

Comparative studies of protein structures often provide insights into their evolution. The HK97 fold is a structural motif used to form the coat protein shells that encapsidate the genomes of many dsDNA phages and viruses. The structure and function of coat proteins based on the HK97 fold are often embellished by the incorporation of I-domains. In the present work we compare I-domains from three phages representative of highly divergent P22-like homology groups. While the three I-domains share a six-stranded β-barrel skeleton, there are differences (i) in structure elements at the periphery of the conserved fold, (ii) in the locations of disordered loops important in capsid assembly and conformational transitions, (iii) in surfaces charges, and (iv) in sequence motifs that are potential ligand-binding sites. These structural modifications on the rudimentary I-domain fold suggest that considerable structural adaptability was needed to fulfill the versatile range of functional requirements for distinct phages.
KEYWORDS: dsDNA viruses, nuclear magnetic resonance, protein evolution, protein structure
ABSTRACT
Despite very low sequence homology, the major capsid proteins of double-stranded DNA (dsDNA) bacteriophages, some archaeal viruses, and the herpesviruses share a structural motif, the HK97 fold. Bacteriophage P22, a paradigm for this class of viruses, belongs to a phage gene cluster that contains three homology groups: P22-like, CUS-3-like, and Sf6-like. The coat protein of each phage has an inserted domain (I-domain) that is more conserved than the rest of the coat protein. In P22, loops in the I-domain are critical for stabilizing intra- and intersubunit contacts that guide proper capsid assembly. The nuclear magnetic resonance (NMR) structures of the P22, CUS-3, and Sf6 I-domains reveal that they are all six-stranded, anti-parallel β-barrels. Nevertheless, significant structural differences occur in loops connecting the β-strands, in surface electrostatics used to dock the I-domains with their respective coat protein core partners, and in sequence motifs displayed on the capsid surfaces. Our data highlight the structural diversity of I-domains that could lead to variations in capsid assembly mechanisms and capsid surfaces adapted for specific phage functions.
IMPORTANCE Comparative studies of protein structures often provide insights into their evolution. The HK97 fold is a structural motif used to form the coat protein shells that encapsidate the genomes of many dsDNA phages and viruses. The structure and function of coat proteins based on the HK97 fold are often embellished by the incorporation of I-domains. In the present work we compare I-domains from three phages representative of highly divergent P22-like homology groups. While the three I-domains share a six-stranded β-barrel skeleton, there are differences (i) in structure elements at the periphery of the conserved fold, (ii) in the locations of disordered loops important in capsid assembly and conformational transitions, (iii) in surfaces charges, and (iv) in sequence motifs that are potential ligand-binding sites. These structural modifications on the rudimentary I-domain fold suggest that considerable structural adaptability was needed to fulfill the versatile range of functional requirements for distinct phages.
INTRODUCTION
Viruses encapsulate their nucleic acids in a protein shell called a capsid that protects the genome until it is delivered to target cells. The capsid is self-assembled from viral coat proteins. Viral lineages fall into four families based on the major capsid coat protein structure: PRD1-like, picorna virus-like, HK97-like, and BTV-like (1, 2). The HK97 fold is found in tailed double-stranded DNA (dsDNA) viruses, including phage P22, as well as some archaeal dsDNA viruses and herpesviruses (3–7). Within the lambda phage supercluster there is a P22-like phage homology cluster that includes over 150 phages. This cluster can be further subdivided into three major groups: P22-like, CUS-3-like, and Sf6-like (8, 9). The coat proteins from the three representative phages, P22, CUS-3, and Sf6, have low amino acid sequence identity (14 to 28%), but all contain a protein domain incorporated within the HK97 fold scaffold, termed the insertion domain (I-domain).
In previous work, we showed that the I-domain is an important modulator of the stability and function of the coat protein from phage P22. Specifically, the I-domain D-loop forms critical electrostatic interactions across the icosahedral 2-fold axes that stabilize and determine the morphology of the phage P22 capsid (10). A second S-loop in the I-domain participates in capsid triangulation number (T) determination (11). In addition to capsid assembly, the I-domain is involved in additional functions, including nucleating the folding of the coat protein and phage incorporation of the portal complex that is used to package the dsDNA genome (11–15).
The nuclear magnetic resonance (NMR) structure of the P22 I-domain contains a six-stranded anti-parallel β-barrel, with a small accessory subdomain comprised of three short β-strands and an α-helix. The previously mentioned D- and S-loops occur between β-barrel strands 1 and 2 and strands 3 and 4, respectively (Fig. 1A; see also Movie S1 in the supplemental material) (13). The D-loop is disordered in the isolated I-domain and in full-length coat protein monomers but becomes structured upon procapsid assembly by forming intercapsomer salt bridges (10, 13, 16). Stronger cryo-electron microscopy (cryo-EM) density for the D-loop segment in the procapsid than in the mature capsid suggests that that the D-loop is less structured in the latter as the salt bridges are replaced with a smaller number of polar contacts upon capsid maturation (13). Thus, the intrinsic flexibility of the P22 I-domain’s D-loop appears to provide functionally important malleability, both for capsid assembly and the conformational transition between the procapsid and the mature capsid.
FIG 1.

(A to C) NMR structures of I-domains of P22 (PDB code 2M5S [13]), CUS-3 (PDB code 6MNT), and Sf6 (PDB code 6MPO). For each structure the conserved six-stranded anti-parallel β-barrel motif is in blue, and the accessory subdomains consisting of three β-strands and a 1.5-turn α-helix are in orange and magenta, respectively. In Sf6, two additional β-strands that cross over the top of the six-stranded β-barrel are shown in green. The top structure in each panel is the structure closest to the mean.
The I-domain protrudes from the capsid surface, above the coat protein core. Here, we refer to the “core” as the HK97 fold portion of the coat protein without the I-domain. The β-barrel of the P22 I-domain has a markedly polar distribution of electrostatic charges. The side of the β-barrel facing the environment is predominantly acidic, giving the capsid surface an overall negative charge. The opposite side of the barrel facing the coat protein core has a high content of positively charged basic residues. The resulting positively charged surface electrostatically docks the I-domain against a negatively charged patch on the core of the coat protein structure. Many amino acid substitutions, located on the positively charged docking side of the I-domain’s β-barrel, are associated with the temperature-sensitive folding (tsf) phenotype in phage P22, suggesting an additional role for the P22 I-domain in coat protein folding and stability (13, 15). Indeed, the I-domain contributes about half of the thermodynamic stability of coat protein monomers (14).
