

SUMMARY
Mammalian chromosomes are partitioned into A/B compartments and topologically associated domains (TADs). The inactive X (Xi) chromosome, however, adopts a distinct conformation without evident compartments or TADs. Here, through exploration of an architectural protein, SMCHD1, we probe how the Xi is reconfigured during X-chromosome inactivation. A/B compartments are first fused into “S1” and “S2” compartments, coinciding with Xist spreading into gene-rich domains. SMCHD1 then binds S1/S2 compartments and merges them to create a compartment-less architecture. Contrary to current views, TADs remain on the Xi but in an attenuated state. Ablating SMCHD1 results in a persistent S1/S2 organization and strengthening of TADs. Furthermore, loss of SMCHD1 causes regional defects in Xist spreading and erosion of heterochromatic silencing. We present a step-wise model for Xi folding, where SMCHD1 attenuates a hidden layer of Xi architecture to facilitate Xist spreading.
Graphical Abstract
INTRODUCTION
X chromosome inactivation (XCI) balances the dosage of X-linked genes between mammalian females and males. For over 50 years, XCI has served as an epigenetic paradigm (Disteche, 2012; Lee, 2011; Starmer and Magnuson, 2009; Wutz, 2011) and, more recently, as a model to study 3D genome organization (Fraser, 2016; Jegu et al., 2017). Molecular mapping of genome topology has revealed salient features of chromosomal architecture (Bonev and Cavalli, 2016; Schmitt et al., 2016). For instance, mammalian chromosomes are organized into alternating “A/B compartments” — megabase-sized spatial compartments harboring chromatin of similar states, with A compartments being actively transcribed and gene-rich, and B compartments being transcriptionally inactive and gene-poor (Bickmore and van Steensel, 2013; Lieberman-Aiden et al., 2009). At a finer scale, mammalian chromosomes are partitioned into “topologically associated domains” (TADs) of ~1 megabase (Mb) average size. Within TADs, genetic elements frequently contact each other, but they rarely do so between TADs (Dixon et al., 2012). Although these chromosome structures are currently thought to partition functional domains for coordinated gene expression, mechanistic and functional relationships are not fully understood (Dekker and Mirny, 2016; Martinez-Jimenez and Odom, 2016; Ong and Corces, 2014).
The mammalian inactive X chromosome (Xi) is unique in its reconfiguration into an unusual chromosome conformation. Whereas the active X (Xa) is similar to autosomes in being organized into A/B compartments and TADs, the ~60 A/B compartments and ~110 TADs originally on the Xa are thought to be eliminated globally during XCI (Giorgetti et al., 2016; Minajigi et al., 2015; Pandya-Jones and Plath, 2016). The Xi is instead folded into two “megadomains” of 73 and 93 Mb in mice, separated by a boundary near the macrosatellite repeat, Dxz4 (Deng et al., 2015; Giorgetti et al., 2016; Minajigi et al., 2015; Rao et al., 2014). The noncoding Xist RNA plays a central role in structural reconfiguration of the Xi (Giorgetti et al., 2016; Minajigi et al., 2015; Splinter et al., 2011), with expression of Xist inducing megadomains, and deleting Xist restoring selective TADs along the Xi. Without A/B compartments and TADs, the Xi is currently considered to be “unstructured”.
In consideration of protein factors that may regulate the structure of the Xi, of particular interest are architectural factors in the family of structural-maintenance-of-chromosomes (SMC) proteins. Two family members, the cohesins and condensins, are known to play key roles in chromosomal architecture (Hirano, 2016; Jeppsson et al., 2014; Merkenschlager and Odom, 2013; Wood et al., 2010). In mammals, cohesins and CCCTC-binding factor (CTCF), another architectural protein, drive TAD formation, with cohesins enabling chromatin looping within TADs and CTCF defining TAD boundaries (Rao et al., 2017; Schwarzer et al., 2017). A role for condensins has been precedented by nematode dosage compensation, where condensins bind at TAD borders to downregulate two X chromosomes in the hermaphrodite (XX) to achieve dosage parity with the XO male (Crane et al., 2015; Wood and Bickmore, 2015). To date, no Xi-specific SMC complex has been identified in mammals. However, previous studies have shown that a variant — the structural-maintenance-of-chromosomes hinge domain containing 1 (SMCHD1; Fig. 1A) — is enriched on the Xi (Blewitt et al., 2008). Mutations of Smchd1 in mice cause defective X chromosome silencing and female embryonic lethality (Blewitt et al., 2008; Blewitt et al., 2005). How SMCHD1 functions mechanistically is not known. Here we probe a potential architectural role for SMCHD1 and investigate the mechanism by which it shapes the Xi and represses gene expression.
Figure 1: SMCHD1-sensitive vs SMCHD1-insensitive genes of the Xi.

(A) Structure of SMCHD1 and canonical SMC dimers. N-terminus, N. C-terminus, C.
(B) Immuno-RNA-FISH for SMCHD1 and Xist RNA. Top two rows: Wild-type (WT) and XaWTXiΔXist female fibroblasts. Fibroblasts are SV40-large-T-immortalized and are therefore polyploid (2n to 8n). Bottom: Male MEF lines harboring WT Xist transgene (♂X+P). Number of cells with SMCHD1 foci co-localizing with the Xist is shown.
(C) An RNA-seq MA plot shows upregulation of Pcdh α cluster (green) and a subset of X-linked genes (red) in Smchd1-/- cells. Gray dots, genes not differentially expressed.
(D) Cumulative distribution plots (CDP) of fold-changes of expressed X-linked genes (red) versus autosomal genes (black) between various WT and Smchd1-/- NPC clones as indicated. P-values by Wilcoxon ranked sum (Wilcox) test (unpaired, one-sided).
(E) Workflow showing classification of X-linked genes into SMCHD1-sensitive vs -insensitive genes.
(F) FPM-normalized RNA-seq coverage profiles showing failed silencing of 2 SMCHD1-sensitive genes. comp, all reads. cas, cas-specific reads (Xa). mus, mus-specific reads (Xi). Scales shown in brackets.
(G) Scatter plots comparing degree of allelic skewing (%mus) between various clones, as indicated. Plotted are 230 genes subject to XCI in NPCs. Red dots, SMCHD1-sensitive genes. Blue dots, SMCHD1-insensitive genes.
(H) Probability density plots of %mus for 73 concordant SMCHD1-sensitive genes.
(I) X-chromosomal locations of SMCHD1-sensitive and -insensitive genes.
(J) Clustering of SMCHD1-sensitive and -insensitive genes. Left: Distribution of distances from a SMCHD1-sensitive gene to the nearest sensitive (red) or insensitive gene (blue). Right: Distribution of distances from a SMCHD1-insensitive gene to the nearest sensitive (red) or insensitive gene (blue). *, P=3.2×10−8. **, P=1.6×10−8 (Wilcox test).
RESULTS
A SMCHD1 loss-of-function system reveals selective sensitivity of Xi genes
Xist RNA has been established as a scaffold upon which epigenetic factors are recruited to the Xi (Chu et al., 2015; McHugh et al., 2015; Minajigi et al., 2015). Among >100 factors identified in a recent proteomic study was SMCHD1 (Minajigi et al., 2015). To test if SMCHD1 recruitment requires Xist, we performed SMCHD1 immunofluorescence in female mouse fibroblasts (MEF) in which Xist is conditionally deleted from the Xi [XaWTXiΔXist (Zhang et al., 2007)]. Whereas wild-type (WT) fibroblasts showed prominent SMCHD1 foci co-localizing with the Xist cloud, XaWTXiΔXist cells showed no SMCHD1 enrichment despite normal SMCHD1 levels (Fig. 1B, S1A). To test sufficiency, we examined autosomes carrying an inducible Xist transgene in male and female MEFs (Jeon and Lee, 2011). Xist induction resulted in appearance of a large colocalizing SMCHD1 focus (Fig. 1B, ♂X+P; S1B, ♀X+P). Murine Xist RNA is therefore both necessary and sufficient to direct SMCHD1 in cis to the Xi, consistent with human studies (Kelsey et al., 2015; Nozawa et al., 2013).
We established a loss-of-function system to explore the role of SMCHD1 in XCI using mouse embryonic stem (ES) cells, an ex vivo model for XCI. Prior studies suggested that only a minority (~10%) of X-linked genes are sensitive to SMCHD1 (Blewitt et al., 2008; Gendrel et al., 2012; Gendrel et al., 2013; Mould et al., 2013). However, these studies could not distinguish between Xa versus Xi expression. To overcome this technical limitation, we utilized a hybrid cell line that carries one Mus musculus X-chromosome (Xmus) and one Mus castaneus X (Xcas). This TsixTST/+ cell line also harbors a mutated mus Tsix allele that thereby forces Xmus to be Xi (Ogawa et al., 2008), enabling usage of >600,000 X-linked sequence polymorphisms to discriminate Xa versus Xi expression. We then used CRISPR-Cas technology to derive Smchd1-/- mutant clones (Fig. S1C–F), with each confirmed as deletions (Fig. S1F, S2A). Below, multiple Smchd1-/- and WT (Smchd1+/+) clones were examined, with biological replicates yielding similar results.
To obtain a population with completed XCI, we differentiated ES cells into neural progenitor cells (NPCs, Fig. S1G–H) and isolated XX subclones to circumvent a tendency for Smchd1-/- cells to become XO during differentiation (Fig. S1I,J). RNA-seq analysis of two WT and two Smchd1-/- clones showed that a majority (70%; 892/1275) of differentially expressed genes were upregulated (Fig. 1C, S2B). Apart from expected autosomal changes (e.g., protocadherin α (Pcdh α) (Gendrel et al., 2013; Mould et al., 2013)(Fig. 1C, S2C)), a fraction of X-linked genes was also upregulated (Fig. 1C, S2B). Cumulative distribution plots showed a significant right shift of X-linked gene expression (red) relative to autosomal profiles (black) in Smchd1-/- cells (Fig. 1D). Approximately 30% (383/1275) of genes were downregulated, but they were not enriched on the X chromosome (Fig. S2B). The X chromosome was therefore disproportionately upregulated by ablating Smchd1.
To determine whether upregulation occurred on the Xa or Xi, we performed allelic analyses and observed an Xi-specific upregulation (Fig. 1E–H, Fig. S2D–G). 230 of >1000 total X-linked genes passed our analysis pipeline and were identified as genes subject to XCI in NPCs (Xi genes)(Fig. 1E). We quantified the extent of allelic skewing by computing percent expression from Xmus (Xi) alleles (%mus) for the 230 Xi genes and then examined the difference between WT and Smchd1−/- clones. For WT clones 1 and 2, the degree of allelic skewing was extreme with values at or near 0% (Fig. 1G, left panel), consistent with XCI. On the other hand, in both Smchd1−/- clones, there was significant upward movement along the y-axis (Fig. 1G, middle panels), indicating a derepression of Xi genes. Two subpopulations were discernible. A subset of 131 genes remained inactivated (blue dots) — henceforth called “SMCHD1-insensitive.” The remaining 99 failed to be silenced (red dots) — henceforth called “SMCHD1-sensitive” (90 in clone 1, 82 in clone 2) — as illustrated by Mecp2 and Ogt (Fig. 1F). This failure of silencing occurred to varying degrees (Fig. 1G,H).
Between two independent Smchd1-/- clones, 73 Xi genes were concordantly dependent on SMCHD1 (P<0.0002 in comparison to a model generated by random sampling; Fig. S2E). The degree of derepression was also well-correlated between clones (r=0.78, Fig. 1G, right panel) and biological replicates (r=0.76 in clone 1, 0.88 in clone 2; Fig. S2D). Probability density plots demonstrated similar propensity towards expression of 73 concordant Xmus alleles in both Smchd1−/- clones (Fig. 1H), while Xa (Xcas) expression was unchanged (Fig. S2F). Increased Xi expression resulted in up to 2-fold upregulation of total expression, consistent with an additive effect of Xi and Xa alleles (Fig. S2G). While SMCHD1-sensitive genes were found throughout the Xi, they showed a tendency to cluster (Fig. 1I,J)(Gendrel et al., 2013). Thus, SMCHD1 loss results in failure to silence up to 43% of Xi genes (99/230)— a much larger fraction than previously reported (Gendrel et al., 2013; Mould et al., 2013).
SMCHD1 regulates spreading of Xi heterochromatin
Previous work suggested that SMCHD1 is not required for Xist spreading or trimethylation of histone H3 at lysine 27 (H3K27me3) by Polycomb repressive complex 2 (PRC2), as SMCHD1-deficient cells still exhibited large cytological foci of Xist and H3K27me3 (Blewitt et al., 2008). Because cytology lacks molecular resolution, we performed allele-specific ChIP-seq to map H3K27me3 and the activating H3K4me3 mark. In WT clones, H3K27me3 was enriched on the Xi, whereas H3K4me3 was enriched on the Xa, as expected (Fig. 2A). In Smchd1-/- clones, the pattern changed significantly, particularly for SMCHD1-sensitive genes. Promoters for SMCHD1-sensitive genes became marked by H3K4me3 and gene bodies became depleted of H3K27me3 (Fig. 2A, red shading). H3K27me3-depleted regions were sharply demarcated, with boundaries coinciding with gene borders — a chromatin signature reminiscent of genes that escape XCI (escapees; e.g., Kdm5c, Fig. 2A, and Eif2s3x, Fig. 2B). Failure of silencing of SMCHD1-sensitive genes correlated with gain of H3K4me3 (Fig. S3A, left) and loss of H3K27me3 (Fig. S3A, right), indicating an erosion of heterochromatin. A total of 126 Xi genes belonged to this class — henceforth called “Class I” — for which SMCHD1 depletion resulted in loss of H3K27me3 and gain of H3K4me3 in NPCs. The total number (126) was defined by chromatin marks and therefore was greater than the 99 defined strictly by Xi-expression. However, the overlap between the two subgroups was high (78 genes), with Class I genes accounting for nearly all of the X-upregulation (Fig. 2C).
Figure 2. Segmental erosion of H3K27me3 domains reveals SMCHD1’s role in spreading heterochromatin.

(A) H3K4me3 and H3K27me3 profiles for representative Class I genes (red-shaded area) and an escapee (Kdm5c). cas, mus, comp as defined in Fig 1.
(B) H3K4me3 and H3K27me3 profiles for representative Class II genes (yellow-shaded area). Zfx, a Class I gene. Eif2s3x, an escapee.
(C) CDPs of fold-changes in gene expression between Smchd1-/- and WT cells for autosomal genes (black), Class I genes (red), and all other X-linked genes (black). P-values by Wilcox test (unpaired, one-sided). Between Class I versus autosomal genes (red), and all other X-linked genes versus autosomal genes (blue).
(D) For each of three gene classes, comparison of allelic skewing in expression (top), allelic skewing of H3K4me3 peaks at promoters (middle), and H3K27me3 enrichment in gene bodies (bottom) between Smchd1-/- (y-axes) versus WT (x-axes) cells.
(E) Failure of H3K27me3 spreading covering intergenic regions and gene bodies of Class I genes (blue-shaded area).
(F) Chromosomal locations of various categories of X-linked genes. We segmented the X into 400-kb bins and plotted the number of genes for each category.
(G) Nearest neighbor analysis: Box plots showing distance relationships of Class I, II, and III genes to each other. E.g., the left panel shows the distribution of distances from a Class I gene to the nearest Class I (red), Class II (yellow), or Class III (blue) gene. NS: not significant (P>0.05). a, P=3.3×10−14. b, P=6.3×10−16. c, P=5.0×10−11. d, P=9.9×10−11(Wilcox test).
(H) Immuno-RNA-FISH for H3K27me3 and Xist RNA on WT and Smchd1-/- NPCs. Number of cells with an Xist cloud and a co-localizing H3K27me3 focus shown.
Interestingly, altered chromatin states were not limited to Class I genes. Another class consisting of 80 genes — henceforth Class II — acquired H3K4me3 at promoters, but remained covered by H3K27me3 in gene bodies (Fig. 2B, yellow shading). Despite acquiring H3K4me3, Class II genes remained silent (Fig. 2D) and were therefore only partially responsive to SMCHD1 depletion. Overall, 58% of Xi genes were responsive to SMCHD1, either at the chromatin and/or gene expression levels. The remaining 42% of Xi genes were unresponsive and categorized as Class III. Loss of SMCHD1 also caused erosion of heterochromatin in nongenic regions (Fig. 2E, blue shading), with ~70% of intergenic regions flanked by Class I genes becoming depleted of H3K27me3 (Fig. S3B), indicating a heterochromatin “spreading” defect around Class I genes (Fig. 2E). Class I genes tend to co-cluster within gene-rich regions (r=0.63; Fig. 2F,G). Nearest neighbor analysis showed that Class I genes have shortest distances to other Class I genes, and longer distances to Class II (partially sensitive) and Class III (resistant) genes (Fig. 2G; left panel). Reciprocally, Class III genes showed shortest distances to other Class III genes, and longer distances to Class I and II genes (Fig. 2G; right panel). Altogether, these data demonstrate that, although H3K27me3 signals appear unaffected cytologically on the Xi (Fig. 2H), ablating Smchd1 significantly disrupts spreading of H3K27me3 at the regional level.
Defective spreading of Xist RNA
Because Xist RNA recruits PRC2 (Zhao et al., 2008), the regional loss of H3K27me3 in Smchd1-/- cells suggested a defect in Xist spreading. To map Xist binding, we performed CHART-seq, a genomic mapping method for RNA in which chromatin is isolated using complementary oligonucleotides for the RNA of interest (Simon et al., 2013). In two biological replicates, Xist RNA was broadly enriched across the X chromosome (Fig. 3A,S3C)(Engreitz et al., 2013; Simon et al., 2013). For both Smchd1-/- and WT cells, Xist showed high correlation with H3K27me3 (r=0.95 for Smchd1-/-; r=0.87 for WT; Fig. 3A). In Smchd1-/- cells, however, regions of Xist depletion were evident throughout the Xi (Fig. 3A,S3C, green shaded regions). Xist depletion strongly correlated with H3K27me3 depletion (r=0.88; Fig. 3A, middle tracks) and anti-correlated with H3K4me3 acquisition (r= −0.40; Fig. 3A, bottom tracks).
Figure 3. SMCHD1 deficiency results in regional Xist spreading defects.

(A) Xist binding patterns (CHART-seq, comp), H3K27me3 enrichment (ChIP-seq, comp), and H3K4me3 enrichment (ChIP-seq, mus) across X chromosome. Enrichment difference determined by subtracting WT from Smchd1-/-. Green-shaded area, Xist-depleted regions. Gray-shaded area, unmappable regions.
