

Alphaherpesviruses are ubiquitous DNA viruses and include the human pathogens herpes simplex virus 1 (HSV-1) and HSV-2 and are significant causes of ulcerative mucosal sores, infectious blindness, encephalitis, and devastating neonatal disease. Successful primary infection and persistent coexistence with host immune defenses are dependent on the ability of these viruses to counter the antiviral response. HSV-1 and HSV-2 and other primate viruses within the Simplexvirus genus encode US11, an immune antagonist that promotes virus production by preventing shutdown of protein translation. Here we investigated the impact of US11 deletion on HSV-1 growth in vitro and pathogenesis in vivo. This work supports a role for US11 in pathogenesis and emergence from latency, elucidating immunomodulation by this medically important cohort of viruses.
KEYWORDS: herpes simplex virus, innate immunity, pathogenesis
ABSTRACT
Herpes simplex virus 1 (HSV-1) cycles between phases of latency in sensory neurons and replication in mucosal sites. HSV-1 encodes two key proteins that antagonize the shutdown of host translation, US11 through preventing PKR activation and ICP34.5 through mediating dephosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α). While profound attenuation of ICP34.5 deletion mutants has been repeatedly demonstrated, a role for US11 in HSV-1 pathogenesis remains unclear. We therefore generated an HSV-1 strain 17 US11-null virus and examined its properties in vitro and in vivo. In U373 glioblastoma cells, US11 cooperated with ICP34.5 to prevent eIF2α phosphorylation late in infection. However, the effect was muted in human corneal epithelial cells (HCLEs), which did not accumulate phosphorylated eIF2α unless both US11 and ICP34.5 were absent. Low levels of phosphorylated eIF2α correlated with continued protein synthesis and with the ability of virus lacking US11 to overcome antiviral immunity in HCLE and U373 cells. Neurovirulence following intracerebral inoculation of mice was not affected by the deletion of US11. In contrast, the time to endpoint criteria following corneal infection was greater for the US11-null virus than for the wild-type virus. Replication in trigeminal ganglia and periocular tissue was promoted by US11, as was periocular disease. The establishment of latency and the frequency of virus reactivation from trigeminal ganglia were unaffected by US11 deletion, although emergence of the US11-null virus occurred with slowed kinetics. Considered together, the data indicate that US11 facilitates the countering of antiviral response of infected cells and promotes the efficient emergence of virus following reactivation.
IMPORTANCE Alphaherpesviruses are ubiquitous DNA viruses and include the human pathogens herpes simplex virus 1 (HSV-1) and HSV-2 and are significant causes of ulcerative mucosal sores, infectious blindness, encephalitis, and devastating neonatal disease. Successful primary infection and persistent coexistence with host immune defenses are dependent on the ability of these viruses to counter the antiviral response. HSV-1 and HSV-2 and other primate viruses within the Simplexvirus genus encode US11, an immune antagonist that promotes virus production by preventing shutdown of protein translation. Here we investigated the impact of US11 deletion on HSV-1 growth in vitro and pathogenesis in vivo. This work supports a role for US11 in pathogenesis and emergence from latency, elucidating immunomodulation by this medically important cohort of viruses.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) and HSV-2 are highly prevalent neurotropic viruses that coexist lifelong with their host (1). During primary infection, HSV-1 replicates in mucosal epithelia in a lytic cycle limited initially by cell-intrinsic and innate immunity. Progeny virions that access underlying sensory neuron axon termini are trafficked in the retrograde direction to the neuronal soma within sensory ganglia (2). Once in the nucleus, the viral genome is driven into latency largely by epigenetic modifications (3). Cellular stresses sporadically trigger reactivation from latency (4), yielding new virions that are transported in the anterograde direction toward axonal termini at the mucosal surface (2). There, lytic infection and lesion development may occur followed by viral dissemination to a new host. A balance between antiviral immunity and viral countermeasures has evolved to ensure that the viral niche and burden are constrained, thus averting neuronal destruction, encephalitis, and death, outcomes that result in a nontransmissible dead end for the virus.
Cellular pattern recognition receptor (PRR) pathways galvanize an antiviral state that curtails viral spread, and, accordingly, alphaherpesviruses encode antagonists to several of these pathways (reviewed in references 5 and 6). Within the alphaherpesviruses, US11 is unique to the primate herpesviruses of the Simplexvirus genus (7). US11 is a γ2 (true late) gene, and US11 protein is expressed late during HSV infection (8). The best-understood function of US11 is the obstruction of pathways that lead to translational arrest, and it does this at three nodes involving PRR sentinels for double-stranded RNA (dsRNA). The first node centers on the dsRNA-activated protein kinase PKR. Upon binding of PKR to dsRNA, or to its dsRNA-bound activator PACT, PKR autoactivates and then phosphorylates its target, the α subunit of eukaryotic initiation factor 2 (eIF2α) (9, 10). eIF2α phosphorylation inactivates the eIF complex, arresting protein translation and promoting autophagy (11, 12). Although herpesviruses are DNA viruses, dsRNA generated during HSV-1 transcription accumulates to small amounts in the cytosol from mid-late infection (13). US11 has a high affinity for dsRNA (14) and PACT (15) and sequesters both from PKR (16). By limiting activation of PKR, US11 indirectly prevents eIF2α phosphorylation, thus maintaining protein translation late in infection (17). In addition to US11, HSV-1 and HSV-2 encode ICP34.5, with which US11 collaborates in modulating eIF2α activity. In contrast and in complement to US11, ICP34.5 acts midway through infection and directly regulates eIF2α through corecruitment of eIF2α and its phosphatase PP1α (18). HSV-1 mutants lacking ICP34.5 are highly attenuated in vivo and in vitro (19–21). However, attenuation is partially rescued by aberrant, early expression of US11, which suffices to inhibit eIF2α phosphorylation and support protein synthesis despite the lack of ICP34.5 (22–26). Sequential roles for ICP34.5 and US11 in the translational arrest pathway are now recognized, with both ICP34.5 and US11 required for resistance of HSV-1 to type I interferon (IFN) (27, 28).
The second node US11 intersects in translational arrest pathways are those of MDA5 and RIG-I, PRRs that bind dsRNA differentially based on oligoduplex length (29). When bound to dsRNA, MDA5 and RIG-I activate TBK1 through MAVS (30). TBK1 phosphorylates the transcription factor IRF3, stimulating transcription of type I interferon genes (31, 32). Interferons, in turn, act in infected and bystander cells to stimulate transcription of the interferon-stimulated genes (ISGs), whose products amplify the antiviral response (33). By scavenging dsRNA, US11, present late in infection when dsRNA is most abundant, prevents activation of MDA5 and RIG-I (34). IRF3 is thus kept inactive, shunting the pathway away from interferon production, ISG transcription, and establishment of the antiviral state (34).
Finally, US11 interferes with the node governed by the PRR 2’-to-5′ (2’-5′) oligoadenylate synthetase (OAS). dsRNA-bound, activated 2’-5′ OAS synthesizes the second messenger, 2’-5′-linked oligoadenylate, which in turn is recognized by RNaseL (35). Activated RNaseL indiscriminately cleaves mRNAs and rRNAs, crippling virus production (35, 36). Following ocular HSV-1 infection, mice lacking RNaseL suffer higher rates of mortality than their wild-type counterparts, likely due to widespread corneal destruction and unimpeded access of the virus to sensory neurons (37). US11 is necessary and sufficient to repress 2’-5′ OAS activity in HSV-1-infected cells through its interaction with 2’-5′ OAS complexed with dsRNA (38).
Like many herpesvirus proteins, US11 has been attributed with multiple functions. Current evidence points to two functions that are distinct from the translation arrest pathways: silencing of proapoptotic signaling and microtubule-based cargo transport. US11 is a small, basic phosphoprotein (39, 40). Its carboxy-terminal half contains several arginine-X-proline (R-X-P) repeats conferring its RNA-binding property and both nucleolar import and nuclear export signals as well as domains involved in all characterized protein interactions (14, 15, 34, 41–47). In the nucleus, US11 interacts with homeodomain-interacting protein kinase 2 (HIPK2) (48). HIPK2 modulates transcription in response to cell surface and cytoplasmic signals, including endoplasmic reticulum (ER) stress (49), regulating cell cycle progression and proapoptotic pathways (50). When coexpressed with HIPK2, US11 antagonizes HIPK2-induced cell growth arrest (48), suggesting that it may lock infected cells into viral production. In the cytosol, US11 interacts with two components of the microtubule motor protein kinesin family. The kinesin heavy chain and light chain-related protein PAT1 were found to interact with US11 by in vitro pulldown and yeast two-hybrid screens, respectively (46, 47). Kinesins are instrumental in rapid axonal transport of proteins in neurons (51) and anterograde transport of mRNAs to distal sites of localized translation in axons as well as distal processes in astrocytes (52–55). Anterograde transport is required for efficient “round-trip” infection of periocular tissue following ocular HSV-1 infection (56), and the idea of a possible link between US11 and axonal transport is intriguing.
Defining the key roles for US11 in HSV-1 infection and pathogenesis has been challenging due to subtle phenotypes for US11 single null mutants in vitro (27, 57). In addition, older in vitro and in vivo studies largely dismissed the importance of US11 (57–59), thereby reducing enthusiasm in the field for more detailed studies. Here we aimed to test the phenotype of US11-null viruses created through targeted mutagenesis of the US11 open reading frame (ORF). This report describes the results of generation of US11 deletion and marker-rescued viruses and compares their abilities to regulate the eIF2α pathway, to replicate in vitro, to replicate and cause disease in vivo, and to emerge from latency.
RESULTS
Construction of Us11-null strain 17 virus.
