

Abstract
Exosomes represent one class of extracellular vesicles that are thought to be shed by all cell types. Although the exact nature of exosome biogenesis and function remains incompletely understood, they are increasingly recognized as a source of intercellular communication in health and disease. Recent observations of RNA exchange via donor cell-derived exosomes that exert genetic regulation in recipient cells have led to a boon into exosome research. The excitement and promise of exosomes as a new therapeutic avenue for human pathologies remain limited by challenges associated with their isolation from culture media and bio-fluids. The introduction of new methodologies to facilitate the isolation of exosomes has simultaneously raised concerns related to the reproducibility of studies describing exosome effector functions. Even high speed ultracentrifugation, the first and long considered gold standard approach for exosome isolation has recently been noted to be subject to uncontrolled variables that could impact functional readouts of exosome preparations. This chapter describes principles and methods that attempt to overcome such limitations by first concentrating exosomes in a liquid cushion and subsequently resolving them using density gradient ultracentrifugation. Our approach avoids possible complications associated with direct pelleting onto plastic tubes and allows for further purification of exosomes from dense protein aggregates.
Keywords: exosomes, density gradient ultracentrifugation, flow cytometry, nanoparticle tracking analysis, iodixanol
1. Introduction
Exosomes are nano-sized membrane vesicles of endocytic origin that are secreted by almost all cell types and are abundantly present in body fluids (1). The study of exosomes has recently attracted immense interest owing to their capacity to carry and transfer a variety of cell-signaling molecules including protein and RNA (2, 3). Exosomes have thus become firmly recognized as vehicles for intercellular communication in health and in disease. Moreover, exosomes represent an ideal source of diagnostic biomarkers and a potential delivery vehicle for therapeutic applications (4, 5).
A major challenge in this rapidly evolving field is the lack of a robust approach to isolate exosomes with high purity and biophysical integrity. Ever since the discovery of exosomes (6), ultracentrifugation has remained a commonly used approach to isolate these extracellular vesicles in a non-biased manner (7). This procedure, however, does not discriminate between exosome subtypes and larger microvesicles, as well as from other nanoparticles, or even large protein aggregates (8–10). Furthermore, the physical and biological integrity of exosomes isolated by ultracentrifugation may be compromised due to nanoparticle aggregation (8), high hydrostatic pressures caused by direct pelleting, and shear forces during dispensing (11). As such, this method that has long been accepted as a gold standard for exosome isolation could be subject to substantial qualitative and quantitative variability. Such limitations could subsequently impact on downstream analyses, making it difficult to compare, reproduce and interpret results of experiments performed across laboratories with exosomes prepared using this method (11). As recently highlighted in a commentary by the EV-TRACK Consortium, there is a pressing need to develop methodologies to isolate and study the function of exosomes in a consistent and reproducible manner (12).
To address this issue, we built on numerous prior reports (13–16) and devised a robust methodological platform to isolate exosomes with high yield, purity and integrity. We termed our method: Cushioned–Density Gradient Ultracentrifugation (C-DGUC). Our approach, illustrated in Figures 1 & 2, offers multiple advantages over traditional ultracentrifugation of biofluids and culture media. First, the use of a 60% iodixanol cushion during the nanoparticle concentration step maximizes exosome recovery and allows for a better preservation of their physical integrity and biological activity by preventing pellet formation in the centrifuge tube. Second, the use of density gradient ultracentrifugation provides for high-quality exosome purification by efficiently removing non-exosome nanoparticles and protein contaminates (~90% of total protein, Figure 3). Lastly, the biological inertness and compatibility of iodixanol with most downstream in vitro functional assays and even use in animals allows for reliable and efficient biochemical and physiological study of purified exosomes without the need for its removal. Fractions taken from iodixanol gradients can be readily analyzed for protein, nanoparticle, and RNA content by SDS-PAGE (Figure 4A), western blotting (Figure 4B), flow cytometry (Figure 5), nanoparticle tracking analysis (Figure 6), microchip capillary electrophoresis (Figure 7) all without a need for dialysis. The protocol detailed below represents a practical example of exosome isolation from mouse bone marrow-derived macrophage culture supernatants using our C–DGUC method. It is noteworthy that this method could be extended to exosome isolation from biofluids including plasma and urine for clinical research and diagnostic applications.