In the current work, we report the nuclear magnetic resonance (NMR) structures of the I-domains from phages CUS-3 and Sf6. Together with the previously determined NMR structure of P22 (13), these represent the three major groups encompassing 150 phages in the P22-like cluster (8, 9). The NMR structures of the three I-domains are complemented with information on protein dynamics derived from NMR relaxation data. We compare the I-domain NMR structures to those from recent cryo-EM reconstructions of phage P22 and Sf6 capsids (17, 18). Finally, we analyze how differences in I-domain structures lead to variations in capsid surfaces that could impact phage functions.
RESULTS
I-domains share a conserved β-barrel structure with differences at the periphery.
A motivation for the present work was that early cryo-EM reconstructions of phages P22, CUS-3, and Sf6 showed differences in surface morphologies for regions corresponding to the I-domains, suggesting that there might be structural differences between these modules in the three representative phages (19). To examine the extent to which structural properties are conserved, we determined the NMR structures of the I-domains from phages CUS-3 and Sf6 (Fig. 1 and Movie S1). These were compared to the previously determined structure of the phage P22 I-domain (13). Statistics pertaining to the NMR structures of CUS-3 and Sf6 are given in Table 1.
TABLE 1.
NMR structure statistics for the CUS3 and Sf6 I-domains
| Parameter | Value for the I-domain of: |
|
|---|---|---|
| CUS-3 | Sf6 | |
| Total no. of NMR restraints | 2,114 | 1,412 |
| Total no. of NOE distance restraints | 1,888 | 1,150 |
| Long range (|i – j| > 4) | 504 | 397 |
| Medium range (|i – j| ≤ 4) | 242 | 36 |
| Sequential (|i – j| = 1) | 558 | 348 |
| Intraresidue | 584 | 369 |
| Total no. of dihedral restraints | 202 | 233 |
| φ/ψ | 156 | 183 |
| χ1 | 46 | 50 |
| No. of H-bond restraints (2/bond) | 48 | 58 |
| RMSD from ideal geometry | ||
| Bonds (Å) | 0.0039 ± 0.00014 | 0.0046 ± 0.00028 |
| Angles (°) | 0.57 ± 0.03 | 0.61 ± 0.04 |
| Improper angles (°) | 1.71 ± 0.12 | 1.90 ± 0.19 |
| RMSD from experimental restraintsa | ||
| NOE distance (Å) | 0.0207 ± 0.00119 | 0.0643 ± 0.01413 |
| Dihedral (°) | 0.807 ± 0.1460 | 0.678 ± 0.2531 |
| RMSD from mean structure (Å) | ||
| Entire structure backbone | 2.15 ± 0.40 | 3.58 ± 0.87 |
| Entire structure heavy atoms | 2.57 ± 0.38 | 4.23 ± 0.84 |
| Ordered region, backboneb | 0.92 ± 0.19 | 0.88 ± 0.21 |
| Ordered region heavy atomsb | 1.53 ± 0.23 | 1.43 ± 0.20 |
| β-barrel, backbonec | 0.66 ± 0.18 | 0.66 ± 0.16 |
| β-barrel, heavy atomsc | 1.16 ± 0.24 | 1.18 ± 0.22 |
| Ramachandran plot-Procheck (%)d | ||
| Most favored | 84.7 | 73.8 |
| Additionally allowed | 12.9 | 25.9 |
| Generously allowed | 1.9 | 0.3 |
| Disallowed | 0.6 | 0.0 |
| PSVS Quality Z-scoresd | ||
| Procheck (φ/ψ)d | –2.60 | –3.86 |
| Molprobity clash | –5.97 | –3.8 |
Structures had no NOE violations of >0.5 Å or dihedral violations of >5°.
Backbone atoms were Cα, C′, N, and O. Ordered regions based on 15N relaxation data (Fig. 5) were the following: for CUS-3, residues 228 to 238, 249 to 275, 286 to 308, and 319 to 336 (79 residues); for Sf6, residues 226 to 240, 243 to 274, 292 to 315, and 334 to 341 (79 residues).
Residues that comprise the conserved six-stranded β-barrel motif are as follows: for CUS-3, residues 226 to 238, 245 to 253, 266 to 271, 292 to 296, 308 to 313, and 335 to 341 (35 residues); for Sf6, residues 226 to 238, 245 to 253, 266 to 271, 292 to 296, 308 to 313, and 335 to 341 (46 residues).
Calculated using the Protein Structure Validation Suite (PSVS)(42).
The structures of all three I-domains adopt a conserved six-stranded anti-parallel Greek key β-barrel motif (in blue in Fig. 1). The overall structures of the three I-domains are precisely defined, with backbone root mean square deviations (RMSDs) ranging between 0.48 and 0.92 Å for ordered regions of the structure (Table 1) (13). While the β-barrels are similar for the three I-domains, there are important differences at the periphery of this conserved motif. These include extensions of the secondary structure elements that comprise the β-barrel. Thus, the segment corresponding to the large flexible D-loop in P22 (Fig. 1A) is replaced by an extension of the hairpin between strands β1 and β2 in the CUS-3 and Sf6 I-domains (Fig. 1B and C). The accessory subdomain in the P22 and CUS-3 I-domains consisting of three short β-strands and an α-helix (Fig. 1 orange and magenta, respectively) is replaced by the tangential loop (T-loop) in the Sf6 I-domain (Fig. 1C). Finally, the Sf6 I-domain has a unique extension of strands β2 and β4 (β2′ and β4′) that goes over the top of the β-barrel motif (shown in green in Fig. 1C). In summary, the three I-domains manifest structural modifications within a conserved six-stranded β-barrel fold.
The three I-domain structures are distinct despite a shared fold.
To gauge the structural agreement between I-domains, we aligned the structures with the DALI/FSSP program (Table 2) (20). The RMSD values between the three NMR structures ranged between 2.5 and 3.8 Å, indicating that while the three I-domains share a six-stranded β-barrel fold, the individual structures are distinct. Differences between the structures included loop regions, the accessory subdomain that is absent in the Sf6 I-domain, and β-strand extensions to the six-stranded β-barrel fold motif, as well as more subtle variations in the β-barrel and segments outside the regular secondary structure. Better structural agreement was seen between the NMR structures of the P22 and CUS-3 I-domains, while the Sf6 I-domain is more distinct (Table 2), consistent with the higher sequence homology between the P22 and CUS-3 I-domains (8, 9).