HSV-1 transcriptional units often overlap, and the Us10-12 locus is complex: the Us11 promoter is located within the Us12 ORF, and a substantial portion of the Us10 ORF and its promoter lies within the Us11 ORF. A deletion or replacement of the Us11 control elements or coding sequence would thus compromise neighboring genes. We therefore designed a null virus by precise targeting of a nonoverlapping region of the US11 ORF, avoiding the coterminal Us12-Us10 transcripts (Fig. 1A). We targeted a unique SphI restriction site in the 5’ portion of the US11 ORF by inserting a 23-bp linker containing stop codons in all three reading frames (shown in red) and a unique BglI site (underlined). This linker was designed to minimally impact the HSV-1 genome and Us11 transcript yet terminate Us11 mRNA translation at codon 29. Linker insertion and marker rescue of the US11-null allele were carried out using standard homologous recombination methods (60). Following screening by PCR, Southern blot analysis was used to confirm the genotypes of the parental strain (strain 17), 17Δ11, and 17Δ11R viruses (Fig. 1B). Hybridization between the SphI-digested products and a radiolabeled probe spanning the targeted region revealed the expected bands of 3,659 and 1,337 bp in strain 17 and strain 17Δ11R DNA and of a single 5,015-bp product in strain 17Δ11.
FIG 1.

Construction of strain 17 Us11 viruses. (A) Genomic map of Us11 and modification strategy. At top is a representation of the HSV-1 genome, indicating the long and short terminal repeats (TRL and TRS, respectively), long and short internal repeats (IRL and IRS, respectively), and unique long and short (UL and US, respectively) regions. At the bottom, a magnified US-TRS region shows the transcripts (bold black arrows), promoters (bent green arrows), and ORFs (hollow boxes) for the indicated US genes. An SphI restriction site at the beginning of the Us11 ORF served as the insertion site of a BglI linker, placing stop codons (in red) in all 3 reading frames. The introduced BglI site is underlined and the linker sequence is shown in uppercase letters. (B) Southern blot analysis of viral genomic DNA digested with SphI and hybridized to a radiolabeled probe derived from pSW1, containing the US11 coding sequence. The presence of the SphI site in strain 17 and 17Δ11R produced bands of 3,659 and 1,337 bp, while the 17Δ11 virus lacking the SphI site has a single 5,015-bp band. M, molecular mass markers.
The role of Us11 in translational arrest is dependent on virus and cell type.
As PKR antagonism is a major activity of US11, we sought to test the phenotype of our US11-null virus using a direct downstream readout of PKR activity: eIF2α phosphorylation. We chose human corneal limbal epithelial cells (HCLEs), a well-established model of the corneal epithelium. Corneal epithelial cells are relevant to HSV-1 pathogenesis as they constitute the first barrier in primary corneal infections, are impacted by ocular herpes stromal keratitis, and have robust innate immune responses (61–65). Additionally, since a Patton HSV-1 strain deleted of US11 was found to be poor at blocking eIF2α phosphorylation in U373 glioblastoma cells (U373s) (27), we also performed our studies in U373s as a comparison. HCLEs or U373s were infected with strain 17 or its US11 mutants, 17Δ11, 17Δ11R, and 17A11, a double deletion mutant lacking US11 and both copies of ICP34.5. A parallel cohort of Patton strain viruses (provided by Ian Mohr, NYU) was also used to address virus strain influence. The original virus designations are listed in Materials and Methods, but for clarity, here these viruses are termed Patton, PatΔ11, PatΔ11R, and ICP34.5−/US11− double deletion mutant PatΔ34Δ11 (27). Lysates were prepared 18 h postinfection (hpi), and immunoblot analyses were performed using antibodies specific to phosphorylated or total eIF2α, US11, and tubulin (Fig. 2A). US11 expression was restricted to the wild-type (strain 17 and Patton) and marker rescue (17Δ11R and PatΔ11R) viruses as expected (US11 blots), and total eIF2α levels were comparable between lysates (eIF2α blots). In HCLEs infected with strain 17 or 17Δ11R, eIF2α phosphorylation remained at its basal (uninfected) level (HCLE P-eIF2α blot). Unexpectedly, in the 17Δ11 lysate, the level of eIF2α phosphorylation was not appreciably higher, yet the additional absence of ICP34.5 in the 17A11 infection increased eIF2α phosphorylation by ∼40%, reflecting the key activity of ICP34.5 late in infection. Compared to the strain 17 and 17Δ11R infections, Patton and PatΔ11R infections yielded elevated levels of eIF2α phosphorylation. The PatΔ11 lysate had even higher phospho-eIF2α levels, equal to that of the double mutant 17A11. The Patton double mutant PatΔ34Δ11 had the highest level of phospho-eIF2α in HCLEs, twice that measured in uninfected cells. The findings in HCLEs indicate greater control of eIF2α phosphorylation by strain 17 than by Patton in late infection, which is independent of US11 and dependent on ICP34.5. The data also suggest a role for US11 in late Patton infection, cooperating with ICP34.5 to limit phosphorylation of eIF2α. The impact of US11 deletion was more substantial in U373s, where a 50% to 80% increase in eIF2α phosphorylation was measured in strain 17Δ11 infections and PatΔ11 infections compared to strain 17 and Patton infections, respectively (U373 P-eIF2α blot, red arrow). Again the infections with the ICP34.5−/US11− double mutants 17A11 and PatΔ34Δ11 resulted in the highest level of phospho-eIF2α, consistent with cooperation between US11 and ICP34.5 in late infection.
FIG 2.

Suppression of protein translation by Us11 is cell and virus strain dependent. (A) Immunoblot analyses of eIF2α phosphorylation following infection of HCLEs (left panels) or U373 cells (right panels) with the indicated viruses for 18 h. Detection antibodies are indicated between the upper panels. In the right panel, the red arrow denotes P-eIF2α and the asterisk marks a nonspecific protein. The infected/mock-infected ratio of phospho-eIF2α is shown below the blots for each lane. (B) Autoradiographs showing incorporation of [35S]amino acids into total protein in HCLE and U373 cells during 1 h of pulse-labeling at 18 hpi with the indicated viruses. Molecular mass markers are at the far right. Red asterisks denote Us11. 35S inc., quantitation of the level of 35S incorporated into nascent proteins from the average of results from three replicates. This is shown as a ratio to infection with the wild-type parental virus. (C) Immunoblot analyses of ICP34.5 and US11 accumulation in U373 cells infected with either strain 17 or Patton HSV-1, as denoted above the panels. The time of infection (or mock infection) is shown above each lane. (D) The kinetics of gene expression of ICP34.5 and US11 from strain 17 or Patton were measured by densitometric analysis of a representative immunoblot (depicted in panel C). Data are displayed as the percentage of expression at each time point relative to that of maximal expression of that protein during the time course.
To correlate the phosphorylation state of eIF2α with total protein synthesis, HCLEs and U373s were infected with the virus cohorts and pulse-labeled with [35S]methionine and [35S]cysteine from 17 to 18 hpi. Autoradiography of whole-cell lysates was used to profile the late infection proteome (Fig. 2B). Compared to mock-infected samples, translation in infected samples had shifted from production of cellular proteins to production of viral late proteins. Overall, conditions marked by suppressed eIF2α phosphorylation (see Fig. 2A) also resulted in sustained protein synthesis, while those marked by elevated phospho-eIF2α led to a corresponding degree of translational arrest (17A11, PatΔ11, and PatΔ11Δ34 lanes). In HCLE cell infections, the correlation between eIF2α phosphorylation and translational arrest was proportional. Moderately elevated levels of phospho-eIF2α (17A11 and PatΔ11) corresponded to moderate translational arrest (∼50%), as did that in 17Δ34.5 infection (data not shown). Moreover, the markedly elevated levels of phospho-eIF2α in the PatΔ11Δ34 lysate corresponded to marked (90%) translational arrest. However, this trend did not hold true in U373s. While the parental virus (strain 17 and Patton) and marker rescue virus (17Δ11R and PatΔ11R) infections showed equal levels of protein synthesis and the double mutant (strains 17A11 and PatΔ11Δ34) infections characterized by the highest levels of eIF2α phosphorylation were profoundly arrested in translation, the US11 single mutants 17Δ11 and PatΔ11 both yielding moderately increased phospho-eIF2α levels showed 5% and 70% levels of translational arrest, respectively. This indicates that the residual U373 pool of eIF2α kept unphosphorylated by ICP34.5 in strain 17 infection suffices to ensure continued viral protein production late in infection, while in Patton infection it does not do so. It seems, therefore, that the Patton strain is more reliant on US11 late in infection than strain 17 to meet the demands of viral protein synthesis.
To determine whether differences in the relative timing of expression and/or accrual of ICP34.5 and US11 differed between strain 17 and Patton may explain the results described above, a time course of protein expression during infection of U373 cells was performed (Fig. 2C). Immunoblot analyses of ICP34.5 and US11 showed both similar maximal amounts of protein expression and accumulation of both proteins over time (Fig. 2C). In both virus strain infections, ICP34.5 expression was evident during mid-infection, while the onset of US11 expression occurred later. What appeared in the immunoblots to be delayed expression kinetics for both Patton ICP34.5 and US11 proteins was confirmed by densitometric analysis (Fig. 2D). Quantitation of the pixel density per band, corrected for background and expressed as a percentage of maximal expression, showed that Patton protein expression was shifted to a later time point than that seen with strain 17. However, the relative expression kinetics revealed by comparisons between Patton ICP34.5 and US11 were nearly identical to those of strain 17. Considered together, these experiments point to virus strain- and cell type-specific differences in the importance of US11 in controlling phosphorylation of eIF2α and protein synthesis in late infection and indicate the differences are not attributable to a strain-dependent difference in gene expression cascades.
US11 is dispensable for growth in vitro and does not contribute to IFN resistance.
