDNA NPs
The assembly into NPs by electrostatic interactions among Ca2+, pDNA, and AlgS was validated in high-resolution transmission electron microscopy (TEM) images (the final concentrations of components were 2.5 μg/mL AlgS, 25 mM Ca2+, and 15 ng/μL pDNA for dry-TEM and 25 μg/mL AlgS, 250 mM Ca2+, and 150 ng/μL pDNA for cryogenic-TEM [cryo-TEM]) (Figure 1). The NP size, measured on images from cryo-TEM, showed particles with a mean diameter of 188 ± 50 (n = 17), much larger than the size observed in the dry-TEM images, indicating that water molecules participate in the assembly and structure of these NPs.
Figure 1.

High-Resolution TEM Images of AlgS-Ca2+-pDNA NPs
(A and B) Dry-TEM micrographs of NPs (2.5 μg/mL AlgS, 25 mM Ca2+, and 15 ng/μL pDNA) with gold-labeled AlgS. (C) Cryo-TEM micrographs of complexes (250 mM Ca2+ and 150 ng/μL pDNA). (D) Cryo-TEM micrographs of NPs (25 μg/mL AlgS, 250 mM Ca2+, and 150 ng/μL pDNA). Scale bars, 500 nm (A) and 100 nm (B–D).
The dynamic light scattering (DLS) analysis of the NPs (diluted 1:50) reveals a mean hydrodynamic diameter of 270 nm (Table 1), slightly larger than the size directly measured on the TEM images. This difference could be due to the different methods used for the analysis; in DLS, the assumption is that particles are spherical, while the TEM images show the NPs are not perfectly that. Most notably, the size of the AlgS-Ca2+-pDNA NPs was nearly twice the size of AlgS-Ca2+-siRNA NPs (∼130 nm15), as expected due to the larger size of pDNA.
Table 1.
Size Distribution and Surface Charge of NPs Prepared with Different Concentrations of Ca2+ over 72 h
| NP Formulationa | Diameterb (nm) T = 0 | Diameterb (nm) T = 24 h | Diameterb (nm) T = 72 h | ζ (mV) T = 0 | ζ (mV) T = 24 h | ζ (mV) T = 72 h |
|---|---|---|---|---|---|---|
| Ca2+, 5 mM pDNA, 3 ng/μL AlgS, 500 ng/mL |
270.5 ± 24.9 | 250.5 ± 25.1 | 257.9 ± 43.1 | −14.5 ± 1.0 | −13.9 ± 1.9 | −15.8 ± 2.0 |
| Ca2+, 2.5 mM pDNA, 3 ng/μL AlgS, 500 ng/mL |
227.4 ± 11.6 | 226.6 ± 5.8 | 232.9 ± 60.8 | −12.0 ± 1.3 | −13.0 ± 1.4 | −13.1 ± 0.7 |
Results are mean ± SD (n = 3). ζ, Zeta potential.
The component concentration is after 1:50 dilution in HEPES.
Diameter is unweighted.
The Zeta potential (ζ) measurements showed a slightly anionic (∼−14 mV) surface charge for the AlgS-Ca2+-pDNA NPs; the surface was more anionic than that recorded for the AlgS-Ca2+-siRNA NPs (∼10 mV13).
As different cells may present different sensitivity to Ca2+,12, 14 we prepared AlgS-Ca2+-pDNA NPs with 2-fold lower Ca2+ in the complex (the final concentration of components was 500 ng/mL AlgS, 2.5 mM Ca2+, and 3 ng/μL pDNA). These NPs had a smaller mean diameter by DLS and lesser surface charge (Table 1). Throughout the study, the NPs with the different Ca2+ concentrations were used in comparison to find the optimal composition.
DLS analysis and ζ potential measurements did not reveal any significant changes in the particle size or surface charge for all NPs examined over a 3-day period, indicating that the NPs form stable suspensions without aggregation.
Nanoparticle tracking analysis (NTA) revealed the size of the 5 mM Ca2+ NPs to be 194 ± 20 nm, in agreement with the size measured by TEM (188 ± 50 nm). When prepared with final concentrations of 12.5 mM Ca2+, 7.5 ng/μL pDNA, and 1.25 μg/mL AlgS, the pDNA NP concentration was found to be 8.4 × 107 ± 1.5 × 106/mL.
An ethidium bromide (EtBr) exclusion assay was performed to determine the entrapment efficiency of AlgS-Ca2+-pDNA NPs, prepared with different Ca2+ concentrations, ranging from 25 to 250 mM, while the concentrations of AlgS (25 μg/mL) and pDNA (150 ng/μL) were maintained constant. The EtBr exclusion assay is based on the decrease in fluorescence of EtBr as a result of the physical masking of pDNA from intercalation with EtBr due to encapsulation in the NPs. The results (Figure S1) represent the percentage of fluorescence intensity emitted from the sample relative to that emitted by free pDNA. An efficiency of 55% ± 0.7% was measured for NPs prepared with 25 mM CaCl2. The efficiencies of NPs prepared with higher Ca2+ concentrations were not statistically significantly different from each other, and the value measured for NPs prepared with 250 mM CaCl2 was 69.6% ± 0.1%. This value was lower compared to the high entrapment efficiency of siRNA, >98%, measured for AlgS-Ca2+-siRNA NPs.15
The ternary composition of the AlgS-Ca2+-pDNA NPs was validated by X-ray photoelectron spectroscopy (XPS) analysis, by comparing the binding energy spectra of the main elements in AlgS-Ca2+-pDNA NPs and Ca2+-pDNA complexes (with no AlgS). The results are summarized in Table S1. In the spectrum of the Ca2+-pDNA complex (no AlgS), the C1 and N1 assigned to the DNA and O1, P2p, and Ca2p are in agreement with values reported for Ca(H2PO4)2.13, 15, 16, 17 In the AlgS-Ca2+-pDNA NPs, an additional peak for S2p binding energy appears, in agreement with that reported for CaSO4, and it is attributed to the binding of Ca2+ to the sulfate group on AlgS. Another confirmation for the participation of AlgS in the NP structure is the shift (+0.9 eV) to higher binding energy of the P2p peak, indicating the formation of a chemical bond between Ca2+-pDNA and AlgS.15
Cytocompatibility of AlgS-Ca2+-pDNA NPs
The cytocompatibility of NPs prepared with different Ca2+ concentrations was evaluated by measuring cell metabolic activity using PrestoBlue reagent. Four cell types were tested: CT26-EGFP, mouse colon carcinoma stably transfected with EGFP plasmid; HepG2, human hepatocellular carcinoma; MDA-MB-231, human breast cancer cell line; and neonatal rat isolated cardiac fibroblasts (CFs). With CT26-EGFP and HepG2 cells, the viability was not affected by NPs prepared with Ca2+ as high as 5 mM (the final concentration in NPs added to cell culture), for as long as 72 h post-transfection (Figure 2). In the primary CF and MDA-MB-231 cultures, the NPs were cytocompatible when composed with up to 3 mM Ca2+ for all tested time points. With the 5 and 4 mM Ca2+ NPs, there was a decrease in cell metabolic activity as soon as 24 h post-exposure, and the cells did not recuperate with time.
Figure 2.

Cytocompatibility of NPs Made with Different Ca2+ Concentrations
(A) 24 h, (B) 48 h, and (C) 72 h post-exposure to NPs (mean ± SD; *p < 0.05,**p < 0.01,***p < 0.005, Tukey’s multiple comparison test compared to control; n = 3).