Figure 1.
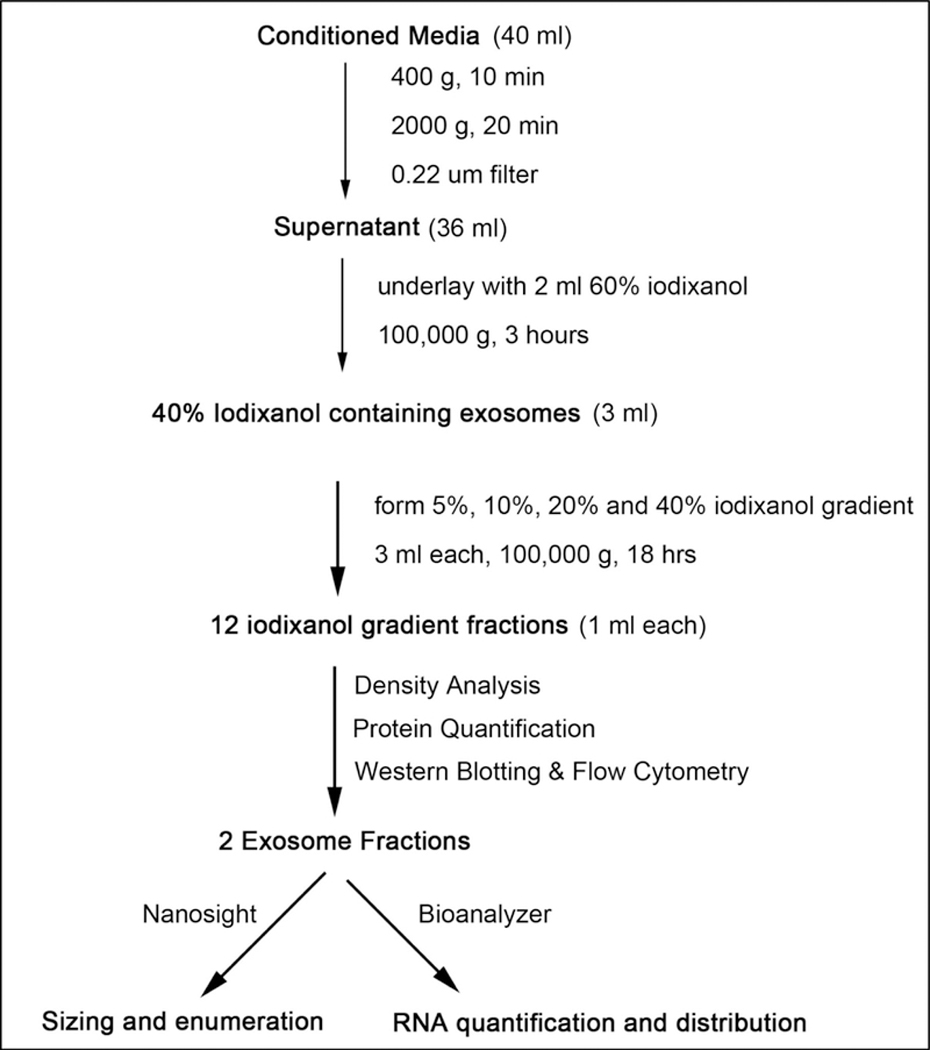
Experimental workflow of cushioned density gradient ultracentrifugation (C–DGUC) for exosome isolation
Figure 2. Schematic illustration of C-DGUC for exosome isolation.
