

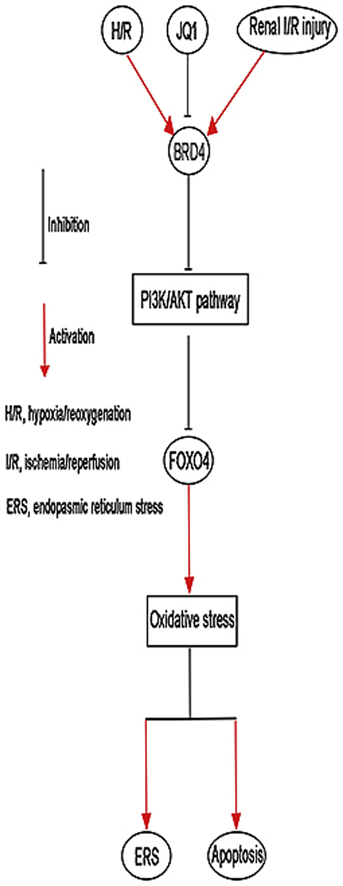
Abstract
Ischemia/reperfusion injury (I/R) is one of the leading causes of acute kidney injury (AKI) that typically occurs in renal surgeries. However, renal I/R still currently lacks effective therapeutic targets. In this study, we proved that inhibition of Brd4 with its selective inhibitor, JQ1, could exert a protective role in renal I/R injury in mice. Inhibiting Brd4 with either JQ1 or genetic knockdown resulted in reduction of endoplasmic reticulum stress (ERS)-associated protein and proapoptotic protein expression both in I/R-induced injury and hypoxia/reoxygenation (H/R) stimulation in HK-2 cells. H/R-induced apoptosis and ERS depended on oxidative stress in vitro. Moreover, FoxO4, which is involved in the generation of hydrogen peroxide, was up-regulated during H/R stimulation-mediated apoptosis and ERS, and this upregulation could be abolished by Brd4 inhibition. Consistently, FoxO4-mediated ROS generation was attenuated upon inhibition of Brd4 with JQ1 or siRNA against Brd4. Further, the transcriptional activity of FoxO4 was suppressed by PI3K and AKT phosphorylation, which are upstream signals of FoxO4 expression, and were enhanced by Brd4 both in vivo and in vitro. In conclusion, our results proved that Brd4 inhibition blocked renal apoptotic and ERS protein expression by preventing FoxO4-dependent ROS generation through the PI3K/AKT pathway, indicating that Brd4 could be a potential therapeutic target for renal I/R injury.
Keywords: Brd4, Renal I/R injury, Oxidative stress, ERS, Apoptosis
Graphical abstract
Highlights
-
•
Brd4 was up-regulated in renal I/R injury.
-
•
Brd4 inhibitor JQ1 alleviated renal I/R injury.
-
•
Brd4 inhibition blocked H/R-induced oxidative stress, apoptosis and ERS through FoxO4.
-
•
Brd4 regulated FoxO4 through the PI3K/AKT pathway.
1. Introduction
In recent years, remarkable progress has been made in the field of therapeutics for acute kidney injury (AKI) and in the emergency treatment of critical and severe AKI cases. Nevertheless, as the incidence and mortality of AKI remain high, there is still lack of effective medications [1,2]. Renal ischemia-reperfusion (I/R) injury is a major cause of acute kidney injury (AKI), which often arises from hypovolemic conditions, septic shock, surgery, and transplantation [3]. I/R injury causes structural and functional damage of renal tubules by directly inducing death of tubular cells, and these dying cells may trigger damaged responses [4]. Studies in recent years have shown that abnormal apoptosis and endoplasmic reticulum stress (ERS) of renal tubular epithelial cells may affect the occurrence and progression of AKI [5]. As the importance of AKI is becoming increasingly evident, it is essential to develop new therapies to prevent renal damage caused by AKI.
The bromodomain and extra-terminal domain (BET) protein family consists of four proteins, namely Brd2, Brd3, Brd4, and tetris-specific Brdt, and are known to read acetylated lysine on histones in the nucleus and change chromatin structure through their bromodomain [6,7]. With the development of numerous BET protein inhibitors, the role of BET proteins have been highlighted to play critical functions in various cellular processes such as cell growth, cell cycle, inflammation, and cancer development [[8], [9], [10]]. Among these inhibitors, JQ1 has been widely used as a potent, relative Brd4-selective, and first generation inhibitor [11]. A previous study indicated that inhibition of Brd4 by JQ1 could reduce the expression of inflammatory mediators in human airway epithelial cells, suggesting that Brd4 may have therapeutic potential in inflammatory diseases [12]. A recent study reported that inhibition of Brd4 suppressed oxidative stress induced by unilateral ureteral obstruction in rats [13]. Another study found that Brd4 could potently reduce cancer cell viability and induce apoptosis of tumor cells through downregulation of E2f1 protein expression, both in vitro and in vivo [14]. However, whether Brd4 could reduce renal I/R injury and its possible mechanism remains unknown.
Oxidative stress plays an important role in renal apoptosis and endoplasmic reticulum stress, especially during reperfusion. Either by acting as signal transduction molecules or by directly causing cellular damage, reactive oxygen species (ROS) activate apoptosis and ERS at multiple stages. The forkhead (Fox) transcription factor O family (FoxO) of proteins are involved in a variety of biological processes, including cell proliferation, oxidative stress response, apoptosis, and metabolism [15]; the most well-known members of this family include FoxO1, FoxO3, FoxO4, and FoxO6. FoxO4, which was initially recognized as a tumor suppressor, has also been linked with various diseases, such as diabetic nephropathy [16], diabetic retinopathy [17], and ischemic limbs [18]. A previous study has already showed that FoxO4 plays a key role in ROS-induced cell apoptosis [19]. However, whether Brd4 is involved in the I/R-induced ROS generation in renal AKI is still unknown.
In the present study, we investigated whether Brd4 inhibition could modulate I/R-induced acute injury in the kidney. We also determined the potential mechanisms involved in the effects of Brd4 inhibition on FoxO4-mediated ROS generation.
2. Methods
2.1. Experimental animals and renal I/R model
Adult male C57 mice (20–25 g) were provided by the Experimental Animal Center of the Medical College of Wuhan University (Wuhan, China). This project was approved by the committee of experimental animals of our university, and the procedures were carried out in accordance with routine animal-care guidelines. All procedures complied with the Guidelines for the Care and Use of Laboratory Animals. Briefly, mice were fully anesthetized with pentobarbital sodium (50 mg/kg, i.p.) and placed on a homeothermic table to maintain core body temperature at 37 °C. All the mice then underwent a midline laparotomy followed by a right nephrectomy. Next, the left kidney was subjected to 30 min of ischemia with a non-traumatic vascular clamp followed by different periods of reperfusion.