NP Uptake and Expression of GFP by Primary CFs and Cell Lines
The uptake of AlgS-Ca2+-pDNA NPs was determined using ImageStreamX imaging flow cytometry, a powerful instrument that combines the statistical power, speed, and sensitivity of flow cytometry with imaging capabilities of high-resolution microscopy. Fluorescein isothiocyanate (FITC)-labeled pDNA-entrapping NPs were created with different Ca2+ concentrations, and they were added to monolayers of HepG2, MDA-MB-231, and CFs; the analysis was performed after 3 h of incubation with the NPs. All cell types showed a significant uptake rate of NPs, prepared with 2.5 mM Ca2+ and above (final concentration in medium), at uptake levels much greater compared to control free FITC-pDNA (Figure 3).
Figure 3.

Uptake of NPs with Different Ca2+ Concentrations by Various Cells and Analysis of AlgS-Ca2+-pDNA Co-localization after Cellular Uptake
(A and B) ImageStream results showing bright-field (BF) and FITC-pDNA in green for NPs with 2.5 mM Ca2+ uptaken by (A) CFs and (B) MDA-MB-231. (C–E) Percent transfected cells counted using ImageStreamX (mean ± SD;*p < 0.05, Sidak multiple comparison test compared to control; n = 3) for (C) MDA-MB-231, (D) CFs, and (E) HepG2. Imaging flow cytometry analysis of AlgS-Ca2+-pDNA uptake in MDA-MB-231 cells, 3 h after NP addition in unfixed live cells, is shown. (F) Representative cell images captured by Amnis ImageStreamX flow cytometer. (G) From total cell population, cells that are double positive or negative for fluorescent signals of AlgS-Cy5 nm and pDNA-FITC. (H) Histogram of Bright Detail Similarity (BDS) score between pDNA (FITC) and AlgS (Cy5) images, showing the cell population exhibiting co-localization of pDNA (FITC) and AlgS (Cy5). A score of >1.0 was considered co-localized.
In addition, the co-localization of AlgS and pDNA was tested in MDA-MB-231 cells to ensure the uptake of AlgS-Ca2+-pDNA NPs as whole particles. Co-localization assay was performed using the ImageStreamX with AlgS labeled with Cy5 and the pDNA with FITC. Co-localization was demonstrated after 3 h of transfection. Quantitative analysis indicated that uptake of the AlgS (Cy5)-Ca2+-pDNA (FITC) NPs occurred in ∼70% of the cells (Figure 3G), as calculated by the percentage of cells double positive for AlgS (Cy5) and pDNA (FITC). Approximately 76% of the double-positive cells demonstrated intracellular co-localization of pDNA (FITC) and AlgS (Cy5) (with a Bright Detail Similarity [BDS] above 1) (Figure 3H). This value was greater compared to the co-localization measured for AlgS-Ca2+-siRNA NPs (30%).15 The results imply that, at this time point, most of the AlgS-Ca2+-pDNA NPs that entered the cells had not disassembled, contrary to results for AlgS-Ca2+-siRNA.
Further, GFP expression by the transfected cells was evaluated using flow cytometry (Figure 4). The cells were treated for 3 h to ensure proper NP uptake, and GFP expression was tested 72 h afterward to ensure GFP generation. Untreated EGFP-transfected CT26 cells were imaged and tested as a control for GFP-expressing cells for calibration of the parameters. GFP expression efficiency reached ∼30% in cell lines of MDA-MB-231 and HepG2, and it was less (∼20%) in the primary CFs. For the lower concentration (2.5 mM) Ca2+ NPs, nearly no GFP expression was noted in all cell types.
Figure 4.

GFP Expression Measured Using Flow Cytometry in Different Cells Transfected with AlgS-Ca2+-pDNA NPs
NPs were prepared with 5 or 2.5 mM [Ca2+] (final concentration in cell medium). Mean + SD; *p < 0.05, Sidak multiple comparison test compared to control for each group.
Uptake Kinetics of AlgS-Ca2+-pDNA in MDA-MB-231 Cells and Mechanism
The uptake profile of AlgS-Ca2+-pDNA NPs was evaluated in MDA-MB-231 cells over 24 h. The NPs were prepared with fluorescein-labeled plasmid, pGL3-GFP, which contains no promoter sequence and so it does not lead to GFP expression. The percentage of cells that internalized the fluorescently labeled plasmid and the mean fluorescence intensity were measured at different time points over the course of 24 h by imaging flow cytometry.
The uptake profile of AlgS-Ca2+-pDNA NPs in MDA-MB-231 cells was found to be relatively rapid; after 30 min, 79% ± 11% of the cells were fluorescent positive, meaning they internalized the fluorescently labeled pDNA (Figure 5). Furthermore, the fluorescent label was found to locate in defined intracellular areas, most likely in endosomes. The maximal percentage of cells that internalized pDNA, 98% ± 1%, was reached after 3 h, and it slightly decreased to 95% ± 0.2% over the next 21 h.
Figure 5.

Uptake Kinetics of AlgS-Ca2+-pDNA NPs by MDA-MB-231 Cells Using ImageStreamX and Inhibition of Endocytosis
(A) Representative images showing bright-field and fluorescein-pDNA (green) accumulation in cells 3 h post-transfection. (B) Percentage of fluorescent-positive cells over 24 h. (C) Mean fluorescence intensity over 24 h, normalized to the peak value (at 3 h post-transfection). Error bars represent SEM (n = 2). (D) Percentage of EGFP-expressing cells treated with inhibitors normalized to cells transfected with pDNA NPs without exposure of the cells to the chemical inhibitors. Error bars represent SD (n = 3); asterisks denote significant difference with *p < 0.05, **p < 0.01, and ***p < 0.005 by one-way ANOVA (Dunnet test).
The mean fluorescence intensity of the fluorescent-positive population at each time point was measured as well, and it provides a relative estimation to the average amount of pDNA internalized at each time point. While equal pDNA concentrations were used in all repetitions, in each repetition a different batch of labeled pDNA was used, so the fluorescence intensity emitted by the same amount of DNA might be different as a result of different labeling efficiencies. To overcome this, the mean fluorescence intensity in each repetition was normalized to its maximal value (which was at time point 3 h for all repetitions). The profile shows a gradual increase in the mean fluorescence intensity up to reaching a maximal peak at 3 h after transfection, followed by a slow decrease in the fluorescence intensity, reaching 69% of maximal intensity at 24 h after transfection.
In an effort to elucidate the endocytic pathway(s) involved in the cellular uptake of AlgS-Ca2+-pDNA NPs, we employed a number of chemical inhibitors specific to these processes: NaN3, an inhibitor for the respiratory chain in the mitochondria, thus impairing ATP production in cells and active uptake; Pitstop2, a Clathrin-dependent inhibitor; Genistein, a caveolae-dependent inhibitor; and CdCl2, a competitive calcium antagonist. MDA-MB-231 cells, after treatment with the different inhibitors, were transfected for 3 h with the NPs carrying plasmid EGFP N1 (pEGFP), and the amount of GFP expression was evaluated 48 h post-transfection. ImageStreamX analysis (Figure 5D) quantitatively revealed that NaN3 and Pitstop2 had a significant inhibitory effect on the entry of the NPs and, consequently, on gene expression by the cells, while with Genistein the inhibitory effect showed a trend of reduction, but it did not reach statistical significance, and CdCl2 had no inhibitory effect. Thus, it appears that the entry mechanism of the plasmid NPs is via an active endocytosis, mainly using the clathrin mechanism of entry.
The Influence of AlgS-Ca2+-pDNA NPs on PBMCs
We investigated the effect of AlgS-Ca2+-pDNA NPs on naive human peripheral blood mononuclear cells (PBMCs), isolated from healthy individuals (n = 5) and incubated for 3 h with NPs composed of a final Ca2+ concentration of 5 or 2.5 mM. The expression of several cytokines, indicating T cell proliferation (interleukin-2 [IL-2]) and activation of Th1 (interferon [IFN]-γ), Th2 (IL-10), and Th17 (IL-17) cells, was measured. In addition, cytokines marking macrophage activation were recorded, such as tumor necrosis factor alpha (TNF-α) and IL-6.