TABLE 2.
RMSD values between I-domain structuresa
| Phage and structure type | RMSD value (Å) or no. of residues included in each alignmenta for: |
||||
|---|---|---|---|---|---|
| P22 NMR | CUS-3 NMR | Sf6 NMR | P22 EM | Sf6 EM | |
| P22 NMR | 2.5 | 3.8 | 2.0 | 2.7 | |
| CUS-3 NMR | 100 | 3.6 | 3.0 | 2.5 | |
| Sf6 NMR | 79 | 74 | 3.8 | 3.6 | |
| P22 EM | 108 | 110 | 74 | 2.2 | |
| Sf6 EM | 101 | 98 | 100 | 105 | |
Pairwise structure alignments and RMSD values were calculated with the DALI/FSSP (20) server (http://ekhidna2.biocenter.helsinki.fi/dali/). Entries above the diagonal are RMSD values. Entries below the diagonal indicate the number of residues included in each alignment. For NMR structures, the conformer closest to the ensemble average was used (entry number 1 in the PDB file). For cryo-EM structures, we used the I-domains from the 3.3-Å-resolution structure (18) of the P22 capsid (PDB code 5UU5, subunit F) and the 2.9-Å-resolution structure (17) of the Sf6 capsid (PDB code 5L35, subunit C). No high-resolution cryo-EM structure is available for the CUS-3 capsid.
For any icosahedral capsid with a triangulation number T of >1, coat protein monomers with identical amino acid sequences must adopt slightly different quasi-equivalent conformations in the asymmetric unit used to build the 20 faces of an icosahedron (21). Thus, in the cryo-EM structures of the T=7 capsids of P22 and Sf6, the seven coat protein monomers show slight differences in structure that extend to differences in their I-domains. The average RMSDs between the seven I-domains in the icosahedral asymmetric units are small: 0.37 Å for the 3.3-Å-resolution structure of P22 and 0.24 Å for the 2.9-Å-resolution structure of Sf6. As of yet there is no high-resolution structure available for the CUS-3 capsid. The I-domains of P22 and Sf6 from the cryo-EM capsid structures aligned with an RMSD of 2.2 Å (Table 2), a value considerably greater than the intrinsic variability of the structures. Thus, the cryo-EM structures also point to genuine differences between the P22 and Sf6 I-domains despite a shared six-stranded β-barrel fold.
We also evaluated the agreement between the I-domain NMR structures and the corresponding modules from the two available high-resolution cryo-EM structures for P22 and Sf6 capsids (Fig. 2 and Table 2). In the case of the P22 I-domains, the NMR and cryo-EM structures gave an RMSD of 2.0 Å; for Sf6 the RMSD was 3.6 Å. Figure 2 illustrates these differences in a superposition of the NMR structures (dark blue) onto the I-domain extracted from the cryo-EM structures (cyan) from pentamer (top) or hexamer (bottom) subunits. Again, differences in the cryo-EM structures of the I-domains from the pentamers and hexamers due to icosahedral quasi-equivalence are small. In both the P22 and Sf6 I-domains, more substantial differences are seen when the cryo-EM (cyan) and NMR (dark blue) structures are compared (Fig. 2). The largest differences were seen at the periphery of the conserved six-stranded β-barrel motif since it is these peripheral sites that are perturbed in the capsids by intramonomer contacts between the I-domain and the coat protein core and by contacts between I-domains in adjacent coat protein monomers.
FIG 2.
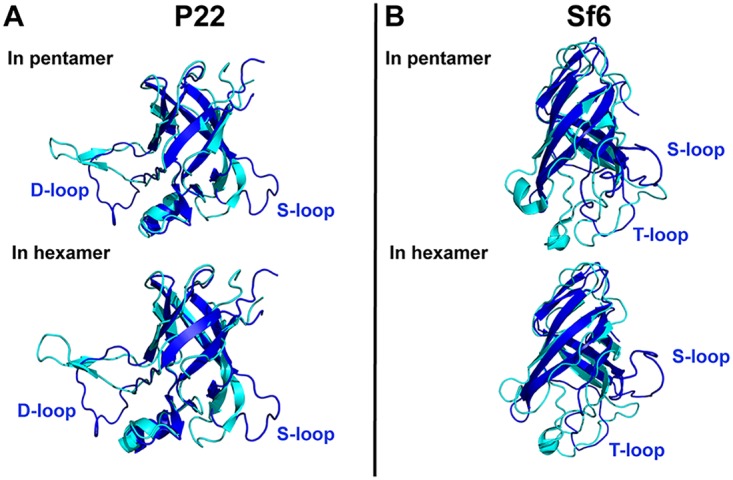
(A and B) Alignment of the P22 and Sf6 I-domain NMR structures (dark blue) to the I-domains from the P22 (PDB code 5UU5 [18]) and Sf6 (PDB code 5L35 [18]) cryo-EM capsid reconstructions (cyan). The I-domains from the cryo-EM structures are extracted from a pentamer and a hexamer subunit, as indicated.
Variations in electrostatic docking of the I-domain to the coat protein core.
The I-domains examined in this study adopt similar but distinct β-barrel structures. We previously noted the marked charge polarity of the P22 I-domain (Fig. 3) (13, 18). The part of the I-domain structure exposed to solution is negatively charged, while the opposite side that docks onto the core of the coat protein is positively charged (13, 18). To explore structural differences in more detail, we calculated electrostatic surface potentials for each of the I-domain NMR structures (Fig. 3) along with the coat protein core (Fig. 4). Since there is no high-resolution structure of the CUS-3 virion, we used I-Tasser to calculate a model of the CUS-3 monomer (22). The electrostatic potential maps show that the positively charged character of the P22 I-domain docking interface (Fig. 3A and Movie S2) switches to nearly neutral in CUS-3 (Fig. 3B and Movie S2) and is predominantly negatively charged in Sf6 (Fig. 3C and Movie S2). The electrostatic differences in the docking interfaces contributed by the I-domains are compensated by changes in charged residues at the cognate binding pockets of the corresponding coat protein cores (Fig. 4). Thus, in P22, the patch from the coat protein core used to dock the corresponding I-domain is negatively charged (Fig. 4A) whereas in CUS-3 it is nearly neutral (Fig. 4B), and in Sf6 it has a slight positively charged character (Fig. 4C).