Alpha/beta interferon (IFN-α/β) induction represents a key form of antiviral defense, and corneal epithelial cells produce IFN-α in response to HSV-1 infection in vivo (61) and rapidly initiate production of IFN-α and IFN-β following in vitro infection (62). The observation that 17Δ11 can produce normal levels of protein even in the presence of phosphorylated eIF2α (Fig. 2) led us to question whether it could overcome natural IFN-stimulated innate defenses. To address this, HCLEs or U373s were infected at a low multiplicity of infection (MOI) and a multistep growth analysis was performed with strain 17, 17Δ11, or 17Δ11R (Fig. 3). Following the eclipse phase, the 17Δ11 virus reached titers similar to those of strain 17 and 17Δ11R by 24 hpi and all viruses replicated comparably at 48 hpi and 72 hpi in both cell lines. Moreover, priming HCLEs with low levels of IFN-β prior to infection limited the replication levels of strain 17, strain 17Δ11, and strain 17Δ11R equally, and there was no additional growth defect for strain 17Δ11 (Fig. 3A). We also examined the replication of strain 17Δ11 in primary fibroblasts from mouse embryos and adult dermis and found no growth defect in these cell types relative to the results seen with wild-type viruses (data not shown). In light of the modest growth defect suffered by the US11-null Patton strain in U373s (1 log PFU/ml at one time point) (27), our findings (Fig. 2 and 3) suggest that US11 in isolation is not a major factor in overcoming the innate immune defenses of the cell during lytic replication.
FIG 3.
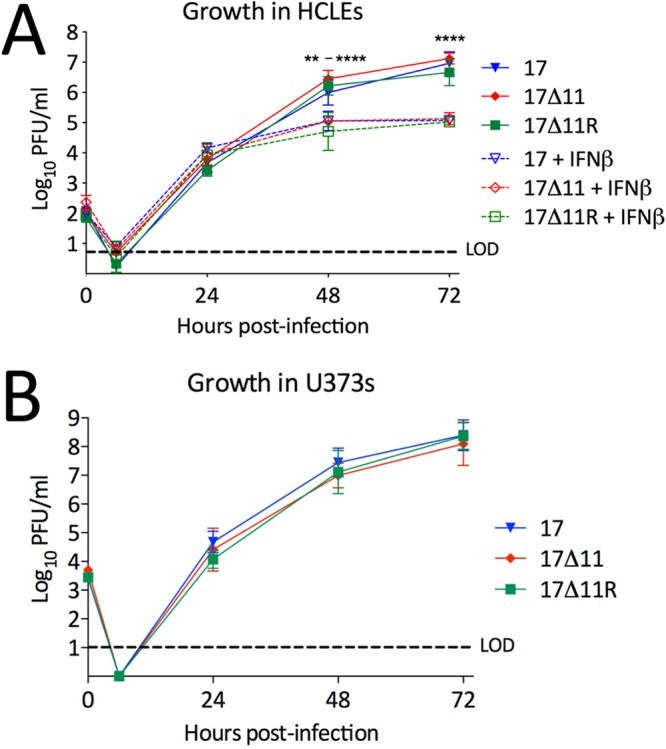
Us11 is not required for growth in vitro. HCLEs, either left untreated or pretreated with 30 IU/ml IFN-β (A), or U373s (B) were infected with the indicated viruses at MOI = 0.001. At 1 h postadsorption, viral inocula were replaced with media and wells (media and cells) harvested at the indicated time points and titers were determined on Vero cells. Data points represent means ± standard deviations (SD) of results from experiments performed three times. P = <0.01 to 0.0001 (two-way ANOVA) where shown. LOD, limit of detection.
The virulence of HSV-1 lacking US11 is dampened following mucosal but not intracranial infection.
To examine the importance of US11 for neurovirulence, the time from infection to endpoint criteria for euthanasia was tested following intracranial infection of wild-type (129SvEv) mice with 100 PFU of strain 17, strain 17Δ11, or strain 17Δ34.5 (Fig. 4A). While ICP34.5 deletion mutant 17Δ34.5 was completely avirulent as expected (19, 20), both strain 17 and 17Δ11 were neurovirulent and all animals succumbed to infection within 8 dpi.
FIG 4.

Us11 contributes to virulence. (A) Wild-type 129SvEv mice were injected intracranially with 100 PFU of strain 17, strain 17Δ11, or strain 17Δ34.5 and euthanized when endpoint criteria were met. (B and C) Wild-type 129SvEv mice were subjected to corneal infection following scarification. Inoculation of either 2 × 106 PFU/eye (B) or 104 PFU/eye (C) of the indicated viruses was performed, and mice were euthanized when endpoint criteria were met. P values determined using the log rank (Mantel-Cox) test and numbers of animals per group are as indicated.
Corneal infections were performed to test neuroinvasion. Following corneal scarification, virus was applied to the corneal surface at a high dose (2 × 106 PFU/eye) (Fig. 4B) or low dose (2 × 104 PFU/eye) (Fig. 4C). Following high-dose infection, all mice reached endpoint criteria at 9 to 10 dpi for strain 17-infected mice at and 9 to 14 dpi for strain 17Δ11-infected mice (Fig. 4B). These modest differences in lethality did not meet statistical significance (P = 0.098). The low dose led to mortality within 21 days in 45% to 50% of mice infected with strain 17 or 17Δ11R and in 20% of mice following strain 17Δ11 infection (Fig. 4C). Strain 17Δ11 was significantly less virulent than strain 17Δ11R (P = 0.03), but its virulence was more modestly attenuated than that of strain 17 (P = 0.136).
Round-trip viral spread is promoted by US11.
“Round-trip” transport of HSV-1 following corneal infection involves sequential infection of the cornea, trigeminal ganglia (TG), and, finally, periocular skin, in a pattern typical of zosteriform spread (56). This process depends on bidirectional axonal transport, and herpesviruses lacking key transport-promoting components or that are pharmacologically blocked in axonal transport are attenuated in virulence in vivo (66). The proposed role for US11 in anterograde transport (46, 47) prompted us to examine the sequential spread of US11-null virus following corneal infection of 129SvEv mice. At 24 hpi with 2 × 106 PFU/eye, the time point at which HSV-1 activity in the cornea peaks (56), tear film contained 105 to 106 PFU/ml virus, irrespective of the genotype (Fig. 5A). By 48 hpi, viral titers had declined equally and were close to the limit of detection in all three virus groups. Replication and reporter activity of HSV-1 peaks in the TG at 3 dpi (56). At that time point, strain 17 and strain 17Δ11R reached higher (P < 0.05 to 0.001) titers than strain 17Δ11. The titers for all viruses were comparable by 5 dpi (Fig. 5B). Peak HSV-1 titers and reporter activity in periocular tissue occur at 3 to 4 dpi, with periocular disease ensuing thereafter (56). Strain 17 and 17Δ11R reached significantly higher titers (P < 0.05 to 0.001) than strain 17Δ11 in periocular tissues at 3 and 5 dpi (Fig. 5C).
FIG 5.
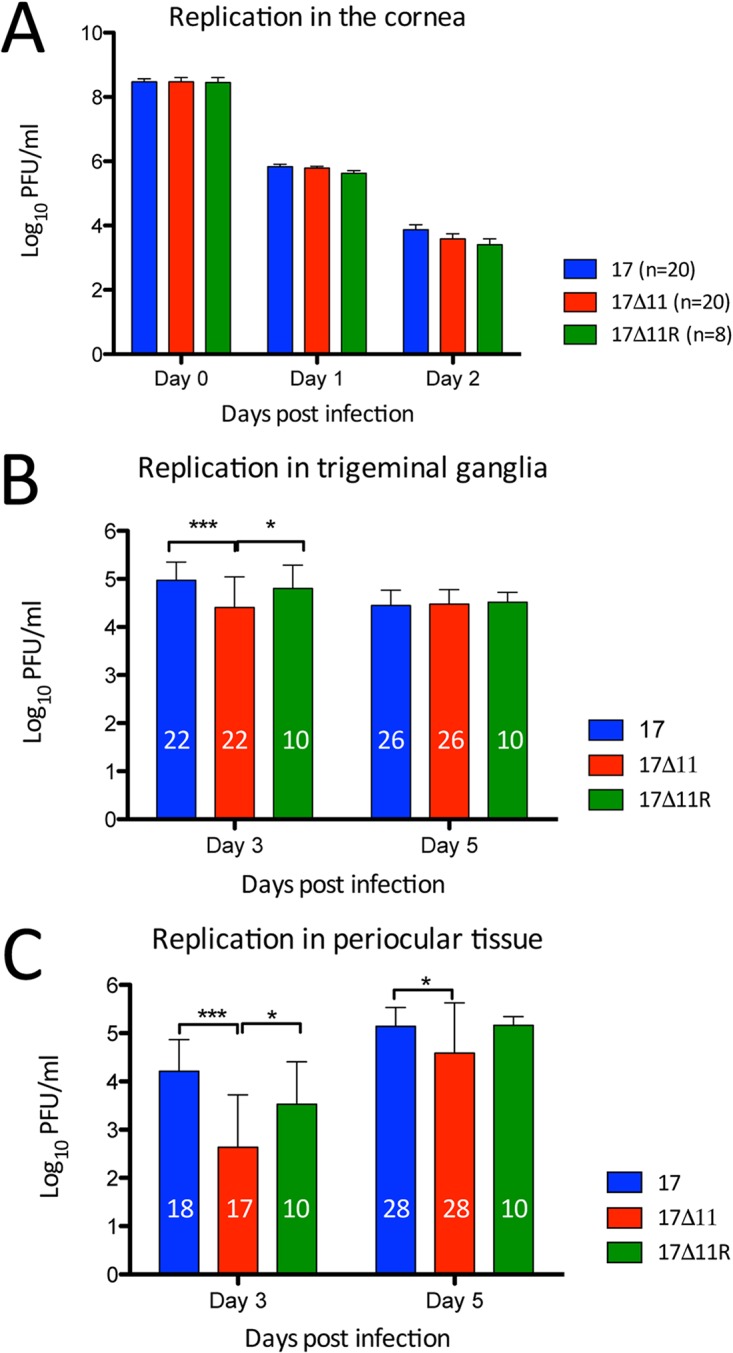
Viral replication in mouse tissues. Corneas of wild-type 129SvEv mice were scarified and infected with 2 × 106 PFU/eye of strain 17, strain 17Δ11, or strain 17Δ11R. (A) At 24 and 48 hpi, eyes were swabbed and titers of virus in tear film were determined. The number of animals per group is indicated in the key. (B and C) Mice were sacrificed on days 3 and 5, and titers of virus were determined in trigeminal ganglia (B) and periocular tissue (C). Numbers within bars indicate the total number of animals in each group. Error bars show the standard deviations. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (two-way ANOVA and Bonferroni posttests).