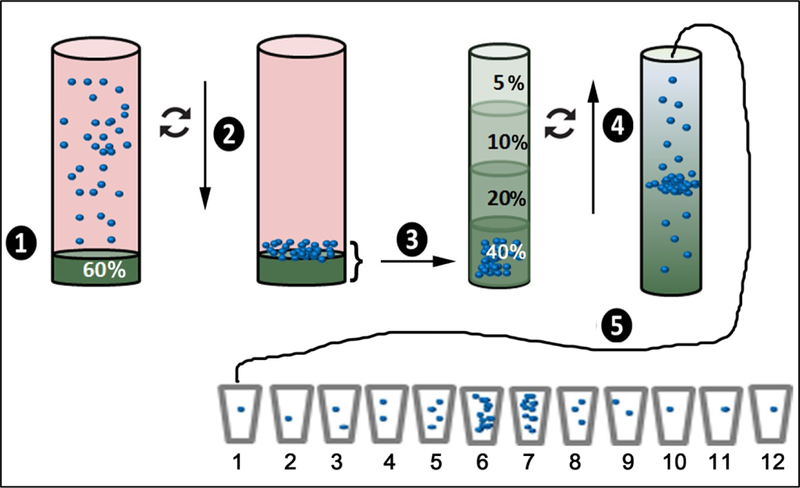
(1) Place a 2 mL solution of 60% iodixanol below the pre-cleared conditioned medium that serves to cushion the nanoparticles during centrifugation; (2) sediment the nanoparticles at 100,000 × g for 3 hrs; (3) remove the 2 mL cushion along with 1 mL of overlaying medium and place it below a pre-formed step density gradient composed of 3 layers of iodixanol solution, 3 mL each; (4) perform density gradient ultracentrifugation at 100,000 × g for a period of 18 hrs; (5) collect the medium from the top of the tube in 1 mL increments. Exosomes derived from cultured primary BMDM typically are isolated in fractions 6 & 7.
Figure 3. Density analysis and protein quantitation of iodixanol gradient fractions.
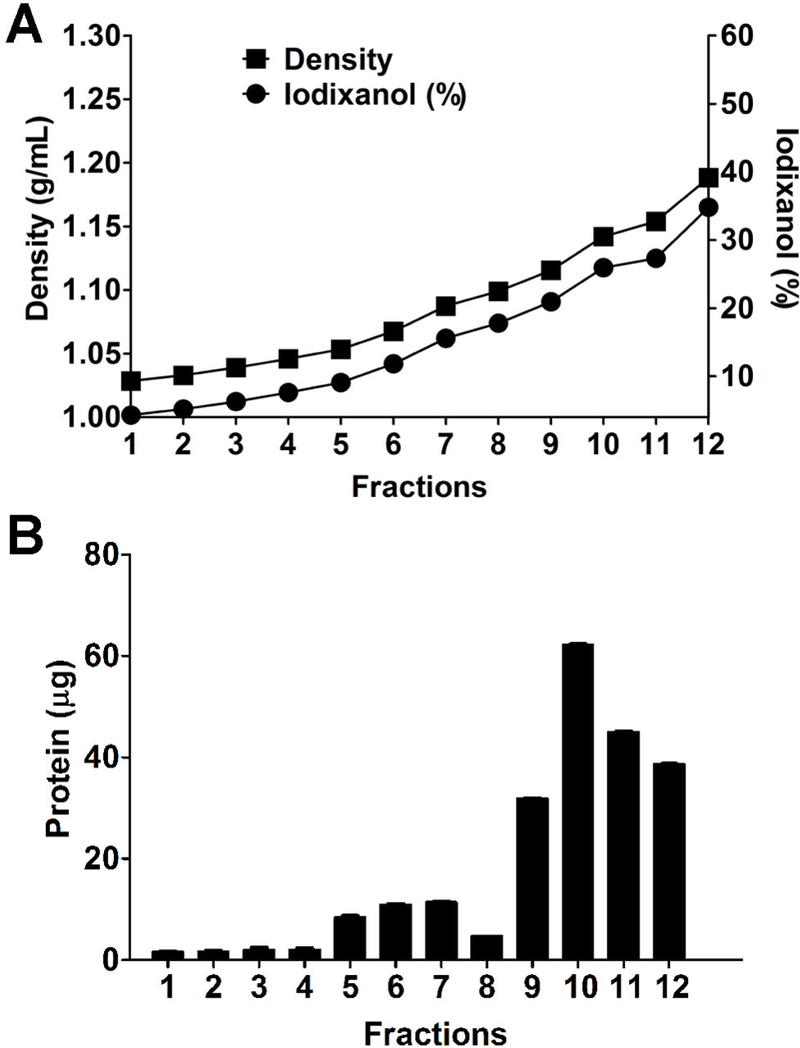
(A) The density and iodixanol percentage gradually increase in the 12 fractions collected from the top to the bottom of the gradient; (B) protein quantification in the 12 gradient fractions was performed by using Qubit Protein Assay.
