

Abstract
The NAA10-related syndrome is a rare X-linked neurodevelopmental condition that was first described in 2011. The disorder is caused by pathogenic variants in the NAA10 gene located on chromosome X at position Xq28. Clinical features typically include severe psychomotor developmental delay, cardiac disease, dysmorphic features, postnatal growth failure, and hypotonia, although there is significant variability in the severity of the phenotype among affected individuals. We describe a 5-year-old female with the syndrome; massively parallel exome sequencing and analysis revealed the c.247C>T (p.Arg83Cys) pathogenic variant that has been previously reported in ten affected individuals. Ocular manifestations of the NAA10-related syndrome are not uncommon, although they have not been well characterized in literature reports. From a systematic review of previously published cases to date, ocular abnormalities are present in more than half of patients with the syndrome. Common ocular findings reported include astigmatism, hyperopia, cortical vision impairment, microphthalmia/anophthalmia, and hypertelorism. Our patient presented with growth restriction, dysmorphic features, and hypotonia. Ocular manifestations identified in this child include downslanting palpebral fissures, myopic astigmatism, nystagmus, and exotropia. We speculate that the type and severity of ocular defects present in individuals with the NAA10-related syndrome are dependent on the specific NAA10 pathogenic variant involved.
1. Introduction
Ogden syndrome (MIM #300855) is a rare X-linked neurodevelopmental disorder caused by pathogenic variants of the NAA10 gene located at Xq28 [1]. The syndrome was originally described in 8 severely affected young boys in two unrelated families by Rope et al. in 2011 [1]. Since the initial report, this description of Ogden syndrome has broadened to include a variety of phenotypes categorized as NAA10-related syndrome [2]. Patients have a variety of clinical findings that include postnatal growth failure, delayed psychomotor development, dysmorphic features, and hypotonia. This categorization of NAA10-related syndrome also includes Lenz microphthalmia syndrome (MIM#309800), a disorder also caused by pathogenic variants in the NAA10 gene which is characterized by abnormalities of the skeletal and urinary systems, teeth, ears, digits, and several ocular defects that may include unilateral or bilateral microphthalmia/anophthalmia, cataracts, nystagmus, coloboma, and glaucoma [3]. A milder NAA10-related intellectual disability phenotype associated with different variants has also been described. NAA10 encodes for the ubiquitously expressed primary protein acetyltransferase in humans, N-terminal-acetyltransferase A (NatA). Thus, a variant and subsequent dysfunctionality of NatA results in profound consequences to the proteome. Thirty-nine individuals with NAA10 pathogenic variants with related phenotypic features have been reported [3–10].
Ocular manifestations of the NAA10-related syndrome have not been well characterized in the literature. In this report, we summarize the ocular features of the syndrome from a systematic review of the literature and additionally present a young female with the syndrome who initially presented with growth restriction, failure to thrive, and hypotonia. Her ocular manifestations identified include downslanting palpebral fissures, myopic astigmatism, nystagmus, and exotropia.
2. Case Report
The patient, now a 5-year-old female, had a gestation period notable for intrauterine growth restriction (IUGR); her G3P2 mother had pre-eclampsia and hyperemesis during the pregnancy. The patient was born to non-consanguineous parents at 38 weeks by cesarean section, weighing 2.49 kg. She required resuscitation at birth. During infancy, she had hypotonia, laryngomalacia requiring supplemental oxygen, aspiration episodes requiring Nissen and g-tube placement, and prolonged growth failure. Her head circumference maintained trajectory at the 50th percentile, although her length/height has been consistently below the 5th percentile. Her dysmorphic features included broad forehead, midface hypoplasia with prognathism, depressed nasal bridge, hypertelorism, synophrys, deep set eyes, downslanting palpebral fissures, tongue protrusion, occipital flattening, and small hands.
MRI at age 3 showed ventricular prominence without hydrocephalus and diminutive geni and corpus callosum. EEG showed moderate generalized slowing and occasional independent left and right lateralized slow waves during sleep (bihemispheric dysfunction) and no epileptiform activity. EKG and echocardiogram were normal.