(B) Xist binding patterns, H3K27me3 and H3K4me3 enrichment across one representative Xist-depleted domain.
(C) Metagene analysis of H3K27me3 and Xist enrichment in WT versus Smchd1-/- NPCs. Different X-linked gene categories are shown. The Xist locus was excluded as an escapee. TSS, transcription start site. TTS, transcription termination site. Gene bodies are “squished” between distances from 1–3 kb. Upstream and downstream regions are in absolute distance (kb).
(D) Two color RNA FISH for Xist and Mecp2 (left) or Atrx (right) in WT versus Smchd1-/- cells. %nuclei shown with nascent Mecp2 or Atrx signal outside or overlapping the edge of Xist cloud. In WT cells, 85% (159/186) of nuclei show monoallelic Mecp2 and 96% (129/134) show monoallelic Atrx. In Smchd1-/- cells, 73% of Mecp2 is biallelic (186/255), and 88% of the Xi allele is outside of Xist cloud; for Atrx, 80% is biallelic (135/168), and 70% of the Xi allele is outside of Xist cloud.
On a regional scale, the spreading defects were accentuated. Broad Xist-depleted regions coincided with domains showing loss of H3K27me3 and gain of H3K4me3 (Fig. 3B,S3D). Metagene analysis comparing Smchd1-/- vs. WT cells showed that Xist and H3K27me3 were strongly depleted across Class I genes (Fig. 3C) and their intergenic regions (Fig. S3E), suggesting that aberrant Xist distribution underlies defective heterochromatin spreading. On the other hand, Xist binding was not reduced across Class II or III genes (Fig. 3C). Furthermore, no changes in Xist binding were observed for escapees and for genes not normally expressed in NPCs. Because Xi genes translocate into the territory enshrouded by Xist RNA (Chaumeil et al., 2006; Namekawa et al., 2010), we asked if aberrant Xist spreading causes a translocation defect in Smchd1-/- cells. Two-color RNA FISH to localize SMCHD1-sensitive genes, Mecp2 and Atrx, showed two nascent transcripts indicative of failed Xi silencing and neither localized within the Xist cloud (Fig. 3D).
Collectively, we demonstrate that Smchd1 loss results in regional defects in Xist spreading, heterochromatin formation, and gene silencing within Class I domains. These findings underscore the idea that a normal cytological appearance of Xist and H3K27me3 (Blewitt et al., 2008)(Fig. 2H,3D) may belie significant underlying molecular defects.
SMCHD1 merges chromatin compartments on the Xi
Depleting SMCHD1 was previously shown to affect Xi morphology at the cytological level (Nozawa et al., 2013). To probe effects at a higher resolution, we performed in situ high-throughput chromosome conformation capture (Hi-C) (Rao et al., 2014) in WT and Smchd1−/-NPCs. Allele-specific analysis revealed two highly reproducible biological replicates (Fig. S4A). Contact heatmaps at 200-kb resolution showed that, while loss of SMCHD1 did not affect Xa structure (Fig. 4A, top panels), the Xi underwent marked topological changes (Fig. 4B, top panels). Whereas the WT Xi exhibited two megadomains separated by Dxz4, the mutant Xi showed altered megadomain appearance, with chromatin interactions becoming heterogeneous within each megadomain. The Pearson correlation map showed a checkerboard pattern for the Smchd1−/- Xi, in sharp contrast to the WT Xi (Fig. 4B, bottom panels), indicating appearance of smaller-scale compartments in Smchd1−/- cells. Importantly, the new compartments did not resemble the finer A/B compartments of the Xa (Fig. 4A, bottom panels).
Figure 4. Smchd1 ablation reveals a hidden layer of Xi organization.

(A) Allele-specific Hi-C analysis: Top: Contact maps of the Xa (Xcas) in WT and Smchd1-/- NPCs analyzed in 200-kb bins. Bottom: Corresponding Pearson correlation maps. Gray-shaded area, unmappable regions.
(B) Allele-specific Hi-C analysis. Top: Contact maps of the Xi (Xmus) in WT and Smchd1-/- NPCs analyzed in 200-kb bins. Bottom: Corresponding Pearson correlation maps.
(C) Principle component analysis (PCA) of the Xa correlated with allele-specific H3K4me3 peaks (black tracks). Positive principle component 1 (PC1) values represent A (active) compartments (red); negative PC1 values represent B (repressed) compartments (blue).
(D) PCA of the Xi correlated with allele-specific H3K4me3 peaks (black tracks), Xist binding (CHART heatmap), and H3K27me3 (ChIP heatmap) in NPCs. Red/blue structures are S1/S2 compartments.
(E) Xist-binding patterns and the distribution of Class I genes correlated with S1/S2 compartments.
To characterize the new compartments, we applied principle component analysis (PCA). In WT NPCs, the Xa harbored ~60 A/B compartments, with the active A compartments correlating with H3K4me3-enrichment (r=0.66); the Xi showed characteristic megadomains (Fig. 4C,D). In Smchd1−/- cells, A/B compartments were unchanged on the Xa (r=0.98), but the Xi looked dramatically different: 26 new compartments appeared with an average size 4.7 Mb (Fig. 4C,D). These compartments were distinct from A/B compartments of the Xa and lacked correlation with H3K4me3 (r=0.20). Their border also did not align with the sharp megadomain transition at Dxz4 (Fig. 4D; light blue-shaded region). Henceforth, we dub the new compartments “S1” (red) and “S2” (blue). The S1/S2 compartments are unique to the Xi.
We investigated the relationship between Xist RNA and S1/S2 compartments and observed that, while Xist density did not correlate with megadomains in WT cells (r=0.26), it showed high correlation with S1 domains of Smchd1-/- cells (r=0.76, Fig. 4D,E). H3K27me3 density was likewise strongly correlated (r=0.85, Fig. 4D). Furthermore, S1 domains had higher gene density, with both Class I and Class III genes enriched in this compartment (Fig. S4B). Class I gene clusters were usually seen where Xist dipped in coverage within S1 compartments (Fig. 4E). SMCHD1 depletion thus revealed a tendency of Xist-rich and Xist-poor chromatin to form distinct spatial compartments, a hidden layer of architectural organization obscured on the normal Xi. We conclude that SMCHD1 plays a critical role in organizing Xi structures by merging chromatin compartments.
SMCHD1 controls Xi TAD strength
The Xi is thought to lack TADs (Giorgetti et al., 2016; Minajigi et al., 2015). Interestingly, however, by employing in situ Hi-C, we observed clearly discernible TADs, albeit weak, across the entire Xi of WT NPCs at 40-kb resolution (Fig. 5A, S4C). By computing insulation scores (Crane et al., 2015; Giorgetti et al., 2016) based on chromatin interactions across a sliding genomic window along the Xi, we observed “waves” reflecting changes in chromatin interaction frequency, with local minima signifying borders between TADs. Although wave amplitudes were smaller on the Xi, the local minima and maxima often coincided with those on the Xa (Fig. 5A, S4C, S5A), implying that the Xi has a similar underlying TAD organization. For Xi TADs, boundary strength was usually lower except at Dxz4 and Firre (Fig. S4D, S5A), two repeat-associated loci that together form a “super-loop” (Darrow et al., 2016; Rao et al., 2014; Yang et al., 2015). Thus, TADs appear attenuated but not lost on the Xi. The higher sensitivity of in situ Hi-C (Nagano et al., 2015) may explain why they were missed in previous studies.
Figure 5. Smchd1 ablation leads to retention of CTCF/cohesin and strong TADs on the Xi.

(A) Hi-C interaction maps (in 40-kb bins), insulation profiles, and the genomic location of Class I and II genes at two representative X-linked regions. TADs (as defined by Dixon et al., 2012) are delineated as blue bars between interaction maps and as dashed lines in the insulation graphs.
(B) CDPs of the insulation scores (in 40-kb bins) on the Xa (blue) and the Xi (red) in WT versus Smchd1-/- NPCs as indicated. WT: p<2.2×10−16. Smchd1-/-: p=1.6×10−13 (KS test).
(C) CDPs of the insulation scores for the Xa and Xi in WT (black) versus Smchd1-/- (KO, red) NPCs. Xa: p=0.31. Xi: p=1.4×10−10 (KS test).
(D) Pearson correlation analysis of the insulation profiles analyzed in B,C.
(E) CDPs of insulation scores in WT (black) versus Smchd1-/- (red) NPCs for bins containing Class I genes versus other bins. Xa and Xi profiles shown, as indicated. Xa: Class I regions, p=0.55; other regions, p=0.28. Xi: Class I regions, p=0.038; other regions, p=4.4×10−11(KS test).
(F) Allelic CTCF and RAD21 binding within a representative X-linked region. Xi, mus. Xa, cas. Significant binding peaks are indicated in green.
(G) Top: Probability density plots of CTCF (top) or RAD21 (bottom) binding to the mus allele (%mus) of chrX or chr13 in WT versus Smchd1-/- NPCs, as indicated. P-values by KS test.
(H) Box plots showing the distribution of differences in allelic skewing (Δ%mus) for CTCF in Smchd1-/- vs WT cells for the categories indicated. Class I vs II genes, P=1. Class I genes vs all other X gene categories, P< 0.03. Chr13 vs escapees, P=1. Chr13 vs all other X gene categories, P<0.001 (one-sided Wilcox test with Bonferroni correction).
Next, we asked if Smchd1 ablation affects TAD strength. Whereas no obvious effects were observed on the Xa, TADs were quantifiably strengthened on the Xi (Fig. 5A,C; S4C,D; S5A). Cumulative distribution plots (CDPs) for insulation scores showed that, while the Xa and Xi of WT cells had distinct profiles, these profiles became more similar in Smchd1-/- cells (Fig. 5B). CDPs comparing WT versus Smchd1−/- cells also showed a statistically significant difference for the Xi (P=1.4×10−10, Fig. 5C), with the slope of Smchd1-/- curve being shallower, indicative of greater fluctuation of insulation scores and stronger TADs. Pearson correlation analysis further suggested a more Xa-like profile in Smchd1-/- cells (Fig. 5D). Deviations in insulation scores were greatest for Class I regions (Fig. 5A,S4C,S5A), as supported by a shallower cumulative distribution curve for Class I regions (Fig. 5E) and greater differences in insulation scores (Δ insulation score, Fig. S4E). Additionally, non-Class I regions also showed strengthened TADs (Fig. 5E,S4E), with the exception of escapees. Thus, depleting SMCHD1 leads to an Xi-specific strengthening of TADs. SMCHD1 therefore controls TAD strength in an Xi-specific manner.
SMCHD1 repels binding of architectural proteins on the Xi
Cohesins and CTCF are critical for TAD formation (Dixon et al., 2012; Ong and Corces, 2014; Rao et al., 2017). To determine if cohesin and CTCF binding is affected by SMCHD1, we performed allele-specific ChIP-seq. In WT cells, a majority of CTCF and RAD21 (a cohesin subunit) peaks were bi-allelic (Berletch et al., 2015; Calabrese et al., 2012; Minajigi et al., 2015), but peaks were typically taller on the Xa (Fig. 5F,S5B). In Smchd1−/- cells, CTCF and RAD21 exhibited increased binding to the Xi (Fig. 5F,S5B). Probability density distributions showed a significant right-shift of Xi-binding (%mus; Fig. 5G) and scatter plots showed an upward shift in Xi-binding in Smchd1−/- cells (Fig. S5C,D). Minimal differences were observed in autosomal binding (chr13 shown, Fig. 5G, S5C,D). Thus, SMCHD1 strongly influences binding of architectural factors to the Xi.
We noted that effects differed for Class I, II, and III regions (p<0.001, one-way ANOVA). Class I genes showed greater change than escapees, Class III, and silent genes (p<0.03)(Fig. 5H), though significant changes could still be observed to varying degrees for all classes of genes (p<0.001), excepting escapees (Fig. 5H). These findings are consistent with general effects on TAD strength for all gene classes (Fig. S4E). TAD strengthening was associated with increased CTCF binding at boundaries (Fig. S4F), consistent with CTCF’s role in demarcating TAD borders (Sanborn et al., 2015). Increased CTCF binding was also detected in most intergenic regions of the Xi (p<0.025)(Fig. S5E). Finally, we integrated Xist CHART and CTCF/RAD21 ChIP datasets and found that Xist/H3K27me3 depletion was not required for increased CTCF/RAD21 binding (Fig. S5F,G). We conclude that SMCHD1 suppresses CTCF and cohesin binding on a pan-Xi scale, with strongest effects seen in regions containing Class I genes.
SMCHD1 bridges S1 and S2 compartments on the Xi
If SMCHD1 regulates Xi organization by merging S1/S2 compartments as we propose, we would predict a binding pattern that bridges S1 and S2. Genomic binding maps are typically produced by ChIP-seq, but SMCHD1 has been refractory to this method, possibly because of the protein’s large size (>220 kDa) and indirect contact with DNA. A recent SMCHD1 ChIP-seq study in male mouse cells reported high background (Chen et al., 2015). After multiple unsatisfactory attempts of our own, we turned to DamID, whereby a protein of interest is fused to bacterial adenine (A) methyltransferase (Dam) and genomic regions in close proximity to the protein of interest become marked by A-methylation, which can subsequently be identified by differential sensitivity of restriction enzymes to A-methylation (Vogel et al., 2006). We fused SMCHD1 to Dam (Dam-SMCHD1, Fig. S6A,B) and compared its DamID pattern to two controls — unfused Dam (Dam alone) or Dam fused to CBX1 (HP1β)(Fig. S6B,C), a protein that binds H3K9me3. To correct for differences in chromatin accessibility and non-targeted methylation, we normalized Dam-SMCHD1 profiles to Dam alone [log2(Dam-SMCHD1/Dam alone)].
SMCHD1 DamID produced high-quality datasets, with the Xi showing striking SMCHD1 enrichment in two biological replicates (Fig. 6A,S6D,E). Neither Dam alone nor CBX1 was enriched on the Xi (Fig.S6D). On the Xa and autosomes, SMCHD1 associated with gene-poor regions (Fig. 6B, S6F), a pattern similar to that of CBX1 (r=0.77)(Fig. 6C), in agreement with interaction between SMCHD1 and H3K9me3 (Nozawa et al., 2013). On the Xi, SMCHD1 enrichment spanned both gene-rich and gene-poor regions (Fig. 6B,D,E). [Two artificial “troughs” (green-shaded regions) arose from dearth of SNPs to call alleles (Fig. 6B,S6G)]. No sharp transitions were seen at S1/S2 borders (Fig. 6D) or at TAD boundaries (Fig. S6H). There were also no obvious differences between Class I and III genes (Fig. 6F). However, SMCHD1 appeared mildly more enriched in S2 compartments, while Xist and H3K27me3 were more enriched in S1, though there was considerable overlap in S1 and S2 distributions (Fig. 6G). We conclude that SMCHD1 bridges S1 and S2 compartments, consistent with a role in merging them.
Figure 6. SMCHD1 bridges S1 and S2 to merge compartments on the Xi.

(A) Genomic SMCHD1 and CBX1 binding determined by DamID. Enrichment profiles were determined by log2(Dam fusion/ Dam alone) of each GATC fragment, with negative values (depleted relative to Dam alone) displayed with gray.
(B) SMCHD1 and CBX1 enrichment profiles across the X chromosome correlated with Class I and II genes, S1/S2 compartments in Smchd1-/- NPCs, Xist CHART, and H3K27me3 ChIP profiles in MEFs (GSE48649). Green-shaded areas, SNP-scarce regions.
(C) Scatter plots comparing SMCHD1 and CBX1 allele-specific binding to autosomes (black dots) and X chromosomes (red dots) at 100-kb bins. r, Pearson correlation coefficient for autosomes (black) and X chromosomes (red).
(D) SMCHD1 DamID profiles at two representative S1/S2 borders.
(E) SMCHD1 DamID profiles at two representative Class I regions.
(F) Box plots comparing SMCHD1 binding for indicated gene categories.
(G) Box plots comparing SMCHD1, Xist, and H3K27me3 enrichment in S1 vs S2 compartments. P-values by Wilcox test (one-sided).
S1/S2 compartments are intermediate Xi structures during de novo XCI
So far, S1/S2 compartments were only observed in the Smchd1-ablated “pathological” state. Because SMCHD1 recruitment is a late-stage event (Gendrel et al., 2012), we investigated whether S1/S2 occurred naturally but only transiently during XCI — prior to SMCHD1 binding. We carried out an allele-specific Hi-C time course on differentiating WT female ES cells (TsixTST/+) undergoing de novo XCI. Contact heatmaps at 200-kb resolution showed that the two Xa’s of D0 ES (pre-XCI) cells had a similar conformation (Xmus, Fig. 7A; Xcas, Fig. S7A), but unique structures appeared during XCI (Fig. 7A). Between D4-D7, Xi compartments became stronger, evidenced by stronger checkerboard patterns on Pearson correlation maps (Fig. 7A, lower panels). The intensified pattern disappeared in post-XCI cells (Fig.7A, right, WT NPC).
Figure 7. S1/S2 compartments are transitional states during XCI.

(A) Allele-specific Hi-C analysis: Top: Contact maps (200-kb resolution) of the Xi (Xmus) in undifferentiated ES cells (D0 ES, pre-XCI), embryoid bodies after 4 or 7 differentiation days (D4 and D7 EB, early-mid XCI), and WT and Smchd1-/- NPCs (post-XCI). Bottom: Pearson correlation maps.
(B) PCA of the D0 Xa (comp), the Xi (Xmus) at different stages of XCI, and correlation with Xist spreading patterns (GSE48649). D3 Xist patterns represent “early domains.” Blue and red arrows indicate fusion of consecutive A/B compartments to form S1/S2.
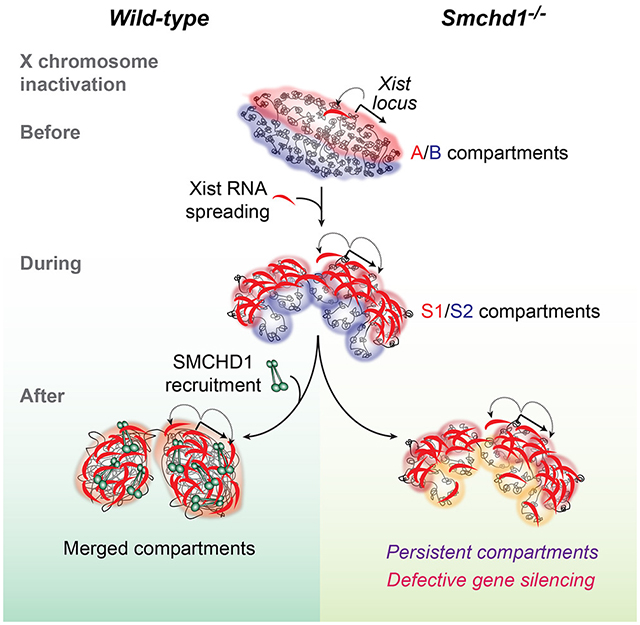
(C) The Origami Model for step-wise Xi folding. Xist RNA is produced from A compartment and initially spreads to co-segregated A compartments (red) through proximity transfer. Xist fuses A/B into S1/S2 compartments, correlating with the two-step Xist spreading into early (gene-rich), then late (gene-poor) domains (Simon et al., 2013). Following SMCHD1 recruitment, S1/S2 compartments are merged (top) to form a compartmentless structure. In Smchd1-/- cells, the final transformation does not occur and S1/S2 compartments persist (bottom), resulting in defective Xist spreading and gene silencing.