Figure 4. Protein characterization of iodixanol gradient fractions.
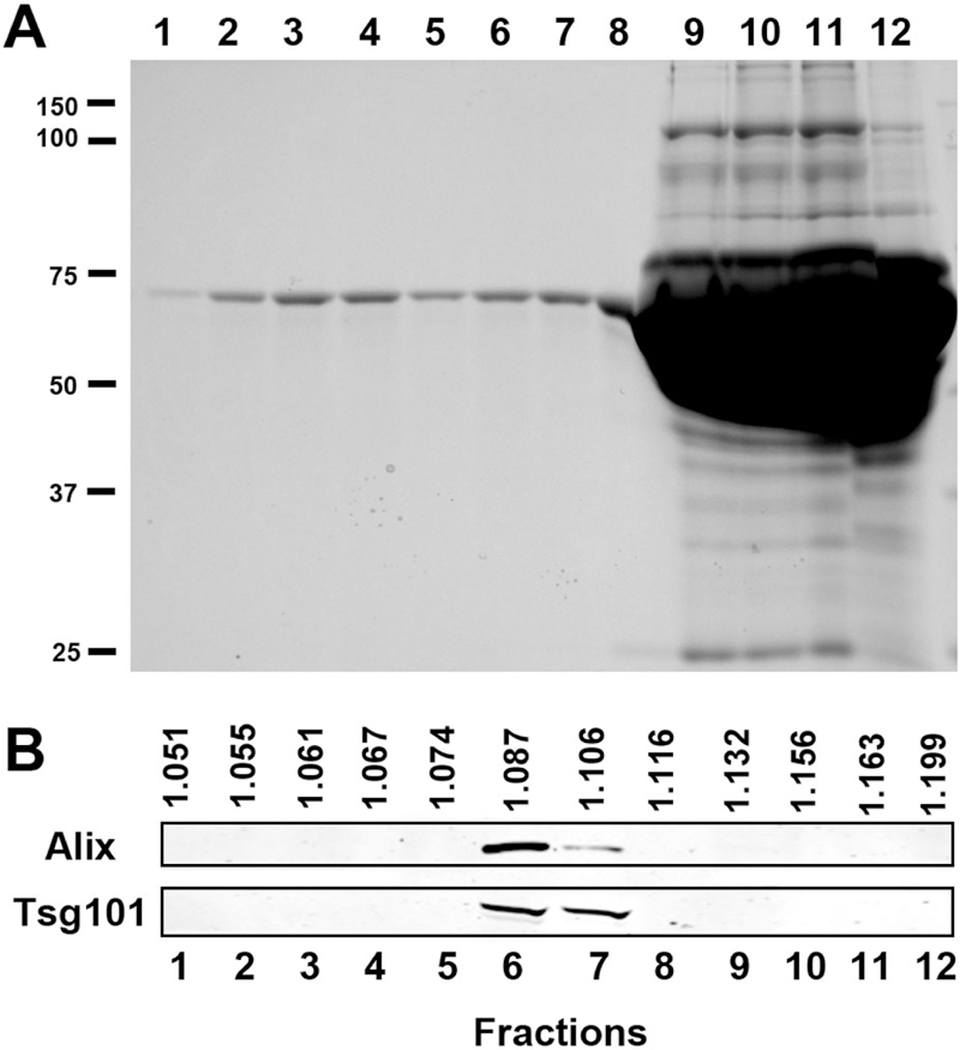
(A) SDS-PAGE analysis of gradient fractions. Fractions 1 to 12 (90 μL) were loaded. (B) Western Blot analysis of gradient fractions for the exosome marker proteins Alix and Tsg101. Exosome proteins were observed in fractions 6 to 7.