At her last examination at 5 years of age (Figure 1), she remains significantly delayed. She smiles and knows 3-5 single words that are used infrequently. She is able to sit, roll, and start to cruise when placed in standing position. Her ocular abnormalities include having a myopic astigmatism in both eyes requiring glasses, an intermittent alternating exotropia, and high frequency, low amplitude horizontal nystagmus. She was unable to cooperate with eye chart testing, but was able to fix and follow an object with each eye.
Figure 1.
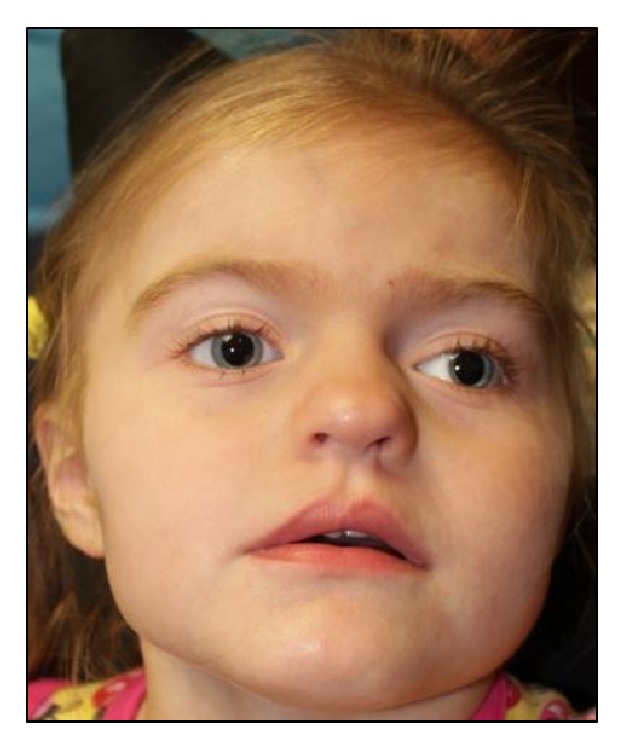
The NAA10-related syndrome. A 5-year-old female with a broad forehead, hypertelorism, downslanting palpebral fissures, exotropia, midface hypoplasia with prognathism, and a depressed nasal bridge.
Her additional medical problems include idiopathic hypertension, precocious puberty, obstructive sleep apnea, eosinophilic gastritis, seizures, hypohydrosis with overheating, recurrent fever of unknown origin, and intellectual and motor disability. Whole exome sequencing conducted on both parents and the patient (trio WES) identified a pathogenic missense c.247C>T p.Arg83Cys de novo variant in the NAA10 gene. X-inactivation testing was conducted by PCR analysis of a polymorphic CAG repeat in the first exon of the androgen receptor (AR) gene. Methylation of sites close to this short tandem repeat has been demonstrated to correlate with X chromosome inactivation [11]. Amplification of the AR gene both before and after digestion with the methylation sensitive HpaII restriction enzyme was used to determine the methylation status of the X chromosome. This testing revealed a highly skewed X-inactivation pattern (100:0).
3. Methods
We performed a systematic review of the literature to summarize ocular disease in individuals with the NAA10-related syndrome. A PubMed/Medline search of “Ogden syndrome” OR “NAA10” led us to find a total of 208 articles after removing duplicates on July 2018. No articles were excluded based on year published or language. Articles describing patients with NAA10 pathogenic variants with a clinical syndrome consistent with the description by Wu et al. (2018) were included. Within the articles, we identified describing cases of NAA10-related syndrome; we reviewed the references to identify other articles that did not appear in our original search.
4. Discussion
From our systematic review, we found 9 articles describing 39 reported cases of NAA10 related syndromes [11]. Including our reported patient, this brings the total number of cases to 40. 50 percent (n=20) of these patients with NAA10-related syndrome had ophthalmologic abnormalities (Table 1). The most common ocular findings were astigmatism (n=6), hyperopia (n=4), cortical vision impairment (n=3), hypertelorism (n=3), exotropia (n=3), myopia (n=3), and anophthalmia/micropthalmia (n=3). The most common NAA10 genetic variant leading to ocular disease, the c.247C>T p.Arg83Cys pathogenetic variant (n=6), results in reduced Acetyl-CoA binding and enzyme activity [5].
Table 1.
Summary of the 20 patients with the NAA10-related syndrome that have reported ocular findings.