FIG 3.
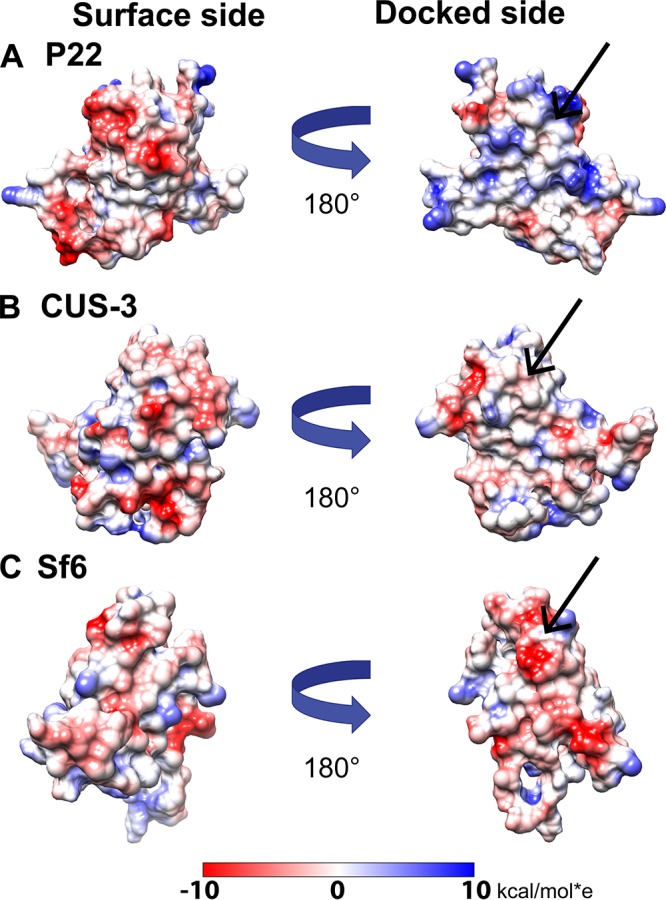
(A to C) Electrostatic properties of the I-domains of P22, CUS-3, and Sf6. Coulombic potential surfaces (red, negative; blue, positive) were calculated with the APBS program (40) on a scale from −10 to +10 kcal/mol·e. The straight arrows indicate the I-domain surface that docks to the coat protein core.
FIG 4.
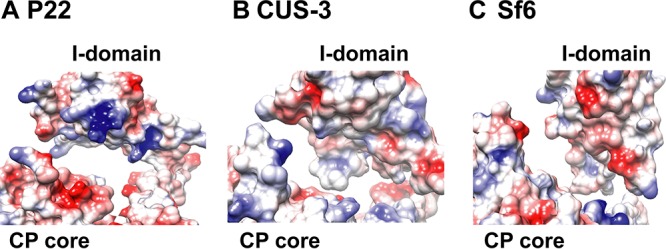
(A to C) Electrostatic surface potentials illustrating the lack of conservation for the interaction interfaces between I-domains and HK97 fold coat protein (CP) cores for P22, CUS-3, and Sf6. Electrostatic surfaces were calculated on a scale from −10 to +10 kcal/mol·e with the APBS program (40). See also Movie S2 in the supplemental material.
Disordered loop regions are poorly conserved.
While the six-stranded β-barrel motif is conserved in the three I-domain NMR structures, loop segments between the β-strands are not. Figure 5 shows backbone dynamics data for the three I-domains calculated from 15N relaxation data (data not shown) using model-free formalism (23). In this approach, the S2 “order” parameter describes the amplitude of backbone 1H-15N bond vibrations on a picosecond-to-nanosecond timescale. Regions with S2 order parameters of ≥0.85, indicative of a rigid structure (blue in Fig. 5), include all of the secondary structure elements in the I-domains, as well as some of the connecting loop and turn segments. Regions with S2 order parameters below 0.85 correspond to flexible or disordered segments (red in Fig. 5). These include the chain termini, the D- and S-loops in the P22 and CUS-3 I-domains, and the S- and T-loops in the Sf6 I-domain. Overall, there is good agreement between regions with low S2 order parameters (Fig. 5) and regions that are imprecisely defined in the NMR structures of the three I-domains (Fig. 1), indicating that the latter reflect flexible protein segments.
FIG 5.

(A to C) S2 order parameters describing backbone dynamics for the I-domains of P22, CUS-3, and Sf6. Rigid regions with an S2 of ≥0.85 are shown in blue, sites that experience dynamics on the picosecond-nanosecond timescale with an S2 of <0.85 are in red. The secondary structures of the I-domains are shown at the bottom of each panel for reference (color coded as described in the legend of Fig. 1), and the flexible loop segments are labeled. The S2 values for P22 were previously published (13) and are shown here for comparison.
DISCUSSION
I-domains evolved embellishments on a conserved folding motif.
The HK97 fold, one of the most abundant protein folds on earth, is found in viruses that infect all three domains of life (1, 3, 7, 24). Often the HK97 fold has elaborations such as the I-domains, which are likely needed to diversify the function of this module for specific adaptations (13, 15). The present work shows that the I-domains themselves exhibit variations on a conserved structural theme that includes differences in secondary structure elements, the locations of flexible loops, and surface electrostatic properties. Variations between I-domains that represent the three main groups of a family of over 150 P22-like phages (8, 9) occur primarily at the periphery and surface of the conserved six-stranded β-barrel motif (Table 3), as seen with other protein folding motifs (25).
TABLE 3.