US11 promotes periocular disease.
The lower 17Δ11 titers in periocular skin were consistent with the hypothesis that US11 promotes round-trip spread and suggest that it may therefore promote periocular disease. To test this hypothesis, 129SvEv or C57BL/6J mice were infected with 2 × 104 PFU/eye (129SvEv) or 2 × 106 PFU/eye (C57BL/6J). These inocula were below the 50% lethal dose (LD50) in each of the respective mouse strains, thereby allowing scoring of periocular symptoms over a 21-day period. Periocular pathogenesis in HSV-1-infected mice is typified by inflammation of the eyelid, muzzle fur ruffling and swelling, and periocular lesions. Disease scores were assigned on a 0 to 5 scale in a blind fashion. All virus genotypes elicited a response, with periocular symptoms arising at 5 dpi, peaking at 7 dpi (for C57BL/6J) or 11 dpi (for 129SvEv), and resolving by 21 dpi. Area under the curve (AUC) analysis of the mean daily C57BL/6J scores through the symptomatic period indicated that strain 17Δ11 infection resulted in less than half the total disease score seen with strain 17 (the AUC score for strain 17 was 20.75 and for strain 17Δ11 was 8.63). Examining the peak mean disease scores, strain 17 and strain 17Δ11R produced the greatest extent of disease, which differed significantly (P < 0.05 to 0.001) from that of the strain 17Δ11 infection in the 129SvEv cohort (Fig. 6A). The same trend was observed in the smaller C57BL/6J cohort, but statistical significance was not achieved. We also examined the percentage of days in which disease scores equaled or exceeded the mean scores over 21 days (0.85 for 129SvEv and 0.25 for C57BL/6J; Fig. 6B and C). In both mouse strains, scores were greater than or equal to the mean during 70% to 80% of the days following infection with strain 17 or strain 17Δ11R. In contrast, infection with strain 17Δ11 resulted in fewer symptomatic days (11% fewer in 129SvEv mice and 44% fewer in C57BL6 mice). The data are therefore consistent with a role for US11 in the pathogenesis of HSV-1 periocular disease.
FIG 6.

Us11 promotes periocular pathogenesis. Corneas of wild-type 129SvEv and C57BL/6J mice were scarified and infected with 2 × 104 PFU/eye (129SvEv) or 2 × 106 PFU/eye (C57BL/6J) of strain 17, strain 17Δ11, or strain 17Δ11R. Mice were scored for disease daily from disease onset until resolution of periocular disease by an observer in a blind fashion. (A) Using the scoring metrics in the table (right), the mean numerical disease score on the day of maximal disease is shown for each virus/host strain combination (left). The number of animals per group is shown inside each bar. **, P < 0.01; ***, P < 0.001 (one-way ANOVA and Bonferroni posttests). (B and C) Data from the same experiment are shown as percentages of total days following infection in which the mean disease score equaled or exceeded 0.85 (129SvEv [SvEv] mice) (B) or 0.25 (C57BL/6J [Bl6] mice) (C).
Emergence following reactivation is delayed for US11-null viruses.
Establishment of latency was determined by quantitating the number of HSV-1 genome copies per TG by real-time PCR after 28 dpi (Fig. 7A). There was no significant difference in genome copy number/TG for the three viruses, all yielding 1.7 to 3 × 103 genomes/TG. These data indicate that US11 is dispensable for the efficient establishment of latency in TG neurons. Having shown this, the time to recrudescence of virus and the levels of viral titers emerging from the TG were quantitated. To measure emergence, 129SvEv or C57BL/6J TG mice were individually subjected to explantation at 28 dpi using permissive cells. TG were moved to and incubated in a new well of Vero cells daily for 3 days. The three “indicator plates” from which the TGs had been removed were then scored daily for cytopathic effect (CPE) in a blind fashion (Fig. 7B). None of the wells in the h 0 to 24 or h 24 to 48 postexplantation (hpe) indicator plates showed any CPE (data not shown). The plate corresponding to 48 to 72 hpe, however, showed some differences in the appearance of CPE. After 48 h of incubation, ∼40% of the wells from the strain 17 and strain 17Δ11R indicator plates had become CPE positive, compared with ∼20% of wells from the strain 17Δ11 indicator plates. This held true for both the 129SvEv and C57BL/6J explantation cultures. At 72, 96, and 120 h, explantation cocultures from 129SvEv mice continued this trend, whereby strain 17Δ11 CPE lagged that of strain 17 and strain 17Δ11R. In contrast, at these later time points, the C57BL/6J explantation cultures had levels of recrudescent virus that were equally detectable between infection groups. To quantitate reactivating virus, tissues were homogenized and assayed in accordance with the studies of Doll and Sawtell (67) (Fig. 7C). At 72 hpe, the level of virus production after strain 17Δ11 reactivation was similar to that seen with the wild-type viruses (∼104 PFU/ml) in all six mouse/virus strain pairings. This titer is in agreement with results reported previously (67). Taken together, these results reveal a modest early delay in the recrudescence of strain 17Δ11 that was unrelated to the establishment of latency.
FIG 7.

Establishment of and reactivation from latency. Trigeminal ganglia were collected from wild-type 129SvEv and C57BL/6J mice 28 dpi with 2 × 104 PFU/eye of strain 17, strain 17Δ11, or strain 17Δ11R. (A) DNA extracted from latently infected 129SvEv TGs was analyzed for viral thymidine kinase copy number by qRT-PCR. Data were normalized by comparison to data from a single-copy mouse adipsin gene. Data are expressed as numbers of genome copies per individual TG. (B) 129SvEv and C57BL/6J TG explants were plated with indicator Vero cells for 24 h during the interval 48 to 72 hpe. Wells were scored as CPE+ or CPE− at 48, 72, and 96, and 120 h. Data shown represent levels of CPE+ wells as a percentage of total wells (shown in legend) within each infection cohort. *, P < 0.05 (two-way ANOVA and Bonferroni posttests). (C) At 72 hpe, TG from 129SvEv (SvEv) and C57BL/6J (Bl6) mice were homogenized and titers in the homogenate were determined on Vero cells. In panels A and C, bars indicate the mean titers, which were not significantly different (P > 0.05 [one-way ANOVA]).
DISCUSSION
Among the antagonists that HSV-1 encodes to thwart cell-intrinsic innate immune mechanisms, ICP34.5 and US11 prevent antiviral cellular translational arrest. While deficits in growth in vitro and neurovirulence in vivo are well documented for ICP34.5 deletion mutants (19–21), the importance of US11 in vivo is less well studied. Here we created a phenotypically US11-null mutant in the background of strain 17 and of a marker-rescued virus. These viruses were used to probe the influence of US11 on protein translation and growth in interferon-responsive cells and on virulence, pathogenesis, and latency in mice. Key findings of this study are as follows. US11 of either strain 17 or Patton inhibited eIF2α phosphorylation in U373 cells, in cooperation with ICP34.5. In HCLE cells, US11 of Patton also limited eIF2α phosphorylation and late protein synthesis was sensitive to even modest levels of phosphorylated eIF2α. In contrast, during late infection with strain 17, eIF2α phosphorylation was solely controlled by ICP34.5, and protein synthesis continued whether or not phosphorylated eIF2α had accumulated. US11 was dispensable for growth in vitro, even when cells were primed with exogenous IFN-β, with strain 17Δ11 achieving titers similar to those seen the wild-type viruses in multistep growth assays. Mice infected intracranially with strain 17Δ11 succumbed with kinetics indistinguishable from those seen with the wild-type strain. Following corneal infection, however, strain 17Δ11-infected mice showed an extended time to onset of morbidity relative to wild-type viruses, pointing to a role for US11 in neuroinvasion. US11 also promoted viral replication in TG and periocular tissue and was a contributing factor in periocular disease. Although US11 was dispensable for the establishment of latency in TG neurons, the data indicated a potential role for US11 in emergence following reactivation.
The conservation of US11 in the Simplexvirus genus suggests that it has a key role in the evolutionary success of the virus and that it most likely predates γ34.5. If a central function of US11 is to dampen translational arrest late in infection, why was γ34.5, a second effector in this pathway, acquired and stabilized within the HSV-1 and -2 genomes? It is thought that ICP34.5 largely supplanted the eIF2α antagonist role of US11, since the hyperphosphorylation of eIF2α by an ICP34.5 null virus is suppressed by immediate early expression of US11 in a spontaneous suppressor mutant (22). Interestingly, “B virus” (Cercopithecine herpesvirus 1), which has conservation with HSV and is derived from a common ancestor, uses US11 naturally in this fashion; it lacks γ34.5 and yet expresses US11 with immediate early kinetics (68). In HSV-1 and HSV-2, leaky-late expression of ICP34.5 renders US11 redundant until ICP34.5 expression wanes. By this line of thinking, assuming that auxiliary functions of US11 are dispensable, it is not surprising that US11-null viruses have subtle phenotypes relative to those deleted for γ34.5.