(D) Model: SMCHD1 bridges Xist-rich and Xist-poor chromatin to promote chromatin mixing and merging of S1/S2 compartments (left). Without SMCHD1, Xist-rich chromatin co-segregates, leading to the formation of S1/S2 compartments (right).
Principle component analysis showed a striking resemblance to S1/S2 compartments (Fig. 7B). Between D4-D7, smaller-scale A/B compartments were merged to create broader S1/S2-like compartments (Fig. 7B, red and blue arrows). The transient compartments in ES cells showed high correlation with S1/S2 structures of Smchd1-/- NPCs (r=0.73)(Fig. 7B). Their resemblance was further supported by hierarchical clustering analysis (Fig. S7C). Importantly, during this intermediate stage (D4-D7), immuno-RNA-FISH showed that the Xist cloud had already formed but SMCHD1 had not yet been recruited to the Xi (Fig. S6I). This intermediate stage thereby simulates the Smchd1−/- state that originally revealed the S1/S2 structures. The S1/S2 transformation was not observed on the Xa (Fig. S7A,B). The large-scale compartment merging to create S1/S2 is therefore distinct from “A/B compartment switching,” a reorganization reported for other chromosomes during differentiation (Dixon et al., 2015).
Interestingly, newly appeared S1 compartments of D4-D7 cells coincided with initial spreading of Xist RNA into gene-rich regions (r=0.91, Fig. 7B). During XCI, Xist is known to spread across the Xi via a two-step mechanism, initially targeting gene-rich regions (“early domains”) before spreading to gene-poor “late domains” (Simon et al., 2013). We wondered if SMCHD1 might facilitate Xist spreading from S1 (early) into S2 (late) domains. By comparing Xist and H3K27me3 coverages between S1 and S2 compartments, we found that Smchd1 ablation caused a significantly widened gap in Xist/H3K27me3 densities between S1 and S2, in two biological replicates (Fig. S6J). Xist and H3K27me3 accumulated more in S1 compartments when SMCHD1 was deficient. Deeper valleys in Xist coverage were observed at S2 regions (Fig. 7B, comparing the Xist track in Smchd1-/- to WT NPCs). Thus, failure to recruit SMCHD1 during XCI compromises the spreading of Xist RNA into S2 or “late domains” of the Xi. We conclude that S1/S2 compartments are natural states during XCI, forming transiently as Xist RNA initially spreads across the Xi prior to SMCHD1 localization.
DISCUSSION
Here we have discovered a hidden layer of Xi organization. During de novo XCI, A/B compartments are first remodeled into S1/S2 compartments as Xist RNA initially spreads through the Xi (Fig. 7C,S7D). When SMCHD1 is recruited at a later stage (Gendrel et al., 2012), S1/S2 compartments are merged together to create a “compartment-less” structure (Fig. 4,7). Such a step-wise mechanism is consistent with how Xist RNA spreads. The Xist locus and the nucleation site reside in an A compartment (Fig. 7B)(Jeon and Lee, 2011). Through “phase separation,” chromatin compartments tend to self-associate and spatially co-segregate (Hnisz et al., 2017; Larson et al., 2017; Strom et al., 2017). In this way, Xist could co-segregate with gene-rich regions located in other A compartments. Through proximity transfer, newly synthesized Xist transcripts could then spread to these “early domains” first (Simon et al., 2013), explaining the strong correlation between A compartments and “early domains” (r=0.79)(Fig. 7B). Interestingly, a majority of A compartments (76–87%) become remodeled into S1 compartments (Fig. 7B). Furthermore, S1/S2 compartments sometimes resemble fusions of several consecutive A/B compartments (Fig. 7B, dotted arrows). We speculate that, when Xist spreads from A to adjoining B compartments, S1 and S2 structures are formed. The creation of S1/S2 structures does not require SMCHD1, as SMCHD1 is not enriched on the Xi during this transitional stage. However, the final architectural transformation cannot be completed without SMCHD1. SMCHD1 binds and bridges S1 and S2 compartments to create the “compartmentless” state (Fig. 4,7D). Thus, whereas Xist RNA fuses A/B into S1/S2 compartments, SMCHD1 merges S1/S2 (Fig. 7D). Failure to merge S1/S2 compartments is functionally significant, as regional Xist spreading becomes compromised (Fig. 3) and ~43% of genes normally subject to XCI cannot be properly heterochromatinized or silenced (Fig. 1,2). Xist spreading may thereby occur in two phases. In phase 1, Xist spreads from early (A compartments) to late domains (B compartments) to form S1/S2 compartments on a global scale. In phase 2, Xist depends on SMCHD1 to maintain binding to local regions within a compartmentless structure.
In addition to merging compartments, SMCHD1 works together with Xist to weaken TADs. Without SMCHD1, CTCF and cohesin binding remains strong on the Xi, coincident with globally strengthened TADs (Fig. 5). The idea that SMCHD1 suppresses TADs by repelling other architectural proteins is especially appealing given suggested antagonism between SMCHD1 and CTCF on chromatin (Chen et al., 2015). Within the context of our step-wise folding model, we posit that the tendency to form compartments and TADs is not erased. These structures appear to be actively attenuated in a multi-step process — akin to “origami”, where layer upon layer of folding mechanisms are superimposed for a desired shape. Interestingly, the human Xi harbors two types of heterochromatin domains — one enriched with XIST RNA/H3K27me3 and the other with H3K9me3 (Chadwick and Willard, 2004; Nozawa et al., 2013) — and they create partitions in a Hi-C map (Darrow et al., 2016). These partitions may correlate with our S1/S2 compartments, and SMCHD1 may indeed bridge them as well on the human Xi (Nozawa et al., 2013).
Finally, the impact of SMCHD1 on chromosome conformation appears distinct from that of canonical architectural proteins. CTCF and cohesins drive formation of TADs but are dispensable for (CTCF) or even interfere with (cohesins) chromosome compartmentalization (Haarhuis et al., 2017; Nora et al., 2017; Rao et al., 2017; Schwarzer et al., 2017). SMCHD1 is unusual both for its role in diminishing (rather than reinforcing) architectural structures and for concordant effects on compartments and TADs. The unmasking of S1/S2 compartments by loss of SMCHD1 is reminiscent of cohesin depletion in revealing finer compartment structures (Rao et al., 2017; Schwarzer et al., 2017). Cohesins may antagonize phase separation by facilitating chromatin looping between different compartments. Given that SMCHD1 resembles SMC proteins, we posit that SMCHD1 promotes chromatin mixing by mediating random chromatin interactions that span the boundaries of existing structures, thereby blurring compartments and TADs (Fig. 7D). This idea is supported by the nearly homogeneous distribution of SMCHD1 on the Xi, contacting both S1 and S2 compartments (Fig. 6). With its ATPase domain (Brideau et al., 2015; Chen et al., 2016), SMCHD1 might serve as Xist’s engine to drive attenuation of both compartments and TADs to complete the final Xi transformation. Future work will test our stepwise folding Origami Model.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Jeannie T. Lee (lee@molbio.mgh.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
XaWTXiΔXist (clone III.3, female), 2lox(Xist+) (clone III.8, female), ♀X+P (female), ♂X+P (male), and EY.T4 (female) fibroblast lines have been described (Jeon and Lee, 2011; Yildirim et al., 2012; Zhang et al., 2007) and were grown in MEF media [DMEM, high glucose, GlutaMAX supplement, pyruvate (10569044, Thermo Fisher Scientific), 10% FBS, 25mM HEPES pH 7.2–7.5 (15630130, Thermo Fisher Scientific), 1× MEM non-essential amino acids (11140076, Thermo Fisher Scientific), 1× Pen/Strep (15140163, Thermo Fisher Scientific), and 0.1mM β-mercaptoethanol (21985023, Thermo Fisher Scientific)]. TsixTST/+ female mouse embryonic stem (ES) cell line has also been described (Ogawa et al., 2008) and were grown on γ-irradiated MEF feeders in ES media [DMEM, high glucose, GlutaMAX supplement, pyruvate (10569044, Thermo Fisher Scientific), 15% Hyclone FBS, 25mM HEPES pH 7.2–7.5 (15630130, Thermo Fisher Scientific), 1× MEM non-essential amino acids (11140076, Thermo Fisher Scientific), 1× Pen/Strep (15140163, Thermo Fisher Scientific), 0.1mM β-mercaptoethanol (21985023, Thermo Fisher Scientific), and 500U/ml LIF (ESG1107, Sigma)].
METHOD DETAILS
Generation of Smchd1-/- female ES cells using CRISPR-Cas9
Smchd1-/- ES cell lines were generated by transfecting TsixTST/+ female ES cells with a mixture of two plasmids (PX461; 48140, Addgene) expressing Cas9 D10A nickase, EGFP, and a pair of guide RNAs (Ran et al., 2013) targeting the first exon of Smchd1 using Lipofectamine LTX & Plus Reagent (15338100, Thermo Fisher Scientific). This “double nicking” strategy has been shown to minimize off-target effects (Ran et al., 2013). To further control for off-target effects, two different pairs of guide RNAs were used (pair 1: CTTGTTTGACCGGCGCGGGA, CACCGTCCTACAGCCGTCGA; pair 2: GGGAGAGATGGCGCCGTCGA, CGAGAGGCCGGCGGGATCGC)(Table S1). Wild-type (WT) ES lines were generated by transfecting TsixTST/+ ES cells with PX458 expressing only Cas9 D10A nickase and EGFP. 24 hours after transfection, ES colonies were dissociated and transfected cells were sorted by EGFP into 96-well plates, with one cell plated in each well. ES clones were screened by Western blot using an antibody against SMCHD1 (Sigma, HPA039441). We validated this antibody in detecting murine SMCHD1 on Western blot by siRNA-mediated knockdown of SMCHD1 in ♀MEFs using Smchd1 siRNA (Dharmacon, L-040501–01-0005) and scramble control siRNA (Dharmacon, D-001810–10-05). Smchd1-/- clones were further verified by the presence of frame-shifting mutations detected by Sanger sequencing of the CRISPR-targeted region, which was PCR amplified from genomic DNA and cloned into a TOPO-TA vector. Primers are listed in Table S1.
Derivation of neural progenitor cells from ES cells
Neural progenitor cells (NPCs) were derived from WT and Smchd1-/- female ES cells and cultured using a protocol from the lab of Dr. Fred H. Gage. Briefly, undifferentiated ES cells were trypsinized and feeders were removed. 4 million ES cells were suspended in 20ml ES media without LIF for 1 day to trigger the formation of embryoid bodies on a 10-cm low-attachment culture dish. The next day, embryoid bodies were spun down at 1000 rpm for 2 minutes and incubated in 10ml N2 media [DMEM/F-12, GlutaMAX™ supplement (10565018, Thermo Fisher Scientific), 1× N2 supplement (10565018, Thermo Fisher Scientific), 1× B27 supplement (17504044, Thermo Fisher Scientific), 1× Pen/Strep (15140163, Thermo Fisher Scientific)] supplemented with 500ng/ml NOGGIN (120–10C, Peprotech) for 4 days, with media changed every other day. Embryoid bodies from ~1/40 of the culture were then plated on 1 well of poly-L-ornithine (0.001%) and laminin (1 μg/ml) coated 6-well plates with 2.5ml N2 media supplemented with 20ng/ml FGF2 (233-FB-025, Fisher Scientific) and 1 μg/ml laminin (23017015, Thermo Fisher Scientific) for 5 days, with media changed every other day. Neural rosettes were then picked manually and maintained in N2 media supplemented with 20ng/ml FGF2, 20ng/ml EGF (AF-100–15, Peprotech), 1 μg/ml laminin, and 10 μg/ml heparin (H3149–100KU, Sigma). The identity and homogeneity of NPCs were verified by RT-PCR and immunostaining for NESTIN (Abcam, ab6142) and SOX2 (Cell Signaling, 2748). PCR primers are listed in Table S1. Primers for Emx2, Olig2, and Blbp have been described (Conti et al., 2005). Because Smchd1-/- NPCs showed a heightened tendency to become XO aneuploidy during differentiation, we isolated subclones that retained two X chromosomes. NPCs were immortalized by SV-40 T-antigen and plated at clonal density. NPC colonies were then picked manually and Xist RNA-FISH was used to identify the XX clones (>95% cells exhibiting one Xist cloud). For RNA-seq, we analyzed two WT (WT clone1 and clone2) and two Smchd1-/- clones (Smchd1-/- clone1, generated by guide RNA pair 1, and Smchd1-/- clone2, generated by guide RNA pair 2). For ChIP-seq, CHART-seq, and in situ Hi-C, WT clone1 and Smchd1-/- clone1 were analyzed.
EB differentiation of ES cells
To determine how the transition of compartment structures occurs during de novo XCI in WT cells, we performed in situ Hi-C on differentiating TsixTST/+ female mouse ES cells undergoing XCI. It is known that female ES cells maintained in traditional serum-containing media initiate XCI asynchronously. Thus, Hi-C performed on these cells represents the aggregate of a population of cells at heterogeneous stages of XCI. To overcome this problem, we adapted our female ES cells to serum-free 2i media, a condition where they exhibit more synchronous XCI (Marks et al., 2015). Prior to adaptation, ES cells maintained on γ-irradiated MEF feeders in serum-containing ES media were passaged at least once after thawing. After removing feeders, 1 million ES cells were washed with serum-free N2B27 media [mixture of 500ml DMEM/F12 media (11330032, Thermo Fisher Scientific), 500ml Neurobasal media (21103–049, Thermo Fisher Scientific), 5ml N2 supplement (17502048, Thermo Fisher Scientific), 10ml B27 supplement (17504044, Thermo Fisher Scientific), 10ml Pen/Strep (15140163, Thermo Fisher Scientific), 5ml GlutaMAX (35050061, Thermo Fisher Scientific), and 2ml 55mM β-mercaptoethanol (21985023, Thermo Fisher Scientific)] once and then plated on a 10-cm tissue culture plate coated with 0.2% gelatin in N2B27 media supplemented with “2i+LIF” [1 M MEK inhibitor PD0325901 (S1036, Selleck Chemicals), 3 M GSK3 inhibitor CHIR-99021(S2924, Selleck Chemicals), and 1000U/ml LIF (ESG1107, Sigma)]. ES cells were passaged at 1:4 to 1:6 ratio in N2B27 media supplemented with 2i+LIF for > 10 days (> 5 passages). To induce differentiation, ES cells grown in 2i condition were dissociated by Accutase (07920, Stemcell Technologies) and 6 million ES cells were grown in suspension in a 10-cm low-attachment plate with 10ml differentiation media [DMEM, high glucose, GlutaMAX supplement, pyruvate (10569044, Thermo Fisher Scientific), 15% Hyclone FBS, 25mM HEPES pH 7.2–7.5 (15630130, Thermo Fisher Scientific), 1× MEM non-essential amino acids (11140076, Thermo Fisher Scientific), 1× Pen/Strep (15140163, Thermo Fisher Scientific), 0.1mM β-mercaptoethanol (21985023, Thermo Fisher Scientific)] to allow embryoid body (EB) formation. Media were changed daily and EBs were settled on two 15-cm tissue culture plates coated with 0.2% gelatin on the fifth day of differentiation (D5). To harvest ES cells at different stages of differentiation, cells were treated with either Accutase for 5 minutes (D0 ES cells and D4 EBs) or 0.25% Trypsin (25200056, Thermo Fisher Scientific) for 10 minutes (D7 EBs) at 37°C. Dissociated ES cells and EBs were passed through a 40 M cell strainer (352340, Falcon) to remove clumps before experiments.
Xist RNA-FISH, DNA-FISH, and SMCHD1 Immunostaining
For sequential SMCHD1 immunostaining and Xist RNA-FISH on ♀fibroblasts, cells were grown overnight on glass coverslips coated with 0.2% gelatin. Cells were rinsed in chilled PBS for 5 minutes three times, pre-extracted with CSKT buffer (100mM NaCl, 300mM sucrose, 10mM PIPES, 3mM MgCl2, 0.5% Triton X-100, pH 6.8) supplemented with 10mM ribonucleoside vanadyl complex (VRC; S1042S, NEB) for 10 minutes on ice, and fixed with chilled 100% methanol at −20°C for 10 minutes. After fixation, c ells were washed with chilled PBS for 5 minutes, further permeabilized by CSKT with 10mM VRC for 10 minutes on ice, followed by three washes with chilled PBS for 5 minutes. After being blocked with 5% normal goat serum (PCN5000, Thermo Fisher Scientific) and 0.6 unit/μl Protector RNase Inhibitor (3335402001, Sigma) in PBS at room temperature for 1 hour, cells were incubated with rabbit polyclonal SMCHD1 antibodies (HPA039441, Sigma, 1:200) in antibody dilution buffer (5% normal goat serum, 0.6U/μl Protector RNase Inhibitor, and 0.2% Tween-20 in PBS) at room temperature for 1 hour, washed with PBST (PBS with 0.2% Tween-20) for 5 minutes three times, and incubated with secondary antibody at room temperature for 1 hour. Three 5-minute PBST washes were then performed, followed by re-fixation in 4% paraformaldehyde (15713, Electron Microscopy Sciences) in PBS at room temperature for 10 minutes and two 5-minute washes in PBS. Cells were then dehydrated by sequential incubation with 70%, 80%, 90%, and 100% ethanol for 3 minutes and air-dried. RNA-FISH was performed with 40ng of Xist probes (nick-translation of the pSx9–3 plasmid) in 10 l hybridization buffer [2× SSC, 50% formamide, 10% dextran sulfate, 100ng/μl mouse Cot-1 DNA (18440016, Thermo Fisher Scientific)] at 37°C overnight. Prior to hybridization, probes were denatured in hybridization buffer at 95°C for 10 minutes and preannealed at 37°C for 30 minutes. After being washed three times in 2× SSC at room temperature for 10 minutes, cells were mounted with Vectashield mounting media with DAPI (H-1200, Fisher Scientific). Images were acquired with a Nikon Eclipse 90i microscope and a Hamamatsu CCD camera. Image analysis was performed using Volocity (Perkin-Elmer).