All mice were randomly divided into different treatment groups (n = 8 each). In the Sham group, only the right kidneys were removed. In the Sham + JQ1 (Selleck, S7110) group, surgical procedure was the same as that of the Sham group, but the mice were treated with JQ1 (25 mg/kg, 50 mg/kg, and 100 mg/kg) by intraperitoneal injection for 7 consecutive days before surgical operation. In I/R group, the left kidney vessels were occluded with a clamp for 30 min; reperfusion was then performed for different time points by unclamping. In the I/R+JQ1 group, surgical procedure was the same as that of the Sham group, and mice were treated with JQ1 (25 mg/kg, 50 mg/kg, and 100 mg/kg) by intraperitoneal injection for 7 consecutive days before the I/R model established. The vehicle control rats (n = 8) were administered an equal amount of DMSO in the carrier solution.
2.2. Cell culture and cell hypoxia/reoxygenation (H/R) model
The human renal proximal tubular epithelial cell line (HK-2) was purchased from American Type Culture Collection (ATCC, USA) and cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, USA) with nonessential amino acids, 0.05 mg/mL bovine pituitary extract, 50 ng/mL human recombinant epidermal growth factor, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum under 5% CO2 and 95% air atmosphere at 37 °C. A cell H/R model was established according to the previously described methods [20]. Briefly, HK-2 cells were cultured for 12 h under hypoxic conditions (1% O2, 94% N2, and 5% CO2) in medium without nutrients (glucose-free, serum-free) to induce hypoxic injury. Then, the medium was refreshed again, and the plates were moved to a normoxic cell incubator (5% CO2 and 95% air) for 2 h, 4 h, and 6 h. Control cells were incubated in complete culture medium in a regular incubator (5% CO2 and 95% air).
2.3. Quantitative real-time PCR
Total RNA was isolated from HK-2 cells or frozen kidney tissues using RNAiso Plus (TaKaRa Biotech, Dalian, China) per the manufacturer's instructions and subjected to reverse transcription into cDNA with a PrimeScript™ RT Reagent Kit (TaKaRa Biotech, Dalian, China). Quantitative real-time PCR analysis was performed using an ABIViiA7DX System (Foster City, CA, USA). GAPDH expression was used in all experiments. The RT-PCR primers designed for specific target genes are listed in Supplemental Table I and were synthesized by TaKaRa Biotech. The PCR reactions were performed using the following cycling conditions: 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s, 60 °C for 30 s, and 72 °C for 20 s.
2.4. Western blot analysis
HK-2 cells and renal tissues were lysed with RIPA buffer containing protease inhibitors (Beyotime, Jiangsu, China) to obtain the total proteins. The protein samples were separated on SDS polyacrylamide gels and transferred to PVDF membranes, and the membranes were blocked with 5% fat-free milk and immunoblotted with primary antibodies. The primary antibodies were used at the following dilutions: Brd4 (1:1000, Abcam, Ab128874); Cleaved Caspase-3 (1:1000, Cell Signaling, 9664); Bax (1:200, Cell Signaling, 2772); Bcl-2 (1:1000, Abcam, Ab196495); CHOP (1:1000, Cell Signaling, 2895); GRP78 (1:1000, Abcam, Ab21685); p-AKT (1:200, Cell Signaling, #4060p); AKT (1:1000, Cell Signaling, 4961); p-PI3K (1:1000, Cell Signaling, 4228); PI3K (1:1000, Cell Signaling, 4257); FoxO4 (1:1000, Abcam, Ab128908); and GAPDH (1:1000, Hangzhou Goodhere Biotechnology Co., Ltd., AB-P-R 001). After incubation with an appropriate secondary antibody, the western blots were visualized using the Chemiluminescent HRP Substrate (Millipore, Billerica, MA, USA). The data analysis was performed using Image J Software (NIH, USA) to quantify the protein levels.
2.5. Histological staining and immunohistochemistry
Kidneys were first fixed and embedded, and then used to prepare 4-μm-thick sections. The sections were then gradually deparaffinized and hydrated and stained with hematoxylin and eosin (H & E) staining. Morphological assessments were observed by two experienced renal pathologists who were blinded to the treatments. An established grading scale of 0–4, outlined by Jablonski et al., was used for the histopathological assessment of I/R-induced damage.
Immunohistochemistry was performed using a Polink-1 one-step polymer detection system (ZSGB-BIO, Beijing, China). Briefly, the kidney sections were stained with anti-caspase-3 (1:200, Abcam, Ab13847) and anti-Brd4 (1:200, Abcam, Ab12887); followed by incubation with secondary antibodies, and then detected with the EnVision/HRP Kit (Dako, Denmark). For quantitation, the relative mean integrated optical density (IOD) of each group was divided by the average IOD of the control, as determined by Image-Pro Plus software (version 6.0). All sections were photographed at a magnification of 400×.
2.6. Serum assays
Assays were carried out using commercial kits and 2 mL of blood. All kits were used in accordance with the manufacturer's instructions (Nanjing Jiancheng Co., China). Serum levels of blood urea nitrogen (BUN) and creatinine (Cr) were calculated using spectrophotometric measurements.
2.7. Flow cytometry
Cell apoptosis were assessed by flow cytometry using the Annexin V-APC apoptosis analysis kit (Sungene Biotech Co., Ltd., # AO2001-11A-G, China) according to the manufacturer's instructions. Briefly, HK-2 cells were washed twice with PBS and stained with 50 μL binding buffers containing 5 μL 7-ADD in the dark and room temperature conditions for 15 min. Then, 450 μL binding buffers and 1 μL Annexin V-APC were added into the above solution for staining under the same conditions. The apoptotic cells were detected with a FACS flow cytometer (BD, Germany).
2.8. Cell viability
Cell viability was assessed using a CCK-8 assay (Beyotime Biotechnology, #C0037) according to the manufacturer's instructions. Briefly, HK-2 cells were seeded into 96-well plates at a density of 5 × 103 cells/well in 96-well plates. After overnight culture, the cells were incubated with different doses of JQ1 (0, 0.1, 1, 10 μM) for 24 h before establishment of the H/R model. After the H/R process, cells were incubated with CCK-8 reagent. Then, cell viability was evaluated by absorbance measurements using a microplate reader (Molecular Devices, USA) at 450 nm.
2.9. Superoxide dismutase (SOD) and malondialdehyde (MDA) measurement
Commercial kits were used in accordance with the manufacturer's instructions (Nanjing Jiancheng Co., China) to measure SOD activity (xanthine oxidase method, catalog#: A001-3) and MDA concentration (thiobarbituric acid method, catalog#: A003-1).
2.10. Terminal deoxynucleotidyl transferase duTP conjugated with fluorescein (TUNEL) assay
Apoptotic cells in kidney tissues were evaluated by TUNEL assay using an In Situ Cell Death Detection Kit, POD (Roche, Germany) according to the manufacturer's instruction, as previously described [21]. The average number of TUNEL-positive cells was calculated in 10 random fields.