A spectrum of host permissivity exists toward HSV-1 viruses lacking the ability to antagonize PKR activity. Primary glioblastoma stem-like cells are restrictive for such viruses (69), and here we have found HCLEs to be relatively permissive for replication of viruses deleted in US11. An intermediate restriction of US11-null Patton strain virus was documented in the U373 glioblastoma cell line (27), likely stemming from decreased protein translation late in infection (Fig. 2B) (27). However, we also found that in the context of strain 17 infection, US11 is dispensable late in infection of U373 cells. This was evidenced by unabated protein synthesis in strain 17Δ11 infection and replication of strain 17Δ11 to an extent indistinguishable from that seen with the wild-type viruses. Moreover, the growth profile of strain 17Δ11 in U373s was strikingly similar to that in HCLEs, with neither kinetic lag nor decreased peak titer. So while differences in the host cell clearly influence the phenotype of HSV-1 lacking US11, it also seems that the virus strain factors into the outcome of infection. The Patton strain is most closely related to the KOS63 strain (70), and yet, unlike KOS63 (71), it is neuroinvasive following corneal infection (26), is comparable in that respect to strain 17, and has no growth defect in vitro. While we cannot exclude the possibility that differences in the US11 loci in strain 17Δ11 and strain PatΔ11 [triple stop codon insertion versus green fluorescent protein (GFP)-poly(A) insertion (27)] were designed to minimally impact the US10-to-US12 region. We instead envisage two possible explanations for the collective findings. The differences between the virus strains in the primary amino acid sequence of ICP34.5 (70) may affect the degree of translational arrest prior to US11 expression. It is also plausible that an accessory/alternative US11 role may be more important in certain host cells or for different strains of HSV-1.
One confounding factor in elucidating the function of US11 has been controversy regarding its presence in the virion tegument. Tegument proteins delivered upon cell-virus membrane fusion are present to function immediately, prior to viral gene expression. While US11 has long been considered a major tegument component (39, 40), a role for tegument-derived US11 has remained obscure. The requirement for ectopic, immediate early expression of US11 in order to rescue the translational arrest during infection with ICP34.5-null viruses (22, 69) is inconsistent with the supposed abundance of US11 in the tegument. In addition, following high-MOI strain 17 infection in the presence of cycloheximide, there was no impact of input tegument proteins on phosphorylation of eIF2α (data not shown). Using a mass spectrometry approach with the sensitivity to detect 12 copies of tegument protein/virion, an analysis of highly purified HSV-1 virions concluded that US11 is not present (72). Moreover, a recent report described US11 as cargo in extracellular vesicles derived from HSV-1-infected cells (73). It is possible that, like ICP4, US11 is preferentially incorporated into capsid-less, fusogenic L particles (74) or extracellular vesicles, both of which are produced naturally during HSV infection (73).
While US11 is a nonessential gene and while there is clear evidence for its strain-to-strain diversity (57, 75–77), the ORF was found to be preserved in all HSV-1 strains that have been sequenced. These findings imply that although US11 variants arise, there is significant selective pressure to retain US11 function. Our data suggest that assertions of the lack of importance of US11 for neurovirulence in mice (59) need to be refined. One study using a Patton strain lacking US9 to US12 found no discrepancy in neurovirulence following IC infection (59) and, surprisingly, ignored various differences in LD50 levels that followed infection by a variety of peripheral routes (59). In agreement with that work, the current study showed that US11 is dispensable for neurovirulence when HSV-1 is introduced directly into the brain. Also in agreement with that work, our study showed that there was a delay in the time to endpoint criteria following peripheral introduction of strain 17Δ11 into mice relative to wild-type virus infection. There was, therefore, a clear and reproducible trend of dampened virulence of strain 17Δ11. This report supports and extends the hypothesis that US11 has a role in promoting access of the virus to the central nervous system (CNS).
The round-trip model of HSV-1 zosteriform spread from the corneal surface likens the TG axon to a conduit conducting internalized virion capsids to the soma and nascent capsids back to the periocular surface (56, 78). While the level of replication of strain 17Δ11 in the cornea was normal, there was a modest and yet significant decrease in the strain 17Δ11 titer in the TG. This finding implies the presence of obstacles with respect to strain 17Δ11 reaching or replicating in the TG neuron. Potential mechanisms include inefficiencies in retrograde transport of capsids to the soma, localized transport or translation of mRNA transcripts, anterograde transport of capsids to nerve termini, and viral replication and/or assembly or budding. The interaction of US11 with plus-end-directed microtubule motor proteins (46, 47) lends credence to the possibility that strain 17Δ11 is defective in anterograde transport. That said, it remains unclear whether the diminished titers of strain 17Δ11 in periocular skin represent a “knock-on” effect from decreased TG titers or an independent consequence of US11 deletion. Regardless, the level of periocular disease was diminished following corneal infection with strain 17Δ11. Both the extent and incidence of periocular disease were affected by deletion of US11, and this held true in two strains of mice. This periocular disease phenotype is in concordance with lessened TG and periocular skin replication. For many HSV mutants, there is a direct proportionality between periocular skin viral titers and inflammation and disease (79). It is unclear whether in this case the HSV-1 burden correlates directly with virus-mediated tissue destruction or with proinflammatory immune responses or both.
Here we uncovered a subtle but reproducible latency phenotype following strain 17Δ11 infection. While older papers showed that US11 had no role in HSV-1 latency (58, 59), there were caveats with respect to those studies. One study inoculated corneas with 100-fold more US11-null virus than wild-type virus, thereby likely increasing the number of genome copies available for reactivation (58). Another study performed corneal infections at 2-fold to 5-fold the LD50 level, and reactivation could therefore be assessed only on the 2 to 4 surviving animals/group (59). Together, the high inoculum and small sample size would tend to mask subtle phenotypes. While we concur with their collective conclusions that US11 is dispensable for latency establishment and virus production upon reactivation, we did find that the early kinetics of viral recrudescence from TG were modestly impacted, in two mouse strains.
In this study, we have attempted to isolate the role of US11 from that of ICP34.5 to illuminate the unique contribution of US11 to the HSV-1 life cycle in vitro and in vivo. Although our attention centered on the impact of US11 on the eIF2α pathway in our in vitro studies, whether the disease phenotypes found in vivo pertain to the interface of US11 with the protein translation pathway or with other host pathways remains to be determined. We also consider it plausible that the central function and other important functions of US11 such as intracellular transport and apoptosis have yet to be defined.
MATERIALS AND METHODS
Cell culture.
hTERT-immortalized human cornea limbal epithelial cells (HCLEs) were obtained from the Gipson laboratory (Schepens Eye Research Institute, Boston, MA) (63). This cell line is nontransformed and can realize the full differentiation potential of the in vivo progenitor (63). HCLEs were routinely cultured in serum-free defined keratinocyte media (Gibco, catalog no. 17005-042) supplemented with 250 U/ml penicillin, 250 μg/ml streptomycin, 0.3 mM CaCl2, epidermal growth factor (0.2 ng/ml), and bovine pituitary extract (20 to 30 μg/ml). Human glioblastoma cells, here termed U373, were originally obtained from ATCC. This line was subsequently reclassified as U251 human glioblastoma and was discontinued by ATCC (www.atcc.org). We refer to them here as the U373 cell line in order to foster continuity between this study and that previously reported by the Mohr laboratory using the same cells (27). Vero cells (African green monkey kidney, used for virus titering) and U373 cells were propagated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 250 U/ml penicillin, and 250 μg/ml streptomycin. For experiments, cells were subjected to passage into multiwell plates and used upon confluence. Routinely, one well was trypsinized and cell density assessed for calculating the needed PFU. All cultures were maintained in a humidified incubator at 37°C and 5% CO2.
Viruses.
The Mohr laboratory (New York University, NY, NY) engineered the Patton strain viruses pAUS11 (here called strain PatΔ11), pAUS11-Rep (here called strain PatΔ11R), and GFPpAUS11 (here called strain PatΔ34Δ11) as described previously (27). Briefly, pAUs11 comprises a Patton strain in which a GFP ORF fused to a poly(A) site lies immediately downstream of the US11 promoter; pAUS11-Rep is derived from pAUs11 with the US11 locus repaired; and GFPpAUS11 is the double mutant in which the pAUS11 mutation was superimposed on the background of the γ34.5 deletion (both γ34.5 loci).
Wild-type HSV-1 strain 17syn+ was the background for all viruses generated in this study. The 17TermA virus lacking both copies of ICP34.5 (here called 17Δ34.5) was described previously (19).
In creating the Us11 deletion strains, we strove to maintain the viral genome structure by targeting Us11 protein expression. Strain 17Δ11 and strain 17A11 were generated by first cloning a 5,035-bp EcoRI/HpaI fragment containing the Us10, Us11, and Us12 genes from either strain 17syn+ infectious DNA (for strain 17Δ11) or 17TermA infectious DNA (for strain 17A11) into plasmid Litmus39 (NEB, Beverly, MA). From this plasmid, pSW1, a 1,911-bp BstBI/BssHII fragment, was further subcloned into plasmid Litmus28 (NEB). The resulting construct, pSW3, contained a unique SphI restriction site located in the 5′ end of the Us11 ORF (strain 17syn+, nucleotide position 145166; GenBank accession no. X14112). To create the mutant allele of Us11, oligonucleotides 5′-GCTTGAGTGAGTGACCATG-3′ and 5′-GTCACTCACTCAAGCCATG-3′ were annealed and ligated into the SphI site. This “stop linker” contains termination codons in all three reading frames (underlined), destroys the SphI site, and generates a BglI restriction site. This construct, called pUS11-STOP, was linearized with HpaI and cotransfected with infectious DNA from strain 17 or strain 17TermA (lacking both copies of ICP34.5 [19]) into Vero cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Virus from the transfections was subjected to plaque purification and screened for homologous recombination by PCR. PCR products spanning the insertion site were analyzed by restriction digestion, and those that were SphI insensitive/BglI sensitive were identified. The corresponding plaques were purified for three rounds prior to isolation of strain 17Δ11 or strain 17A11 viral DNA. “17Δ11R,” the marker rescue virus with a repaired Us11 locus, was generated by cotransfection of strain 17Δ11 infectious DNA with pSW3 and was isolated using criteria opposite those described above (SphI sensitive/BglI insensitive). Southern blot analysis confirming the genotypes of strain 17Δ11, strain 17A11, and strain 17Δ11R was performed using a [α-32P]dCTP-labeled probe generated from pSW1 through random priming (Prime-a-Gene System, Promega, Madison WI). The recombinant viruses were further verified by Sanger sequencing of the Us11 locus.