The secretion profiles of the different cytokines marking lymphocyte activation (IL-2, IFN-γ, IL-10, and IL-17) revealed that the AlgS-Ca2+-pDNA NPs had no significant activation effect on naive PBMCs compared with the activation of these PBMCs with CD3 and CD28 Dynabeads (Figure 6). As expected, there was slight activation of macrophages, possibly due to phagocytosis of the NPs by these cells, as reflected by TNF-α secretion in two PBMC samples and IL-6 secretion in 3 PBMC samples.
Figure 6.

The Effect of AlgS-Ca2+-pDNA NPs on PBMC Activation
Human PBMCs, isolated from five healthy individuals, were incubated for 3 h with NPs composed of a final Ca2+ concentration of 5 or 2.5 mM and 3 ng/μL pDNA and 500 nM/mL AlgS (NP concentration of ∼3.2 × 107/mL). Cytokine secretion was detected using ELISA. Anti-CD3 and anti-CD28 Dynabead activation was used as a positive control. (A) IL-2 secretion measured 24 h after activation; (B) IFN-γ secretion measured 48 h after activation; (C) IL-10 secretion measured 48 h after activation; (D) IL-6 secretion measured 24 h after activation; and (E) TNF-α secretion measured 24 h after activation. Data are presented as means ± SEM, and y axis is in logarithmic scale (n = 5; *p < 0.05, **p < 0.01, ****p < 0.0001, one-way ANOVA, Tukey’s multiple comparison test).
Inhibition of GFP Expression by Transgenic Expression of DT-A
To further demonstrate the potential of AlgS-Ca2+-pDNA NPs, a therapeutic gene encoding Diphtheria Toxin Fragment A (DT-A) was used. DT-A efficiently inactivates the elongation factor 2 (EF-2), resulting in the inhibition of protein synthesis, which eventually leads to cell death.18 DT-A lacks its membrane-binding domain (fragment b) and so it is non-toxic in its extracellular form.
The effect of delivering the DT-A-expressing plasmid, pDT-A N1, on protein synthesis inhibition was examined by co-transfecting MDA-MB-231 cells with pDT-A N1 and the reporter gene plasmid pEGFP N1, by the commercial reagent Lipofectamine 2000 or by AlgS-Ca2+-pDNA NPs. In both cases, both plasmids were pre-mixed and incorporated into the same carrier. The percentage of GFP-expressing cells and the mean fluorescence intensity in co-transfected cells were measured 24 h after transfection, and they were compared to those of cells transfected only with pEGFP N1. From the results presented (Figure 7), it can be seen that co-transfection of pDT-A N1 and pEGFP N1 led to significantly lower percentages of GFP-expressing cells compared to transfection with pEGFP N1 alone, for both transfection methods, thus validating the capability of pDT-A N1 to inhibit GFP synthesis.
Figure 7.

Reduced Percentage of GFP-Expressing Cells, 24 h after Being Transfected with Both pEGFP N1 and pDT-A N1, Delivered in Lipofectamine 2000 or AlgS-Ca2+-pDNA NPs
Results are mean ± SEM, n = 3. Asterisks denote significant difference with ***p < 0.0001.
Evaluation of the Effect of pDT-A N1 Delivery on Cell Proliferation
The final goal, the killing of cancer cells by AlgS-Ca2+-pDNA-mediated delivery of the DT-A gene, was demonstrated by measuring DNA content in cultures over 3 days after transfection. MDA-MB-231 cells were transfected with pDT-A N1-loaded NPs, pEGFP N1-loaded NPs, or pDT-A N1-loaded Lipofectamine 2000. The DNA content of each group was measured every 24 h over a 3 day period, and the results are presented in Figure 8 as the percentage DNA in the treated group from the DNA content of the control group (untreated cells) measured on the same day. Cells transfected with pEGFP N1 served as an additional control for testing possible toxicity originating from the AlgS-Ca2+-pDNA NPs themselves.
Figure 8.

Proliferation of MDA-MB-231 Cells Transfected with Either pEGFP N1 in AlgS-Ca2+ NPs, pDT-A N1 in AlgS-Ca2+ NPs, or by pDT-A N1 in Lipofectamine 2000
Control group represents untreated cells. Data are presented as the percentage DNA in treated cells from DNA content of the control group measured on the same day. Results are mean ± SEM, n = 3. Asterisks denote significant difference with *p < 0.05, **p < 0.01, and ***p < 0.005 by one-way ANOVA (Tukey post hoc test).
At 24 h post-transfection, no significant change in DNA content was observed for the cells transfected with AlgS-Ca2+-pDNA NPs, while the DNA content in cultures treated with pDT-A N1 in Lipofectamine dropped to 55% compared to the control cultures, i.e., untreated cells and cells treated with pEGFP N1-loaded NPs. At 48 h post-transfection, the relative DNA content in cultures treated with pDT-A N1 loaded in AlgS-Ca2+-pDNA NPs dropped to 55% compared to untreated cells; at this time point a slight decrease in DNA content was noted in cells treated with pEGFP N1-loaded NPs, although not significantly different compared to untreated cells. At 72 h post-transfection, an additional decrease in DNA content was noted in the pDT-A N1 AlgS-Ca2+-pDNA group (∼36%) compared to control untreated cells. In cultures treated with pEGFP N1 in AlgS-Ca2+-pDNA, there was also a decrease in DNA content, yet the decrease was much more pronounced in those treated with the pDT-A N1 in AlgS-Ca2+-pDNA NPs.
Discussion
The development of simple, safe, and efficient nanocarriers for DNA delivery is a prerequisite for the wide implementation of gene therapy.3 Herein we investigated the potential application of electrostatically driven self-assembled NPs as a transfecting agent for pDNA. These NPs, due to the complexation of AlgS with the nucleic acid mediated by calcium ion bridges, present several potential advantages, such as a simple and “green” fabrication method, formulation stability, cytocompatibility, and most of all the NPs were shown to be very effective for intracellular delivery and the induction of target gene silencing. As pDNA presents additional challenges compared to siRNA delivery, including its size and site of action, it was important to investigate, optimize, and test the implementation of this type of nanocarrier for the intracellular delivery of pDNA.
TEM images confirmed the formation of distinct NPs, while the XPS analysis verified their ternary composition and elucidated the molecular interactions between Ca2+ and the other two components, the pDNA and the AlgS. The AlgS-Ca2+-pDNA NPs were 2-fold larger than the siRNA NPs, most likely due to the larger size of the entrapped pDNA compared to the siRNA molecule, and they had a more negative surface charge (ζ∼−14 mV) due to the use of a greater amount (50×) of AlgS in the NPs, but which is still in the range allowing sufficient internalization of the particles by cells. The pDNA entrapment efficiency of nearly 70% was lower compared to the nearly 100% siRNA entrapment in similar Ca2+ composition NPs. Increasing [Ca2+] in the NPs slightly improved the entrapment efficiency of pDNA, up to a certain concentration.
Efficient transgene expression is one of the most important properties desired from a non-viral vector. In our study, we initially evaluated the transfection efficiency of a model pDNA as the percentage of cells expressing the reporter gene GFP of the total population. We found the AlgS-Ca2+-pDNA NPs to be a relatively efficient transfection agent compared to other non-viral carriers used to transfect MDA-MB-231 cells.19, 20, 21, 22, 23 The transfection efficiency was dependent on the Ca2+ concentration used in preparing the NPs. Those prepared with 250 mM Ca2+ (final concentration in culture media 5 mM) showed a transfection efficiency of 34%, 48 h post-transfection, while almost none was seen for those NPs prepared with 125 mM Ca2+ (final concentration in culture media 2.5 mM). Of note, these two NP types revealed the same particle size and surface charge and showed similar levels of cell internalization (80%–100% uptake). We relate the observed difference in transfection efficiencies to the alterations in Ca2+ contents of these NPs, in particular to the loosely associated Ca2+, which can participate in the cellular processes leading to plasmid expression, such as particle internalization, endosomal escape, and plasmid transport to the nucleus.