2.11. Small interfering RNA (siRNA) transfection
HK-2 cells were transfected with siRNAs specific to Brd4 or FoxO4 or with non-targeting siRNAs (Santa Cruz, CA, USA) as a negative control for 48 h using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA). All siRNAs were used at a concentration of 100 nM. After 6 h of transfection, the cells were incubated in DMEM/F12 containing 0.2% FBS for 48 h. Western blotting and RT-PCR were conducted to confirm the effects of siRNA transfection.
2.12. Adenoviral infection
HK-2 cells at 70–80% confluence were infected with adenovirus to overexpress human FoxO4 at an MOI of 50 in DMEM without serum or antibiotics for 6 h before switching to DMEM/F12 containing 10% FBS for 72 h.
2.13. Luciferase reporter assays
The FoxO4 promoter reporter vector was designed and synthesized by Sangon Biotech (Shanghai, China). The reporter construct was transiently transfected along with a Renilla control plasmid and either Brd4 siRNA or non-targeting siRNAs using Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer's instructions. At 6 h after transfection, the culture medium was replaced with DMEM/F12 supplemented with 0.2% FBS. After 48 h of transfection, HK-2 cells were subjected to H/R. The luciferase activity was detected using a dual-luciferase reporter assay system (Promega, Madison, WI, USA). Three independent experiments were performed, with six replicates for each condition.
2.14. Measurement of ROS production
Intracellular ROS levels were determined using Reactive Oxygen Species Assay Kit (Beyotime Biotechnology, #S0063) according to the instruction. Briefly, cells pre-treated with different reagents were incubated with 20 μM dichlorodihydrofluorescein diacetate (DCFH-DA) in Hanks' balanced salt buffer for 30 min at 37 °C. The ROS level was quantified using a FACS flow cytometer (Becton Dickinson, USA).
2.15. Amplex Red assay for H2O2 production
H2O2 production was assessed by Amplex Red assay using the established techniques [22]. The HK-2 cells were pretreated with JQ1, Brd4 siRNA, or FOXO4 siRNA and then treated with or without H/R. The H2O2 levels in kidney tissue were detected as previously described [23]. The H2O2 in the homogenate was measured according to the manufacturer's suggestions (Molecular Probes). Fluorescent readings were obtained from the rat kidneys after 1 h of incubation at 37 °C, and the values were normalized to the protein amount as measured by Bradford assay. The Amplex Red reagent is a colorless substrate that reacts with H2O2 with a 1:1 stoichiometry to produce the highly fluorescent resorufin (excitation/emission maxima = 570/585 nm).
2.16. Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). The means of the different groups were compared using one-way analysis of variance (ANOVA) and the Student–Newman–Keuls test. Differences were considered statistically significant when P < 0.05.
3. Results
3.1. Up-regulation of Brd4 in the progression of renal I/R injury
We first detected the expression of Brd4 in renal tissues. As shown in Fig. 1A and B, both the mRNA and protein levels of Brd4 were increased in the renal I/R group when compared with the Sham group. Moreover, the expression of Brd4 was markedly enhanced as reperfusion time increased (6 h vs. 12 h vs. 24 h). To determine the localization of Brd4 protein, immunohistochemical analysis was performed to detect its localization. Brd4-positive staining was easily observed in the I/R group, and the density was enhanced along with the increase in reperfusion time (Fig. 1C and D). These results implied that Brd4 might be involved in the development of renal I/R.
Fig. 1.

Brd4 was up-regulated in the kidney after mice were suffered from IRI. (A) Brd4 protein levels were detected by western blotting analysis at various reperfusion time points, such as 6 h, 12 h, and 24 h, and bar graph showing the fold changes of Brd4 relative to sham group from three independent samples. (B) Brd4 mRNA levels were detected by real-time RT-PCR at 6 h, 12 h, 24 h of reperfusion time. (C–D) Bar graph showing the fold changes of Brd4 positive areas relative to sham group, and immunohistochemical staining of Brd4 in renal tissues at 6 h, 12 h, 24 h of reperfusion time. Values are expressed as the mean ± SEM. *P < 0.05, relative to the sham group, n = 3.
3.2. Brd4 inhibitor JQ1 attenuated renal I/R injury
JQ1, a Brd4 inhibitor, was employed to investigate whether Brd4 inhibition attenuated renal I/R. First, mice were subjected to Sham operation with various doses of JQ1 (25 mg/kg, 50 mg/kg, 100 mg/kg) to avoid the nephrotoxicity of JQ1 itself. As shown in the Supplementary Figs. S1A and 1B, as the dose of JQ1 reached 100 mg/kg, no obvious nephrotoxicity was found, which was evidenced by no significant differences in Cr and BUN levels, as well as morphological changes, when compared with the Sham group.
In addition, mice pretreated with different doses of JQ1 showed decreased levels of Brd4, with more obvious inhibitory effects on Brd4 expression observed at a concentration of 100 mg/kg (Fig. 2A). Moreover, mice subjected to I/R showed obvious renal dysfunction when compared with the Sham group, and mice treated with JQ1 showed significantly improved renal function, with more obvious effects observed at a concentration of 100 mg/kg (Fig. 2B and C). Renal tissue morphology is also shown in Fig. 2D and E. Renal tissues were normal in the Sham group. However, kidneys in the I/R group exhibited acute tubular damage in the proximal tubules, including tubular dilatation and loss of the brush border. JQ1 protected the tubular epithelium from swelling and from loss of the brush border, with more protective effects observed at the concentration of 100 mg/kg. Taken together, these results indicated that JQ1 treatment could attenuate renal I/R and reduce the expression of Brd4 in a dose-dependent manner. Accordingly, we chose the dose of 100 mg/kg to carry out all following experiments.
Fig. 2.

JQ1 treatment protected kidney against I/R. (A) Western blots of Brd4 protein expression after treatment of mice with JQ1 at doses of 25 mg/kg, 50 mg/kg, 100 mg/kg, and bar graph showing the fold changes of Brd4 relative to sham group from three independent samples. (B–C) Protective effect of JQ1 at doses of 25 mg/kg, 50 mg/kg, 100 mg/kg on renal function of mice exposed to renal IRI. (D–E) Protective effect of JQ1 at various doses of 25 mg/kg, 50 mg/kg, 100 mg/kg on renal tissue damage detected by H&E (×400), and randomly selected image fields from eight independent kidney samples were used for quantification renal tubular injury scores. Values are expressed as the mean ± SEM. *P < 0.05, relative to sham group; **P < 0.05, relative to vehicle control; #P < 0.05, relative to I/R+JQ1 (25 mg/kg); & P < 0.05, relative to I/R+JQ1 (50 mg/kg).