Figure 5. Exosome characterization by flow cytometry.
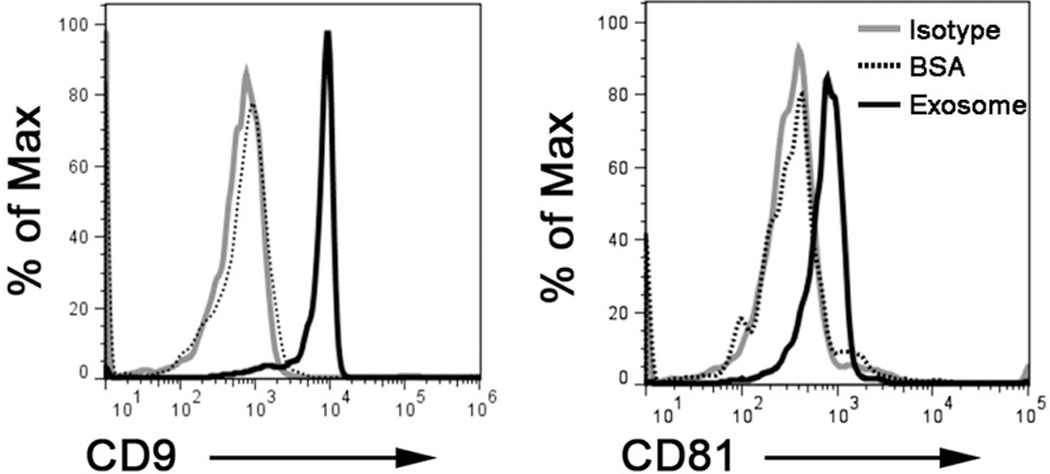
Exosomes (10 ug) were isolated by C-DGUC and non-specifically captured on latex beads, immune-stained and analyzed by flow cytometry. Flow cytometry dot plots show positivity for exosomes markers CD9 and CD81.
Figure 6. Exosome enumeration and sizing by nanoparticle tracking analysis.
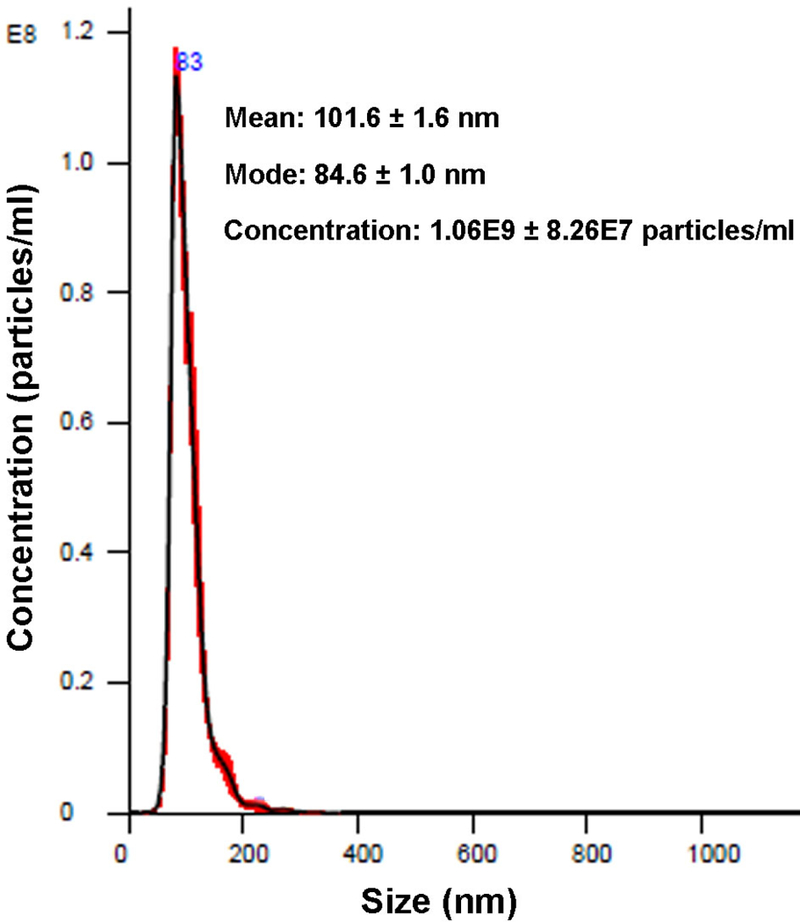
Exosomes-containing Fractions 6 and 7 were pooled and diluted at 1:200 before analysis using the Nanosight LM14 instrument. A sharp peak at 84 nm was observed.