The critical role of calcium ions in cell internalization has been previously substantiated for Ca-siRNA complexes14 and AlgS-Ca2+-siRNA NPs15 and in the present study for AlgS-Ca2+-pDNA NPs. In these NP formulations, a portion of Ca2+ ions are present as a cationic cloud surrounding the nucleic acid (verified by XPS analysis in the present study), and this cloud of ions can interact with cell receptors, thus inducing receptor-mediated endocytosis events.24, 25 Further, our investigation with chemical inhibitors specific to the different endocytic pathways supported the mechanism for cell internalization of the AlgS-Ca2+-pDNA NPs mainly by clathrin-mediated endocytosis, a pathway known to be affected by calcium ion binding clathrin.26, 27 Thus, the treatment with the inhibitor of the clathrin-dependent endocytosis pathway showed the most significant reduction in GFP expression in MDA-MB-231 cells, while that of the inhibitor of the caveolin-mediated endocytosis showed only a trend, and CdCl2, a competitive antagonist of Ca2+, showed no effect at all on gene expression.
After internalization, the pDNA was seen to be located in distinct intracellular areas, most likely in the endosomes. The mean fluorescence intensity, indicating the relative amount of internalized DNA per cell, reached a maximal value after 3 h, and then it slowly decreased to 70% of its maximal value within 24 h. These kinetics indicate a slow process of endosomal release for pDNA compared to siRNA (which was shown to decrease to 34% of the initial value within 18 h14). The slow release rate from endosomes could be the result of (1) the larger size of the releasing molecule and/or (2) a weaker proton sponge effect because of a relatively lesser amount of loosely bound Ca2+ ions in the pDNA NPs. This together with the increased stability of pDNA in the cytoplasm compared to siRNA28, 29 explains the prolonged persistence of pDNA after cell internalization.
The pDNA should be transported to the nucleus and pass the nuclear membrane for its expression. It can reach the nucleus as associated with the NPs after their release from the endosome to the cytoplasm or as a pDNA, disassembled from the NPs and released to the cytoplasm. Both the pDNA and the assembled NPs in this study lack an active mechanism of nuclear internalization, and thus their entry into the nucleus, if it happens, can occur mainly following dissolution and reorganization of the nuclear envelope during cell division.30, 31, 32 Cell cycle has been known for years to be related to Ca2+ concentration and uptake.33, 34, 35, 36, 37 Therefore, the high concentration of Ca2+ in our NPs may induce cell proliferation and, thus, the entry of the plasmid into the nucleus. Supporting this mechanism is the finding that the relatively low proliferating primary CFs exhibited a lower rate of plasmid expression. At present, we still remain with the questions of whether and how much of the pDNA reaches the nucleus as associated to the NPs, and future studies will explore these issues.
Since it has been shown that very low concentrations of DT-A result in the abrogation of protein synthesis and apoptosis of cells, DT-A might serve as an efficient killer in cancer gene therapy.38, 39 Co-transfection studies of pEGFP N1 and pDT-A N1, delivered in either Lipofectamine 2000 or AlgS-Ca2+-pDNA NPs, confirmed the inhibitory effect of our constructed plasmid on protein synthesis, resulting in cell death as measured by DNA content. Cell death rate was more rapid for the Lipofectamine 2000 compared to that observed with AlgS-Ca2+-pDNA NPs; it took 48 h for the AlgS-Ca2+-pDNA NPs to reduce the DNA content to 55%, and it further decreased to 35% at 72 h. These kinetics are in agreement with the slower rate of endosomal escape of these NPs and the absence of active guidance of pDNA to the nucleus. It is expected that, with the addition of active guidance to pDNA, i.e., a nuclear localization signal (NLS), the delivery to nucleus would be more efficient,40, 41, 42 thus increasing the gene expression.
The most effective AlgS-Ca2+-pDNA NPs with the highest Ca2+ concentration (5 mM) were non-toxic to CT26-GFP and HepG2 cells for as long as several days after transfection, while they reduced cell viability in primary CFs and MDA-MB-231 cultures at 24 h. We relate this behavior to the cell capability to cope with Ca2+ bursts by employing designated pumps on the plasma and mitochondrion membranes. Similar results were reported previously for nanoparticles made of lipid, calcium, and phosphate.43 Our results thus indicate that a careful optimization of Ca2+ content in NPs should be performed per cell type to minimize this phenomenon.
Importantly, we show that, in contrast to anti-CD3 and anti-CD28 stimulation, the AlgS-Ca2+-pDNA NPs, composed of either 2.5 or 5 mM Ca2+, did not induce T cell proliferation (no elevation in IL-2 secretion) or activation of Th1, Th2, or Th17 phenotypes, in five PBMC samples from healthy adult individuals. A mild innate response was, however, triggered by the AlgS-Ca2+-pDNA NPs, especially those composed of the 2.5 mM Ca2+, in a few PBMC samples, possibly due to the NP uptake by phagocytes. Our results show, that AlgS-Ca2+-pDNA NPs do not activate T cells, especially (allergic) Th2 responses, may be important in rendering them ideal agents as carriers for gene and drug delivery systems or for diagnostic application.
Conclusions
The results of this work provide evidence for the development of an efficient non-viral vector for gene therapy with a potential use for the treatment of cancer. The NPs showed cytocompatibility and did not induce lymphocyte activation. While the AlgS-Ca2+-pDNA NPs provide a decent delivery rate of the plasmid to the nucleus (34%), there is still room for improvement. One such improvement could be the addition of an NLS to the plasmid itself, allowing for improved mobility of the plasmid from the cytosol to the nucleus.42 We believe that through such improvement we could achieve a simple, stable, and effective non-viral vector for the delivery of pDNA both in vitro and in vivo for future gene therapy.
Materials and Methods
Materials and Cells
The plasmids pEGFP N1 (4,733 bp, GenBank: U55762) and pGL3 (4,818 bp, GenBank: U47298) were kindly provided by Professor Ziv Reich (Weizmann Institute of Science, Israel). Labeling of plasmids with fluorescein or Cy5, using Label IT Tracker (fluorescein or Cy5) Nucleic Acid Labeling Kit (Mirus Bio, Madison WI), was performed according to the manufacturer’s instructions. The DT-A- (UniProtKB: Q6NK15) encoding plasmid, pDT-A N1 (4,671 bp), was designed by replacing the GFP gene from pEGFP N1 with DT-A. Based on the sequence provided by us, the DT-A gene was synthesized by Syntezza Bioscience (Jerusalem, Israel) and sub-cloned by Bio Basic (Markham, ON, Canada). All plasmids were propagated in E. coli and purified by QIAGEN Midiprep kits according to the manufacturer’s instructions (Hilden, Germany). Dynabeads Human T-Activator CD3 and CD28 were used according to the manufacturer’s instructions (Thermo Fisher Scientific, MA, USA). All antibodies used for ELISA were purchased from BioLegend (CA, USA) unless stated otherwise. Sodium alginate (LVG, >65% guluronic acid content) was from NovaMatrix FMC Biopolymers (Drammen, Norway). AlgS was prepared as previously described.44 Cell culture reagents (DMEM, RPMI 1640, L-glutamine, penicillin and streptomycin, and heat-inactivated fetal bovine serum [FBS]) were from Biological Industries (Kibbutz Beit-Haemek, Israel). All salts and other reagents were from Sigma-Aldrich (Rehovot, Israel) unless specified otherwise.