For RNA- and DNA-FISH, 1–2×105 mouse ES cells or NPCs were affixed by cytospinning (“cytospun”) onto glass slides at 1000 rpm for 10 minutes. After air-drying the slides, cells were rinsed with chilled PBS for 5 minutes, pre-extracted with CSKT for 10 minutes on ice, followed by fixation with 4% paraformaldehyde in PBS at room temperature for 10 minutes, and stored at 4°C in 70% ethanol. To prepare slides for FISH, cel ls were dehydrated with graded series of ethanol and air-dried. For RNA-FISH, cells were incubated with 40ng probes at 37°C overnight. For DNA-FISH, cells were treated with 400 g/ml Purelink RNaseA (12091021, Thermo Fisher Scientific) in PBS at 37°C for 40 minutes and rinse d once with PBS. To denature DNA, cells were incubated with 70% formamide in 2× SSC at 80°C for 10 minutes and quenched in 70% ethanol, which had been pre-chilled at −20°C, for 5 minutes. Cells were then dehydrated again with ethanol series before 40ng probes were added for hybridization. Probes for Xist, Mecp2, Atrx and Tmem39a were generated by nick translation of pSx9–3, fosmid WIBR1–1417J09, BAC RP23–377P8 and fosmid WIBR1–2543D17, respectively. Prior to hybridization, probes were denatured in hybridization buffer at 95°C for 10 mi nutes and pre-annealed at 37°C for 30 minutes. Following hybridization, cells were washed with 2× SSC plus 50% formamide at 37°C for 20 minutes once, 2× SSC for 5 minutes at 37°C t hree times, and mounted with Vectashield mounting media with DAPI.
For sequential H3K27me3 immunostaining and Xist RNA-FISH on NPCs, 1.5×105 dissociated NPCs were cytospun onto glass slides at 1000 rpm for 10 minutes. After air-drying the slides, cells were rinsed with chilled PBS for 5 minutes, pre-extracted with chilled CSKT for 2 minutes on ice, followed by fixation with 4% paraformaldehyde in PBS at room temperature for 10 minutes, and stored at 4°C in 70% ethanol. To prepa re slides for immunostaining, cells were dehydrated with graded series of ethanol, air-dried, rehydrated with PBS for 5 minutes and permeabilized with 0.5% Triton in PBS for 10 minutes. Permeabilized cells were washed with PBS for 5 minutes three times and blocked with 5% normal goat serum and 0.6 unit/μl RNasin Plus RNase Inhibitor (N2615, Promega) in PBS at room temperature for 30 minutes. Blocked cells were incubated with rabbit monoclonal H3K27me3 antibodies (GTX60892, GENETEX, 1:3000) in antibody dilution buffer at room temperature for 1 hour, washed with PBST for 5 minutes three times, and incubated with secondary antibodies at room temperature for 1 hour. Three 5-minute washes with PBST were then performed, followed by re-fixation in 4% paraformaldehyde in PBS at room temperature for 10 minutes and two washes in PBS for 5 minutes. Cells were then dehydrated by sequential incubation with 70%, 80%, 90%, and 100% ethanol for 3 minutes and air-dried. Xist RNA-FISH was performed using nick translation of pSx9–3 as described above.
For sequential SMCHD1 immunostaining and Xist RNA-FISH on EBs, 2×105 dissociated cells were cytospun onto glass slides at 1000 rpm for 10 minutes. After air-drying the slides, cells were rinsed with chilled PBS for 5 minutes, pre-extracted with chilled CSKT for 2 minutes on ice, followed by fixation with 4% paraformaldehyde in PBS at room temperature for 10 minutes, and stored at 4°C in 70% ethanol. To prepare slides for immunostaining, cells were dehydrated with graded series of ethanol, air-dried, rehydrated with PBS for 5 minutes, incubated with 100% methanol at −20°C for 10 minutes, washed with PBS f or 5 minutes twice, and permeabilized with 0.5% Triton in PBS for 10 minutes. Permeabilized cells were washed with PBS for 5 minutes three times, blocked, incubated with rabbit polyclonal SMCHD1 antibodies (HPA039441, Sigma, 1:200) for 1 hour, and washed with PBST for 5 minutes three times. Cells were then incubated with secondary antibodies at room temperature for 1 hour, washed again with PBST three times for 5-minutes, followed by re-fixation in 4% paraformaldehyde in PBS at room temperature for 10 minutes, and two washes in PBS for 5 minutes. Cells were then dehydrated by graded series of ethanol and air-dried. Xist RNA-FISH was performed as described (Del Rosario et al., 2017), with 10ng Xist oligonucleotide probes in 10 l hybridization buffer [2× SSC pH 7.0, 30% formamide, 10% dextran sulfate, 100ng/μl mouse Cot-1 DNA (18440016, Thermo Fisher Scientific)] at room temperature in a dark humidified chamber for 2 hours. Xist probes are listed in Table S1 and have been described (Del Rosario et al., 2017). Prior to hybridization, probes were denatured in hybridization buffer at 80°C for 5 minutes and pre-annealed at 37°C for 30 minutes. Following hybridization, Slides were washed with 2× SSC, 30% formamide for 5 minutes on a shaker four times, followed by washes with 2× SSC for 5 minutes three times. After washes, slides were air-dried and mounted with Vectashield mounting media with DAPI.
For sequential V5-tag immunostaining and Xist RNA-FISH on MEFs expressing Dam fusion proteins, cells were grown overnight on glass coverslips coated with 0.2% gelatin. Cells were rinsed in chilled PBS for 5 minutes three times, pre-extracted with CSKT buffer supplemented with 10mM VRC for 10 minutes on ice, and fixed with chilled 4% paraformaldehyde in PBS at room temperature for 10 minutes. After fixation, cells were washed with chilled PBS for 5 minutes, further permeabilized by CSKT with 10mM VRC for 10 minutes on ice, followed by three washes with chilled PBS for 5 minutes. After being blocked with 5% normal goat serum and 0.6U/μl Protector RNase Inhibitor in PBS at room temperature for 1 hour, cells were incubated with mouse monoclonal polyclonal V5-tag antibodies (MCA1360, AbD Serotec, 1:200) in antibody dilution buffer at room temperature for 1 hour. Secondary antibody incubation and Xist RNA-FISH were performed as described above for fibroblasts.
RNA-sequencing analysis of wild-type and Smchd1-/- NPCs
RNA-seq libraries were generated as described previously (Minajigi et al., 2015). Briefly, total RNA was extracted using TRIzol (15596018, Thermo Fisher Scientific), with ribosomal RNA depleted with RiboMinus™ Eukaryote Kit v2 (A15020, Thermo Fisher Scientific) and genomic DNA depleted with TURBO DNase (AM2239, Thermo Fisher Scientific). 30–100ng ribosomal RNA-depleted RNA were fragmented in RT buffer at 94°C for 10 minutes, followed by random-primed first strand cDNA synthesis with SuperScript III Reverse Transcriptase (18080085, Thermo Fisher Scientific) in the presence of actinomycin D (11805017, Thermo Fisher Scientific) at 50°C for 30 minutes. Secondary stran d synthesis was performed using NEBNext mRNA Second Strand Synthesis Module (E6111S, NEB) supplemented with dUTP. Double-strand cDNAs were purified with 1.8× Agencourt AMPure XP beads (A63881, Beckman Coulter). Libraries were constructed with NEBNext ChIP-Seq Library Prep Master Mix Set for Illumina (E6240S, NEB). RNA-seq was performed in two biological replicates. All libraries were sequenced with Illumina HiSeq, generating 43–59 millions paired-end 50 nucleotide reads per sample.
Analysis of RNA-seq datasets in wild-type and Smchd1-/- NPCs
RNA-seq reads were aligned allele-specifically to 129S1/SvJm (mus) and CAST/Eih (cas) genome using TopHat2 (Kim et al., 2013) as previously described (Minajigi et al., 2015). After removal of PCR duplicates, all unique reads mapped to the exons of each gene were quantified by Homer (Heinz et al., 2010). The normalized differential expression analyses were performed by DESeq (Anders and Huber, 2010). Differentially expressed genes were called if the associated adjusted p value was less than 0.05. To compare the fold changes of autosomal and X-linked genes, only genes with fragments per kilobase of transcript per million mapped exonic reads (FPKM) >0.1 were considered. For allele-specific analysis, we defined %mus as the percentage of mus-specific exonic reads in all allele-specific (mus-specific + cas-specific) exonic reads of each transcript. For classification of X-linked genes, we defined active genes as genes having non-zero FPKM in all samples. Allelically-distinguishable genes were defined as active genes that had a minimum of 13 allele-specific reads in all samples (Pinter et al., 2015). It has been described that a small fraction of genes overlap with incorrectly annotated SNPs and produce unexpected allelic skewing (Calabrese et al., 2012; Pinter et al., 2015). These genes were identified by analyzing a published RNA-seq dataset of ♀ tail-tip fibroblasts (TTF) from pure Mus castaneous background (GSE58524)(Pinter et al., 2015) and the RNA-seq data of an XcasO NPC clone we derived in this study. Allelically-distinguishable genes having %mus greater than 9.09% in either the pure cas TTFs or the XcasO NPC clone were considered as genes with miscalled SNPs. After excluding these genes, we defined escapees as allelically-distinguishable genes whose expression from the Xi was greater than 10% of the expression from the Xa in at least one biological replicate of at least one WT clone. Genes subject to XCI (Xi genes) were defined as allelically-distinguishable genes that were not genes with miscalled SNPs or escapees. SMCHD1-sensitive genes were defined as Xi genes with %mus in an Smchd1-/- clone that was 3-fold higher than the %mus in both WT clones and greater than 9.09% in both biological replicates. SMCHD1-insensitive genes were defined as Xi genes that were not SMCHD1-sensitive genes.
To analyze the genomic distribution of different classes of X-linked genes, we segmented the X into 500-kb bins and assigned each X-linked gene to these bins based on their TSS. The number of genes in each bin was counted and plotted as histograms. To visualize RNA-seq coverage, we generated strand-resolved fragments per million (fpm)-normalized bigWig files for all (comp), mus-specific (mus), and cas-specific (cas) reads separately, which were displayed using Integrative Genomics Viewer (IGV) with scales indicated in each track.
ChIP-sequencing of H3K4me3, H3K27me3, CTCF, and RAD21
NPCs were cross-linked in PBS with 1% formaldehyde at room temperature for 10 minutes at 2 million cells/ml and quenched with 0.125M glycine at room temperature for 5 minutes. Cross-linked cells were washed three times with chilled PBST (1× PBS with 0.05% Tween-20) before snap-frozen in liquid nitrogen. 20 million cross-linked cells were thaw on ice for 15 minutes, resuspended in 2ml Buffer 1 [50mM HEPES pH7.5, 150mM NaCl, 1mM EDTA, 0.5% Nonidet P-40, 0.2% Triton X-100, 1× cOmplete EDTA-free Protease Inhibitor Cocktail (PIC; 11873580001, Sigma)], and rotated at 4°C for 10 minutes. Nuclei were pelleted by centrifugation at 1700g for 5 minutes at 4°C, resuspended with 2ml Buffer 2 (10mM Tris pH 8.0, 200mM NaCl, 5mM EDTA, 2.5mM EGTA, 1× PIC) supplemented with 40 l Purelink RNaseA (12091021, Thermo Fisher Scientific), and incubated for 15 minutes with rotation at 4°C. Nuclei were then pelleted again by centrifugation at 1700g for 5 minutes at 4°C, resus pended with 980 l Buffer 3 (10mM Tris pH 8.0, 5mM EDTA, 2.5mM EGTA, 1× PIC) supplemented with 10 l 20% SDS, and incubated for 15 minutes with rotation at 4°C. Following incubati on, nuclei were transferred to a 1ml Covaris milliTUBE with AFA fiber and sonicated for 21 minutes using Covaris E220e (140W peak incident power, 5% duty factor, 200 cycles/burst). Sonicated chromatin was mixed at 1:1 ratio with 2× IP buffer (30mM Tris pH 8.0, 300mM NaCl, 2% Triton X-100, 0.5% N-lauroylsarcosine, 1× PIC) and centrifuged at full speed for 30 minutes at 4°C to remove insoluble debris. 40 l supernatant was set aside as the input sample. For ChIP, 400 l supernatant was further diluted by mixing with 400 l 1× IP buffer (1:1 mixture of Buffer 3 and 2× IP buffer), before incubating with antibodies overnight with rotation at 4°C. Ant ibodies used were: α-H3K4me3 (2.5 l; Millipore 17–614), α-H3K27me3 (5 l; Millipore 07–449), α-CTCF (5 l; Cell Signaling, 2899S), α-RAD21 (5 l; Abcam ab992), and normal rabbit IgG (2729S, Fisher Scientific), which served as a negative control. Following overnight incubation, chromatin was mixed with 30 l Dynabeads Protein G (10004D, Thermo Fisher Scientific), which had been washed three times in 1× IP buffer (15mM Tris pH8.0, 150mM NaCl, 1% Triton X-100, 0.25% N-lauroylsarcosine, 2.5mM EDTA, 1.25mM EGTA, 1× PIC), and incubated for 2 hours with rotation at 4°C. Beads were then washed with 1ml 1× IP buffer once, 1ml wash buffer [for H3K4me3, H3K27me3, and CTCF: 50mM HEPES pH 7.5, 500mM LiCl, 1mM EDTA, 1% Nonidet P-40, 0.7% sodium deoxycholate, 1× PIC; for RAD21: 50mM HEPES pH 7.5, 250mM LiCl, 1mM EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1× PIC] six times, and 1× TEN buffer (10mM Tris pH 8.0, 50mM NaCl, 1mM EDTA) once. Beads were then incubated with 200 l TES buffer (50mM Tris pH 8.0, 1% SDS, 10mM EDTA) at 65°C for 30 minutes w ith intermittent vortex. The supernatant, which contained ChIP-enriched chromatin, was separated from the beads by a magnet and incubated at 65°C overnight, together with input co ntrols. After cross-link reversal, input and ChIP-enriched chromatin was mixed with 200 l TE and 4 l RNase A (12091021, Thermo Fisher Scientific) at 37°C for 2 hours, followed by additi on of 4 l Proteinase K (03115844001, Sigma) and incubation at 55°C for 2 hours. Input and ChIP- enriched DNA was purified with phenol-chloroform and quantified with Quant-iT™ PicoGreen® dsDNA Reagent (P7581, Thermo Fisher Scientific). To verify successful ChIP, ChIP-qPCR was performed for the following loci: Actb promoter (positive control for H3K4me3), Gata6 promoter (positive control for H3K27me3), H19 (positive control for CTCF and RAD21), and an intergenic region on chromosome 5 (negative control). Primers are listed in Table S1. ChIP samples with the positive control locus at least 20-fold more enriched than the normal rabbit IgG control and the negative control locus were subject to sequencing. ChIP-seq was performed in two biological replicates. Input and ChIP-seq libraries were constructed using NEBNext ChIP-Seq Library Prep Master Mix Set for Illumina (E6240S, NEB) and sequenced on Illumina HiSeq, generating 27–48 millions paired-end 50 nucleotide reads per sample.
Analysis of H3K4me3, H3K27me3, CTCF, and RAD21 ChIP-seq datasets
Input and ChIP-seq reads were aligned allele-specifically to 129S1/SvJm (mus) and CAST/Eih (cas) genome using NovoAlign as described (Minajigi et al., 2015). After removing PCR duplicates, uniquely mapped reads were used for further analysis. To visualize ChIP-seq coverage, we generated fpm-normalized bigWig files from ChIP-seq reads for all uniquely mapped (comp), mus-specific (mus), and cas-specific (cas) reads separately, which were displayed using IGV with scales indicated in each track. To compare the distribution of Xi heterochromatin between WT and Smchd1-/-, H3K27me3 ChIP-seq tracks were further scaled based on the total number of ChIP-seq reads mapped to the X.
The %mus of H3K4me3 at the promoter of each gene was defined as the percentage of mus-specific ChIP-seq reads in all allele-specific ChIP-seq reads mapped to the promoter [-1kp to +2kb from the transcription start site (TSS)]. Only genes with a minimum of 13 allele-specific ChIP-seq reads mapped to the promoter in both WT and Smchd1-/- NPCs were considered allelically-distinguishable and included in the analysis. H3K27me3 enrichment of each gene was calculated by dividing the number of ChIP-seq reads (comp) by input reads (comp) mapped to the gene body in count-normalized input and ChIP datasets. To compare H3K27me3 enrichment on the X chromosome between WT and Smchd1-/-, we scaled them based on the total number of H3K27me3 ChIP-seq reads mapped to the X.
Using the expression profiles and chromatin signatures, we defined 6 classes of X-linked genes. We first filtered out X-linked genes that were too short (<200bp) and genes embedded within the gene body of a longer gene. Silent genes were defined as X-linked genes having zero FPKM in at least one RNA-seq sample. Constitutive escapees were defined as active (not silent) X-linked genes with bi-allelic (%mus>7%) allelically-distinguishable H3K4me3 peaks at promoters and H3K27me3-depleted gene bodies (H3K27me3 enrichment < 1) in WT cells.
Class I genes were defined as active X-linked genes with allelically-distinguishable H3K4me3 peaks at promoters and depletion of H3K27me3 in gene bodes in Smchd1-/- cells (H3K27me3 enrichment in Smchd1-/- < 0.3 × H3K27me3 enrichment in WT). Class II genes were defined as active X-linked genes with bi-allelic allelically-distinguishable H3K4me3 peaks at promoters in Smchd1-/- but not WT cells (%mus >7% in Smchd1-/-, %mus ≤ 7% in WT, and %mus in Smchd1-/- > 3-fold %mus in WT) and without depletion of H3K27me3 in gene bodies in Smchd1-/- cells (H3K27me3 enrichment in Smchd1-/- ≥ 0.3 × H3K27me3 enrichment in WT). Class III genes were defined as X-linked genes that were active, had allelically-distinguishable H3K4me3 peaks at promoters, but did not belong to escapees, Class I, or Class II genes. All other genes, such as active X-linked genes whose H3K4me3 peaks at promoters were not allelically-distinguishable, were considered as unclassified genes.