3.3. Inhibition of Brd4 alleviated apoptosis and ER stress in vivo
To investigate whether JQ1 had protective effects on apoptosis of kidney tissues, TUNEL staining was performed. As shown in Fig. 3A, renal I/R resulted in more TUNEL-positive cells than the Sham group, which was partially abolished by JQ1 treatment. Western blot also indicated that I/R enhanced the expression of Bax and caspase-3 and reduced Bcl-2 expression compared with the Sham group, and mice pretreated with JQ1 showed obvious reduction of Bax and caspase-3 expression and enhancement of Bcl-2 expression compared with the I/R group (Fig. 3B). In addition, the results of Caspase-3 detected by immunohistochemistry were consistent with that of western blotting (Fig. 3C). Next, we examined whether JQ1 regulated ERS on mice subjected to I/R. The expression of ERS-associated proteins in the I/R group, such as GRP78 and CHOP, were obviously up-regulated (Fig. 4D). Furthermore, it was found that mice pretreated with JQ1 had lower expression of GRP78 and CHOP than the I/R group. Taken together, these results suggest that JQ1 could alleviate I/R-induced apoptosis and ERS.
Fig. 3.

JQ1 inhibited apoptosis and ERS induced by renal IRI in mice. (A) Representative images of TUNEL staining on kidney sections (×400). (B) Western blots of Bax,Bcl-2, Caspse-3 at 24 h of reperfusion time, and bar graph showing the fold changes of Bax,Bcl-2, Caspse-3 relative to sham group from three independent samples. (C) Immunohistochemical staining of Caspse-3 in renal tissues at 24 h of reperfusion time. (D) Western blots of GRP78, CHOP at 24 h of reperfusion time, and bar graph showing the fold changes of GRP78, CHOP. Values are expressed as the mean ± SEM. *P < 0.05, relative to sham group; #P < 0.05, relative to vehicle control.
Fig. 4.

Oxidative mediated apoptosis and ERS in a H/R cell model. HK-2 cells were pretreated with 5 mM N-acetyl-cysteine (NAC) for 1 h, and then subjected to H/R. (A) Brd4 protein levels were detected by western blot analysis at various reoxygenation time, including 2 h, 4 h, and 6 h, and bar graph showing the fold changes of Brd4 relative to sham group from three independent samples. (B–C) ROS and H2O2 production were measured and bar graph quantification of ROS accumulation from three independent experiments. (D) Apoptotic rates of normal cells and cells exposed to H/R were detected and bar graphs represent three independent experiments, each performed in triplicates. (E) Western blot analysis for the protein expression of Bax, BCL-2, caspase-3, and protein levels were quantified by densitometry and normalized to the expression of GAPDH from three independent experiments. (F) Western blot analysis for the protein expression of GRP78, CHOP, and protein levels were quantified by densitometry and normalized to the expression of GAPDH from three independent experiments. Values are expressed as the mean ± SEM. *P < 0.05, relative to control group; #P < 0.05, relative to the H/R+DMSO.
3.4. H/R-induced apoptosis and ERS depends on oxidative stress in vitro
First, we explored whether the time of reoxygenation affected cell viability and the expression level of Brd4. As shown in Fig. 4A and Supplementary Fig. S2A, cell viability was reduced and Brd4 levels were up-regulated when cells were exposed to H/R. Moreover, the prolonged reoxygenation time (2 h, 4 h, 6 h) further reduced cell viability and enhanced the expression of Brd4. Therefore, we choose 6 h as the reoxygenation time to perform the following experiments.
ROS is a mediator in the development of renal I/R injury. In this experiment, N-acetyl-cysteine (NAC), a highly efficient ROS inhibitor, was used to investigate the role of oxidative stress in H/R-induced apoptosis and ERS. The total ROS detected with the fluorescent dye DCFH-DA showed that H/R caused ROS accumulation in the HK-2 cells (Fig. 4B and Supplementary Fig. S2B). Hydrogen peroxide, one type of ROS, was markedly increased in HK-2 cells subjected to H/R process (Fig. 4C). Simultaneously, the results of flow cytometry showed that H/R significantly resulted in cell apoptosis (Fig. 4D and Supplementary Fig. S2C). Concomitantly, the expression of apoptotic and ERS markers such as Bax, Caspase-3, GRP78, and CHOP was significantly up-regulated after H/R (Fig. 4E and F). Further, the ablation of ROS with NAC inhibited cellular apoptosis (Fig. 4D and Supplementary Fig. S2C) and decreased H/R-induced proapoptotic and ERS protein expression (Fig. 4E and E). Therefore, H/R induced renal apoptosis and ERS through oxidative stress.
3.5. Brd4 inhibition alleviated H/R-induced oxidative stress, apoptosis, and ERS in vitro
CCK8 was used to determine the optimal drug concentration of JQ1. As shown in the Supplementary Fig. S3A, the concentration of 10 μM JQ1 showed no obvious toxicity on HK-2 cell under normoxic conditions. We next treated HK-2 cells with various concentrations of JQ1 before being subjected to H/R. The results showed that 10 μM JQ1 showed the most optimal protective effect on cells against H/R (Fig. 5A). In addition, cells exposed to H/R indicated that Brd4 was markedly up-regulated, and JQ1 resulted in a reduction of Brd4 expression in a dose-dependent manner, with more obvious inhibitory effects observed at the concentration of 10 μM (Fig. 5B). Therefore, we chose the concentration of 10 μM to perform the following experiments.
Fig. 5.

JQ1 alleviated oxidative stress, apoptosis and ERS in vitro. (A–B) HK-2 cells were pretreated with or without JQ1 at different doses (0.1, 1, 10 μM) for 1 h, and then were exposed to H/R. (A) Effect of JQ1 with different concentrations on cell viability detected by CCK8 upon H/R stimulation in the indicated groups, and (B) representative bands of Western blot analysis for the expression of Brd4 and bar graph quantification as indicated from three independent experiments, Values are expressed as the mean ± SEM; *P < 0.05, relative to control; **P < 0.05, relative to H/R+DMSO; #P < 0.05, relative to H/R+JQ1 (0.1 μM); & P < 0.05, relative to H/R+JQ1 (1 μM). (C–G) HK-2 cells were transfected with a siRNA against Brd4 or a negative control siRNA (si-NC) for 48 h and were pretreated with or without JQ1 at dose of 10 μM for 1 h before being exposed to H/R. (C) Representative bands of Western blot analysis for the expression of Brd4 and bar graph quantification as indicated from three independent experiments. (D) Apoptotic cells exposed to H/R were detected by flow cytometry, and bar graphs represent three independent experiments, each performed in triplicates. (E) Representative Western blot analysis of Bax, Bcl-2, Caspase-3 and bar graphs from three independent experiments, each performed in triplicates. (F) Representative bands of Western blot analysis for the expression of GRP78 and CHOP, and bar graphs from three independent experiments. (G–H) ROS production was measured by flow cytometry and bar graph showed the quantification of ROS in the indicated groups from three independent experiments, and H2O2 production measured by Amplex Red in HK-2 cells, and bar graphs represent three independent experiments, each performed in triplicates. Values are expressed as the mean ± SEM; *P < 0.05, relative to H/R group; #P < 0.05, relative to H/R+si-NC.