(a).
| Rope, et al. [2011] | Esmailpour, et al. [2014] | Casey, et al. [2015] | |||||||
|
| |||||||||
| NAA10 gene pathogenic variant | c.109T>C p.Ser37Pro | c.109T>C p.Ser37Pro | c.109T>C p.Ser37Pro | c.109T>C p.Ser37Pro | c.109T>C p.Ser37Pro | Intron 7 splice c.471+2T→A | Intron 7 splice c.471+2T→A | Intron 7 splice c.471+2T→A | c.128A>C p.(Tyr43Ser) |
|
| |||||||||
| Inheritance | inherited | inherited | inherited | inherited | inherited | inherited | inherited | inherited | inherited |
|
| |||||||||
| Gender | M | M | M | M | M | M | M | M | M |
|
| |||||||||
| Eye findings | Hypertelorism | Prominent eyes, downslanted palpebral fissures, ocular hypertelorism | Prominent eyes, downslanted palpebral fissures, ocular hypertelorism | Large eyes, bilateral ptosis | Mild lagophthalmos, infraorbital creases | Anophthalmia/ microphthalmia |
Anophthalmia/ microphthalmia |
Anophthalmia/ microphthalmia |
Moderate right convergent squint, hyperopic astigmatism, dense right amblyopia |
|
| |||||||||
| Dysmorphic features | + | + | + | + | + | + | + | + | |
|
| |||||||||
| Cardiac anomalies | + | + | + | + | + | ||||
|
| |||||||||
| Renal anomalies | + | - | - | ||||||
|
| |||||||||
| Neuro | MRI brain: bilateral globus pallidus T2 prolongation without diffusion restriction, ventricular dilation | Hyper/ hypotonia |
MRI- arterial sclerosis | Seizures | Hypotonia. MRI- mild dialation of ventricles nd mild cerebral atrophy | ||||
|
| |||||||||
| Feeding issues/FTT | - | + | + | + | - | ||||
|
| |||||||||
| Developmental delay | + | + | + | + | + | + | + | ||
|
| |||||||||
| Motor delay | + | + | + | + | |||||
(b).
| Saunier, et al. [2016] | Valentine, et al. [2018] | Gupta, et al. [2019] | |||||||||
|
| |||||||||||
| NAA10 gene pathogenic variant | c.319G>T p.(Val107Phe) | c.384T>A p.(Phe128Leu) | c.384T>A p.(Phe128Leu) | c.382T>A p.(Phe128Ile) | c.247C>T p.(Arg83Cys) | c.247C>T p.(Arg83Cys) | c.247C>T p.(Arg83Cys) | c.247C>T p.(Arg83Cys) | c.247C>T p.(Arg83Cys) | c.346C>T (p.Arg116Trp) | c.247C>T p.(Arg83Cys) |
|
| |||||||||||
| Inheritance | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | inherited | de novo | de novo |
|
| |||||||||||
| Gender | F | F | F | F | F | F | F | F | F | F | F |
|
| |||||||||||
| Eye findings | Astigmatism, strabismus, mild optic atrophy | Cortical vision impairment | Cortical vision impairment, ambylopia | Hyperopia, astigmatism, exotropia | Myopia, astigmatism | Alternating exotropia, cortical vision impairment | Astigmatism, hyperopia | Hyperopia | Myopia | Ptosis, eyelid myoclonus | Myopic astigmatism, nystagmus, exotropia |
|
| |||||||||||
| Dysmorphic features | + | - | + | + | + | + | + | ||||
|
| |||||||||||
| Cardiac anomalies | + | + | - | + | + | - | + | + | - | - | - |
|
| |||||||||||
| Renal anomalies | - | - | - | - | - | - | + | - | + | - | |
|
| |||||||||||
| Neuro | Hypotonia. MRI- thin corpus callosum | Hypotonia. MRI- parenchymal atrophy, thin corpus callosum. Pre and postnatal vetriculomegaly | Hypotonia. Seizures. MRI normal at age 1 | Hyper/ hypotonia. MRI- Supraventricular cyst without hyocephalus |
Hypotonia. MRI- Periventricular white matter loss | Hyper/ hypotonia. MRI- IVH occipital horn, periventricular leukomalacia, hypoxia ischemic encephalopathy |
Hypotonia. MRI normal at age 3 | Hypertonia | Hypo/ hypertonia. Absence seizures |
Hypotonia, seizures, MRI at age 3- ventricular prominence without hydrocephalus and diminutive geni and corpus callosum | |
|
| |||||||||||
| Feeding issues/FTT | + | + | + | + | + | + | + | + | - | + | |
|
| |||||||||||
| Developmental delay | + | + | + | + | + | + | + | + | + | + | + |
|
| |||||||||||
| Motor delay | + | + | + | + | + | + | + | + | + | ||
The nature of ophthalmologic abnormalities in patients was dependent on the NAA10 variant involved (Table 1). Rope et al. reported eight patients in two different families with Ogden disease with the c.109T>C p.Ser37Pro variant. No significant visual abnormalities were reported in these patients, although facial dysmorphisms involving the ocular adnexa were present that included hypertelorism, prominent eyes, and downslanting palpebral fissures [1]. Another three patients with a novel missense variant of NAA10 (c.128A>C; p.Tyr43Ser) had significant findings on eye exam including moderate right convergent squint, hyperopic astigmatism, and dense right amblyopia [4].