Immunoblot analyses.
HCLEs or U373s in 12-well dishes were mock infected or infected in their respective media at an MOI of 5 for 1 h at 37°C. Following adsorption, media were replaced and cells were incubated for a total infection time of 18 h or a time course from 1 to 18 h. The cells were washed gently in phosphate-buffered saline (PBS) that included Ca2+ and Mg2+ (PBS+) and were lysed in 30 μl of 1× SDS-PAGE sample buffer containing 0.1 M dithiothreitol directly in culture wells. Whole-cell lysates were boiled at 100°C for 5 min and loaded directly onto 12.5% SDS-PAGE gels. Following electrophoretic separation, proteins were transferred to polyvinylidene difluoride (PVDF) in a wet-transfer chamber at 100 V for 1 h. Blots were blocked for 30 min at room temperature in 2% (wt/vol) bovine serum albumin diluted in TBS-T (50 mM Tris-Cl [pH 8.0], 0.2 M NaCl, 0.1% [vol/vol] Tween 20). Blocked blots were probed for 1 h at room temperature by the use of the concentration of primary antibody suggested by the provider. Blots were washed twice in TBS-T for 5 min each time, probed for 1 h at room temperature with horseradish peroxidase (HRP)-conjugated secondary antibodies, washed again, and imaged using Pierce ECL Western blotting substrate (Thermo Fisher) and an Alpha Innotech FluorChemQ imaging station (ProteinSimple, San Jose, CA). The rabbit anti-phospho eIF2α antibody (447728G) used was from Thermo Fisher/Invitrogen (Waltham, MA), mouse anti-eIF2α (L57A5) was from Cell Signaling Technologies (Danvers, MA), and mouse anti-tubulin (DM1) was from Sigma (St. Louis, MO). The mouse monoclonal anti-Us11 antibody was kindly provided by the Roizman laboratory (University of Chicago, Chicago, IL) (40). The rabbit polyclonal anti-ICP34.5 antibody was a gift from Ian Mohr (NYU, New York, NY) (27). HRP-conjugated secondary (anti-rabbit and anti-mouse) antibodies were from Jackson ImmunoResearch (West Grove, PA).
For the phosphorylation blots, following image acquisition, the average pixel intensity in bands from the P-eIF2α blot was measured using AlphaView software and the ratio of the values from each infected sample/uninfected sample calculated. For the expression time course, the values of the (background-corrected) bands at each time point were compared to the value at the time point of maximal expression of that protein and expressed as a percentage.
Metabolic labeling and autoradiography.
Infected (MOI = 5) or mock-infected cells in 24-well dishes were radiolabeled 17 hpi as follows. Complete medium was aspirated from wells, cells were rinsed gently with PBS+, the medium was changed to DMEM without cysteine/without methionine (DMEM–cyst/−meth) supplemented with 0.1 mCi/ml of 35S-EasyTag Express (Perkin Elmer, Boston, MA) in a 0.25-ml volume for 1 h at 37°C. After labeling, wells were washed gently in PBS+ and scraped into 30 μl 1× SDS-PAGE sample buffer containing 0.1 M dithiothreitol, boiled, and loaded onto 4% to 20% Tris-glycine gradient gels (Bio-Rad, Hercules, CA). After electrophoretic separation, proteins were transferred to PVDF in a wet-transfer chamber at 100 V for 1 h. Blots were stained with Ponceau Red S to visualize lanes, dried, and exposed to BioMax MR film (Kodak, Rochester, NY). After autoradiography, the lanes were individually excised and placed into EcoScint scintillant (National Diagnostics, Atlanta, GA) and counts per minute per lane were measured with a liquid scintillation counter. The average of results from three replicates was used to obtain a ratio of 35S incorporation between parental strain (strain 17 or Patton) infection and the mock or derivative mutant strain infections.
In vitro replication competence assays.
Multistep growth assays were performed essentially as described previously (19). Briefly, HCLEs or U373s were seeded into 12-well plates at a density of 1 × 105 cells/well and allowed to grow to confluence. HCLE cultures treated with IFN-β were treated for 16 h with 30 IU/ml of human IFN-β prior to infection. Wells were infected with each virus strain using an MOI of 0.001 in a minimal volume of media. After a 1-h adsorption, virus was removed and replaced with complete growth medium. At the desired time points, cells were scraped into the media and disrupted by sonication and the titers of the liberated virus were determined on Vero cells. Titers of the inocula were determined to measure virus concentration for the 0 h time point and to ensure the accuracy of the MOI in all experiments.
In vivo infections.
Mice were cared for in accordance with all federal and institutional policies. Mice were housed in the biosafety level 2 (BSL2) suite of the animal facilities at The Geisel School of Medicine at Dartmouth or at Washington University School of Medicine. The following two HSV-1-susceptible strains of inbred mice were used in these studies: 129SvEvTac (Taconic Biosciences, Germantown, NY) and C57BL/6J (Jackson Laboratory, Bar Harbor, ME). Adult mice (6 to 8 weeks of age, with the genders equally represented in each experimental group) were anesthetized by intraperitoneal injection of ketamine (87 mg/kg of body weight) and xylazine (13 mg/kg). In intracranial infections, mice were injected intracranially with 100 PFU of virus in a volume of 20 μl. In corneal infections, corneas were bilaterally scarified with a 25-gauge needle and inoculated with the indicated dose of virus in a volume of 5 μl. The weight and body temperature of mice were monitored daily. For virulence studies, the time to endpoint criteria for euthanasia was recorded over 21 days. The endpoint criteria used were as follows: loss of ≥15% starting body weight and/or body temperature drop of ≥10%. For virus replication studies, corneas were swabbed daily for 5 days or mice were sacrificed on day 3 or day 5 postinfection for tissue harvesting. TG and 6-mm biopsy punches of periocular skin were placed into tubes containing 1-mm-diameter glass beads and 1 ml of media. Tissue homogenates were prepared by freeze-thawing the samples and mechanical disruption in a Mini-Beadbeater-8 instrument (Biospec Products, Bartlesville, OK) for 1.5 min followed by sonication for 30 s (repeated three times). Homogenates and eye swab samples were processed by standard plaque assay, and viral content was expressed as PFU level per milliliter of tissue homogenate or eye swab sample. For periocular disease progression studies, mice were scored daily by an observer in a blind fashion using the following scale: 0 = no symptoms; 1 = minimal eyelid swelling; 2 = moderate swelling, ruffled muzzle fur, crusty discharge; 3 = eyes crusted and swollen shut, periocular hair loss, skin lesions; 4 = severe muzzle swelling and hair loss, skin lesions; 5 = neurological manifestations and/or endpoint criteria. Sacrificed mice were censored from the graphs, and the numbers of animals (indicated inside each bar in Fig. 6A) reflect only survivors with scoreable or no eye disease.
Reverse transcription-quantitative PCR (qRT-PCR) quantitation of latent genomes.
At 28 days after corneal infection, the mice were sacrificed and TG removed. TG DNA was prepared using a DNeasy kit (Qiagen, Valencia, CA). Real-time PCR to determine the number of latent HSV-1 genomes per TG was performed as described previously (80). Briefly, a 70-bp fragment of the thymidine kinase (tk) gene was amplified from TG DNA and a 10-fold dilution series of HSV-1 bacterial artificial chromosome (BAC) DNA (17–49, 81). BAC DNA was used to generate a standard curve of numbers of genome copies per trigeminal ganglion, since BAC DNA models the episomal, latent genome. To control for the total DNA content of each sample, real-time PCR for the single-copy mouse adipsin gene and a standard curve of mouse genomic DNA were performed for each TG sample. The values corresponding to the tk copy numbers were normalized to the lowest value of the mouse adipsin copy number to yield the normalized genome copy numbers per ganglion.
Ex vivo latency reactivation assays.
At 28 days after corneal infection, mice were sacrificed and TG removed. TG were placed individually in wells of a 48-well plate containing Vero cells. TG were moved to a new plate of Vero cells 24 h postexplantation (hpe) and again at 48 hpe. These three “indicator plates” were scored daily for 7 days for cytopathic effect (CPE+/−). TG explants were plated in a blinded fashion, so wells were scored without bias. At 72 hpe, the TG were combined with 250 μl of media and 1-mm-diameter glass beads and subjected to freezing-thawing at −80°C. Thawed samples were mechanically disrupted in a Mini-Beadbeater-8 instrument for 1.5 min followed by sonication for 30 s (repeated three times). Tissue debris was pelleted by centrifugation at 1,200 × g for 3 min, and the levels of virus present in supernatants were measured by plaque assay on Vero cells.
Statistics.
Statistical analysis was performed using Graph Pad Prism 5. For assay of the multiple-group conditions used in the experiments described here, one- or two-way analysis of variance (ANOVA) was performed followed by Bonferroni posttests. Kaplan-Meier (time to endpoint criteria) graphs were subjected to analysis using the log rank test. Asterisk notations are as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
ACKNOWLEDGMENTS
We are grateful to the Center for Comparative Medical Research at The Geisel School of Medicine at Dartmouth and the animal care facility at Washington University School of Medicine for excellent animal husbandry. Ilene Gipson (Schepens Eye Research Institute, Boston, MA) kindly provided the HCLE cell line. We thank Ian Mohr (New York University, New York, NY) for the Patton strain viruses used here.