In HK-2 cells, consistent with the inhibitory effect of JQ1 on Brd4 expression, siRNA could also reduce its mRNA and protein expression level (Fig. 5C and Supplementary Fig. S3B). Moreover, JQ1 reduced H/R-induced apoptotic and ERS gene expression (Fig. 5E and F) and decreased cellular apoptosis (Fig. 5D and Supplementary Fig. S3C). siRNA was used to genetically inhibit Brd4. Similarly, cellular apoptosis was significantly reduced, and the expression of apoptotic and ERS proteins was markedly suppressed after Brd4 knockdown. Furthermore, we investigated whether Brd4 inhibition could suppress H/R-induced ROS generation. JQ1 significantly decreased H/R-mediated induction of total ROS and hydrogen peroxide (Fig. 5G and H and Supplementary Fig. S3D). Brd4 knockdown also resulted in reduced ROS and hydrogen peroxide generation compared with the control group (Fig. 5G and H Supplementary Fig. S3D). These results indicated that the protective effects of Brd4 inhibition could be attributed to the reduction of oxidative stress, apoptosis, and ERS.
3.6. Brd4 inhibition blocked FoxO4-mediated oxidative stress, apoptosis, and ERS in HK-2 cells
The FoxO family includes FoxO1, FoxO3a, FoxO4, and FoxO6, and FoxO6 expression is restricted to the central nervous system. The result showed that HK-2 cells exposed to H/R hardly affected FoxO1 and FoxO3a levels (Supplementary Fig. S4A). H/R-induced FoxO4 expression in HK-2 cells and si-RNA against FoxO4 obviously decreased its protein and mRNA levels (Fig. 6A and Supplementary Fig. S4B). We also found lower levels of the apoptotic and ERS proteins after FoxO4 knockdown (Fig. 6D and E) as well as reduced hydrogen peroxide generation and total ROS (Fig. 6B and C and Supplementary Fig. S4C). These data demonstrated the critical role of FoxO4 in H/R-induced oxidative stress, apoptosis, and ERS. Next, we examined the effects of Brd4 inhibition on FoxO4 expression. As shown in Fig. 6F and H, JQ1 blocked H/R-induced FoxO4 expression at both the mRNA and protein levels. Likewise, Brd4 knockdown resulted in reduced FoxO4 expression (Fig. 6F and H). To further investigate whether Brd4 regulates oxidative stress, apoptosis, and ERS through FoxO4, we compensated the JQ1-mediated FoxO4 reduction by delivering an adenovirus carrying human FoxO4 to HK-2 cells. The compensation of FoxO4 blunted the JQ1-induced reduction of apoptosis and ERS protein levels in HK-2 cells subjected to H/R (Fig. 6I and J). Moreover, compensation of FoxO4 also reversed reduction of total ROS and hydrogen peroxide (Fig. 6K and L and Supplementary Fig. S4D). Therefore, these results demonstrated that Brd4 inhibition exerts an anti-oxidative stress, anti-apoptotic and anti-ERS role through the regulation of FoxO4.
Fig. 6.

Brd4 inhibition ameliorated apoptosis and ERS through FoxO4. (A) Representative bands of Western blot analysis for the expression of FoxO4 and bar graphs from three independent experiments. (B–C) ROS production measured by flow cytometry in the indicated groups from three independent experiments and bar graph showing the of quantification ROS, and H2O2 production measured by Amplex Red in HK-2 cells, and bar graphs represent three independent experiments, each performed in triplicates. (D) Western Blot analysis for the protein expression of Bax, Bcl-2, Caspase-3 and bar graph quantification as indicated from three independent experiments. (E) Western Blot analysis for the protein expression of GRP78 and CHOP, and bar graph quantification as indicated from three independent experiments. Values are expressed as the mean ± SEM; *P < 0.05, relative to control; #P < 0.05, relative to H/R+si-NC. (F) Western Blot analysis for the protein expression of FoxO4, and bar graphs represent three independent experiments. (G) FoxO4 mRNA levels were detected by real-time RT-PCR and bar graph quantification as indicated from three independent experiments, *P < 0.05, relative to H/R; #P < 0.05, relative to H/R+si-NC. (H–L) HK-2 cells were pretreated with JQ1 (10 μm) for 1 h before H/R model established, and HK-2 cells were infected with adenovirus carrying the human FoxO4 for 48 h before being exposed to H/R. (H) Representative Western blot analysis of FoxO4 in the indicated groups. (I) Representative Western blot analysis of Bax, Bcl-2, Caspase-3 in the indicated groups. (J) Western blot analysis for the protein expression of GRP78 and CHOP in the indicated groups. (K–L) ROS and H2O2 production were measured. Values are expressed as the mean ± SEM; *P < 0.05 versus H/R+JQ1.
3.7. Brd4 regulated FoxO4 expression via the PI3K/AKT pathway
To further explore the underlying mechanisms responsible for the regulation of FoxO4 by Brd4, we examined the possible pathway involved. PI3K/AKT pathways have been reported to play a key role in regulation of FoxO4 [24]. As shown in Fig. 7A and B, the level of p-PI3K and p-AKT was down-regulated after being subjected to H/R, which was partially reversed by JQ1 treatment. Consistently, knockdown Brd4 with si-RNA resulted in obvious enhancement in the level of p-PI3K and p-AKT compared with the negative control (Fig. 7A and B). In addition, combination of JQ1 and PI3K/AKT inhibitor, LY294002, led to an induction in the level of FoxO4 in HK-2 cells upon H/R stimulation (Fig. 7C).
Fig. 7.

Brd4 regulated FoxO4 expression via the PI3K/AKT pathway. (A–B) Western blot analysis for the protein expression of PI3K, p-PI3K, AKT, p-AKT in the indicated groups and quantitative analysis of p-PI3K and p-AKT. *P < 0.05 versus control, #P < 0.05 versus H/R+JQ1, & P < 0.05 versus H/R+si-NC. (C–D) HK-2 cells were pretreated with JQ1 (10 μM) for 1 h following treatment with LY294002 (PI3K inhibitor, 20 μM) and then exposed to H/R. (C) Western blot analysis for the protein expression of FoxO4 in the indicated groups and quantification, *P < 0.05 versus H/R+JQ1. (D) Luciferase assay of FoxO4 promoter activity in the presence of JQ1 or Brd4 knockdown with siRNA or combination of JQ1 and LY294002 from three independent experiments, each performed in six replicates. *P < 0.05 versus H/R, #P < 0.05 versus H/R+si-NC, & P < 0.05 versus H/R+si-Brd4.
Next, we investigated whether Brd4 inhibition significantly decreased FoxO4 promoter activity by luciferase reporter assays. Briefly, HK-2 cells were transfected with a luciferase reporter plasmid containing the human FoxO4 promoter region. The promoter assay proved that either JQ1 or Brd4 siRNA inhibited FoxO4 promoter activity, and combination of Brd4 si-RNA and LY294002 reversed the reduction in the level of FoxO4, collectively indicating that Brd4 regulated FoxO4 through the upstream PI3K/AKT pathway and then transcriptionally decreased the FoxO4 promoter activity (Fig. 7D).