To analyze the genomic distribution of different classes of X-linked genes, we segmented the X into 416 400-kb bins and assigned each X-linked gene to these bins based on their TSS. The number of genes in each bin was counted and plotted as histograms. The Pearson’s correlation coefficient (r) was then computed to determine the relationship between the occurrence of Class I genes and gene density. Bins with no X-linked genes were excluded in the correlation analysis.
To analyze intergenic regions, we defined H3K27me3-depleted intergenic regions as intergenic regions at which the H3K27me3 enrichment in Smchd1-/- < 0.3 × H3K27me3 enrichment in WT. Intergenic regions that were too short (<100bp) or unmappable (no reads in at least one input or H3K27me3 ChIP-seq libraries) were excluded from the analysis.
For CTCF and RAD21 ChIP-seq, peaks were called by MACS2 using 0.01 as the q value cutoff. The peaks identified in WT and Smchd1-/- NPCs were merged and the %mus of each peak was computed. Only peaks with a minimum of 13 allele-specific ChIP-seq reads in both WT and Smchd1-/- were considered allelically-distinguishable and included in the analysis. To identify TAD boundary-associated CTCF peaks, we used TAD boundaries defined in a previous study (Dixon et al., 2012). Peaks were considered as boundary-associated if located within a ±20kb window centered at the midpoint of each TAD boundary, as previously described (Dixon et al., 2012). TsixTST/+ mESC line is not fully hybrid. Therefore, for the autosomal control, we analyzed chromosome 13, which is fully hybrid.
Xist CHART-sequencing in wild-type and Smchd1-/- NPCs
Xist CHART-seq was performed as described (Simon et al., 2013) with modifications incorporated from Chromatin Isolation by RNA Purification (ChIRP) protocol (Chu et al., 2011; Chu et al., 2017; Zovoilis et al., 2016). NPCs were cross-linked in PBS with 1% formaldehyde at room temperature for 10 minutes at 2 million cells/ml and then quenched with 0.125M glycine at room temperature for 5 minutes. Cross-linked cells were washed three times with chilled PBST (1× PBS with 0.05% Tween-20) before being snap-frozen in liquid nitrogen. 25 million cross-linked cells were thaw on ice for 10 minutes with 1ml chilled sucrose buffer (10mM HEPES pH7.5, 0.3M sucrose, 1% Triton X-100, 100mM potassium acetate, 0.1mM EGTA) supplemented with 0.5mM spermidine, 0.15mM spermine, 1mM DTT, 1× Protease Inhibitor Cocktail (11873580001, Sigma), 10U/ml SUPERaseIN RNase Inhibitor (AM2694, Thermo Fisher Scientific) freshly. After thawing, cells were resuspended by pipetting and incubated at 4°C for 10 minutes with rotation. 1ml cell suspensi on were then mixed with 2ml chilled sucrose buffer in a 15ml glass dounce tissue grinder (357544, Wheaton), which had been chilled on ice and rinsed once with RNaseZAP RNase Decontamination Solution (AM9782, Thermo Fisher Scientific), three times with DEPC-treated water, and once with 2ml sucrose buffer. To release nuclei, cells were dounced 20 times with a tight pestle. 10 l dounced cells were mixed with 10 l 0.4% Trypan blue (15250061, Thermo Fisher Scientific) on a glass slide and examined by a microscope to ensure complete release of nuclei. The nuclear suspension was then carefully layered on top of a cushion of 7.5ml chilled glycerol buffer (10mM HEPES pH7.5, 25% glycerol, 1mM EDTA, 0.1mM EGTA, 100mM potassium acetate) freshly supplemented with 0.5mM spermidine, 0.15mM spermine, 1mM DTT, 1× PIC, and 5U/ml SUPERaseIN, and centrifuged at 1500g at 4°C for 10 minutes. Purified nuclear pelle ts were then resuspended with 3ml PBS and further cross-linked with 3% formaldehyde by mixing with 3ml cross-linking solution (1× PBS, 6% formaldehyde), followed by incubation at room temperature for 30 minutes with rotation. After the second cross-linking step, nuclei were pelleted again by centrifuge at 1000g for 5 minutes at 4°C and washed three times with 1ml chil led PBST. Washed nuclei were resuspended in 1ml chilled nuclear extraction buffer (50mM HEPES pH 7.5, 250mM NaCl, 0.1mM EGTA, 0.5% N-lauroylsarcosine, 0.1% sodium deoxycholate, 5mM DTT, 10U/ml SUPERaseIN) and incubated at 4°C for 10 minutes wit h rotation. To harvest extracted nuclei, nuclear suspension was centrifuged at 400g for 5 minutes at 4°C. Nuclei were then resuspended in 230 l chilled sonication buffer (50mM HEPES pH 7.5, 75mM NaCl, 0.1mM EGTA, 0.5% N-lauroylsarcosine, 0.1% sodium deoxycholate, 0.1% SDS, 5mM DTT, 10U/ml SUPERaseIN) to reach approximately 270 l final volume. Nuclei were sonicated in two 130 l batches within a Covaris microTUBE, which had been rinsed with 100 l sonication buffer and pre-chilled on ice, for 5 minutes with Covaris E220e (140W peak incident power, 10% duty factor, 200 cycles/burst). Sonicated chromatin was pooled and centrifuged at full speed for 20 minutes at 4°C. The supernatant (~220 l) was then mixed with 110 l sonication buffer to reach ~330 l final volume, which was then split into two CHART reactions (Xist capturing CHART and a negative control using sense probes).
For two CHART reactions, 600 l MyOne™ Streptavidin C1 beads (65001, Thermo Fisher Scientific) were used, which were first rinsed with 1ml DEPC-treated H2O twice, blocked with 600 l blocking buffer [33% sonication buffer, 67% 2× hybridization buffer, 500ng/μl yeast RNA (AM7118, Thermo Fisher Scientific), 1% BSA] at room temperature for 1 hour, washed with 1ml DEPC-treated H2O twice, and resuspended in 610 l 1× hybridization buffer [33% sonication buffer, 67% 2× hybridization buffer (see below for the composition)]. For each CHART, 160 l chromatin was mixed with 320 l 2× hybridization buffer (50mM Tris-HCl pH 7.0, 750mM NaCl, 1% SDS, 1mM EDTA, 15% formamide, 1mM DTT, 1mM PMSF, 1× PIC, and 100U/ml SUPERaseIN) and precleared with 60 l pre-blocked MyOne™ Streptavidin C1 beads at room temperature for 1 hour with rotation. Pre-cleared chromatin was separated form the beads, with two 5 l aliquots saved as the input samples. The rest of chromatin (~500 l) was mixed with 36pmol of either biotinylated Xist-targeting capture probes or sense control probes (antisense to the capture probes). We used a pool of 10 Xist-targeting capture probes, which were identical to the 11 Xist-targeting capture probes described previously (Simon et al., 2013) excluding probe X.8, which has sequences similar to a region on chromosome 1. Probes are listed in Table S1. Hybridization was carried out at 37°C for 4 h follo wed by incubation with 240 l blocked MyOne™ Streptavidin C1 beads at 37°C for 1 h. The beads w ere washed once with 1ml 1× hybridization buffer (33% sonication buffer, 67% 2× hybridization buffer) at 37°C for 10 minutes, five times with 1ml wash buffer (10mM HEPES pH 7.6, 150mM NaCl, 2% SDS, 2mM EDTA, 2mM EGTA, and 1mM DTT) at 37°C for 5 minutes, and t wice with 1ml elution buffer (10mM HEPES pH7.6, 150mM NaCl, 0.5% NP-40, 3mM MgCl2, and 10mM DTT) at 37°C for 5 minutes.
In the final wash, a 10 l aliquot was saved from 1ml wash reactions as the “RNA sample,” which was later used to estimate the degree of Xist RNA enrichment by CHART. CHART-enriched DNA was eluted by 20 l RNase H (5U/μl; M0297S, NEB) in 200 l elution buffer twice at 37°C for 30 minutes. One input sample and the eluent were treated with 10 l Purelink RNase A (20mg/ml; 12091–021, Thermo Fisher Scientific) at 37°C for 1h and then with 1% SDS, 10mM EDTA, and 10 l proteinase K (20mg/ml; 03115844001, Sigma) at 55°C for 1h. Reversal of cross-link was performed by supplementing the eluent with 0.3M NaCl and incubation at 65°C overnight.
CHART-enriched DNA was then extracted by phenol-chloroform, resuspended in 160 l H2O, and quantified with Quant-iT™ PicoGreen® dsDNA Reagent (P7581, Thermo Fisher Scientific). Quantitative PCR for the Xist (positive control) and Gapdh promoter (negative control) was then conducted to verify successful CHART. Primers are listed in Table S1. Xist CHART experiments with the Xist promoter at least 50-fold more enriched than Gapdh promoter and at least 5-fold more enriched than the control CHART (using sense probes) were sequenced. Input and CHART-enriched RNA (the “RNA sample”) were mixed with 240 l PK buffer (10mM Tris pH7.0, 100mM NaCl, 1mM EDTA, 0.5% SDS) and 10 l proteinase K (20mg/ml; 03115844001, Sigma), incubated at 55°C for 1 hour, and then 65°C for 1 h our to reverse cross-link. RNA was then extracted by 750 l TRIzol LS (10296010, Thermo Fisher Scientific). RT-qPCR was performed to detect Xist RNA and Gapdh mRNA to ensure specific enrichment of Xist RNA. Primers are listed in Table S1. To generate sequencing libraries, 100 l input and CHART-enriched DNA were mixed with 30 l TE buffer and further sheared to below 500bp in a Covaris microTUBE for 2 minutes with Covaris E220e (140W peak incident power, 10% duty factor, 200 cycles/burst). Sonicated DNA was purified using 1.8× Agencourt AMPure XP beads (A63881, Beckman Coulter). Libraries were prepared by NEBNext® Ultra™ DNA Library Prep Kit for Illumina® (E7370S, NEB), with double size selection (0.6X-1.2X Agencourt AMPure XP beads) performed after the adapter ligation step. Experiments were performed in two biological replicates. Input and CHART-seq libraries were sequenced on Illumina HiSeq, generating 26–42 millions paired-end 50 nucleotide reads per sample.
Analysis of Xist CHART-seq datasets
Input and CHART-seq reads (enriched by either the Xist capture probes or the sense control probes) were aligned allele-specifically as described (Simon et al., 2013). After removal of PCR duplicates, uniquely aligned reads were used to generate input-subtracted coverage profiles using SPP (Kharchenko et al., 2008), with smoothing using 1-kb windows recorded every 500bp, as described previously (Simon et al., 2013). Given that Xist RNA only coats the Xi (Xmus), we considered the number of Xcas reads as an estimate of the background noise of each CHART experiment. Therefore, to compare the distribution of Xist between WT and Smchd1-/- cells, we scaled the Xist coverage profiles produced by SPP based on the total number of Xmus reads minus the total number of Xcas reads. Scaled Xist coverage profiles were visualized by IGV, with scales indicated in each track. For metagene analysis, we generated SPP-normalized coverage profiles with smoothing using 2-kb windows recorded every 50bp, as described previously (Simon et al., 2013) and calculated average density from the scaled coverage profiles using CEAS (Ji et al., 2006).
To investigate the correlation of between Xist, H3K27me3, and H3K4me3 binding patterns, we segmented the X into 500-kb bins and computed their coverage at each bin. Pearson’s correlation coefficients (r) were then calculated to compare their distributions. Unmappable regions (bottom ~15% coverage, defined using Hi-C data) were excluded in the correlation analysis.
In situ Hi-C in wild-type NPCs, Smchd1-/- NPCs, and differentiating female wild-type ES cells
In situ Hi-C was performed essentially as described (Nagano et al., 2015; Rao et al., 2014) with minor modifications outlined below. Briefly, 5 million cells were cross-linked with 1% formaldehyde for 10 minutes at room temperature. Cross-linked cells were lysed and the nuclei were digested with 15 l HindIII (100 U/μl; R0104M, NEB) at 37°C overnight. We noticed that some clumps formed following overnight digestion, which were removed by brief spinning, with the supernatant transferred to a new tube for the fill-in reaction with biotin-14-dATP (19524016, Thermo Fisher Scientific) and subsequent ligation. Re-ligated DNA was purified and then sheared to 300–500bp in a Covaris microTUBE for 80 seconds with Covaris E220e (140W peak incident power, 10% duty factor, 200 cycles/burst). Double size selection was performed on sheared DNA using 0.55X-0.75X Agencourt AMPure XP beads (A63881, Beckman Coulter). Biotinylated ligation products were purified with 30 l MyOne™ Streptavidin C1 beads (65001, Thermo Fisher Scientific), end-repaired, dA-tailed, ligated to 3 l 15 M NEBNext Adaptor for Illumina [enclosed in NEBNext® Multiplex Oligos for Illumina® (Index Primers Set 1); E7335S, NEB], and digested with 3 l USER enzyme [also enclosed in NEBNext® Multiplex Oligos for Illumina® (Index Primers Set 1)] at 37°C for 15 min utes. Hi-C libraries were then amplified with 12–16 cycles of PCR. In situ Hi-C on WT and Smchd1-/- NPCs was performed in two biological replicates, which were sequenced on Illumina HiSeq, generating 188–220 millions paired-end 50 nucleotide reads per sample. Hi-C libraries generated from D0 ES cells, D4 embryoid bodies, and D7 embryoid bodies were sequenced on Illumina HiSeq in a rapid run mode, generating 105–128 millions paired-end 50 nucleotide reads per sample.
Analysis of in situ Hi-C datasets in wild-type NPCs, Smchd1-/- NPCs, and differentiating female wild-type ES cells
Hi-C reads were aligned allele-specifically as described (Minajigi et al., 2015). Briefly, each end of the read-pairs was aligned separately as single-end reads to 129S1/SvJm (mus) and CAST/Eih (cas) genome using NovoAlign and allele-specificity was assigned. The single-end alignments were joined to generate a Hi-C summary file. Allele-specific reads were filtered and Hi-C contact matrices were constructed using Homer (Heinz et al., 2010). To increase depth, two biological replicates of WT and Smchd1-/- Hi-C reads were pooled. Our WT and Smchd1-/- Hi-C libraries had similar fraction of interchromosomal reads (17.7% for both libraries), indicating that the difference in Hi-C profiles was not caused by differing qualities of Hi-C experiments. The numbers of filtered read pairs mapped to the Xa and Xi in WT and Smchd1-/-libraries were also similar, arguing against different sequencing depths as the origin of different Hi-C profiles between WT and Smchd1-/-. Iterative correction of Hi-C matrices and construction of Pearson correlation matrices were performed using HiTC (Servant et al., 2012). Unmappable regions, defined as bins with low read coverage (bottom ~15%), were set to NA before iterative correction. Principle component analysis (PCA) was conducted using HiTC at 200-kb resolution and the compartment profiles (PC1 values) were visualized using IGV. Insulation scores and insulation boundary strength were generated from iteratively corrected 40-kb binned Hi-C matrices using the script matrix2insulation.pl (publically available on Github: https://github.com/dekkerlab/giorgetti-nature-2016) with parameters −is 480000 −ids 320000 −im iqrMean −nt 0 −ss 160000 −yb 1.5 −nt 0 −bmoe 0, as described in a recent study (Giorgetti et al., 2016).
To compare the Hi-C profiles of WT and Smchd1-/- NPCs with those of D0 ES, D4 EB, and E7 EB, we randomly sampled 50% interactions from the Xa and Xi from replicate 1 and reconstructed the Hi-C contact maps, Pearson correlation maps, and PC1 profiles to account for different sequencing depths between libraries. We noted that the compartmentalization in the Hi-C dataset of D0 ES cells was less distinct, as shown by the weaker checkerboard pattern on the Pearson correlation maps constructed using allele-specific reads (Xcas and Xmus maps), which led to less well-defined A/B compartment profiles derived from PCA. Therefore, although we displayed the contact maps and Pearson correlation maps of D0 Xcas and Xmus, we used the compartment profiles of D0 ES cells generated using all reads mapped to the X chromosomes (the Xcomp map) in analyses. The D0 Xcomp compartment profiles provided better-defined A/B compartment borders to investigate the relationships between A/B and S1/S2 compartments. Importantly, the Xcomp compartment profiles of D0 ES cells should be representative of either the Xcas or the Xmus chromosome, as they are both active and folded similarly in D0 ES cells. For comparison, we also reanalyzed a deeply sequenced Hi-C dataset of undifferentiated female ES cells (Giorgetti et al., 2016). Reads were downloaded from GEO (GSE48649) and aligned to the mm9 genome using bowtie2. The A/B compartment profiles were then similarly obtained at 200-kb resolution using HiTC. The resulting X chromosomal compartment profile is highly similar to our D0 ES Xcomp profile (r=0.92). The hierarchical clustering analysis on compartment profiles was performed using the dist (method=“euclidean”) and hclust (method=“complete”) functions in R.
Xist binding patterns in undifferentiated (D0), day 3 (D3) and day 7 (D7) differentiating female mouse ES cells were downloaded from GEO (GSE48649). To investigate the correlation between PC1 values, Xist, H3K27me3, and H3K4me3 binding patterns, we segmented the X into 500-kb bins and computed the coverage at each bin. Pearson’s correlation coefficients (r) were then calculated. Unmappable regions and regions where PC1 values were not computed were excluded in the correlation analysis. To investigate the relationships between the early domains of Xist spreading and the A compartments on the X chromosome in pre-XCI cells, we compared D3 Xist binding pattern with the PC1 values of D0 ES cells using three different datasets, which all showed a high correlation (D0 Xcomp, r=0.79; D0 Xmus, r=0.70, Giogetti et al, r=0.82).
To calculate the genomic space (% length of the X chromosome) of each compartment, we used the PC1 values at 200-kb resolution, and defined each 200-kb bin with positive PC1 value as A/S1-compartments, with unmappable regions and regions where PC1 values were not computed excluded in the analysis. To investigate the relationship between X-linked gene classes and changes in chromosome conformation, the associated PC1 value and insulation score of each X-linked gene were determined by the bin containing the TSS of each gene. Genes associated with positive PC1 values were considered as residing in the A (Xa) or S1 (Smchd1-/- Xi) compartments. Genes located in the left megadomains were defined as genes located in the first 73 Mb of the X chromosome.
To investigate the effect of Smchd1 ablation on spreading of Xist RNA from S1 (early domains) to S2 compartments (late domains), we computed the coverage density of scaled Xist CHART (comp) and H3K27me3 ChIP-seq (comp) profiles in WT and Smchd1-/- at each 200-kb bin on the X chromosome. As regions harboring Class I genes showed Xist and H3K27me3 depletion, we excluded these regions by removing bins overlapping with the gene bodies of Class I genes and the centrometric end of Xist (0–30 Mb) from this analyses. We then computed the median coverage densities of S1 (bins with positive PC1 values on the Smchd1-/- Xi) and S2 (bins with negative PC1 values on the Smchd1-/- Xi), with unmappable regions and bins where PC1 values were not computed excluded. The S1-S2 gap was then computed by subtracting median Xist/H3K27me3 density of S2 from that of S1. Two biological replicates were analyzed to compute statistical significance using paired t-test (one-sided).