Summary of differences found between P22, CUS-3, and Sf6 I-domains
| Phage | Dock interface |
D-loop region |
S-loop region |
T-loop region |
||||
|---|---|---|---|---|---|---|---|---|
| Residues | CP core interactiona | Residues | Interaction | Residues | Interaction | Residues | Interaction | |
| P22 | R268–F275, A293–V298, I308–P310 | Electrostatic (I-domain, +; core, −) | A233–N254 | Intercapsomer (across 2-fold axes) | L281–Q291 | Intracapsomer with spine helix and A domain of neighboring subunit | E323–N331 | Intrasubunit, close to E-loop |
| CUS-3 | V264–I270, Q288–V292, I302–P304 | Neutral | H236–T254 | NAb | V276–Q287 | NA | R317–D323 | NA |
| Sf6 | A264–F270, F292–V297, L313–G315 | Electrostatic (I-domain, −; core, +) | N239–A254 | Intracapsomer, interacts with the N-terminal arm of an adjacent subunit | L276–N286 | Intracapsomer with spine helix and I-domain of neighboring subunit | T321–S330 | Intrasubunit, close to E-loop |
CP, coat protein. Plus and minus indicate positive and negative charges, respectively.
NA, not available since there is as yet no high-resolution cryo-EM structure for the CUS-3 capsid.
Functionally important loops are not well conserved.
The flexibility of the P22 I-domain D-loop is critically important for its function, since it participates in intercapsomer salt bridge interactions across the icosahedral 2-fold axis and undergoes a conformational change during the maturation transition from procapsid to dsDNA genome-packaged phage (10, 11, 13). Moreover, the intrinsic flexibility of the D-loop could be important in allowing coat protein monomers to adjust to the necessarily nonequivalent structures required for the asymmetric unit of an icosahedral particle with a triangulation number greater than T=1 (21). Yet despite the connection between the flexibility of the I-domain D-loop (Fig. 5A) and its biological functions in phage P22, the flexibility of the region corresponding to the D-loop is greatly reduced in the I-domain from phage CUS-3 (Fig. 5B) and is entirely absent from the I-domain of phage Sf6 (Fig. 5C). In both the CUS-3 and Sf6 I-domains, the segments corresponding to the D-loop in the P22 I-domain are part of an extension of the β1-β2 hairpin from the I-domain’s six-stranded β-barrel fold (Fig. 1).
The P22 S-loop has a role in modulating capsid size through maintaining the correct flexibility and orientation of the A and I-domains in phage P22 (11). Steric clashes between the two domains can affect the conformation and flexibility of the coat protein. These clashes likely cause a change in the curvature of the capsomers, which influences capsid size (11). The S-loop is disordered in both of the isolated P22 and Sf6 I-domains but becomes structured in the capsids (Fig. 2) due to interactions with other regions of the coat protein. The disorder of the S-loop is greatly diminished in the isolated CUS-3 I-domain, with only one residue showing marginal flexibility (Fig. 5B).
Finally, in the Sf6 I-domain a new loop we term the tangential loop (T-loop) occurs in a region corresponding to the accessory subdomain present in the P22 and CUS-3 structures (Fig. 5C). Native-state hydrogen exchange experiments on the P22 I-domain indicated that the accessory subdomain has a smaller stability to unfolding (~6.5 kcal/mol) than the conserved β-barrel structure (~8.3 kcal/mol) (26). The dynamics data indicate that in contrast to the β-barrel scaffold of the I-domain, flexible regions are poorly conserved and are sometimes replaced by elements or extensions of secondary structure.
Sequence conservation profiles reveal nonconserved loop regions between the I-domains.
We compared our structural results to amino acid sequence conservation profiles for the coat protein I-domains of the P22-like cluster (Fig. 6). We used the Guidance server to align the sequences with CLUSTALW and a Perl script to calculate the conservation profile, as described in Materials and Methods (27, 28). We separately analyzed the P22-like (Fig. 6A), CUS-3-like (Fig. 6B), and Sf6-like (Fig. 6C) groups, as well as the group of 78 nonredundant sequences covering the entire P22-like cluster (Fig. 6D) (8). A larger score indicates lower conservation. We mapped the secondary structures from our P22, CUS-3, and Sf6 NMR structures at the bottom of each of the plots for reference. In the P22-like group (Fig. 6A), the least conserved regions include the D-loop. Since the residues at the tip of the D-loop are critically important for proper capsid assembly and stability in the P22 phage, this observation is unexpected and suggests that the phages in the P22-like group must be evolving alternate stabilizing interactions (11). Within the P22-like group the S-loop, which is important for size determination of the assembly product, is more conserved than the D-loop (11). Overall, in the CUS-3-like group, the I-domain appears to be more conserved than either the P22- or Sf6-like group though it is unclear why this should be. In this instance, the D-loop is well conserved, and the S-loop is somewhat less so. In the Sf6-like group the S-loop was more conserved than the new T-loop. When all 78 phages are compared, the D-loop region shows lower conservation than the S-loop region. However, the S-loop region, while more conserved within the P22- and Sf6-like groups, does not show high conservation in the P22-like phage cluster. Thus, it appears that loop regions, which for P22 are shown to have important roles in capsid stability and morphology, are not conserved. This suggests that the different groups of phages evolved either alternate functions or different interactions for the same convergent functional purpose for each of the loops.
FIG 6.

Sequence conservation of I-domains in the P22-like phage cluster. Large and small scores indicate weak and strong sequence conservation, respectively. (A) P22 family. (B) CUS-3 family. (C) Sf6 family. (D) Overall comparison of I-domains from a database of 78 P22-like phages. The gap in panel D around position 340 is due to an insertion in 2 of the 78 phages. The secondary structures of the P22, CUS-3, and Sf6 I-domains are shown at the bottom of each plot. In panel D, the secondary structures of all represented families are indicated, and the locations of the flexible D-, S-, and T-loops are labeled.
Diversity in surface features.
Differences in structures are also manifested in the surfaces of the I-domains, which in turn affect the surface properties of the capsids. The high-resolution cryo-EM structure of the P22 capsid (18) is shown in Fig. 7A, with the hexons and pentons of the I-domains shown in blue and cyan, respectively. The D-loops are shown in red and black to highlight the noncovalent interactions formed between the I-domains of P22. While the D-loop is disordered in the isolated I-domain, and when the I-domain is part of the full-length coat protein, it becomes structured to stabilize intercapsomer interactions across the icosahedral 2-fold axes (13, 18). This gives the pentons the starfish-like appearance in the P22 capsid structure (Fig. 7A) but not in the Sf6 capsid structure where the pentons have a pinwheel-like shape (Fig. 7B). The region corresponding to the D-loop in Sf6 forms an ordered extension of the β1-β2 hairpin (Fig. 7B) that tucks in toward the coat protein core structure, in contrast to the intercapsomer interaction present in phage P22.