This research was funded by RO1 EY09080 to D.A.L.
REFERENCES
- 1.Virgin HW, Wherry EJ, Ahmed R. 2009. Redefining chronic viral infection. Cell 138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 2.Smith G. 2012. Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol 66:153–176. doi: 10.1146/annurev-micro-092611-150051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knipe DM. 2015. Nuclear sensing of viral DNA, epigenetic regulation of herpes simplex virus infection, and innate immunity. Virology 479-480:153–159. doi: 10.1016/j.virol.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cliffe AR, Wilson AC. 3 January 2017. Restarting lytic gene transcription at the onset of herpes simplex virus reactivation. J Virol doi: 10.1128/JVI.01419-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. 2011. Recognition of herpesviruses by the innate immune system. Nat Rev Immunol 11:143–154. doi: 10.1038/nri2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma Y, He B. 2014. Recognition of herpes simplex viruses: Toll-like receptors and beyond. J Mol Biol 426:1133–1147. doi: 10.1016/j.jmb.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tohme S, Cukier CD, Severini A. 2011. RNA binding properties of the US11 protein from four primate simplexviruses. Virol J 8:504. doi: 10.1186/1743-422X-8-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson PA, MacLean C, Marsden HS, Dalziel RG, Everett RD. 1986. The product of gene US11 of herpes simplex virus type 1 is expressed as a true late gene. J Gen Virol 67:871–883. doi: 10.1099/0022-1317-67-5-871. [DOI] [PubMed] [Google Scholar]
- 9.Feng GS, Chong K, Kumar A, Williams BR. 1992. Identification of double-stranded RNA-binding domains in the interferon-induced double-stranded RNA-activated p68 kinase. Proc Natl Acad Sci U S A 89:5447–5451. doi: 10.1073/pnas.89.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomis DC, Doohan JP, Samuel CE. 1992. Mechanism of interferon action: cDNA structure, expression, and regulation of the interferon-induced, RNA-dependent P1/eIF-2 alpha protein kinase from human cells. Virology 188:33–46. doi: 10.1016/0042-6822(92)90732-5. [DOI] [PubMed] [Google Scholar]
- 11.Benne R, Edman J, Traut RR, Hershey JW. 1978. Phosphorylation of eukaryotic protein synthesis initiation factors. Proc Natl Acad Sci U S A 75:108–112. doi: 10.1073/pnas.75.1.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Talloczy Z, Jiang W, Virgin HWt, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. 2002. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A 99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burgess HM, Mohr I. 17 July 2018. Defining the role of stress granules in innate immune suppression by the herpes simplex virus 1 endoribonuclease VHS. J Virol doi: 10.1128/JVI.00829-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khoo D, Perez C, Mohr I. 2002. Characterization of RNA determinants recognized by the arginine- and proline-rich region of Us11, a herpes simplex virus type 1-encoded double-stranded RNA binding protein that prevents PKR activation. J Virol 76:11971–11981. doi: 10.1128/JVI.76.23.11971-11981.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peters GA, Khoo D, Mohr I, Sen GC. 2002. Inhibition of PACT-mediated activation of PKR by the herpes simplex virus type 1 Us11 protein. J Virol 76:11054–11064. doi: 10.1128/JVI.76.21.11054-11064.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kew C, Lui PY, Chan CP, Liu X, Au SW, Mohr I, Jin DY, Kok KH. 2013. Suppression of PACT-induced type I interferon production by herpes simplex virus 1 Us11 protein. J Virol 87:13141–13149. doi: 10.1128/JVI.02564-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lussignol M, Queval C, Bernet-Camard MF, Cotte-Laffitte J, Beau I, Codogno P, Esclatine A. 2013. The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J Virol 87:859–871. doi: 10.1128/JVI.01158-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He B, Gross M, Roizman B. 1997. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A 94:843–848. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolovan CA, Sawtell NM, Thompson RL. 1994. ICP34.5 mutants of herpes simplex virus type 1 strain 17syn+ are attenuated for neurovirulence in mice and for replication in confluent primary mouse embryo cell cultures. J Virol 68:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou J, Kern ER, Whitley RJ, Roizman B. 1990. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 250:1262–1266. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 21.Chou J, Roizman B. 1992. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc Natl Acad Sci U S A 89:3266–3270. doi: 10.1073/pnas.89.8.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mohr I, Gluzman Y. 1996. A herpesvirus genetic element which affects translation in the absence of the viral GADD34 function. EMBO J 15:4759–4766. doi: 10.1002/j.1460-2075.1996.tb00853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cassady KA, Gross M, Roizman B. 1998. The second-site mutation in the herpes simplex virus recombinants lacking the gamma134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2alpha. J Virol 72:7005–7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mulvey M, Poppers J, Ladd A, Mohr I. 1999. A herpesvirus ribosome-associated, RNA-binding protein confers a growth advantage upon mutants deficient in a GADD34-related function. J Virol 73:3375–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cassady KA, Gross M, Roizman B. 1998. The herpes simplex virus US11 protein effectively compensates for the gamma1(34.5) gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J Virol 72:8620–8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ward SL, Scheuner D, Poppers J, Kaufman RJ, Mohr I, Leib DA. 2003. In vivo replication of an ICP34.5 second-site suppressor mutant following corneal infection correlates with in vitro regulation of eIF2 alpha phosphorylation. J Virol 77:4626–4634. doi: 10.1128/JVI.77.8.4626-4634.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mulvey M, Poppers J, Sternberg D, Mohr I. 2003. Regulation of eIF2alpha phosphorylation by different functions that act during discrete phases in the herpes simplex virus type 1 life cycle. J Virol 77:10917–10928. doi: 10.1128/JVI.77.20.10917-10928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mulvey M, Camarena V, Mohr I. 2004. Full resistance of herpes simplex virus type 1-infected primary human cells to alpha interferon requires both the Us11 and gamma(1)34.5 gene products. J Virol 78:10193–10196. doi: 10.1128/JVI.78.18.10193-10196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berke IC, Li Y, Modis Y. 2013. Structural basis of innate immune recognition of viral RNA. Cell Microbiol 15:386–394. doi: 10.1111/cmi.12061. [DOI] [PubMed] [Google Scholar]
- 30.Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, Chen ZJ. 2015. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347:aaa2630. doi: 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- 31.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 32.Pomerantz JL, Baltimore D. 1999. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J 18:6694–6704. doi: 10.1093/emboj/18.23.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hiscott J, Pitha P, Genin P, Nguyen H, Heylbroeck C, Mamane Y, Algarte M, Lin R. 1999. Triggering the interferon response: the role of IRF-3 transcription factor. J Interferon Cytokine Res 19:1–13. doi: 10.1089/107999099314360. [DOI] [PubMed] [Google Scholar]
- 34.Xing J, Wang S, Lin R, Mossman KL, Zheng C. 2012. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J Virol 86:3528–3540. doi: 10.1128/JVI.06713-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silverman RH. 2007. Viral encounters with 2',5'-oligoadenylate synthetase and RNase L during the interferon antiviral response. J Virol 81:12720–12729. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drappier M, Michiels T. 2015. Inhibition of the OAS/RNase L pathway by viruses. Curr Opin Virol 15:19–26. doi: 10.1016/j.coviro.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng X, Silverman RH, Zhou A, Goto T, Kwon BS, Kaufman HE, Hill JM. 2001. Increased severity of HSV-1 keratitis and mortality in mice lacking the 2-5A-dependent RNase L gene. Invest Ophthalmol Vis Sci 42:120–126. [PubMed] [Google Scholar]
- 38.Sanchez R, Mohr I. 2007. Inhibition of cellular 2'-5' oligoadenylate synthetase by the herpes simplex virus type 1 Us11 protein. J Virol 81:3455–3464. doi: 10.1128/JVI.02520-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Masse T, Garcin D, Jacquemont B, Madjar JJ. 1990. Ribosome and protein synthesis modifications after infection of human epidermoid carcinoma cells with herpes simplex virus type 1. Mol Gen Genet 220:377–388. doi: 10.1007/BF00391742. [DOI] [PubMed] [Google Scholar]
- 40.Roller RJ, Roizman B. 1992. The herpes simplex virus 1 RNA binding protein US11 is a virion component and associates with ribosomal 60S subunits. J Virol 66:3624–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roller RJ, Monk LL, Stuart D, Roizman B. 1996. Structure and function in the herpes simplex virus 1 RNA-binding protein U(s)11: mapping of the domain required for ribosomal and nucleolar association and RNA binding in vitro. J Virol 70:2842–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roller RJ, Roizman B. 1991. Herpes simplex virus 1 RNA-binding protein US11 negatively regulates the accumulation of a truncated viral mRNA. J Virol 65:5873–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Attrill HL, Cumming SA, Clements JB, Graham SV. 2002. The herpes simplex virus type 1 US11 protein binds the coterminal UL12, UL13, and UL14 RNAs and regulates UL13 expression in vivo. J Virol 76:8090–8100. doi: 10.1128/JVI.76.16.8090-8100.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacLean CA, Rixon FJ, Marsden HS. 1987. The products of gene US11 of herpes simplex virus type 1 are DNA-binding and localize to the nucleoli of infected cells. J Gen Virol 68:1921–1937. doi: 10.1099/0022-1317-68-7-1921. [DOI] [PubMed] [Google Scholar]