The human breast cancer MDA-MB-231 cell line was from the American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were cultivated in RPMI 1640 medium, supplemented with 10% FBS (v/v), 1% penicillin and streptomycin (v/v), and 1% L-glutamine (v/v). The HepG2 cell line was from the ATCC. The cells were cultivated in high-glucose DMEM supplemented with 10% FBS (v/v), 1% penicillin and streptomycin (v/v) and 1% L-glutamine (v/v). Neonatal CFs were generously donated by Yoram Etzion’s lab (Ben-Gurion University). The cells were cultivated in high-glucose DMEM supplemented with 10% FBS (v/v), 1% penicillin and streptomycin (v/v) and 1% L-glutamine (v/v). All cells were incubated at 37°C, in a humidified air atmosphere containing 5% CO2. CT26 cells stably transfected with EGFP were cultured in DMEM supplemented with 10% FBS (v/v), 1% L-glutamine (v/v), G418 (500 μg mL−1), and 1% penicillin and streptomycin (v/v).
Preparation of AlgS-Ca2+-pDNA NPs
Solutions of pDNA (600 ng/μL), CaCl2 (1 M stock unless specified otherwise), and AlgS (50 μg/mL) were freshly prepared in deionized water. Equal volumes of pDNA and CaCl2 were mixed by gentle vortexing for 30 s and incubated for 30 min at room temperature (RT). Then, an equal volume of AlgS was added and mixed by gentle vortexing for 30 s, followed by incubation for 30 min at RT, resulting in NPs with a final concentration of 250 mM Ca2+, 150 ng/μL pDNA, and 25 μg/mL AlgS. The NPs were freshly prepared before analysis or use and diluted to the final concentration indicated, unless stated otherwise. All assays involving AlgS-Ca2+-pDNA NPs were performed on the same day of preparation.
Size Distribution and Zeta Potential of the NPs
The mean diameter of AlgS-Ca2+-pDNA NPs was measured on a CGS-3 (ALV, Langen, Germany) instrument. Samples were diluted 1:50 in 10 mM HEPES solution (final concentrations of 2.5 or 5 mM Ca2+, 3 ng/μL pDNA, and 500 ng/mL AlgS); analyzed by scattered laser light (Helium-Neon [He-Ne] laser, 20 mW, 632.8 nm); and detected under an angle of 90° for the duration of 10 s, 20 times, at 25°C. Correlograms were calculated by ALV/LSE 5003 correlator and fitted with a version of the CONTIN program.
The size of AlgS-Ca2+-pDNA NPs was also determined by NTA. Samples were diluted 1:20 in 10 mM HEPES solution (final concentrations of 12.5 mM Ca2+, 7.5 ng/μL pDNA, and 1.25 μg/mL AlgS), and they were analyzed on a NanoSight NS300 instrument (Malvern Instruments, UK). Samples were analyzed under a 20× objective and 60-s video clips were taken. The software version NTA 2.3 was used for capture and analysis.
The Zeta potential of NPs (final concentrations of 2.5 or 5 mM Ca2+, 3 ng/μL pDNA, and 500 ng/mL AlgS) was measured on a Zetasizer Nano ZS (Malvern Instruments, UK), using electrophoretic cells (DTS 1060, produced by Malvern Instruments, UK). Zeta potentials were recorded 3 times with 10–100 measurements in each run (depending on SD).
TEM
5 μL AlgS-Ca2+-pDNA NPs (using gold-labeled AlgS) and Ca2+-pDNA were placed on carbon-coated film on copper EM grids hydrophilized by glow discharge. The excess liquid was blotted, and the grids were allowed to dry at RT for 4 h. The samples were imaged at RT using an FEI Tecnai 12G2 TWIN TEM (Gatan model 794 charge coupled device [CCD], bottom mounted) at an acceleration voltage of 120 kV. Specimens were studied in a low-dose imaging mode to minimize beam exposure and electron beam radiation damage. Images were recorded digitally using the Digital Micrograph 3.6 software (Gatan, Munich, Germany).
For cryo-TEM analysis, thin (∼0.25-μm) specimens of NPs were deposited, under controlled humidity and temperature, on perforated carbon films supported on copper grids. The excess liquid was blotted, and the specimen was vitrified by rapid plunging into liquid ethane precooled with liquid nitrogen in a controlled-environment vitrification system. The samples were examined at −178°C using an FEI Tecnai 12 G2TWIN TEM (Gatan model 794 CCD, bottom mounted) equipped with a Gatan 626 cryo-holder. Specimens were studied in a low-dose imaging mode to minimize beam exposure and electron beam radiation damage. Images were recorded digitally using the Digital Micrograph 3.6 software (Gatan). Particle size measurements were performed using Adobe Photoshop CS 5.1 (San Jose, CA, USA).
EtBr Exclusion Assay
The efficiency of pDNA entrapment in AlgS-Ca2+-pDNA NPs prepared with a range of Ca2+ concentrations was determined by EtBr exclusion assay. The NPs were mixed with EtBr solution and incubated at RT for 30 min. Fluorescence intensity was measured using a Synergy Mx microplate reader (BioTek Instruments) at an excitation wavelength of 535 nm and emission wavelength of 595 nm. Efficiency was calculated as the percentage of fluorescence intensity of each sample from the fluorescence intensity of free pDNA.
XPS
Ca2+-pDNA complexes (250 mM Ca2+ and 150 ng/μL pDNA) and AlgS-Ca2+-pDNA NPs (250 mM Ca2+, 150 ng/μL pDNA, and 25 μg/mL AlgS) were prepared, and a 5-μL droplet of each was placed directly on a silicon oxide wafer and dried before conducting the XPS measurements. XPS data were collected using an X-ray photoelectron spectrometer ESCALAB 250 ultrahigh vacuum (1 × 10−9 bar) apparatus with an AlKα X-ray source and a monochromator. The X-ray beam size was 500 μm, and survey spectra were recorded with pass energy (PE) 150 eV and high-energy resolution spectra were recorded with PE 20 eV. The spectral components of C1s, O1s, N1s, Ca2p, S2p, and P2p signals were found by fitting a sum of single component lines to the experimental data by means of nonlinear least-squares curve-fitting.
NP Cytocompatibility
Cytocompatibility of the anionic AlgS-Ca2+-pDNA NPs was assessed using PrestoBlue cell viability assay (Life Technologies, Carlsbad, CA, USA). This assay is based on the live cell’s ability to reduce resazurin (non-fluorescent) to resorufin (fluorescent). PrestoBlue working solution was prepared by dilution of PrestoBlue reagent 1:10 in cell culture medium. At 3 h post-transfection, the medium was aspirated and cells were washed, supplemented with complete medium, and incubated for 24–72 h at 37°C. PrestoBlue working solution was added to the cells for 1 h at 37°C and 5% CO2. Fluorescence intensity was measured using the Synergy Mx microplate reader at an excitation wavelength of 560 nm and emission wavelength of 590 nm. The percentage of cell viability was obtained after normalizing the data to untreated cells.
Cell Studies
Cell Uptake of pDNA NPs by Confocal Microscopy
The cells (CFs, HepG2, and MDA-MB-231) were seeded on 24-well plates at a density of 50,000 cells/well. At 24 h post-seeding, the cells were washed once with transfection medium and treated with NPs (125 or 250 mM Ca2+, 150 ng/μL pDNA, and 25 μg/mL AlgS) that were freshly diluted in transfection medium, at a ratio of 1:50. At 3 h post-transfection, the medium was removed and the cells were washed and supplemented with culture medium. At 72 h post-transfection, the medium was aspirated and cells were supplemented with phenol red-free DMEM. Nuclei were stained with NucBlue (for live, unfixed cells) (Life Technologies). Images were taken using a Nikon C1si laser-scanning confocal microscope (LSCM).