Saunier et al. reported 13 total patients, 9 of which had ocular disease. One patient with a c.319G>T p.Val107Phe variant had astigmatism, strabismus, and mild optic atrophy. Another two patients with the c.384T>A p.Phe128Leu variant had cortical vision impairment. A patient with the c.382T>A p.Phe128Ile variant had hyperopia, astigmatism, exotropia. The remaining five patients had a c.247C>T p.Arg83Cys pathogenic variants similar to our patient; these five individuals displayed a range of clinical findings including myopia, astigmatism, alternating exotropia, cortical vision impairment, and hyperopia.
Three individuals with a severe pathogenic variant in NAA10 at the intron 7 splice donor site (c.471+2T→A) presented with anophthalmia or microphthalmia, prenatal onset of growth deficiency, and significant dysmorphic features [3].
Valentine et al. described a patient with a c.346C>T, p.Arg116Trp variant with mild ptosis and downslanting palpebral fissures with concurrent absence epilepsy with eyelid myoclonus [5].
Four other studies that reported on individuals with the NAA10-related syndrome did not describe any significant ocular findings [6, 7, 9, 10].
Because NAA10 is a ubiquitous protein acetylase, it is not surprising that the phenotype in the NAA10-related syndrome involves multiple organ systems, including the eye. The associated ocular abnormalities in the NAA10-related syndrome appear dependent on the nature of the pathogenic variant. Patients with more severe ocular phenotypes associated with variants in the NAA10 intron 7 splice donor site (c.471+2T>A) described by Esmailpour et al. have a resultant dysgenesis of ocular development associated with the Lenz microphthalmia syndrome. Previous studies inducing knockdown of NAA10 in zebrafish have resulted in similar abnormalities; zebrafish with NAA10 knockdown had anophthalmia or deformed eyes [12]. This could be because splice mutations affect NAA10's ability to interact with other proteins such as TSC2 [3], while missense variants only affect enzyme activity. NAA10 itself may also play a role in retinoic acid signaling, which is critical for eye development [3]. Further, NAA10 has been reported to acetylate and activate beta-catenin in lung cancer cell lines; and wnt/beta-catenin signaling is known to be crucial for eye development [13]. In the central nervous system specifically, after deletion of NAA10 myelin basic protein is unstable due to degradation of encephalin, proteins regulating cell survival are affected as well [14, 15]. In the developing brain where NAA10 is highly expressed, this could be the cause of the cerebral atrophy, cortical vision impairment, and severe neurodevelopmental consequences seen in the NAA10-related syndrome.
In summary, this case report and review contributes to our understanding of the relationship of NAA10 pathogenic variants and ocular abnormalities. Further studies on the role of NAA10 in eye development can help elucidate the mechanisms underlying ophthalmologic disease in the NAA10-related syndrome.
Consent
Consent has been obtained.
Conflicts of Interest
Angela S. Gupta, Hind Al Saif, M.D., and Jennifer M. Lent, M.S., had no conflicts of interests. Natario L. Couser, M.D., M.S., is Principal Investigator at the Virginia Commonwealth University site for Retrophin, Inc., “An Observational, Multicenter Study of the Prevalence of Cerebrotendinous Xanthomatosis (CTX) in Patient Populations Diagnosed with Early-Onset Idiopathic Bilateral Cataracts”.