FIG 7.

Comparison of D-loop interactions in P22 and Sf6 and their effects on capsid surface morphology. (A) When mature P22 capsids assemble (18), the dynamic D-loop becomes structured to form salt bridges across the capsid 2-fold symmetry axis (10, 13). On the surface of the capsid, the D-loop interaction (red in subunits A, D, E, and F and black in subunits B, C, and G for the P22 asymmetric unit) gives the pentons (cyan) a starfish-like appearance. (B) The D-loops are not present in Sf6 capsid (17), and the corresponding region that forms an extension of the β1-β2 hairpin tucks into the HK97 core of the coat protein (red in subunits A, B, C, and D and black in subunits E, F, and G in the Sf6 asymmetric unit). Consequently, the pentons in Sf6 have a pinwheel-like shape compared to the starfish in P22.
Another difference between the three I-domains is in the number of TUT (threonine-hydrophobic-threonine) sequence motifs. The TUT motif is found once in P22 (Fig. 8A), twice in CUS-3 (Fig. 8B), and seven times in the Sf6 I-domain (Fig. 8C). TUT motifs are presented on the capsid surface and have been suggested to be involved in carbohydrate binding (17). Although a carbohydrate-binding function still remains to be verified experimentally, perhaps binding to glycans on target cell surfaces is one driver of I-domain evolution.
FIG 8.
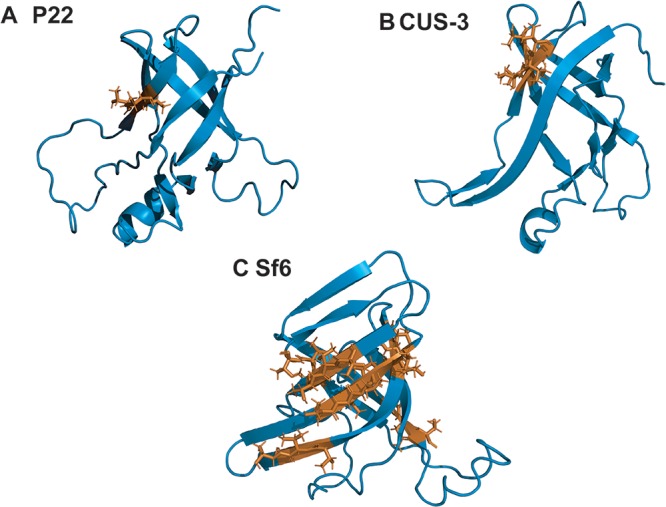
(A to C) TUT (threonine-hydrophobic-threonine) sequence motifs in the I-domains of P22, CUS-3, and Sf6. The TUT motifs are shown in orange.
In conclusion, this study highlights that the six-stranded anti-parallel β-barrel protein module that occurs in the HK97 folds of coat proteins from the P22-like phage cluster has important differences in structural properties. These differences emphasize the complex coevolution of structure and function of phage and virus capsid proteins. These include the following: (i) variations in elements of secondary structure and functionally important flexible loop regions that participate in capsid assembly, (ii) differences in charges at the interface used to dock the I-domain to the coat protein core, and (iii) differences in TUT sequence motifs displayed on the capsid surface that could comprise carbohydrate binding moieties. Thus, while the HK97 fold is one of the most ubiquitous structural motifs in nature, the many embellishments that have been added to the structure suggest that a significant degree of structural adaptability is required to fulfill its versatile range of specific functional requirements.
MATERIALS AND METHODS
NMR structure determination.
Cloning, expression, and purification protocols, along with NMR assignments for the three I-domains from P22, CUS-3, and Sf6 were published previously (29–31). NMR data were recorded on 600 and 800 MHz Varian Inova NMR spectrometers equipped with cryogenic probes. Samples for NMR contained 1.7 mM CUS-3 or 0.9 mM Sf6 I-domain in 20 mM sodium phosphate, pH 6.0, at 25° C. NMR spectra were processed with the programs FELIX and NMRPipe and analyzed with the CcpNMR Analysis suite of programs on the NMRbox platform (32–34). Distance restraints for NMR structures were obtained from three-dimensional (3D) 15N and 13C heteronuclear single quantum coherence-nuclear Overhauser effect spectroscopy (HSQC-NOESY) experiments, acquired with NOE mixing times of 150 and 100 ms, respectively. Dihedral restraints (ϕ, ψ, and χ1) were calculated from Cα, Cβ, Hα, Hβ, NH, and C′ chemical shifts with the program TALOS-N (35). H-bond donors and acceptors were identified using long-range HNCO spectra recorded in transverse relaxation-optimized spectroscopy (TROSY) mode on perdeuterated 2H, 13C, and 15N samples of 1.3 mM CUS-3 and 1.5 mM Sf6. NMR structure calculations were performed with the programs XPLOR-NIH and ARIA (36, 37). The 25 lowest-energy NMR structures for each protein were kept for analysis (Fig. 1).
NMR characterization of protein dynamics.
R1 and R2 (where R1 and R2 are longitudinal and transverse relaxation rate constants, respectively) and 1H-15N NOE 15N relaxation experiments were collected on a Varian INOVA 600 MHz instrument. Interleaved relaxation delays of 0.05, 0.13, 0.21, 0.49, 0.57, 0.71, 0.99, and 1.1 s were used for R1 experiments. For R2 experiments, interleaved relaxation delays of 0.01, 0.03, 0.05, 0.07, 0.09, 0.11, 0.15, and 0.25 s were used. Relaxation data were fit to exponential decay functions to determine relaxation time constants. 1H-15N NOE data were obtained from an experiment in which the proton spectrum was saturated for 3 s, and a control experiment in which the saturation was replaced by a preacquisition delay of equivalent length. Errors for the 15N relaxation values were calculated as previously described (38). The R1 and R2 relaxation times, along with 1H-15N NOE cross-relaxation data were used to determine S2 order parameters and global correlation times according to model-free formalism with the program TENSOR2 (23, 39). The global correlation times were 11.0 ns for CUS-3 and 12.8 ns for Sf6 while the previously reported value for P22 was 11.0 ns (13).
Electrostatic potential maps.