Cell Uptake of pDNA NPs by Imaging Flow Cytometry
The cells (CFs, HepG2, and MDA-MB-231) were seeded on 24-well plates at a density of 50,000 cells/well. At 24 h post-seeding, the cells were washed once with transfection medium and treated with FITC-labeled NPs (125 or 250 mM Ca2+, pDNA 150 ng/μL, and AlgS 25 μg/mL) that were freshly diluted in transfection medium at a ratio of 1:50. At 3 h post-transfection, the cells were trypsinized, resuspended in culture medium, washed twice by resuspension in fluorescence-activated cell sorting (FACS) buffer (PBS containing 2% FBS, v/v), and recovered by centrifugation (650 × g for 5 min at 4°C). The collected cells were then resuspended in FACS buffer and analyzed using the ImageStreamX Mark II (Amnis, Seattle, WA, USA). Cell acquisition and analysis were performed using IDEAS Application, version 6.0. A population of single cells was chosen, and a minimum threshold of fluorescence was decided by negative control samples every experiment. All results that were higher than the minimum were considered positive.
Flow Cytometry Analysis of GFP Expression
The expression of GFP by cells after the uptake of NPs (final concentrations of 2.5 or 5 mM Ca2+, 3 ng/μL pDNA, and 500 ng/mL AlgS) was assessed by flow cytometry at 72 h post-transfection. CT26-EGFP, CFs, HepG2, and MDA-MB-231 cells were seeded on 24-well plates at a density of 50,000 cells/well. Cells were treated with the AlgS-Ca2+-pDNA NPs diluted at a ratio of 1:50 in transfection medium for 3 h. Afterward, the cells were washed and supplemented with culture medium. At specific time points, the cells were trypsinized, washed twice by resuspension in FACS buffer (PBS containing 2% FBS, v/v), and recovered by centrifugation (650 × g for 5 min at 4°C). The collected cells were then resuspended in FACS buffer and transferred to a flow cytometer tube (BD Biosciences, San Jose, CA, USA). Cell acquisition and analysis (10,000 events) were performed using a FACS Calibur machine (BD Biosciences), using Cellquest Pro software (BD Biosciences). Results were calculated using the control population as a gate for negative results, and all cells that exhibited higher GFP fluorescence were considered positive.
Evaluation of EGFP Expression after Endocytosis Inhibition
The effect of chemically inhibiting endocytosis on the cellular uptake of AlgS-Ca2+-pDNA NPs was studied using Pitstop2,45 NaN3,46 genistein,47 and CdCl2.48 MDA-MB-231 cells were seeded in a 12-well plate at a density of 300,000 cells/well. At 24 h post-seeding, the cells were incubated with inhibitors in a transfection medium for 30 min, washed twice with PBS, and treated with AlgS-Ca2+-pDNA NPs (final concentrations of 5 mM Ca2+, 3 ng/μL pDNA, and 500 ng/mL AlgS), diluted 1:50 in transfection medium with addition of the inhibitor. At 3 h post-transfection, the cells were washed and supplemented with culture medium. At 48 h post-transfection, cells were trypenised, resuspended in culture medium, and centrifuged (650 × g for 5 min). Cells were washed twice by resuspension in FACS buffer (PBS containing 2% FBS, v/v) and recovered by centrifugation (650 × g for 5 min). The collected cells were then resuspended in FACS buffer and analyzed using the ImageStreamX Mark II (Amnis, Seattle, WA, USA). Cell acquisition and analysis were performed using IDEAS Application, version 6.0.
Effects of NPs on Human PBMCs
Ethics Statement
All experiments were performed and conducted in accordance with good clinical practices and Helsinki approval of the Soroka Medical Center.
PBMCs from five healthy donors were isolated from heparinized venous blood over Ficoll Paque Plus (Sigma), at a ratio of blood:Ficoll of 10:8. Samples were centrifuged for 30 min at 0°C at 500 × g. From the layers generated, PBMCs were collected using a Pasteur pipette. Cells were then washed twice with RPMI (centrifuged for 10 min at 500 × g). Cells were resuspended (107 cells/mL) in complete RPMI 1640 medium supplemented with 10% FBS, 4 mM L-glutamine, 25 mM HEPES buffer, 50 U/mL penicillin, and 50 μg/mL streptomycin, and they were seeded in a 96-well plate coated with anti-CD3 (BioLegend, San Diego, CA, USA) at a density of 1 × 105 cells/well. Post-seeding, the cells were treated with AlgS-Ca2+-pDNA NPs, diluted 1:50 in a transfection medium (final concentrations of the NP components were 5 or 2.5 mM Ca2+, 3 ng/μL pDNA, and AlgS 500 ng/mL). Dynabeads Human T-Activator CD3 and CD28 were used as positive controls for PBMC activation, according to the manufacturer’s instructions. The medium with the pDNA NPs was removed after 3 h, and it was replaced with full RPMI culture medium for the remainder of the experiment. At 24, 48, and 72 h later, the supernatants were analyzed by ELISA (for the cytokines IL-2, TNF-α, IL-10, IFN-γ, IL-17, and IL-6), according to the manufacturer’s instructions.
Inhibition of GFP Expression by Transgenic Expression of DT-A
Co-transfection of the pEGFP N1 plasmid and pDT-A N1, a DT-A-expressing plasmid, was utilized to examine the effects of DT-A expression on protein synthesis. Co-transfection was performed using either AlgS-Ca2+-pDNA NPs or Lipofectamine 2000 containing the same amount of pDNA. To account for the increased amount of pDNA in the formulation, a control of cells transfected with the same amount of pEGFP N1 and the non-expressing plasmid pGL3 GFP was used.
MDA-MB-231 cells were seeded in 12-well culture plates at a cell density of 100,000 cells/well. At 24 h post-seeding, cells were treated with AlgS-Ca2+-pDNA NPs, diluted 1:50 in transfection medium (final concentrations of 5 mM Ca2+; 3 ng/μL pEGFP N1, pDT-A, or pGL3 GFP; and 500 ng/mL AlgS). Other cells were treated with Lipofectamine 2000, mixed according to the manufacturer’s instructions with the same amount of pDNA used in the preparation of AlgS-Ca2+-pDNA NPs. The medium was removed after 3 h and replaced with full culture medium. The cells were grown for an additional 24 h, after which they were trypsinized, resuspended in culture medium, and centrifuged (650 × g for 5 min). Cells were washed twice by resuspension in FACS buffer and recovered by centrifugation (650 × g for 5 min). The collected cells were then resuspended in FACS buffer and analyzed using the ImageStreamX Mark II (Amnis, Seattle, WA, USA). Cell acquisition and analysis were performed using IDEAS Application, version 6.0.
Evaluation of pDT-A Delivery on Cell Proliferation by DNA Content
The ability of AlgS-Ca2+-pDNA NPs to deliver pDT-A and kill cells was evaluated by estimating the DNA content in transfected cells over 72 h, using the fluorescent dye bisbenzimidazole Hoechst 33258 (Sigma). MDA-MB-231 cells were seeded in 24-well plates at a density of 25,000 cells/well. After 24 h, the cells were transfected with AlgS-Ca2+-pDNA containing pDT-A N1, diluted 1:50 in transfection medium (final concentrations of 5 mM Ca2+, 3 ng/μL pDNA, and 500 ng/mL AlgS) or with Lipofectamine 2000 containing the same amount of pDT-A N1. The medium was removed after 3 h and replaced with culture medium, and the cells were grown for the duration of 1–3 additional days. Every 24 h, cells were trypsinized, resuspended in culture medium, and centrifuged (650 × g for 5 min). The cells were washed twice and recovered by additional centrifugation (650 × g for 5 min). The collected cells were then suspended in 100 μL lysis buffer (0.02% SDS in saline-sodium citrate [SSC] buffer [pH 7.0]) and incubated for 1 h at 37°C. Then, 100 μL Hoechst 33258 assay solution (2 μg/mL in SCC buffer) was added, followed by an additional 10-min incubation at 37°C. 180 μL of each sample was transferred to a 96-well plate, and fluorescence intensity was measured using a Synergy Mx microplate reader (BioTek Instruments) at an excitation wavelength of 485 nm and emission wavelength of 530 nm.