References
- 1.Rope A., Wang K., Evjenth R., et al. Using VAAST to Identify an X-Linked Disorder Resulting in Lethality in Male Infants Due to N-Terminal Acetyltransferase Deficiency. American Journal of Human Genetics. 2011;89(1):28–43. doi: 10.1016/j.ajhg.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu Y., Lyon G. J. NAA10-related syndrome. Experimental & Molecular Medicine. 2018;50(7) doi: 10.1038/s12276-018-0098-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esmailpour T., Riazifar H., Liu L., et al. A splice donor mutation in NAA10 results in the dysregulation of the retinoic acid signalling pathway and causes Lenz microphthalmia syndrome. Journal of Medical Genetics. 2014;51(3):185–196. doi: 10.1136/jmedgenet-2013-101660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casey J. P., Støve S. I., McGorrian C., et al. NAA10 mutation causing a novel intellectual disability syndrome with Long QT due to N-terminal acetyltransferase impairment. Scientific Reports. 2015;5 doi: 10.1038/srep16022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saunier C., Støve S. I., Popp B., et al. Expanding the phenotype associated with NAA10-related N-terminal acetylation deficiency. Human Mutation. 2016;37(8):755–764. doi: 10.1002/humu.23001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sidhu M., Brady L., Tarnopolsky M., Ronen G. M. Clinical manifestations associated with the N-terminal-acetyltransferase NAA10 gene mutation in a girl: ogden syndrome. Pediatric Neurology. 2017;76:82–85. doi: 10.1016/j.pediatrneurol.2017.07.010. [DOI] [PubMed] [Google Scholar]
- 7.Popp B., Støve S. I., Endele S., et al. De novo missense mutations in the NAA10 gene cause severe non-syndromic developmental delay in males and females. European Journal of Human Genetics. 2015;23(5):602–609. doi: 10.1038/ejhg.2014.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valentine V., Sogawa Y., Rajan D., Ortiz D. A case of de novo NAA10 mutation presenting with eyelid myoclonias (AKA Jeavons syndrome) Seizure. 2018;60:120–122. doi: 10.1016/j.seizure.2018.06.008. [DOI] [PubMed] [Google Scholar]
- 9.Støve S. I., Blenski M., Stray-Pedersen A., et al. A novel NAA10 variant with impaired acetyltransferase activity causes developmental delay, intellectual disability, and hypertrophic cardiomyopathy. European Journal of Human Genetics. 2018;26(9):1294–1305. doi: 10.1038/s41431-018-0136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McTiernan N., Støve S. I., Aukrust I., et al. NAA10 dysfunction with normal NatA-complex activity in a girl with non-syndromic ID and a de novo NAA10 p.(V111G) variant - a case report. BMC Medical Genetics. 2018;19(1) doi: 10.1186/s12881-018-0559-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allen R. C., Zoghbi H. Y., Moseley A. B., Rosenblatt H. M., Belmont J. W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. American Journal of Human Genetics. 1992;51(6):1229–1239. [PMC free article] [PubMed] [Google Scholar]
- 12.Ree R., Myklebust L. M., Thiel P., Foyn H., Fladmark K. E., Arnesen T. The N-terminal acetyltransferase Naa10 is essential for zebrafish development. Bioscience Reports. 2015;35(5) doi: 10.1042/BSR20150168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim J., Park J., Chun Y. Human arrest defective 1 acetylates and activates -catenin, promoting lung cancer cell proliferation. Cancer Research. 2006;66(22):10677–10682. doi: 10.1158/0008-5472.CAN-06-3171. [DOI] [PubMed] [Google Scholar]
- 14.De Haan E. C., Wauben M. H. M., Wagenaar-Hilbers J. P. A., et al. Stabilization of peptide guinea pig myelin basic protein 72-85 by N-terminal acetylation - Implications for immunological studies. Molecular Immunology. 2004;40(13):943–948. doi: 10.1016/j.molimm.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 15.Jayawardene D. S., Dass C. The effect of N-terminal acetylation and the inhibition activity of acetylated enkephalins on the aminopeptidase M-catalyzed hydrolysis of enkephalins. Peptides. 1999;20(8):963–970. doi: 10.1016/S0196-9781(99)00089-3. [DOI] [PMC free article] [PubMed] [Google Scholar]