Calculations of electrostatic potentials were done with the PDB2PQR server (http://nbcr-222.ucsd.edu/pdb2pqr_2.0.0/) using the Adaptive Poisson-Boltzmann Solver (APBS) (40, 41). The structure closest to the NMR ensemble average was used for each I-domain. Electrostatic potentials for the coat protein cores were calculated using the structure of P22 (PDB code 5UU5) (18) and the 2.9-Å-resolution structure of Sf6 (PDB code 5L35) (17). For CUS-3, a high-resolution structure of the capsid is currently not available, so the coat protein was modeled using Iterative Threading ASSEmbly Refinement (I-TASSER) with its closest homolog, the P22 coat protein structure (22).
Comparison of conservation profiles within the P22-like cluster.
To analyze the sequence conservation profiles, the Guidance server was used to align full-length coat protein sequences of P22-like phages with CLUSTALW using a database provided by Sherwood Casjens (University of Utah) (27). Out of an initial database of coat proteins from 151 phages, nearly identical sequences were removed, leaving the 78 sequences used in this study. The conservation profiles were calculated with 39 sequences for P22-like, 18 sequences for CUS-3-like, and 18 sequences for Sf6-like phages. Three sequences were left out of the individual groups as they were too distantly related (9). However, these three sequences were included in the calculation of the 78 phages as they are considered to belong to the P22-like cluster. The alignment results were run through two scripts to first linearize the sequence alignment data and then quantify conservation between sequences (28). The conservation profiles were plotted using a smoothing window of 7 residues. Gaps that were found in 50% of the sequences were removed during conservation profile calculations. From the full-length coat protein sequence alignments and conservation calculations, residues 220 to 345 that correspond to the I-domains were reported in the results.
Data availability.
NMR structures for the I-domains have been deposited in the Protein Data Bank under accession numbers 6MNT for CUS-3, 6MPO for Sf6, and 2M5S for P22. The structure closest to the ensemble average is denoted as structure 1 in the PDB-deposited NMR bundles.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grant R01 GM076661 to C.M.T. and A.T.A.
We thank Vitaliy Gorbatyuk (University of Connecticut [UConn]) and Mark Maciejewski (UConn Health) for help with NMR experiments, J. Peter Gogarten (UConn), Nikhil Ram Mohan (Boston College), and Sherwood Casjens (University of Utah) for helpful discussions on sequence analysis, and Kristin Parent for the Sf6 and CUS-3 phages.
We declare that we have no conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00007-19.
REFERENCES
- 1.Abrescia NG, Bamford DH, Grimes JM, Stuart DI. 2012. Structure unifies the viral universe. Annu Rev Biochem 81:795–822. doi: 10.1146/annurev-biochem-060910-095130. [DOI] [PubMed] [Google Scholar]
- 2.Bamford DH, Grimes JM, Stuart DI. 2005. What does structure tell us about virus evolution? Curr Opin Struct Biol 15:655–663. doi: 10.1016/j.sbi.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 3.Huet A, Makhov AM, Huffman JB, Vos M, Homa FL, Conway JF. 2016. Extensive subunit contacts underpin herpesvirus capsid stability and interior-to-exterior allostery. Nat Struct Mol Biol 23:531–539. doi: 10.1038/nsmb.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dai X, Zhou ZH. 2018. Structure of the herpes simplex virus 1 capsid with associated tegument protein complexes. Science 360:eaao7298. doi: 10.1126/science.aao7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Homa FL, Huffman JB, Toropova K, Lopez HR, Makhov AM, Conway JF. 2013. Structure of the pseudorabies virus capsid: comparison with herpes simplex virus type 1 and differential binding of essential minor proteins. J Mol Biol 425:3415–3428. doi: 10.1016/j.jmb.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuan S, Wang J, Zhu D, Wang N, Gao Q, Chen W, Tang H, Wang J, Zhang X, Liu H, Rao Z, Wang X. 2018. Cryo-EM structure of a herpesvirus capsid at 3.1 Å. Science 360:eaao7283. doi: 10.1126/science.aao7283. [DOI] [PubMed] [Google Scholar]
- 7.Pietila MK, Laurinmaki P, Russell DA, Ko CC, Jacobs-Sera D, Hendrix RW, Bamford DH, Butcher SJ. 2013. Structure of the archaeal head-tailed virus HSTV-1 completes the HK97 fold story. Proc Natl Acad Sci U S A 110:10604–10609. doi: 10.1073/pnas.1303047110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casjens SR, Grose JH. 2016. Contributions of P2- and P22-like prophages to understanding the enormous diversity and abundance of tailed bacteriophages. Virology 496:255–276. doi: 10.1016/j.virol.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casjens SR, Thuman-Commike PA. 2011. Evolution of mosaically related tailed bacteriophage genomes seen through the lens of phage P22 virion assembly. Virology 411:393–415. doi: 10.1016/j.virol.2010.12.046. [DOI] [PubMed] [Google Scholar]
- 10.D'Lima NG, Teschke CM. 2015. A molecular staple: D-Loops in the I-domain of bacteriophage P22 coat protein make important intercapsomer contacts required for procapsid assembly. J Virol 89:10569–10579. doi: 10.1128/JVI.01629-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suhanovsky MM, Teschke CM. 2011. Bacteriophage P22 capsid size determination: roles for the coat protein telokin-like domain and the scaffolding protein amino-terminus. Virology 417:418–429. doi: 10.1016/j.virol.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suhanovsky MM, Teschke CM. 2015. Nature's favorite building block: deciphering folding and capsid assembly of proteins with the HK97-fold. Virology 479-480:487–497. doi: 10.1016/j.virol.2015.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rizzo AA, Suhanovsky MM, Baker ML, Fraser LC, Jones LM, Rempel DL, Gross ML, Chiu W, Alexandrescu AT, Teschke CM. 2014. Multiple functional roles of the accessory I-domain of bacteriophage P22 coat protein revealed by NMR structure and cryoEM modeling. Structure 22:830–841. doi: 10.1016/j.str.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suhanovsky MM, Teschke CM. 2013. An intramolecular chaperone inserted in bacteriophage P22 coat protein mediates its chaperonin-independent folding. J Biol Chem 288:33772–33783. doi: 10.1074/jbc.M113.515312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teschke CM, Parent KN. 2010. “Let the phage do the work”: using the phage P22 coat protein structures as a framework to understand its folding and assembly mutants. Virology 401:119–130. doi: 10.1016/j.virol.