Statistics
Statistical analysis was performed with GraphPad Prism version 6.01 for Windows (GraphPad, San Diego, CA, USA). All variables are expressed as mean ± SEM from at least 2 independent experiments unless stated otherwise. Analyzing statistical difference between the means of several groups was done by one-way ANOVA with Tukey’s post hoc test. p < 0.05 was considered statistically significant unless stated otherwise.
Author Contributions
M.G. and S.S. conducted the experiments. E.V. and M.G. conducted the PBMC experiment. M.G., E.V., S.C., and A.M. designed the PBMC experiment. M.G., S.S., and S.C. designed the experiments, and M.G., E.V., A.M., and S.C. wrote the paper.
Acknowledgments
The Focal Technological Area Program of the Israel National Nanotechnology Initiative (INNI2012) (Bio-inspired Nano-carriers for Sub-cellular Targeted Therapeutics) supported this work. M.G. and S.S. gratefully acknowledge the Darom fellowship from Kreitman School. This work was done in partial fulfillment of the requirements for a PhD degree (M.G.) at the Avram and Stella Goldstein-Goren Department of Biotechnology Engineering, Ben-Gurion University of the Negev, Israel. S.C. holds the Claire and Harold Oshry Professor Chair in Biotechnology.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2019.03.006.
Supplemental Information
References
- 1.Ibraheem D., Elaissari A., Fessi H. Gene therapy and DNA delivery systems. Int. J. Pharm. 2014;459:70–83. doi: 10.1016/j.ijpharm.2013.11.041. [DOI] [PubMed] [Google Scholar]
- 2.Wirth T., Parker N., Ylä-Herttuala S. History of gene therapy. Gene. 2013;525:162–169. doi: 10.1016/j.gene.2013.03.137. [DOI] [PubMed] [Google Scholar]
- 3.Rosenblum D., Joshi N., Tao W., Karp J.M., Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018;9:1410. doi: 10.1038/s41467-018-03705-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones C.H., Chen C.-K., Ravikrishnan A., Rane S., Pfeifer B.A. Overcoming nonviral gene delivery barriers: perspective and future. Mol. Pharm. 2013;10:4082–4098. doi: 10.1021/mp400467x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindberg M.F., Le Gall T., Carmoy N., Berchel M., Hyde S.C., Gill D.R., Jaffrès P.A., Lehn P., Montier T. Efficient in vivo transfection and safety profile of a CpG-free and codon optimized luciferase plasmid using a cationic lipophosphoramidate in a multiple intravenous administration procedure. Biomaterials. 2015;59:1–11. doi: 10.1016/j.biomaterials.2015.04.024. [DOI] [PubMed] [Google Scholar]
- 6.Martínez-Negro M., Barrán-Berdón A.L., Aicart-Ramos C., Moyá M.L., de Ilarduya C.T., Aicart E., Junquera E. Transfection of plasmid DNA by nanocarriers containing a gemini cationic lipid with an aromatic spacer or its monomeric counterpart. Colloids Surf. B Biointerfaces. 2018;161:519–527. doi: 10.1016/j.colsurfb.2017.11.024. [DOI] [PubMed] [Google Scholar]
- 7.Cao X., Wang J., Deng W., Chen J., Wang Y., Zhou J., Du P., Xu W., Wang Q., Wang Q. Photoluminescent Cationic Carbon Dots as efficient Non-Viral Delivery of Plasmid SOX9 and Chondrogenesis of Fibroblasts. Sci. Rep. 2018;8:7057. doi: 10.1038/s41598-018-25330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kauffman A.C., Piotrowski-Daspit A.S., Nakazawa K.H., Jiang Y., Datye A., Saltzman W.M. Tunability of Biodegradable Poly(amine- co-ester) Polymers for Customized Nucleic Acid Delivery and Other Biomedical Applications. Biomacromolecules. 2018;19:3861–3873. doi: 10.1021/acs.biomac.8b00997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chintakunta R., Buaron N., Kahn N., Moriah A., Lifshiz R., Goldbart R., Traitel T., Tyler B., Brem H., Kost J. Synthesis, characterization, and self-assembly with plasmid DNA of a quaternary ammonium derivative of pectic galactan and its fluorescent labeling for bioimaging applications. Carbohydr. Polym. 2016;150:308–318. doi: 10.1016/j.carbpol.2016.05.015. [DOI] [PubMed] [Google Scholar]
- 10.Fröhlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomedicine. 2012;7:5577–5591. doi: 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodman C.M., McCusker C.D., Yilmaz T., Rotello V.M. Toxicity of gold nanoparticles functionalized with cationic and anionic side chains. Bioconjug. Chem. 2004;15:897–900. doi: 10.1021/bc049951i. [DOI] [PubMed] [Google Scholar]
- 12.Ruvinov E., Kryukov O., Forti E., Korin E., Goldstein M., Cohen S. Calcium-siRNA nanocomplexes: what reversibility is all about. J. Control. Release. 2015;203:150–160. doi: 10.1016/j.jconrel.2015.02.029. [DOI] [PubMed] [Google Scholar]
- 13.Forti E., Kryukov O., Elovic E., Goldshtein M., Korin E., Margolis G., Felder S., Ruvinov E., Cohen S. A bridge to silencing: Co-assembling anionic nanoparticles of siRNA and hyaluronan sulfate via calcium ion bridges. J. Control. Release. 2016;232:215–227. doi: 10.1016/j.jconrel.2016.04.033. [DOI] [PubMed] [Google Scholar]
- 14.Goldshtein M., Forti E., Ruvinov E., Cohen S. Mechanisms of cellular uptake and endosomal escape of calcium-siRNA nanocomplexes. Int. J. Pharm. 2016;515:46–56. doi: 10.1016/j.ijpharm.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 15.Korin E., Bejerano T., Cohen S. GalNAc bio-functionalization of nanoparticles assembled by electrostatic interactions improves siRNA targeting to the liver. J. Control. Release. 2017;266:310–320. doi: 10.1016/j.jconrel.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 16.Korin E., Froumin N., Cohen S. Surface Analysis of Nanocomplexes by X-ray Photoelectron Spectroscopy (XPS) ACS Biomater. Sci. Eng. 2017;3:882–889. doi: 10.1021/acsbiomaterials.7b00040. [DOI] [PubMed] [Google Scholar]
- 17.Hanawa T., Ota M. Calcium phosphate naturally formed on titanium in electrolyte solution. Biomaterials. 1991;12:767–774. doi: 10.1016/0142-9612(91)90028-9. [DOI] [PubMed] [Google Scholar]
- 18.Yamaizumi M., Mekada E., Uchida T., Okada Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell. 1978;15:245–250. doi: 10.1016/0092-8674(78)90099-5. [DOI] [PubMed] [Google Scholar]
- 19.Gargouri M., Sapin A., Bouli S., Becuwe P., Merlin J.L., Maincent P. Optimization of a new non-viral vector for transfection: Eudragit nanoparticles for the delivery of a DNA plasmid. Technol. Cancer Res. Treat. 2009;8:433–444. doi: 10.1177/153303460900800605. [DOI] [PubMed] [Google Scholar]
- 20.Li J., He Y.Z., Li W., Shen Y.Z., Li Y.R., Wang Y.F. A novel polymer-lipid hybrid nanoparticle for efficient nonviral gene delivery. Acta Pharmacol. Sin. 2010;31:509–514. doi: 10.1038/aps.2010.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin W.J., Chien W.H. Peptide-conjugated micelles as a targeting nanocarrier for gene delivery. J. Nanopart. Res. 2015;17:349. [Google Scholar]