DamID-sequencing analysis of SMCHD1
Full length Smchd1 was PCR amplified (Smchd1-BamHI-F, CGCGGATCCGCCACCATGGCAGCGGAGGGCGCCA; Smchd1-NotI-R, AAGGAAAAAAGCGGCCGCCGTTTGGTACGTCACCTTTTGGCCTTC) from oligo-dT-primed cDNA of J1 male mouse ES cells and cloned into an in-house vector, with missense mutations corrected by QuikChange II XL Site-Directed Mutagenesis Kit (200521, Agilent Technologies). The Smchd1 cDNA was subsequently cloned into pLgw EcoDam-V5-RFC1 (Vogel et al., 2006)(59209, Addgene) to derive pLgw EcoDam-V5-SMCHD1. The expression and localization of EcoDam-V5-SMCHD1 fusion protein (Dam-SMCHD1) were confirmed by western and immunostaining for the V5 epitope on immortalized female MEFs transiently transfected with pLgw EcoDam-V5-SMCHD1. To generate stable cell lines for DamID, pLgw EcoDam-V5-SMCHD1, pLgw V5-EcoDam (Vogel et al., 2006)(59210, Addgene), and pLgw CBX1-EcoDam (Vogel et al., 2006)(59211, Addgene) were double-digested with AhdI and either NotI [pLgw EcoDam-V5-SMCHD1 replicate 1 (SMCHD1-Dam rep1)] or AleI [pLgw V5-EcoDam (Dam alone), pLgw CBX1-V5-EcoDam (CBX1-Dam), and pLgw EcoDam-V5-SMCHD1 (Dam-SMCHD1 rep2)] to remove the strong CMV promoter. The fragments harboring the Dam fusion protein driven by the HSP70 promoter were isolated by electrophoresis and electroelution, purified by phenol-chloroform, and transfected into EY.T4, an immortalized female MEF line using Lipofectamine LTX & Plus Reagent (15338100, Thermo Fisher Scientific). Similar to NPCs, MEFs have also completed XCI and exhibit merged compartments and attenuated TADs (Minajigi et al., 2015). The EY.T4 MEF line is a clonal F1 hybrid where the active X chromosome (Xa) is of Mus casteneus (cas) origin and the Xi of Mus musculus (mus) origin (Yildirim et al., 2012). Thus, >600,000 X-linked sequence polymorphisms between the Xcas and Xmus enabled us to generate allele-specific SMCHD1-binding maps of the Xa and the Xi. Starting one day after transfection, stably transfected cells were selected with 100ng/ml Zeocin (R25001, Thermo Fisher Scientific) for ~10 days. Genomic DNA of stable lines was purified by DNeasy Blood and Tissue Kits (69506, Qiagen), with the RNase A digestion step included. The presence of transgenes was confirmed by genomic PCR using primers listed in Table S1.
Methylated DNA was PCR-amplified as described (Vogel et al., 2006). Briefly, 2.5 g genomic DNA was digested by 10 units DpnI (R0176S, NEB) in 1× CutSmart buffer (B7204S, NEB) overnight at 37°C, followed by heat-inactivation of DpnI for 20 minutes at 80°C. 2.5 g DpnI-digested DNA was ligated with 2 M DamID adaptor [slowly annealed AdRt (CTAATACGACTCACTATAGGGCAGCGTGGTCGCGGCCGAGGA) and AdRb (TCCTCGGCCG)] using 5 units T4 ligase in 20 l 1× T4 ligation buffer at 16°C overnight, followed by heat-inactivation of T4 ligase for 10 minutes at 65°C. 2.5 g adator-ligated DNA was digested with 10 units DpnII (R0543S, NEB) in 50 l 1× DpnII buffer for 1 hour at 37°C. Methylated DNA was PCR amplified using Advantage®2 PCR enzyme system (639206, Takara Bio Corporation Div Clontech). Reaction consisted of 10 l DpnII digested DNA, 5 l 10× Advantage 2 PCR buffer, 0.625 l 100 M AdR_PCR (GGTCGCGGCCGAGGATC), 1 l 50× dNTP mix, 1 l Advantage 2 Polymerase mix, and 32.375 l H2O, with the reaction program: 1 cycle of 68°C (10 minutes), 95°C (1 minute), 65°C (5 minutes), 68°C (15 minutes); 4 cycles of 95°C (1 minute), 65°C (1 minutes), 68°C (10 minutes); 18 cycles of 95°C (1 minute), 65°C (1 minutes), 68°C (2 minutes). 5 l PCR products were run on 1% agarose gel to verify successful amplification, with no ligase, no DpnI, and non-transfected cell controls yielding no visible PCR products. The remaining PCR products were purified using QIAquick PCR Purification Kit (28106, Qiagen). To remove DamID adaptors, 15 g purified PCR products were digested with 10 units DpnII in 150 l 1× DpnII buffer at 37°C for 3 hours, followed by heat-inactivation of DpnII at 65°C for 20 minutes. DpnII-digested PCR pr oducts were purified again with QIAquick PCR Purification Kit, and 3 g PCR products were sonicated in a Covaris microTUBE for 270 seconds with Covaris E220e (140W peak incident power, 10% duty factor, 200 cycles/burst). DamID-seq libraries were generated from 500ng sonicated PCR products using NEBNext® Ultra™ DNA Library Prep Kit for Illumina® (E7370S, NEB), with double size selection [0.6X-1.2X Agencourt AMPure XP beads (A63881, Beckman Coulter)] performed after the adapter ligation step. Adaptor-ligated DNA was enriched using 5 cycles of PCR and purified with 1.2× Agencourt AMPure XP beads. Libraries for Dam alone, CBX1-Dam, and Dam-SMCHD1 rep1) were sequenced on Illumina HiSeq in a rapid run mode, generating 14–16 millions paired-end 69 nucleotide reads per sample. The library for Dam-SMCHD1 rep2 was sequenced on Illumina HiSeq in a rapid run mode, generating ~38 millions paired-end 50 reads.
Analysis of SMCHD1 DamID-seq datasets
DamID-seq reads were aligned allele-specifically to 129S1/SvJm (mus) and CAST/Eih (cas) genome using NovoAlign as described (Minajigi et al., 2015). After removing PCR duplicates, uniquely mapped read pairs were used for further analysis. Reads mapped to chromosome 3 and X were quantified by samtools. To normalize the signals generated by Dam-fusion proteins over background, we computed the log2(Dam-fusion/Dam alone) values of each GATC fragments using damidseq_pipeline (Marshall and Brand, 2015). The damidseq_pipeline software offers two methods of normalization: one (the kde mode) uses kernel density estimation of log2 GATC fragment ratio and the other (the rpm mode) uses read counts per million reads. These two methods generated similar profiles when analyzing CBX1-Dam and Dam-SMCHD1 rep1. However, the kde mode generated a normalized Dam-SMCHD1 rep2 profile where the average SMCHD1 signal seemed to be higher than the rpm mode, likely because Dam-SMCHD1 rep2 was sequenced at much higher depth. To overcome this issue, we randomly sampled 45% reads from Dam-SMCHD1 rep2 libraries (command: samtools view -bs 42.45) to generate the kde-mode normalized profile of down-sampled Dam-SMCHD1-rep2. As the kde-mode normalized profiles show high correlation between Dam-SMCHD1 rep1 and down-sampled rep2, we displayed the kde-mode normalized Dam-SMCHD1 rep1 profile in the main figures using IGV, with scales indicated in each track.
To investigate the relationships between Dam-SMCHD1 replicates and CBX1-Dam profiles, we computed their degree of enrichment at each 100-kb bin across the genome and computed Pearson correlation coefficients (r). To compare the degree of SMCHD1, Xist, and H3K27me3 enrichment in S1 v.s. S2 compartment, Xist CHART-seq and H3K27me3 ChIP-seq data in female EY.T4 MEFs were downloaded from GEO (GSE48649). Xist, H3K27me3, and SMCHD1 enrichment [log2(Dam-SMCHD1/Dam alone)] (comp tracks) were quantified at each 200-kb bin on the X chromosome, with bins having positive PC1 values in PCA on Smchd1-/- NPC Xi Hi-C data at 200-kb resolution assigned as S1 compartments, and the other bins as S2 compartments. To compare SMCHD1 enrichment at different classes of X-linked genes, we quantified average SMCHD1 enrichment in the gene body of each gene using Homer. To determine if SMCHD1 showed enrichment at TAD boundaries, we used previously defined TAD boundaries (Dixon et al., 2012) and computed the average enrichment profiles for each 10-kb bin spanning a 1-Mb window centered at TAD boundaries. CTCF ChIP-seq in WT NPCs (replicate 1) was similarly analyzed as a positive control.
Western blot analysis of SMCHD1
Cells were washed once with PBS and lysed in chilled lysis buffer [10mM Tris pH 8.0, 1M NaCl, 1% Triton, 0.5% sodium deoxycholate, 0.1% SDS, 1mM EDTA, 0.5mM EGTA, 1× cOmplete EDTA-free Protease Inhibitor Cocktail (PIC; 11873580001, Sigma)]. Lysates were sonicated at 4°C for 15 minutes (30 seconds ON; 30 seconds OFF) using Bioruptor®XL (Diagenode). Proteins were quantified using Bio-Rad Protein Assay (500–0006, Bio-Rad). 20–50 g lysates were denatured in 1X Laemmli sample buffer at 95°C for 5 minutes and resolved by SDS-PAGE (7% resolving, pH 8.8, 29:1 acrylamide:bisacrylamide) performed in 11× Running Buffer (25mM Tris pH 8.3, 192mM glycine, 0.1% SDS) with an XCell SureLock™ Mini-Cell Electrophoresis system (Thermo Fisher Scientific). Proteins were transferred from the gels to PVDF membranes (Immobilon Transfer Membrane; IPVH00010, EMD Millipore) in 11× TBE in a TE77XP Semi-dry Blotter (Hoefer) at 0.8mA/cm2 for 45 minutes. PVDF membranes were blocked with Blocking Buffer (5% milk and 0.1% Tween-20 in PBS) at room temperature for 1 hour, incubated with antibodies against SMCHD1 (HPA039441, Sigma; 1:1000) or β-tubulin (T5201, Sigma, 1:2000) diluted in Blocking buffer at 4°C overnight. Membra nes were then washed with PBST (PBS with 0.1% Tween-20) for 5 minutes three times, incubated with Anti-Rabbit IgG (H+L), HRP Conjugate (W4011, Promega; 1:5000) or Anti-Mouse IgG (H+L), HRP Conjugate (W4021, Promega; 1:10000) diluted in Blocking Buffer at room temperature for 1 hour, washed with PBST for 5 minutes three times, and developed using Western Lightning® Plus-ECL (NEL105001EA, PerkinElmer).
RT-PCR
RNA was isolated from cells using TRIzol (15596018, Thermo Fisher Scientific) according to manufacturer instructions. Genomic DNA was removed from 10 μg RNA in a 50 l reactivation with 2 units TURBO DNase using TURBO DNA-free™ Kit (AM1907, Thermo Fisher Scientific). After inactivating TURBO DNase by DNase Inactivation Reagent, 1.6 μg DNase-treated RNA was reverse transcribed using SuperScript III Reverse Transcriptase (18080–085, Thermo Fisher Scientific) with 250ng random primers (C118A, Promega) at 25°C for 5 minutes, 50°C for 1 hour, and 85°C for 15 minutes. cDNA was 5~10-fold diluted with H2O. Conventional PCR was performed using 21× PCR MasterMix (M7505, Promega). Quantitative PCR was performed using iTaq™ Universal SYBR® Green Supermix (1725125, Bio-Rad) in a CFX96 Real-Time PCR Detection System (Bio-Rad).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters including the statistical tests used and the exact value of p and Pearson’s r are reported in the figures, figure legends, or associated main texts. Statistical significance is determined by the value of p<0.05 by the indicated tests.
DATA AND SOFTWARE AVAILABILITY
Raw next-Gen sequencing data and processed files for RNA-seq, ChIP-seq, CHART-seq, Hi-C, and DamID-seq have been deposited in Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under accession number GEO: GSE99991.
Supplementary Material
(A) Western blot for SMCHD1 in WT and XaWTXiΔXist fibroblasts.
(B) SMCHD1 immunostaining and Xist RNA-FISH in wild-type female MEF (♀WT) and a female MEF clone carrying an inducible Xist transgene (♀X+P). In induced ♀X+P cells, a large transgenic Xist cloud was detected, in addition to two Xi clouds (Jeon and Lee, 2011). SMCHD1 was enriched on all three (♀X+P, top panel). This demonstrates that Xist RNA is sufficient for SMCHD1 recruitment. In some cells, the transgene “squelched” endogenous Xist RNA from the Xi, leaving only the prominent transgenic Xist cloud (Jeon and Lee, 2011). Interestingly, in these cells, SMCHD1 was also lost from the Xi (♀X+P, bottom panel), consistent with localization being dependent on Xist RNA. Arrowheads, transgenic Xist cloud. Fibroblasts have been immortalized with SV40 large T-antigen and are therefore tetraploid.
(C) Validation of the α-SMCHD1 antibody used in this study by siRNA-mediated knockdown of SMCHD1 in MEFs.
(D) Western blot confirmed the absence of SMCHD1 protein in Smchd1-/- ES cell clones.
(E) Two different pairs of guide RNAs (pairs 1 and pairs 2) targeting a region immediately downstream of the start codon (ATG) in the first exon of Smchd1.
(F) Sanger sequencing confirmed the presence of frame-shifting mutations. Regions containing the microdeletions were PCR amplified and cloned into a TOPO-TA vector. Numbers within parentheses indicate the number of clones sequenced. Sequences targeted by the guide RNA pairs are colored in red or blue.
(G) Confirmation of the identity of NPCs by RT-PCR. Mouse ES cell clones expressed pluripotency markers Pou5f1 (Oct4) and Nanog, but not NPC markers Blbp, Oligo2, and Emx2. In contrast, NPCs derived from these ESC clones expressed NPC markers, but not pluripotency markers. Actb (β-actin) and Pax6 served as controls, as they were expressed in both cell types. RT+, reverse transcriptase plus. RT-, reverse transcriptase minus. E, ES cells. N, NPCs.
(H) Immunostaining confirmed that the NPCs we derived expressed NPC markers NESTIN and SOX2.
(I) (from left to right) 1. Xist RNA-FISH showed a greater fraction of Smchd1-/- NPCs without a Xist cloud. 2. DNA-FISH for Atrx, an X-linked gene, showed a greater fraction of Smchd1-/- NPCs displaying only one Atrx locus, indicating that XO cells were enriched, resulting in fewer cells with a Xist cloud. 3. DNA-FISH for Tmem39a, an autosomal gene on chr16, showed that most WT and Smchd1-/- NPCs had two Tmem39a loci, indicating that the effect of Smchd1 ablation on chromosome number is specific to the X. 4. Xist DNA-FISH showed that the majority of mESCs used to derive these NPCs harbored two Xist loci, suggesting that XO cells became enriched in Smchd1-/- NPCs during differentiation.
(J) Derivation of clonal WT and Smchd1-/- NPC lines with homogeneous XX genotypes.
{kind=link}
(A) RNA-seq coverage profiles (comp) at the Smchd1 locus demonstrating microdeletions in Smchd1-/- NPC clones. Smchd1-/- clones 1 and 2 were independently generated, with distinct microdeletions.
(B) A bar plot displaying the distribution of differentially expressed genes on each chromosome. Up: genes upregulated in Smchd1-/- cells. Down: genes downregulated in Smchd1-/- cells.
(C) RNA-seq coverage profiles (comp) at the Pcdh locus demonstrating upregulation of the Pcdhα gene cluster in both Smchd1-/- NPC clones.
(D) Degree of allelic skewing of 244 allele-assessable X-linked genes was highly concordant between two biological replicates. r, Pearson correlation coefficient of 244 allele-assessable X-linked genes (black) or 73 concordant SMCHD1-sensitive genes (red).
(E) A probability density plot of the number of overlapping genes between two independent random samplings of 90 and 82 genes from 230 genes for 10,000 times.
(F) Smchd1 ablation did not alter transcription from the Xcas (Xa). Shown are the cas-specific fragments per kilobase of transcript per million exonic reads (cas fpkm) of 73 concordant SMCHD1-sensitive genes. Solid diagonal, 1:1 ratio. Dashed diagonal, 1:2 ratio.
(G) In Smchd1-/- NPCs, increased expression of the Xmus alleles caused 1~2 fold upregulation of the 73 concordant SMCHD1-sensitive genes. Shown are the comp fpkm values of the 73 concordant SMCHD1-sensitive genes. Solid diagonal, 1:1 ratio. Dashed diagonal, 1:2 ratio.
{kind=link}
(A) Xi expression (%mus) of SMCHD1-sensitive genes showed moderate positive correlation with fraction Xi-enrichment (%mus) of H3K4me3 peaks at promoters (left panel) and negative correlation with H3K27me3 enrichment in gene bodies (right panel). r, Pearson correlation coefficient.
(B) H3K27me3 enrichment in Xi intergenic regions in WT (x-axis) versus Smchd1-/- (y-axis) cells. Three categories of intergenic regions are shown, as indicated above each scatter plot (e.g., between Class I and Class I genes, “Class I – Class I”).
(C) Two biological replicates for Xist CHART experiments show high reproducibility. Also shown are H3K4me3 enrichment (ChIP-seq, mus) across the entire X chromosome. Enrichment difference is determined by subtracting WT values from Smchd1-/-. Green-shaded area, Xist-depleted regions. Gray-shaded area, unmappable regions.
(D) Related to Figure 3B: Xist binding patterns (CHART-seq, comp), H3K27me3 enrichment (ChIP-seq, comp), and H3K4me3 enrichment (ChIP-seq) across a second representative Xist-depleted region.
(E) Xist enrichment in Xi intergenic regions in WT (x-axis) versus Smchd1-/- (y-axis) cells. Three categories of intergenic regions are shown, as indicated above each scatter plot (e.g., between Class I and Class I genes, “Class I – Class I”).
(F) Summary of the epigenetic states for different categories of active X-linked genes.
{kind=link}
(A) Scatter plots comparing normalized interaction frequencies in two Hi-C biological replicates. Each dot represents a 200-kb binned region on the Xa or Xi in WT or Smchd1-/- cells, as indicated. r, Pearson correlation coefficient.