3.8. JQ1 attenuated FoxO4-mediated oxidative stress and enhanced PI3K/AKT pathway in vivo
To recapitulate the in vitro findings, we tested the effects of JQ1 on I/R-induced oxidative stress. JQ1 significantly elevated SOD activity and suppressed MDA content and hydrogen peroxide production in mice subjected to I/R injury (Fig. 8A–C). JQ1 treatment alleviated the expression of FoxO4 induced by I/R injury (Fig. 8B). Additionally, the PI3K/AKT signaling pathway was inactivated after I/R and elevated after JQ1 pretreatment (Fig. 8D and E). Together, these results supported the hypothesis that Brd4 inhibition prevented I/R-induced apoptosis and ERS through activating PI3K/AKT signaling and blocking FoxO4-dependent ROS generation.
Fig. 8.

JQ1 attenuated FoxO4-mediated oxidative stress via the PI3K/AKT pathway in mice. (A–C) MDA, SOD and H2O2 production in mice subjected to I/R with or without JQ1. (D) Western blot analysis for the protein expression of FoxO4 in mice treated with or without JQ1 at 24 h of reperfusion time and quantification. (E–F) Western blot analysis for the protein expression of PI3K, p-PI3K, AKT, p-AKT with or without JQ1 at 24 h of reperfusion time and quantification. *P < 0.05 versus Sham, #P < 0.05 versus I/R.
4. Discussion
This study aimed to provide evidence to whether Brd4 could alleviate renal I/R injury and suggested the possible mechanisms that mediate this effect. We first evaluated the effect of Brd4 using a classical animal model of I/R. The results demonstrated that Brd4 inhibition attenuated I/R-induced apoptotic and ERS gene expression in vivo. Meanwhile, H/R-induced apoptosis and ERS depended on oxidative stress in vitro, and FoxO4-mediated ROS generation was blocked by Brd4 knockdown or JQ1. Further, Brd4 modulated the transcriptional activity of FoxO4, and this was regulated through PI3K/AKT. Therefore, our study showed that Brd4 might be a therapeutic target for renal I/R injury and that JQ1 might be a potential agent for renal ischemic injury.
Recently, a number of BET inhibitors have been developed and administered to patients in clinical trials with certain diseases. Accumulating evidence indicated that Brd4 might be a potential therapeutic target in a wide range of diseases including renal diseases [25]. Renal ischemia-reperfusion injury is a major cause of acute kidney injury (AKI), which often arises from hypovolemic conditions, septic shock, surgery, and transplantation. I/R injury causes structural and functional damage of renal tubules. Studies have indicated that Brd4 is involved in inflammatory disease and fibrosis. It was reported that Brd4 suppression reduced interleukin-1β (IL-1β)–induced inflammatory mediators such as IL-6, suggesting that Brd4 might have therapeutic potential in inflammatory diseases [12]. Another recent study reported that inhibition of Brd4 suppressed unilateral ureteral obstruction-induced renal fibrosis [13]. In this study, we first demonstrated that I/R injury could enhance the expression of Brd4 in vivo. Moreover, treatment with the Brd4 inhibitor, JQ1, resulted in mice kidneys that had undergone I/R injury to display characteristic protection of morphological changes, as well as renal function. Consistent with in vivo findings, our in vitro finding indicated that JQ1 protected HK-2 cells against damage induced by H/R.
Apoptosis, a form of cell death, has been demonstrated to occur after renal I/R [26] and is characterized by the activation of either extrinsic or intrinsic pathways. The endoplasmic reticulum (ER) is characterized as an organelle that participates in the folding of the membrane and secretory proteins [27]. ER and apoptosis are important for cell function and survival. Numerous evidences have indicated that ER stress and apoptosis are an essential step in the pathogenesis of a wide variety of renal diseases, including renal I/R injury [28]. Besides, when cells are stimulated by harmful stimuli, large numbers of ROS are produced and the balance between the oxidation and anti-oxidation system is destroyed, eventually leading to oxidative stress. Exacerbated ROS production leads to AKI through ROS-induced abnormal signal pathway, inflammatory infiltration, cellular dysfunction, and renal cell death [29]. The link between ROS, apoptosis, and ERS has been demonstrated. ROS generation may induce cell death through apoptosis and ER stress. Consistent with previous reports [13], we found that H/R-induced apoptosis and ERS depended on oxidative stress in this study. The results showed that the expression of apoptotic and ERS proteins such as Bax, Caspase-3, GRP78, and CHOP, was significantly up-regulated after H/R. The ablation of ROS with NAC could inhibit H/R-induced pro-apoptotic and ERS proteins' expressions. We further demonstrated that JQ1 dampened H/R-induced ROS levels. Therefore, the renal protective effects of JQ1 might occur through the inhibition of ROS generation induced by H/R injury.
The FoxO family, including FoxO1, FoxO3, and FoxO4, are abundantly expressed in all mammalian tissues involved in a variety of biological processes. FoxO4 is highly expressed in the kidney, muscles, and colorectal tissues [30]. It exerts tumor-suppressive effects in cholangiocarcinoma [31] and gastric cancer development [32]. Nakayoshi et al. found that FoxO4 knockdown prevented early pro-angiogenic cells from oxidative stress-induced apoptosis through downregulation of cleaved-caspase3 [18]. Yu et al. reported that FoxO4 knockdown reduced oxidative stress induced by myocardial I/R injury [33]. Consistent with previous reports, FoxO4 knockdown significantly reduced apoptotic and ERS protein, as well as hydrogen peroxide generation, in HK-2 cells after H/R. Besides, Brd4 inhibition suppressed FoxO4 protein levels and decreased its transcriptional activity and also resulted in a reduction in ROS and H2O2 production. Furthermore, we compensated the JQ1-mediated FoxO4 reduction by delivering an adenovirus carrying human FoxO4 to HK-2 cells. The inhibitory effects of JQ1 on H/R-induced apoptosis, ERS and hydrogen peroxide production were blunted. Therefore, we speculated that Brd4 inhibition attenuates oxidative stress induced by I/R injury via regulating FoxO4 expression.
The phosphoinositide 3-kinase (PI3K)/AKT pathway has been shown to play a critical role in regulating mitogenic signaling, apoptosis, cell proliferation, and survival in different systems [34]. In the kidney, the PI3K/Akt pathway regulates renal repair after I/R injury [35]. A number of studies revealed that upregulation of PI3K/Akt phosphorylation could ameliorate IR-induced renal injury [36]. Therefore, we further explored the underlying mechanisms responsible for the regulation of FoxO4 by Brd4. A previous study reported that downregulation of BRD4 inhibits tumor proliferation via PI3K/AKT pathway [37]. Another study found that PI3K/AKT pathways have been reported to play a key role in the regulation of FoxO4 [24].
Consistent with previous reports, we found that knockdown of Brd4 with si-RNA resulted in enhanced p-PI3K and p-AKT levels. In addition, combination of JQ1 and PI3K/AKT inhibitors, LY294002, led to an induction in the level of FoxO4 in HK-2 cells upon H/R stimulation. Moreover, either JQ1 or Brd4 si-RNA inhibited FoxO4 promoter activity, and combination of Brd4 si-RNA and LY294002 reversed the reduction in the level of FoxO4, collectively indicating that Brd4 regulated FoxO4 through the upstream PI3K/AKT and then transcriptionally decreased FoxO4 promoter activity.