- 45.Greco A, Arata L, Soler E, Gaume X, Coute Y, Hacot S, Calle A, Monier K, Epstein AL, Sanchez JC, Bouvet P, Diaz JJ. 2012. Nucleolin interacts with US11 protein of herpes simplex virus 1 and is involved in its trafficking. J Virol 86:1449–1457. doi: 10.1128/JVI.06194-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benboudjema L, Mulvey M, Gao Y, Pimplikar SW, Mohr I. 2003. Association of the herpes simplex virus type 1 Us11 gene product with the cellular kinesin light-chain-related protein PAT1 results in the redistribution of both polypeptides. J Virol 77:9192–9203. doi: 10.1128/JVI.77.17.9192-9203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diefenbach RJ, Miranda-Saksena M, Diefenbach E, Holland DJ, Boadle RA, Armati PJ, Cunningham AL. 2002. Herpes simplex virus tegument protein US11 interacts with conventional kinesin heavy chain. J Virol 76:3282–3291. doi: 10.1128/JVI.76.7.3282-3291.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giraud S, Diaz-Latoud C, Hacot S, Textoris J, Bourette RP, Diaz JJ. 2004. US11 of herpes simplex virus type 1 interacts with HIPK2 and antagonizes HIPK2-induced cell growth arrest. J Virol 78:2984–2993. doi: 10.1128/JVI.78.6.2984-2993.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee S, Shang Y, Redmond SA, Urisman A, Tang AA, Li KH, Burlingame AL, Pak RA, Jovicic A, Gitler AD, Wang J, Gray NS, Seeley WW, Siddique T, Bigio EH, Lee VM, Trojanowski JQ, Chan JR, Huang EJ. 2016. Activation of HIPK2 promotes ER stress-mediated neurodegeneration in amyotrophic lateral sclerosis. Neuron 91:41–55. doi: 10.1016/j.neuron.2016.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Calzado MA, Renner F, Roscic A, Schmitz ML. 2007. HIPK2: a versatile switchboard regulating the transcription machinery and cell death. Cell Cycle 6:139–143. doi: 10.4161/cc.6.2.3788. [DOI] [PubMed] [Google Scholar]
- 51.Elluru RG, Bloom GS, Brady ST. 1995. Fast axonal transport of kinesin in the rat visual system: functionality of kinesin heavy chain isoforms. Mol Biol Cell 6:21–40. doi: 10.1091/mbc.6.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kanai Y, Dohmae N, Hirokawa N. 2004. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43:513–525. doi: 10.1016/j.neuron.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 53.Gaspar I, Sysoev V, Komissarov A, Ephrussi A. 2017. An RNA-binding atypical tropomyosin recruits kinesin-1 dynamically to oskar mRNPs. EMBO J 36:319–333. doi: 10.15252/embj.201696038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakers K, Lake AM, Khazanchi R, Ouwenga R, Vasek MJ, Dani A, Dougherty JD. 2017. Astrocytes locally translate transcripts in their peripheral processes. Proc Natl Acad Sci U S A 114:E3830–E3838. doi: 10.1073/pnas.1617782114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pilaz LJ, Lennox AL, Rouanet JP, Silver DL. 2016. Dynamic mRNA transport and local translation in radial glial progenitors of the developing brain. Curr Biol 26:3383–3392. doi: 10.1016/j.cub.2016.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Summers BC, Margolis TP, Leib DA. 2001. Herpes simplex virus type 1 corneal infection results in periocular disease by zosteriform spread. J Virol 75:5069–5075. doi: 10.1128/JVI.75.11.5069-5075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Longnecker R, Roizman B. 1986. Generation of an inverting herpes simplex virus 1 mutant lacking the L-S junction a sequences, an origin of DNA synthesis, and several genes including those specifying glycoprotein E and the alpha 47 gene. J Virol 58:583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meignier B, Longnecker R, Mavromara-Nazos P, Sears AE, Roizman B. 1988. Virulence of and establishment of latency by genetically engineered deletion mutants of herpes simplex virus 1. Virology 162:251–254. doi: 10.1016/0042-6822(88)90417-5. [DOI] [PubMed] [Google Scholar]
- 59.Nishiyama Y, Kurachi R, Daikoku T, Umene K. 1993. The US 9, 10, 11, and 12 genes of herpes simplex virus type 1 are of no importance for its neurovirulence and latency in mice. Virology 194:419–423. doi: 10.1006/viro.1993.1279. [DOI] [PubMed] [Google Scholar]
- 60.Rader KA, Ackland-Berglund CE, Miller JK, Pepose JS, Leib DA. 1993. In vivo characterization of site-directed mutations in the promoter of the herpes simplex virus type 1 latency-associated transcripts. J Gen Virol 74:1859–1869. doi: 10.1099/0022-1317-74-9-1859. [DOI] [PubMed] [Google Scholar]
- 61.Conrady CD, Jones H, Zheng M, Carr DJ. 2011. A functional type I interferon pathway drives resistance to cornea herpes simplex virus type 1 infection by recruitment of leukocytes. J Biomed Res 25:111–119. doi: 10.1016/S1674-8301(11)60014-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H, Zhang J, Kumar A, Zheng M, Atherton SS, Yu FS. 2006. Herpes simplex virus 1 infection induces the expression of proinflammatory cytokines, interferons and TLR7 in human corneal epithelial cells. Immunology 117:167–176. doi: 10.1111/j.1365-2567.2005.02275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gipson IK, Spurr-Michaud S, Argueso P, Tisdale A, Ng TF, Russo CL. 2003. Mucin gene expression in immortalized human corneal-limbal and conjunctival epithelial cell lines. Invest Ophthalmol Vis Sci 44:2496–2506. doi: 10.1167/iovs.02-0851. [DOI] [PubMed] [Google Scholar]
- 64.Kumar A, Zhang J, Yu FS. 2006. Toll-like receptor 3 agonist poly(I:C)-induced antiviral response in human corneal epithelial cells. Immunology 117:11–21. doi: 10.1111/j.1365-2567.2005.02258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Albertsmeyer AC, Kakkassery V, Spurr-Michaud S, Beeks O, Gipson IK. 2010. Effect of pro-inflammatory mediators on membrane-associated mucins expressed by human ocular surface epithelial cells. Exp Eye Res 90:444–451. doi: 10.1016/j.exer.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Richards AL, Sollars PJ, Pitts JD, Stults AM, Heldwein EE, Pickard GE, Smith GA. 2017. The pUL37 tegument protein guides alpha-herpesvirus retrograde axonal transport to promote neuroinvasion. PLoS Pathog 13:e1006741. doi: 10.1371/journal.ppat.1006741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Doll JR, Sawtell NM. 27 July 2017. Analysis of herpes simplex virus reactivation in explant reveals a method-dependent difference in measured timing of reactivation. J Virol doi: 10.1128/JVI.00848-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vasireddi M, Hilliard JK. 2017. Regulation of PI3K/Akt dependent apoptotic markers during b virus infection of human and macaque fibroblasts. PLoS One 12:e0178314. doi: 10.1371/journal.pone.0178314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peters C, Paget M, Tshilenge KT, Saha D, Antoszczyk S, Baars A, Frost T, Martuza RL, Wakimoto H, Rabkin SD. 2018. Restriction of gamma34.5-deleted oncolytic herpes simplex virus replication in glioblastoma stem-like cells. J Virol doi: 10.1128/JVI.00246-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pourchet A, Copin R, Mulvey MC, Shopsin B, Mohr I, Wilson AC. 2017. Shared ancestry of herpes simplex virus 1 strain Patton with recent clinical isolates from Asia and with strain KOS63. Virology 512:124–131. doi: 10.1016/j.virol.2017.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thompson RL, Cook ML, Devi-Rao GB, Wagner EK, Stevens JG. 1986. Functional and molecular analyses of the avirulent wild-type herpes simplex virus type 1 strain KOS. J Virol 58:203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Deschamps T, Kalamvoki M. 29 August 2018. Extracellular vesicles released by herpes simplex virus 1 infected cells block virus replication in recipient cells in a STING-dependent manner. J Virol doi: 10.1128/JVI.01102-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Szilagyi JF, Cunningham C. 1991. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J Gen Virol 72:661–668. doi: 10.1099/0022-1317-72-3-661. [DOI] [PubMed] [Google Scholar]
- 75.Szpara ML, Gatherer D, Ochoa A, Greenbaum B, Dolan A, Bowden RJ, Enquist LW, Legendre M, Davison AJ. 2014. Evolution and diversity in human herpes simplex virus genomes. J Virol 88:1209–1227. doi: 10.1128/JVI.01987-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Umene K, Yoshida M, Fukumaki Y. 2015. Genetic variability in the region encompassing reiteration VII of herpes simplex virus type 1, including deletions and multiplications related to recombination between direct repeats. Springerplus 4:200. doi: 10.1186/s40064-015-0990-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Umene K. 1986. Conversion of a fraction of the unique sequence to part of the inverted repeats in the S component of the herpes simplex virus type 1 genome. J Gen Virol 67:1035–1048. doi: 10.1099/0022-1317-67-6-1035. [DOI] [PubMed] [Google Scholar]
- 78.Smith GA, Pomeranz L, Gross SP, Enquist LW. 2004. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc Natl Acad Sci U S A 101:16034–16039. doi: 10.1073/pnas.0404686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smith TJ, Ackland-Berglund CE, Leib DA. 2000. Herpes simplex virus virion host shutoff (vhs) activity alters periocular disease in mice. J Virol 74:3598–3604. doi: 10.1128/JVI.74.8.3598-3604.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Strand SS, Leib DA. 2004. Role of the VP16-binding domain of vhs in viral growth, host shutoff activity, and pathogenesis. J Virol 78:13562–13572. doi: 10.1128/JVI.78.24.13562-13572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gierasch WW, Zimmerman DL, Ward SL, Vanheyningen TK, Romine JD, Leib DA. 2006. Construction and characterization of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J Virol Methods 135:197–206. doi: 10.1016/j.jviromet.2006.03.014. [DOI] [PubMed] [Google Scholar]