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harprecht C, Okifo O, Robbins KJ, Motwani T, Alexandrescu AT, Teschke CM. 2016. Contextual role of a salt bridge in the phage P22 coat protein I-domain. J Biol Chem 291:11359–11372. doi: 10.1074/jbc.M116.716910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao H, Li K, Lynn AY, Aron KE, Yu G, Jiang W, Tang L. 2017. Structure of a headful DNA-packaging bacterial virus at 2.9 A resolution by electron cryo-microscopy. Proc Natl Acad Sci U S A 114:3601–3606. doi: 10.1073/pnas.1615025114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hryc CF, Chen DH, Afonine PV, Jakana J, Wang Z, Haase-Pettingell C, Jiang W, Adams PD, King JA, Schmid MF, Chiu W. 2017. Accurate model annotation of a near-atomic resolution cryo-EM map. Proc Natl Acad Sci U S A 114:3103–3108. doi: 10.1073/pnas.1621152114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parent KN, Tang J, Cardone G, Gilcrease EB, Janssen ME, Olson NH, Casjens SR, Baker TS. 2014. Three-dimensional reconstructions of the bacteriophage CUS-3 virion reveal a conserved coat protein I-domain but a distinct tailspike receptor-binding domain. Virology 464-465:55–66. doi: 10.1016/j.virol.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holm L, Laakso LM. 2016. Dali server update. Nucleic Acids Res 44:W351–W355. doi: 10.1093/nar/gkw357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caspar DL, Klug A. 1962. Physical principles in the construction of regular viruses. Cold Spring Harbor Symp Quant Biol 27:1–24. doi: 10.1101/SQB.1962.027.001.005. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lipari G, Szabo A. 1982. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and validity. J Am Chem Soc 104:4546–4559. doi: 10.1021/ja00381a009. [DOI] [Google Scholar]
- 24.Bamford DH, Burnett RM, Stuart DI. 2002. Evolution of viral structure. Theor Popul Biol 61:461–470. doi: 10.1006/tpbi.2002.1591. [DOI] [PubMed] [Google Scholar]
- 25.Guardino KM, Sheftic SR, Slattery RE, Alexandrescu AT. 2009. Relative stabilities of conserved and non-conserved structures in the OB-fold superfamily. Int J Mol Sci 10:2412–2430. doi: 10.3390/ijms10052412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newcomer RL, Fraser LC, Teschke CM, Alexandrescu AT. 2015. Mechanism of protein denaturation: partial unfolding of the P22 coat protein I-domain by urea binding. Biophys J 109:2666–2677. doi: 10.1016/j.bpj.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Penn O, Privman E, Ashkenazy H, Landan G, Graur D, Pupko T. 2010. GUIDANCE: a web server for assessing alignment confidence scores. Nucleic Acids Res 38:W23–W28. doi: 10.1093/nar/gkq443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swithers KS, Senejani AG, Fournier GP, Gogarten JP. 2009. Conservation of intron and intein insertion sites: implications for life histories of parasitic genetic elements. BMC Evol Biol 9:303. doi: 10.1186/1471-2148-9-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tripler TN, Maciejewski MW, Teschke CM, Alexandrescu AT. 2015. NMR assignments for the insertion domain of bacteriophage CUS-3 coat protein. Biomol NMR Assign 9:333–336. doi: 10.1007/s12104-015-9604-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tripler TN, Teschke CM, Alexandrescu AT. 2017. NMR assignments for the insertion domain of bacteriophage Sf6 coat protein. Biomol NMR Assign 11:35–38. doi: 10.1007/s12104-016-9716-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rizzo AA, Fraser LC, Sheftic SR, Suhanovsky MM, Teschke CM, Alexandrescu AT. 2013. NMR assignments for the telokin-like domain of bacteriophage P22 coat protein. Biomol NMR Assign 7:257–260. doi: 10.1007/s12104-012-9422-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maciejewski MW, Schuyler AD, Gryk MR, Moraru II, Romero PR, Ulrich EL, Eghbalnia HR, Livny M, Delaglio F, Hoch JC. 2017. NMRbox: a resource for biomolecular NMR computation. Biophys J 112:1529–1534. doi: 10.1016/j.bpj.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293. [DOI] [PubMed] [Google Scholar]
- 34.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED. 2005. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 35.Shen Y, Bax A. 2013. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J Biomol NMR 56:227–241. doi: 10.1007/s10858-013-9741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rieping W, Habeck M, Bardiaux B, Bernard A, Malliavin TE, Nilges M. 2007. ARIA2: automated NOE assignment and data integration in NMR structure calculation. Bioinformatics 23:381–382. doi: 10.1093/bioinformatics/btl589. [DOI] [PubMed] [Google Scholar]
- 37.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. 2003. The Xplor-NIH NMR molecular structure determination package. J Magn Reson 160:65–73. doi: 10.1016/S1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 38.Alexandrescu AT, Shortle D. 1994. Backbone dynamics of a highly disordered 131 residue fragment of staphylococcal nuclease. J Mol Biol 242:527–546. doi: 10.1006/jmbi.1994.1598. [DOI] [PubMed] [Google Scholar]
- 39.Dosset P, Hus JC, Blackledge M, Marion D. 2000. Efficient analysis of macromolecular rotational diffusion from heteronuclear relaxation data. J Biomol NMR 16:23–28. doi: 10.1023/A:1008305808620. [DOI] [PubMed] [Google Scholar]
- 40.Jurrus E, Engel D, Star K, Monson K, Brandi J, Felberg LE, Brookes DH, Wilson L, Chen J, Liles K, Chun M, Li P, Gohara DW, Dolinsky T, Konecny R, Koes DR, Nielsen JE, Head-Gordon T, Geng W, Krasny R, Wei GW, Holst MJ, McCammon JA, Baker NA. 2018. Improvements to the APBS biomolecular solvation software suite. Protein Sci 27:112–128. doi: 10.1002/pro.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. 2004. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res 32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhattacharya A, Tejero R, Montelione GT. 2007. Evaluating protein structures determined by structural genomics consortia. Proteins 66:778–795. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NMR structures for the I-domains have been deposited in the Protein Data Bank under accession numbers 6MNT for CUS-3, 6MPO for Sf6, and 2M5S for P22. The structure closest to the ensemble average is denoted as structure 1 in the PDB-deposited NMR bundles.