- 22.Mangraviti A., Tzeng S.Y., Kozielski K.L., Wang Y., Jin Y., Gullotti D., Pedone M., Buaron N., Liu A., Wilson D.R. Polymeric nanoparticles for nonviral gene therapy extend brain tumor survival in vivo. ACS Nano. 2015;9:1236–1249. doi: 10.1021/nn504905q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu D., Wang A., Huang H., Chen Y. PEG-PBLG nanoparticle-mediated HSV-TK/GCV gene therapy for oral squamous cell carcinoma. Nanomedicine (Lond.) 2008;3:813–821. doi: 10.2217/17435889.3.6.813. [DOI] [PubMed] [Google Scholar]
- 24.Gerasimenko J.V., Tepikin A.V., Petersen O.H., Gerasimenko O.V. Calcium uptake via endocytosis with rapid release from acidifying endosomes. Curr. Biol. 1998;8:1335–1338. doi: 10.1016/s0960-9822(07)00565-9. [DOI] [PubMed] [Google Scholar]
- 25.Lariccia V., Fine M., Magi S., Lin M.J., Yaradanakul A., Llaguno M.C., Hilgemann D.W. Massive calcium-activated endocytosis without involvement of classical endocytic proteins. J. Gen. Physiol. 2011;137:111–132. doi: 10.1085/jgp.201010468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Näthke I., Hill B.L., Parham P., Brodsky F.M. The calcium-binding site of clathrin light chains. J. Biol. Chem. 1990;265:18621–18627. [PubMed] [Google Scholar]
- 27.Marsh M., McMahon H.T. The structural era of endocytosis. Science. 1999;285:215–220. doi: 10.1126/science.285.5425.215. [DOI] [PubMed] [Google Scholar]
- 28.Seternes T., Tonheim T.C., Løvoll M., Bøgwald J., Dalmo R.A. Specific endocytosis and degradation of naked DNA in the endocardial cells of cod (Gadus morhua L.) J. Exp. Biol. 2007;210:2091–2103. doi: 10.1242/jeb.003186. [DOI] [PubMed] [Google Scholar]
- 29.Hirsch M., Helm M. Live cell imaging of duplex siRNA intracellular trafficking. Nucleic Acids Res. 2015;43:4650–4660. doi: 10.1093/nar/gkv307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dean D.A., Strong D.D., Zimmer W.E. Nuclear entry of nonviral vectors. Gene Ther. 2005;12:881–890. doi: 10.1038/sj.gt.3302534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Escriou V., Carrière M., Bussone F., Wils P., Scherman D. Critical assessment of the nuclear import of plasmid during cationic lipid-mediated gene transfer. J. Gene Med. 2001;3:179–187. doi: 10.1002/jgm.174. [DOI] [PubMed] [Google Scholar]
- 32.Brunner S., Sauer T., Carotta S., Cotten M., Saltik M., Wagner E. Cell cycle dependence of gene transfer by lipoplex, polyplex and recombinant adenovirus. Gene Ther. 2000;7:401–407. doi: 10.1038/sj.gt.3301102. [DOI] [PubMed] [Google Scholar]
- 33.Berridge M.J. Calcium signalling and cell proliferation. BioEssays. 1995;17:491–500. doi: 10.1002/bies.950170605. [DOI] [PubMed] [Google Scholar]
- 34.Capiod T. Cell proliferation, calcium influx and calcium channels. Biochimie. 2011;93:2075–2079. doi: 10.1016/j.biochi.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 35.Pinto M.C., Kihara A.H., Goulart V.A., Tonelli F.M., Gomes K.N., Ulrich H., Resende R.R. Calcium signaling and cell proliferation. Cell. Signal. 2015;27:2139–2149. doi: 10.1016/j.cellsig.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 36.Resende R.R., Andrade L.M., Oliveira A.G., Guimarães E.S., Guatimosim S., Leite M.F. Nucleoplasmic calcium signaling and cell proliferation: calcium signaling in the nucleus. Cell Commun. Signal. 2013;11:14. doi: 10.1186/1478-811X-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sperti G., Colucci W.S. Calcium influx modulates DNA synthesis and proliferation in A7r5 vascular smooth muscle cells. Eur. J. Pharmacol. 1991;206:279–284. doi: 10.1016/0922-4106(91)90110-4. [DOI] [PubMed] [Google Scholar]
- 38.Chen H. Exploiting the Intron-splicing Mechanism of Insect Cells to Produce Viral Vectors Harboring Toxic Genes for Suicide Gene Therapy. Mol. Ther. Nucleic Acids. 2012;1:e57. doi: 10.1038/mtna.2012.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y., Schulte W., Pink D., Phipps K., Zijlstra A., Lewis J.D., Waisman D.M. Sensitivity of cancer cells to truncated diphtheria toxin. PLoS ONE. 2010;5:e10498. doi: 10.1371/journal.pone.0010498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo R., Nan J. Ultrasound-Targeted Microbubbles Combined with a Peptide Nucleic Acid Binding Nuclear Localization Signal Mediates the Transfection of Exogenous Genes by Improving both Cytoplasmic and Nuclear Import. Ultrasound Med. Biol. 2017;43:S11. doi: 10.3892/mmr.2017.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoare M., Greiser U., Schu S., Mashayekhi K., Aydogan E., Murphy M., Barry F., Ritter T., O’Brien T. Enhanced lipoplex-mediated gene expression in mesenchymal stem cells using reiterated nuclear localization sequence peptides. J. Gene Med. 2010;12:207–218. doi: 10.1002/jgm.1426. [DOI] [PubMed] [Google Scholar]
- 42.Nagasaki T., Myohoji T., Tachibana T., Futaki S., Tamagaki S. Can nuclear localization signals enhance nuclear localization of plasmid DNA? Bioconjug. Chem. 2003;14:282–286. doi: 10.1021/bc025602h. [DOI] [PubMed] [Google Scholar]
- 43.Tseng Y.C., Yang A., Huang L. How does the cell overcome LCP nanoparticle-induced calcium toxicity? Mol. Pharm. 2013;10:4391–4395. doi: 10.1021/mp400028m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freeman I., Kedem A., Cohen S. The effect of sulfation of alginate hydrogels on the specific binding and controlled release of heparin-binding proteins. Biomaterials. 2008;29:3260–3268. doi: 10.1016/j.biomaterials.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 45.Gourlaouen M., Welti J.C., Vasudev N.S., Reynolds A.R. Essential role for endocytosis in the growth factor-stimulated activation of ERK1/2 in endothelial cells. J. Biol. Chem. 2013;288:7467–7480. doi: 10.1074/jbc.M112.446401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeng X., Zhang Y., Nyström A.M. Endocytic uptake and intracellular trafficking of bis-MPA-based hyperbranched copolymer micelles in breast cancer cells. Biomacromolecules. 2012;13:3814–3822. doi: 10.1021/bm301281k. [DOI] [PubMed] [Google Scholar]
- 47.Li Z., Li J., Mo B., Hu C., Liu H., Qi H., Wang X., Xu J. Genistein induces cell apoptosis in MDA-MB-231 breast cancer cells via the mitogen-activated protein kinase pathway. Toxicol. In Vitro. 2008;22:1749–1753. doi: 10.1016/j.tiv.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 48.Fan L.Z., Cherian M.G. Potential role of p53 on metallothionein induction in human epithelial breast cancer cells. Br. J. Cancer. 2002;87:1019–1026. doi: 10.1038/sj.bjc.6600549. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.