(B) Number of various X-linked gene categories in different chromatin compartments. Left, the centromeric megadomain. Right, the telomeric megadomain.
(C) Related to Figure 5A: Hi-C interaction maps (binned at 40-kb resolution), insulation profiles, and the genomic location of Class I and Class II genes. Two additional representative X-linked regions are shown. TADs (as defined by Dixon et al., 2012) are delineated as blue bars between interaction maps and as dashed lines in the insulation graphs.
(D) Box plots comparing the boundary strength (defined as the differences in insulation scores between insulation minimum to the largest neighboring insulation maximum in the insulation profiles) of the Xa and Xi in WT and Smchd1-/- NPCs. The Xi boundary strength of Smchd1-/- tends to be higher than that of WT (P=0.013), but lower than that of Smchd1-/- Xa (P=4.6×10−4). The Xa boundary strength of Smchd1-/- is similar to that of WT (P=0.83). Green arrow, the TAD boundary near Firre. Black arrow, the TAD boundary near Dxz4. Boundaries near Firre and Dxz4 were included in the analysis. NS, not significant (P>0.05). *, P=0.013 (Wilcox test).
(E) Box plots comparing change in insulation scores (Δ) between Smchd1-/- and WT NPCs. The insulation score of each gene was determined by the insulation score of the bin containing the TSS. Xa and Xi values are shown. All classes of X-linked genes, except escapees, showed a wider spread of Δ on the Xi. IQR, interquartile range (Xa, blue; Xi, red).
(F) Box plots showing the distribution of differences in allelic skewing (Δ%mus) in Smchd1-/- relative to WT cells for CTCF peaks that are TAD boundary-associated (+) versus not associated (-). CTCF peaks on chr13 and chrX were shown. P-values from Wilcox test. NS, not significant (p>0.05). *, P=3.5×10−12. **, P=2.2×10−16 (Wilcox test).
{kind=link}
(A) Top four panels: Pair-wise comparison of the insulation profiles across the entire X chromosome for the Xa and Xi in WT versus Smchd1-/- (Mut) cells, as indicated. Bottom two panels: Locations of Class I and Class II genes. Dashed lines, TAD boundaries as defined by Dixon et al., 2012. Green-shaded area, Firre. Yellow-shaded area, Dxz4. Gray-shaded area, unmappable regions.
(B) Allelic patterns of CTCF and RAD21 binding within a representative X-linked region. Xi, mus. Xa, cas. Significant binding peaks are indicated in green.
(C) Skewing of CTCF binding expressed as %mus in WT versus Smchd1-/- cells for chrX or chr13. Dashed diagonal, 1:1 ratio.
(D) Skewing of RAD21 binding expressed as %mus in WT versus Smchd1-/- cells for chrX or chr13. Dashed diagonal, 1:1 ratio.
(E) Box plots showing differences in allelic skewing (Δ%mus) for CTCF binding in Smchd1-/- relative to WT cells for intergenic regions (e.g., between Class I and Class I genes, “Class I – Class I”). All measurements are for X-linked regions, except Chr13 – Chr13 measurements. Chr13 versus unclassified-unclassified, P=1. Chr13 versus all other categories of intergenic regions, P<0.025 (Wilcox test with Bonferroni correction, one sided).
(F) Scatter plot analyses correlating change (Δ) in Xist binding to change in allelic CTCF and RAD21 binding at Class I genes (red), or at all other X-linked genes (black). Xi binding shown (%mus). r, Pearson correlation coefficient.
(G) Scatter plot analyses correlating change (Δ) in H3K27me3 enrichment to change in allelic CTCF and RAD21 binding at Class I genes (red), or at all other X-linked genes (black). Xi binding shown (%mus).
{kind=link}
(A) Detection of Dam-SMCHD1 fusion protein (tagged with the V5 epitope) by western blot. In this experiment, MEFs were transiently transfected with plasmids overexpressing the fusion proteins to verify the fusion is expressed at the expected size.
(B) Immuno-RNA-FISH for overexpressed Dam-fusion proteins (V5-tagged) and Xist RNA demonstrating that fusions share similar subnuclear localization as wild-type proteins. Top: CBX1-Dam colocalizes with DAPI-dense foci, consistent with its recruitment to pericentromeric heterochromatin. Bottom: Dam-SMCHD1 is recruited to the Xi. Note that in the actual cell lines used for DamID-seq, Dam fusion proteins were expressed by the uninduced HSP70 promoter at low levels (see STAR Methods).
(C) Methylation-specific PCR on cells expressing Dam alone, CBX1-Dam, Dam-SMCHD1 (rep1 & 2), and non-transfected cells. Lane 1, DpnI-minus control. Lane 2, ligase-minus control. Land 3 and 4, duplicates. Note that DNA adenine methylation is only detected in transfected cells.
(D) Left, fraction of total unique read pairs mapped to the X and Chr3, an autosome (159 Mb) with similar length as the X (166 Mb). Middle, fraction of cas- and mus-specific pairs in total Chr3 allele-specific read pairs. Right, fraction of cas- and mus-specific pairs in total ChrX allele-specific read pairs. Dam, Dam alone. CBX1, CBX1-Dam. SMCHD1, Dam-SMCHD1.
(E) Scatter plots comparing two biological replicates of allele-specific SMCHD1 binding profiles for each 100-kb bin on the autosomes (black dots) and the X chromosomes (red dots). r, Pearson correlation coefficient for autosomes (black) and X chromosomes (red).
(F) SMCHD1 and CBX1 enrichment profiles across chromosome 3.
(G) A zoom-in at one of the SNP-scarce regions on the X chromosome. Shown are fragment-per-million normalized DamID coverage tracks for Dam alone and Dam-SMCHD1, as well as the log2 ratio [log2(Dam-SMCHD1/Dam alone)] tracks. Note that the scarcity of allelic reads (cas and mus) at the green-shaded regions in Dam alone and Dam-SMCHD tracks, which led to a “trough” in the log2 ratio tracks. SMCHD1 is actually not depleted at this region, as evident by the robust enrichment of SMCHD1 shown in the comp tracks.
(H) Average enrichment plots of SMCHD1 (left) and CTCF (right) at 10-kb resolution centered at the TAD boundary. Black, TAD boundaries on autosomes. Red, TAD boundaries on the X chromosome.
(I) SMCHD1 immunostaining and Xist RNA-FISH in D4 and D7 wild-type embryoid bodies (EB). In D4 EB, 70% cells exhibited an Xist cloud and none of them showed SMCHD1 recruitment on the Xi. Instead, SMCHD1 formed multiple nuclear foci colocalizing with DAPI-dense pericentrometric heterochromatin, consistent with previous reports (Brideau et al., 2015; Gendrel et al., 2012). In D7 EB, 84% cells harbored an Xist cloud, and SMCHD1 was observed to be enriched on the Xi in 4% of these cells.
(J) Box plots comparing the gap between Xist/H3K27me3 coverage densities of S1 and S2 compartments in WT versus Smchd1-/- NPCs. Red dots, S1 compartments. Blue dots, S2 compartments. Median, median coverage densities of S1 (red) or S2 (blue). S1-S2 gap, median S1 density – median S2 density. The S1-S2 gap was significantly widened when Smchd1 is ablated (Xist, p=0.041; H3K27me3, p=0.035, paired t-test, one-sided). Regions harboring Class I genes and the centromeric Xist-depleted regions were excluded in this analysis.
{kind=link}
(A) Allele-specific Hi-C analysis: Top: Contact maps (200-kb resolution) of the Xa (Xcas) in undifferentiated ES cells (D0 ES, pre-XCI), embryoid bodies formed after 4 or 7 differentiation days (D4 and D7 EB, early-mid XCI), and post-XCI wild-type (WT) and Smchd1-/- NPCs. Bottom: Pearson correlation maps. Gray-shaded area, unmappable regions.
(B) Principle component analysis of the Xa (D0, Xcomp; all others, Xcas) at different stages of XCI. Regions with positive principle component 1 (PC1) values represent A compartments; regions with negative PC1 values represent B compartments (blue).
(C) Hierarchical clustering analysis of the compartment profiles (PC1) of the X chromosomes in cells at different stages of XCI.
(D) Summary: Left: The Xa is organized into A/B compartments and TADs. Middle: In WT cells, the Xi is organized into two megadomains, where A/B compartments are absent. Xi TADs are weakened, coinciding with attenuated binding of CTCF and cohesins. Right: In Smchd1-/- cells, the Xi is further segregated into S1/S2 compartments. Xi TADs are strengthened as CTCF and cohesins bind stronger to the Xi.
{kind=link}
Table S1. Related to STAR Methods - Oligonucleotides used in this study
Highlights.
The inactive X (Xi) is folded in a step-wise manner to achieve silencing.
S1 and S2 compartments form when Xist spreading restructures A/B compartments
Upon its recruitment, SMCHD1 merges S1/S2 to create a “compartment-less” Xi.
SMCHD1 loss causes S1/S2 persistence, defective Xist spreading, and failed silencing.
ACKNOWLEDGEMENTS
We thank D. Colognori, A. Saltzman, J.E. Froberg, and A. Kriz for critical reading of the manuscript, and all lab members for support. We thank B. Kesner for bioinformatics advice and Y. Jeon for Xist transgenic MEFs. J. Erwin and F. H. Gage provided advice for NPC derivation. M. Kuroda, S. Buratowski, and A. Gimelbrant provided valuable advice during C-Y.W’s dissertation work. This work was supported by grants to J.T.L. from the National Institutes of Health (RO1-GM090278) and the Rett Syndrome Research Trust. J.T.L is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
Mammalian X inactivation relies on stepwise folding of chromosome compartments
Bibliography
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome biology 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berletch JB, Ma W, Yang F, Shendure J, Noble WS, Disteche CM, and Deng X (2015). Escape from X inactivation varies in mouse tissues. PLoS genetics 11, e1005079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickmore WA, and van Steensel B (2013). Genome architecture: domain organization of interphase chromosomes. Cell 152, 1270–1284. [DOI] [PubMed] [Google Scholar]
- Blewitt ME, Gendrel AV, Pang Z, Sparrow DB, Whitelaw N, Craig JM, Apedaile A, Hilton DJ, Dunwoodie SL, Brockdorff N, et al. (2008). SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nature genetics 40, 663–669. [DOI] [PubMed] [Google Scholar]
- Blewitt ME, Vickaryous NK, Hemley SJ, Ashe A, Bruxner TJ, Preis JI, Arkell R, and Whitelaw E (2005). An N-ethyl-N-nitrosourea screen for genes involved in variegation in the mouse. Proc Natl Acad Sci U S A 102, 7629–7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B, and Cavalli G (2016). Organization and function of the 3D genome. Nat Rev Genet 17, 661–678. [DOI] [PubMed] [Google Scholar]
- Brideau NJ, Coker H, Gendrel AV, Siebert CA, Bezstarosti K, Demmers J, Poot RA, Nesterova TB, and Brockdorff N (2015). Independent Mechanisms Target SMCHD1 to Trimethylated Histone H3 Lysine 9-Modified Chromatin and the Inactive X Chromosome. Molecular and cellular biology 35, 4053–4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese JM, Sun W, Song L, Mugford JW, Williams L, Yee D, Starmer J, Mieczkowski P, Crawford GE, and Magnuson T (2012). Site-specific silencing of regulatory elements as a mechanism of X inactivation. Cell 151, 951–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick BP, and Willard HF (2004). Multiple spatially distinct types of facultative heterochromatin on the human inactive X chromosome. Proc Natl Acad Sci U S A 101, 17450–17455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaumeil J, Le Baccon P, Wutz A, and Heard E (2006). A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes & development 20, 2223–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Dobson RC, Lucet IS, Young SN, Pearce FG, Blewitt ME, and Murphy JM (2016). The epigenetic regulator Smchd1 contains a functional GHKL-type ATPase domain. The Biochemical journal 473, 1733–1744. [DOI] [PubMed] [Google Scholar]
- Chen K, Hu J, Moore DL, Liu R, Kessans SA, Breslin K, Lucet IS, Keniry A, Leong HS, Parish CL, et al. (2015). Genome-wide binding and mechanistic analyses of Smchd1-mediated epigenetic regulation. Proceedings of the National Academy of Sciences of the United States of America 112, E3535–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Qu K, Zhong FL, Artandi SE, and Chang HY (2011). Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell 44, 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Zhang QC, da Rocha ST, Flynn RA, Bharadwaj M, Calabrese JM, Magnuson T, Heard E, and Chang HY (2015). Systematic discovery of xist RNA binding proteins. Cell 161, 404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HP, Cifuentes-Rojas C, Kesner B, Aeby E, Lee HG, Wei C, Oh HJ, Boukhali M, Haas W, and Lee JT (2017). TERRA RNA Antagonizes ATRX and Protects Telomeres. Cell 170, 86–101 e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti L, Pollard SM, Gorba T, Reitano E, Toselli M, Biella G, Sun Y, Sanzone S, Ying QL, Cattaneo E, et al. (2005). Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS biology 3, e283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J, and Meyer BJ (2015). Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrow EM, Huntley MH, Dudchenko O, Stamenova EK, Durand NC, Sun Z, Huang SC, Sanborn AL, Machol I, Shamim M, et al. (2016). Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proceedings of the National Academy of Sciences of the United States of America 113, E4504–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, and Mirny L (2016). The 3D Genome as Moderator of Chromosomal Communication. Cell 164, 1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rosario BC, Del Rosario AM, Anselmo A, Wang PI, Sadreyev RI, and Lee JT (2017). Genetic Intersection of Tsix and Hedgehog Signaling during the Initiation of X-Chromosome Inactivation. Dev Cell 43, 359–371 e356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Ma W, Ramani V, Hill A, Yang F, Ay F, Berletch JB, Blau CA, Shendure J, Duan Z, et al. (2015). Bipartite structure of the inactive mouse X chromosome. Genome biology 16, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disteche CM (2012). Dosage compensation of the sex chromosomes. Annu Rev Genet 46, 537–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, Kadri S, Xing J, Goren A, Lander ES, et al. (2013). The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 341, 1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser P (2016). Turning the tide on 3D nuclear organization. Nat Rev Mol Cell Biol 17, 738. [DOI] [PubMed] [Google Scholar]
- Gendrel AV, Apedaile A, Coker H, Termanis A, Zvetkova I, Godwin J, Tang YA, Huntley D, Montana G, Taylor S, et al. (2012). Smchd1-dependent and -independent pathways determine developmental dynamics of CpG island methylation on the inactive X-chromosome. Developmental cell 23, 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendrel AV, Tang YA, Suzuki M, Godwin J, Nesterova TB, Greally JM, Heard E, and Brockdorff N (2013). Epigenetic functions of smchd1 repress gene clusters on the inactive x chromosome and on autosomes. Molecular and cellular biology 33, 3150–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgetti L, Lajoie BR, Carter AC, Attia M, Zhan Y, Xu J, Chen CJ, Kaplan N, Chang HY, Heard E, et al. (2016). Structural organization of the inactive X chromosome in the mouse. Nature 535, 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarhuis JHI, van der Weide RH, Blomen VA, Yanez-Cuna JO, Amendola M, van Ruiten MS, Krijger PHL, Teunissen H, Medema RH, van Steensel B, et al. (2017). The Cohesin Release Factor WAPL Restricts Chromatin Loop Extension. Cell 169, 693–707 e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T (2016). Condensin-Based Chromosome Organization from Bacteria to Vertebrates. Cell 164, 847–857. [DOI] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, and Sharp PA (2017). A Phase Separation Model for Transcriptional Control. Cell 169, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegu T, Aeby E, and Lee JT (2017). The X chromosome in space. Nat Rev Genet 18, 377–389. [DOI] [PubMed] [Google Scholar]
- Jeon Y, and Lee JT (2011). YY1 tethers Xist RNA to the inactive X nucleation center. Cell 146, 119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppsson K, Kanno T, Shirahige K, and Sjogren C (2014). The maintenance of chromosome structure: positioning and functioning of SMC complexes. Nat Rev Mol Cell Biol 15, 601–614. [DOI] [PubMed] [Google Scholar]
- Ji X, Li W, Song J, Wei L, and Liu XS (2006). CEAS: cis-regulatory element annotation system. Nucleic Acids Res 34, W551–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsey AD, Yang C, Leung D, Minks J, Dixon-McDougall T, Baldry SE, Bogutz AB, Lefebvre L, and Brown CJ (2015). Impact of flanking chromosomal sequences on localization and silencing by the human non-coding RNA XIST. Genome biology 16, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, and Park PJ (2008). Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nature biotechnology 26, 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, and Narlikar GJ (2017). Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature 547, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT (2011). Gracefully ageing at 50, X-chromosome inactivation becomes a paradigm for RNA and chromatin control. Nature reviews Molecular cell biology 12, 815–826. [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks H, Kerstens HH, Barakat TS, Splinter E, Dirks RA, van Mierlo G, Joshi O, Wang SY, Babak T, Albers CA, et al. (2015). Dynamics of gene silencing during X inactivation using allele-specific RNA-seq. Genome biology 16, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall OJ, and Brand AH (2015). damidseq_pipeline: an automated pipeline for processing DamID sequencing datasets. Bioinformatics 31, 3371–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Jimenez CP, and Odom DT (2016). The mechanisms shaping the single-cell transcriptional landscape. Curr Opin Genet Dev 37, 27–35. [DOI] [PubMed] [Google Scholar]
- McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A, et al. (2015). The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkenschlager M, and Odom DT (2013). CTCF and cohesin: linking gene regulatory elements with their targets. Cell 152, 1285–1297. [DOI] [PubMed] [Google Scholar]
- Minajigi A, Froberg JE, Wei C, Sunwoo H, Kesner B, Colognori D, Lessing D, Payer B, Boukhali M, Haas W, et al. (2015). Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mould AW, Pang Z, Pakusch M, Tonks ID, Stark M, Carrie D, Mukhopadhyay P, Seidel A, Ellis JJ, Deakin J, et al. (2013). Smchd1 regulates a subset of autosomal genes subject to monoallelic expression in addition to being critical for X inactivation. Epigenetics & chromatin 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Varnai C, Schoenfelder S, Javierre BM, Wingett SW, and Fraser P (2015). Comparison of Hi-C results using in-solution versus in-nucleus ligation. Genome Biol 16, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekawa SH, Payer B, Huynh KD, Jaenisch R, and Lee JT (2010). Two-step imprinted X inactivation: repeat versus genic silencing in the mouse. Mol Cell Biol 30, 3187–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944 e922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa RS, Nagao K, Igami KT, Shibata S, Shirai N, Nozaki N, Sado T, Kimura H, and Obuse C (2013). Human inactive X chromosome is compacted through a PRC2-independent SMCHD1-HBiX1 pathway. Nature structural & molecular biology 20, 566–573. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Sun BK, and Lee JT (2008). Intersection of the RNA interference and X-inactivation pathways. Science 320, 1336–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CT, and Corces VG (2014). CTCF: an architectural protein bridging genome topology and function. Nature reviews Genetics 15, 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya-Jones A, and Plath K (2016). The “lnc” between 3D chromatin structure and X chromosome inactivation. Semin Cell Dev Biol 56, 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinter SF, Colognori D, Beliveau BJ, Sadreyev RI, Payer B, Yildirim E, Wu CT, and Lee JT (2015). Allelic Imbalance Is a Prevalent and Tissue-Specific Feature of the Mouse Transcriptome. Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nature protocols 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. (2017). Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320 e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. (2015). Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A 112, E6456–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AD, Hu M, and Ren B (2016). Genome-wide mapping and analysis of chromosome architecture. Nat Rev Mol Cell Biol 17, 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, H.H. C, Mirny L, et al. (2017). Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Lajoie BR, Nora EP, Giorgetti L, Chen CJ, Heard E, Dekker J, and Barillot E (2012). HiTC: exploration of high-throughput ‘C’ experiments. Bioinformatics 28, 2843–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MD, Pinter SF, Fang R, Sarma K, Rutenberg-Schoenberg M, Bowman SK, Kesner BA, Maier VK, Kingston RE, and Lee JT (2013). High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature 504, 465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splinter E, de Wit E, Nora EP, Klous P, van de Werken HJ, Zhu Y, Kaaij LJ, van Ijcken W, Gribnau J, Heard E, et al. (2011). The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes & development 25, 1371–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starmer J, and Magnuson T (2009). A new model for random X chromosome inactivation. Development 136, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, and Karpen GH (2017). Phase separation drives heterochromatin domain formation. Nature 547, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel MJ, Guelen L, de Wit E, Peric-Hupkes D, Loden M, Talhout W, Feenstra M, Abbas B, Classen AK, and van Steensel B (2006). Human heterochromatin proteins form large domains containing KRAB-ZNF genes. Genome Res 16, 1493–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AJ, and Bickmore WA (2015). A TAD Closer to Understanding Dosage Compensation. Dev Cell 33, 498–499. [DOI] [PubMed] [Google Scholar]
- Wood AJ, Severson AF, and Meyer BJ (2010). Condensin and cohesin complexity: the expanding repertoire of functions. Nature reviews Genetics 11, 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutz A (2011). Gene silencing in X-chromosome inactivation: advances in understanding facultative heterochromatin formation. Nat Rev Genet 12, 542–553. [DOI] [PubMed] [Google Scholar]
- Yang F, Deng X, Ma W, Berletch JB, Rabaia N, Wei G, Moore JM, Filippova GN, Xu J, Liu Y, et al. (2015). The lncRNA Firre anchors the inactive X chromosome to the nucleolus by binding CTCF and maintains H3K27me3 methylation. Genome Biol 16, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim E, Sadreyev RI, Pinter SF, and Lee JT (2012). X-chromosome hyperactivation in mammals via nonlinear relationships between chromatin states and transcription. Nature structural & molecular biology 19, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LF, Huynh KD, and Lee JT (2007). Perinucleolar targeting of the inactive X during S phase: evidence for a role in the maintenance of silencing. Cell 129, 693–706. [DOI] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song JJ, and Lee JT (2008). Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovoilis A, Cifuentes-Rojas C, Chu HP, Hernandez AJ, and Lee JT (2016). Destabilization of B2 RNA by EZH2 Activates the Stress Response. Cell 167, 1788–1802 e1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Western blot for SMCHD1 in WT and XaWTXiΔXist fibroblasts.