In conclusion, we identified the protective effect of a Brd4 inhibitor during renal I/R injury. Further, we showed that Brd4 inhibition blocked renal apoptotic and ERS protein expression by preventing FoxO4-dependent ROS generation through PI3K/AKT pathway. Overall, these results revealed that Brd4 is a potential therapeutic target in the treatment of renal I/R injury.
Disclosures
None.
Acknowledgments
This study was supported by the Application and Basic Research Project Of Wuhan City (No. 2018060401011321), Wuhan Morning Light Plan of Youth Science and Technology (2017050304010281), Natural Science Foundation of Hubei Province (No. 2016CFB114, 2017CFB181), and Research Project of Wuhan University (No. 2042017kf0097).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101195.
Contributor Information
Zhiyuan Chen, Email: chenzhiyuan163@163.com.
Xiuheng Liu, Email: drliuxh@hotmail.com.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Fig. S1.

(A) Effect of JQ1 at doses of 25 mg/kg, 50 mg/kg, 100 mg/kg on renal fuction of mice exposed to Sham operation. (B) Effect of JQ1 at various doses of 25 mg/kg, 50 mg/kg, 100 mg/kg on renal tissue damage detected by H&E (×400).
Fig. S2.

(A) Effect of different reoxygenation time (2h, 4h, 6h) on cell viability detected by CCK8, *P versus control. (B) ROS production was measured by flow cytometry. (C) Cell apoptosis was evaluated using flow cytometry.
Fig. S3.

(A) Effect of JQ1 at doses of 0.1, 1, 10 μM on cell viability detected by CCK8. (B) Real-time PCR analyses for mRNA expression of Brd4. (C) Cell apoptosis was evaluated using flow cytometry. (D) ROS production was measured by flow cytometry.
Fig. S4.

(A) Western Blot analyses for protein expression of FoxO1 and FoxO3a in indicated groups (n = 3). (B) Real-time PCR analyses for expression of FOXO4. (C) ROS production was measured by flow cytometry. (D) Adenovirus carrying human FoxO4 compensates the reduction of ROS induced by JQ1. Flow cytometry was performed to measure ROS production.
References
- 1.Devarajan P. Update on mechanisms of ischemic acute kidney injury. J. Am. Soc. Nephrol. 2006;17(6):1503–1520. doi: 10.1681/ASN.2006010017. [DOI] [PubMed] [Google Scholar]
- 2.Ikeda M., Prachasilchai W., Burne-Taney M.J., Rabb H., Yokota-Ikeda N. Ischemic acute tubular necrosis models and drug discovery: a focus on cellular inflammation. Drug Discov. Today. 2006;11(7–8):364–370. doi: 10.1016/j.drudis.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 3.Wang L., Chen H., Liu X.H., Chen Z.Y., Weng X.D., Qiu T., Liu L., Zhu H.C. Ozone oxidative preconditioning inhibits renal fibrosis induced by ischemia and reperfusion injury in rats. Exp. Ther. Med. 2014;8(6):1764–1768. doi: 10.3892/etm.2014.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li B., Haridas B., Jackson A.R., Cortado H., Mayne N., Kohnken R., Bolon B., Mchugh K.M., Schwaderer A.L., Spencer J.D., Ching C.B., Hains D.S., Justice S.S., Partida-Sanchez S., Becknell B. Inflammation drives renal scarring in experimental pyelonephritis. Am. J. Physiol. Renal. Physiol. 2017;312(1):F43–F53. doi: 10.1152/ajprenal.00471.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Havasi A., Borkan S.C. Apoptosis and acute kidney injury. Kidney Int. 2011;80(1):29–40. doi: 10.1038/ki.2011.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Owen D.J., Ornaghi P., Yang J.C., Lowe N., Evans P.R., Ballario P., Neuhaus D., Filetici P., Travers A.A. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J. 2000;19(22):6141–6149. doi: 10.1093/emboj/19.22.6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ren C., Zeng L., Zhou M.M. Preparation, biochemical analysis, and structure determination of the bromodomain, an acetyl-lysine binding domain. Methods Enzymol. 2016;573:321–343. doi: 10.1016/bs.mie.2016.01.018. [DOI] [PubMed] [Google Scholar]
- 8.Mele D.A., Salmeron A., Ghosh S., Huang H.R., Bryant B.M., Lora J.M. BET bromodomain inhibition suppresses TH17-mediated pathology. J. Exp. Med. 2013;210(11):2181–2190. doi: 10.1084/jem.20130376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asangani I.A., Dommeti V.L., Wang X., Malik R., Cieslik M., Yang R., Escara-Wilke J., Wilder-Romans K., Dhanireddy S., Engelke C., Iyer M.K., Jing X., Wu Y.M., Cao X., Qin Z.S., Wang S., Feng F.Y., Chinnaiyan A.M. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510(7504):278–282. doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia P.L., Miller A.L., Kreitzburg K.M., Council L.N., Gamblin T.L., Christein J.D., Heslin M.J., Arnoletti J.P., Richardson J.H., Chen D., Hanna C.A., Cramer S.L., Yang E.S., Qi J., Bradner J.E., Yoon K.J. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene. 2016;35(7):833–845. doi: 10.1038/onc.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W.B., Fedorov O., Morse E.M., Keates T., Hickman T.T., Felletar I., Philpott M., Munro S., Mckeown M.R., Wang Y., Christie A.L., West N., Cameron M.J., Schwartz B., Heightman T.D., La Thangue N., French C.A., Wiest O., Kung A.L., Knapp S., Bradner J.E. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khan Y.M., Kirkham P., Barnes P.J., Adcock I.M. Brd4 is essential for IL-1 beta-induced inflammation in human airway epithelial cells. PLoS One. 2014;9(4) doi: 10.1371/journal.pone.0095051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou B., Mu J., Gong Y., Lu C., Zhao Y., He T., Qin Z. Brd4 inhibition attenuates unilateral ureteral obstruction-induced fibrosis by blocking TGF-beta-mediated Nox4 expression. Redox. Biol. 2017;11:390–402. doi: 10.1016/j.redox.2016.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi X., Liu C., Liu B., Chen J., Wu X., Gong W. JQ1: a novel potential therapeutic target. Pharmazie. 2018;73(9):491–493. doi: 10.1691/ph.2018.8480. [DOI] [PubMed] [Google Scholar]