(B) SMCHD1 immunostaining and Xist RNA-FISH in wild-type female MEF (♀WT) and a female MEF clone carrying an inducible Xist transgene (♀X+P). In induced ♀X+P cells, a large transgenic Xist cloud was detected, in addition to two Xi clouds (Jeon and Lee, 2011). SMCHD1 was enriched on all three (♀X+P, top panel). This demonstrates that Xist RNA is sufficient for SMCHD1 recruitment. In some cells, the transgene “squelched” endogenous Xist RNA from the Xi, leaving only the prominent transgenic Xist cloud (Jeon and Lee, 2011). Interestingly, in these cells, SMCHD1 was also lost from the Xi (♀X+P, bottom panel), consistent with localization being dependent on Xist RNA. Arrowheads, transgenic Xist cloud. Fibroblasts have been immortalized with SV40 large T-antigen and are therefore tetraploid.
(C) Validation of the α-SMCHD1 antibody used in this study by siRNA-mediated knockdown of SMCHD1 in MEFs.
(D) Western blot confirmed the absence of SMCHD1 protein in Smchd1-/- ES cell clones.
(E) Two different pairs of guide RNAs (pairs 1 and pairs 2) targeting a region immediately downstream of the start codon (ATG) in the first exon of Smchd1.
(F) Sanger sequencing confirmed the presence of frame-shifting mutations. Regions containing the microdeletions were PCR amplified and cloned into a TOPO-TA vector. Numbers within parentheses indicate the number of clones sequenced. Sequences targeted by the guide RNA pairs are colored in red or blue.
(G) Confirmation of the identity of NPCs by RT-PCR. Mouse ES cell clones expressed pluripotency markers Pou5f1 (Oct4) and Nanog, but not NPC markers Blbp, Oligo2, and Emx2. In contrast, NPCs derived from these ESC clones expressed NPC markers, but not pluripotency markers. Actb (β-actin) and Pax6 served as controls, as they were expressed in both cell types. RT+, reverse transcriptase plus. RT-, reverse transcriptase minus. E, ES cells. N, NPCs.
(H) Immunostaining confirmed that the NPCs we derived expressed NPC markers NESTIN and SOX2.
(I) (from left to right) 1. Xist RNA-FISH showed a greater fraction of Smchd1-/- NPCs without a Xist cloud. 2. DNA-FISH for Atrx, an X-linked gene, showed a greater fraction of Smchd1-/- NPCs displaying only one Atrx locus, indicating that XO cells were enriched, resulting in fewer cells with a Xist cloud. 3. DNA-FISH for Tmem39a, an autosomal gene on chr16, showed that most WT and Smchd1-/- NPCs had two Tmem39a loci, indicating that the effect of Smchd1 ablation on chromosome number is specific to the X. 4. Xist DNA-FISH showed that the majority of mESCs used to derive these NPCs harbored two Xist loci, suggesting that XO cells became enriched in Smchd1-/- NPCs during differentiation.
(J) Derivation of clonal WT and Smchd1-/- NPC lines with homogeneous XX genotypes.
(A) RNA-seq coverage profiles (comp) at the Smchd1 locus demonstrating microdeletions in Smchd1-/- NPC clones. Smchd1-/- clones 1 and 2 were independently generated, with distinct microdeletions.
(B) A bar plot displaying the distribution of differentially expressed genes on each chromosome. Up: genes upregulated in Smchd1-/- cells. Down: genes downregulated in Smchd1-/- cells.
(C) RNA-seq coverage profiles (comp) at the Pcdh locus demonstrating upregulation of the Pcdhα gene cluster in both Smchd1-/- NPC clones.
(D) Degree of allelic skewing of 244 allele-assessable X-linked genes was highly concordant between two biological replicates. r, Pearson correlation coefficient of 244 allele-assessable X-linked genes (black) or 73 concordant SMCHD1-sensitive genes (red).
(E) A probability density plot of the number of overlapping genes between two independent random samplings of 90 and 82 genes from 230 genes for 10,000 times.
(F) Smchd1 ablation did not alter transcription from the Xcas (Xa). Shown are the cas-specific fragments per kilobase of transcript per million exonic reads (cas fpkm) of 73 concordant SMCHD1-sensitive genes. Solid diagonal, 1:1 ratio. Dashed diagonal, 1:2 ratio.
(G) In Smchd1-/- NPCs, increased expression of the Xmus alleles caused 1~2 fold upregulation of the 73 concordant SMCHD1-sensitive genes. Shown are the comp fpkm values of the 73 concordant SMCHD1-sensitive genes. Solid diagonal, 1:1 ratio. Dashed diagonal, 1:2 ratio.
(A) Xi expression (%mus) of SMCHD1-sensitive genes showed moderate positive correlation with fraction Xi-enrichment (%mus) of H3K4me3 peaks at promoters (left panel) and negative correlation with H3K27me3 enrichment in gene bodies (right panel). r, Pearson correlation coefficient.
(B) H3K27me3 enrichment in Xi intergenic regions in WT (x-axis) versus Smchd1-/- (y-axis) cells. Three categories of intergenic regions are shown, as indicated above each scatter plot (e.g., between Class I and Class I genes, “Class I – Class I”).
(C) Two biological replicates for Xist CHART experiments show high reproducibility. Also shown are H3K4me3 enrichment (ChIP-seq, mus) across the entire X chromosome. Enrichment difference is determined by subtracting WT values from Smchd1-/-. Green-shaded area, Xist-depleted regions. Gray-shaded area, unmappable regions.
(D) Related to Figure 3B: Xist binding patterns (CHART-seq, comp), H3K27me3 enrichment (ChIP-seq, comp), and H3K4me3 enrichment (ChIP-seq) across a second representative Xist-depleted region.
(E) Xist enrichment in Xi intergenic regions in WT (x-axis) versus Smchd1-/- (y-axis) cells. Three categories of intergenic regions are shown, as indicated above each scatter plot (e.g., between Class I and Class I genes, “Class I – Class I”).
(F) Summary of the epigenetic states for different categories of active X-linked genes.
(A) Scatter plots comparing normalized interaction frequencies in two Hi-C biological replicates. Each dot represents a 200-kb binned region on the Xa or Xi in WT or Smchd1-/- cells, as indicated. r, Pearson correlation coefficient.
(B) Number of various X-linked gene categories in different chromatin compartments. Left, the centromeric megadomain. Right, the telomeric megadomain.
(C) Related to Figure 5A: Hi-C interaction maps (binned at 40-kb resolution), insulation profiles, and the genomic location of Class I and Class II genes. Two additional representative X-linked regions are shown. TADs (as defined by Dixon et al., 2012) are delineated as blue bars between interaction maps and as dashed lines in the insulation graphs.
(D) Box plots comparing the boundary strength (defined as the differences in insulation scores between insulation minimum to the largest neighboring insulation maximum in the insulation profiles) of the Xa and Xi in WT and Smchd1-/- NPCs. The Xi boundary strength of Smchd1-/- tends to be higher than that of WT (P=0.013), but lower than that of Smchd1-/- Xa (P=4.6×10−4). The Xa boundary strength of Smchd1-/- is similar to that of WT (P=0.83). Green arrow, the TAD boundary near Firre. Black arrow, the TAD boundary near Dxz4. Boundaries near Firre and Dxz4 were included in the analysis. NS, not significant (P>0.05). *, P=0.013 (Wilcox test).
(E) Box plots comparing change in insulation scores (Δ) between Smchd1-/- and WT NPCs. The insulation score of each gene was determined by the insulation score of the bin containing the TSS. Xa and Xi values are shown. All classes of X-linked genes, except escapees, showed a wider spread of Δ on the Xi. IQR, interquartile range (Xa, blue; Xi, red).
(F) Box plots showing the distribution of differences in allelic skewing (Δ%mus) in Smchd1-/- relative to WT cells for CTCF peaks that are TAD boundary-associated (+) versus not associated (-). CTCF peaks on chr13 and chrX were shown. P-values from Wilcox test. NS, not significant (p>0.05). *, P=3.5×10−12. **, P=2.2×10−16 (Wilcox test).
(A) Top four panels: Pair-wise comparison of the insulation profiles across the entire X chromosome for the Xa and Xi in WT versus Smchd1-/- (Mut) cells, as indicated. Bottom two panels: Locations of Class I and Class II genes. Dashed lines, TAD boundaries as defined by Dixon et al., 2012. Green-shaded area, Firre. Yellow-shaded area, Dxz4. Gray-shaded area, unmappable regions.
(B) Allelic patterns of CTCF and RAD21 binding within a representative X-linked region. Xi, mus. Xa, cas. Significant binding peaks are indicated in green.
(C) Skewing of CTCF binding expressed as %mus in WT versus Smchd1-/- cells for chrX or chr13. Dashed diagonal, 1:1 ratio.
(D) Skewing of RAD21 binding expressed as %mus in WT versus Smchd1-/- cells for chrX or chr13. Dashed diagonal, 1:1 ratio.
(E) Box plots showing differences in allelic skewing (Δ%mus) for CTCF binding in Smchd1-/- relative to WT cells for intergenic regions (e.g., between Class I and Class I genes, “Class I – Class I”). All measurements are for X-linked regions, except Chr13 – Chr13 measurements. Chr13 versus unclassified-unclassified, P=1. Chr13 versus all other categories of intergenic regions, P<0.025 (Wilcox test with Bonferroni correction, one sided).
(F) Scatter plot analyses correlating change (Δ) in Xist binding to change in allelic CTCF and RAD21 binding at Class I genes (red), or at all other X-linked genes (black). Xi binding shown (%mus). r, Pearson correlation coefficient.
(G) Scatter plot analyses correlating change (Δ) in H3K27me3 enrichment to change in allelic CTCF and RAD21 binding at Class I genes (red), or at all other X-linked genes (black). Xi binding shown (%mus).
(A) Detection of Dam-SMCHD1 fusion protein (tagged with the V5 epitope) by western blot. In this experiment, MEFs were transiently transfected with plasmids overexpressing the fusion proteins to verify the fusion is expressed at the expected size.
(B) Immuno-RNA-FISH for overexpressed Dam-fusion proteins (V5-tagged) and Xist RNA demonstrating that fusions share similar subnuclear localization as wild-type proteins. Top: CBX1-Dam colocalizes with DAPI-dense foci, consistent with its recruitment to pericentromeric heterochromatin. Bottom: Dam-SMCHD1 is recruited to the Xi. Note that in the actual cell lines used for DamID-seq, Dam fusion proteins were expressed by the uninduced HSP70 promoter at low levels (see STAR Methods).
(C) Methylation-specific PCR on cells expressing Dam alone, CBX1-Dam, Dam-SMCHD1 (rep1 & 2), and non-transfected cells. Lane 1, DpnI-minus control. Lane 2, ligase-minus control. Land 3 and 4, duplicates. Note that DNA adenine methylation is only detected in transfected cells.
(D) Left, fraction of total unique read pairs mapped to the X and Chr3, an autosome (159 Mb) with similar length as the X (166 Mb). Middle, fraction of cas- and mus-specific pairs in total Chr3 allele-specific read pairs. Right, fraction of cas- and mus-specific pairs in total ChrX allele-specific read pairs. Dam, Dam alone. CBX1, CBX1-Dam. SMCHD1, Dam-SMCHD1.
(E) Scatter plots comparing two biological replicates of allele-specific SMCHD1 binding profiles for each 100-kb bin on the autosomes (black dots) and the X chromosomes (red dots). r, Pearson correlation coefficient for autosomes (black) and X chromosomes (red).
(F) SMCHD1 and CBX1 enrichment profiles across chromosome 3.
(G) A zoom-in at one of the SNP-scarce regions on the X chromosome. Shown are fragment-per-million normalized DamID coverage tracks for Dam alone and Dam-SMCHD1, as well as the log2 ratio [log2(Dam-SMCHD1/Dam alone)] tracks. Note that the scarcity of allelic reads (cas and mus) at the green-shaded regions in Dam alone and Dam-SMCHD tracks, which led to a “trough” in the log2 ratio tracks. SMCHD1 is actually not depleted at this region, as evident by the robust enrichment of SMCHD1 shown in the comp tracks.
(H) Average enrichment plots of SMCHD1 (left) and CTCF (right) at 10-kb resolution centered at the TAD boundary. Black, TAD boundaries on autosomes. Red, TAD boundaries on the X chromosome.
(I) SMCHD1 immunostaining and Xist RNA-FISH in D4 and D7 wild-type embryoid bodies (EB). In D4 EB, 70% cells exhibited an Xist cloud and none of them showed SMCHD1 recruitment on the Xi. Instead, SMCHD1 formed multiple nuclear foci colocalizing with DAPI-dense pericentrometric heterochromatin, consistent with previous reports (Brideau et al., 2015; Gendrel et al., 2012). In D7 EB, 84% cells harbored an Xist cloud, and SMCHD1 was observed to be enriched on the Xi in 4% of these cells.
(J) Box plots comparing the gap between Xist/H3K27me3 coverage densities of S1 and S2 compartments in WT versus Smchd1-/- NPCs. Red dots, S1 compartments. Blue dots, S2 compartments. Median, median coverage densities of S1 (red) or S2 (blue). S1-S2 gap, median S1 density – median S2 density. The S1-S2 gap was significantly widened when Smchd1 is ablated (Xist, p=0.041; H3K27me3, p=0.035, paired t-test, one-sided). Regions harboring Class I genes and the centromeric Xist-depleted regions were excluded in this analysis.
(A) Allele-specific Hi-C analysis: Top: Contact maps (200-kb resolution) of the Xa (Xcas) in undifferentiated ES cells (D0 ES, pre-XCI), embryoid bodies formed after 4 or 7 differentiation days (D4 and D7 EB, early-mid XCI), and post-XCI wild-type (WT) and Smchd1-/- NPCs. Bottom: Pearson correlation maps. Gray-shaded area, unmappable regions.
(B) Principle component analysis of the Xa (D0, Xcomp; all others, Xcas) at different stages of XCI. Regions with positive principle component 1 (PC1) values represent A compartments; regions with negative PC1 values represent B compartments (blue).
(C) Hierarchical clustering analysis of the compartment profiles (PC1) of the X chromosomes in cells at different stages of XCI.
(D) Summary: Left: The Xa is organized into A/B compartments and TADs. Middle: In WT cells, the Xi is organized into two megadomains, where A/B compartments are absent. Xi TADs are weakened, coinciding with attenuated binding of CTCF and cohesins. Right: In Smchd1-/- cells, the Xi is further segregated into S1/S2 compartments. Xi TADs are strengthened as CTCF and cohesins bind stronger to the Xi.
Table S1. Related to STAR Methods - Oligonucleotides used in this study
Data Availability Statement
Raw next-Gen sequencing data and processed files for RNA-seq, ChIP-seq, CHART-seq, Hi-C, and DamID-seq have been deposited in Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under accession number GEO: GSE99991.