- 15.Eijkelenboom A., Burgering B.M. FOXOs: signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013;14(2):83–97. doi: 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- 16.Chuang P.Y., Dai Y., Liu R., He H., Kretzler M., Jim B., Cohen C.D., He J.C. Alteration of forkhead box O (foxo4) acetylation mediates apoptosis of podocytes in diabetes mellitus. PLoS One. 2011;6(8) doi: 10.1371/journal.pone.0023566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L., Dong L., Liu X., Jiang Y., Zhang L., Zhang X., Li X., Zhang Y. alpha-Melanocyte-stimulating hormone protects retinal vascular endothelial cells from oxidative stress and apoptosis in a rat model of diabetes. PLoS One. 2014;9(4) doi: 10.1371/journal.pone.0093433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakayoshi T., Sasaki K., Kajimoto H., Koiwaya H., Ohtsuka M., Ueno T., Chibana H., Itaya N., Sasaki M., Yokoyama S., Fukumoto Y., Imaizumi T. FOXO4-knockdown suppresses oxidative stress-induced apoptosis of early pro-angiogenic cells and augments their neovascularization capacities in ischemic limbs. PLoS One. 2014;9(3) doi: 10.1371/journal.pone.0092626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang H., Tindall D.J. Dynamic FoxO transcription factors. J. Cell Sci. 2007;120(Pt 15):2479–2487. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- 20.Tang T.T., Lv L.L., Pan M.M., Wen Y., Wang B., Li Z.L., Wu M., Wang F.M., Crowley S.D., Liu B.C. Hydroxychloroquine attenuates renal ischemia/reperfusion injury by inhibiting cathepsin mediated NLRP3 inflammasome activation. Cell Death Dis. 2018;9(3):351. doi: 10.1038/s41419-018-0378-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Q., Gong X., Kuang G., Jiang R., Xie T., Tie H., Chen X., Li K., Wan J., Wang B. Ferulic acid protected from kidney ischemia reperfusion injury in mice: possible mechanism through increasing adenosine generation via HIF-1 alpha. Inflammation. 2018;41(6):2068–2078. doi: 10.1007/s10753-018-0850-3. [DOI] [PubMed] [Google Scholar]
- 22.Tong X., Khandelwal A.R., Qin Z., Wu X., Chen L., Ago T., Sadoshima J., Cohen R.A. Role of smooth muscle Nox4-based NADPH oxidase in neointimal hyperplasia. J. Mol. Cell. Cardiol. 2015;89(Pt B):185–194. doi: 10.1016/j.yjmcc.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 23.Babelova A., Avaniadi D., Jung O., Fork C., Beckmann J., Kosowski J., Weissmann N., Anilkumar N., Shah A.M., Schaefer L., Schroder K., Brandes R.P. Role of Nox4 in murine models of kidney disease. Free Radic. Biol. Med. 2012;53(4):842–853. doi: 10.1016/j.freeradbiomed.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 24.Liou A.T., Chen M.F., Yang C.W. Curcumin induces p53-null hepatoma cell line Hep3B apoptosis through the AKT-PTEN-FOXO4 pathway. Evid. Based Complement Alternat. Med. 2017;2017:4063865. doi: 10.1155/2017/4063865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sahai V., Redig A.J., Collier K.A., Eckerdt F.D., Munshi H.G. Targeting BET bromodomain proteins in solid tumors. Oncotarget. 2016;7(33):53997–54009. doi: 10.18632/oncotarget.9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolfs T.G., de Vries B., Walter S.J., Peutz-Kootstra C.J., van Heurn L.W., Oosterhof G.O., Buurman W.A. Apoptotic cell death is initiated during normothermic ischemia in human kidneys. Am. J. Transplant. 2005;5(1):68–75. doi: 10.1111/j.1600-6143.2004.00657.x. [DOI] [PubMed] [Google Scholar]
- 27.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012;13(2):89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 28.Gu Y., Huang F., Wang Y., Chen C., Wu S., Zhou S., Hei Z., Yuan D. Connexin32 plays a crucial role in ROS-mediated endoplasmic reticulum stress apoptosis signaling pathway in ischemia reperfusion-induced acute kidney injury. J. Transl. Med. 2018;16(1):117. doi: 10.1186/s12967-018-1493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chien C.T., Lee P.H., Chen C.F., Ma M.C., Lai M.K., Hsu S.M. De novo demonstration and co-localization of free-radical production and apoptosis formation in rat kidney subjected to ischemia/reperfusion. J. Am. Soc. Nephrol. 2001;12(5):973–982. doi: 10.1681/ASN.V125973. [DOI] [PubMed] [Google Scholar]
- 30.Li H., Liang J., Castrillon D.H., Depinho R.A., Olson E.N., Liu Z.P. FoxO4 regulates tumor necrosis factor alpha-directed smooth muscle cell migration by activating matrix metalloproteinase 9 gene transcription. Mol. Cell Biol. 2007;27(7):2676–2686. doi: 10.1128/MCB.01748-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee M.J., Yu G.R., Yoo H.J., Kim J.H., Yoon B.I., Choi Y.K., Kim D.G. ANXA8 down-regulation by EGF-FOXO4 signaling is involved in cell scattering and tumor metastasis of cholangiocarcinoma. Gastroenterology. 2009;137(3):1138–1150. doi: 10.1053/j.gastro.2009.04.015. 1150-1151. [DOI] [PubMed] [Google Scholar]
- 32.Su L., Liu X., Chai N., Lv L., Wang R., Li X., Nie Y., Shi Y., Fan D. The transcription factor FOXO4 is down-regulated and inhibits tumor proliferation and metastasis in gastric cancer. BMC Canc. 2014;14:378. doi: 10.1186/1471-2407-14-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu L., Zhang W., Huang C., Liang Q., Bao H., Gong Z., Xu M., Wang Z., Wen M., Cheng X. FoxO4 promotes myocardial ischemia-reperfusion injury: the role of oxidative stress-induced apoptosis. Am. J. Transl. Res. 2018;10(9):2890–2900. [PMC free article] [PubMed] [Google Scholar]
- 34.Kandel E.S., Hay N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp. Cell Res. 1999;253(1):210–229. doi: 10.1006/excr.1999.4690. [DOI] [PubMed] [Google Scholar]
- 35.Liu H.B., Meng Q.H., Huang C., Wang J.B., Liu X.W. Nephroprotective effects of polydatin against ischemia/reperfusion injury: a role for the PI3K/akt signal pathway. Oxid. Med. Cell Longev. 2015;2015:362158. doi: 10.1155/2015/362158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu H., Ye M., Yang J., Ding J., Yang J., Dong W., Wang X. Nicorandil protects the heart from ischemia/reperfusion injury by attenuating endoplasmic reticulum response-induced apoptosis through PI3K/akt signaling pathway. Cell. Physiol. Biochem. 2015;35(6):2320–2332. doi: 10.1159/000374035. [DOI] [PubMed] [Google Scholar]
- 37.Hao J., Yang Z., Wang L., Zhang Y., Shu Y., Jiang L., Hu Y., Lv W., Dong P., Liu Y. Downregulation of BRD4 inhibits gallbladder cancer proliferation and metastasis and induces apoptosis via PI3K/AKT pathway. Int. J. Oncol. 2017;51(3):823–831. doi: 10.3892/ijo.2017.4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.