Figure 7. Exosomal RNA characterization by Agilent bioanalyzer.
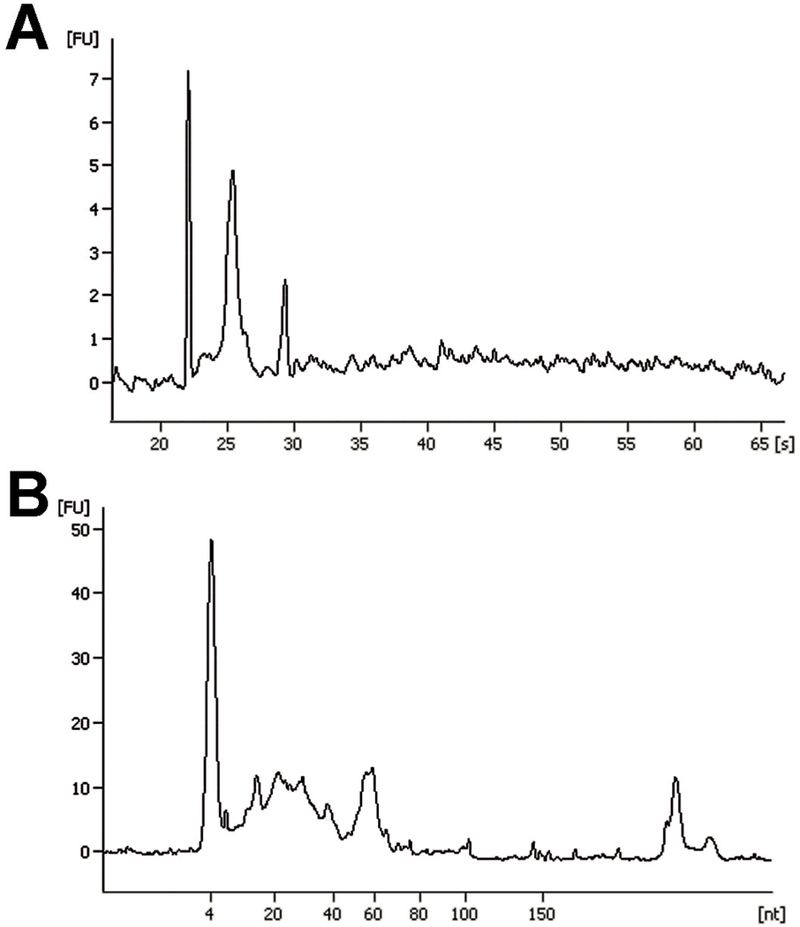
Representative images of Agilent RNA 6000 Pico chip (A) and Small RNA chip (B) assays of total RNA isolated from macrophages-derived exosomes.
2. Materials
2.1. Macrophage culture
Complete DMEM media: Dulbecco’s modified Eagle’s medium (DMEM) (Corning, 10–014), fetal bovine serum (Gibco), GlutaMax (Gibco), Penicillin-Streptomycin (Gibco)
Recombinant murine M-CSF (Peprotech)
Cell strainer, 70 μm (BD)
Non-treated cell culture dish (Corning)
D-PBS (Corning)
ACK lysing buffer (Lonza)
Cellstripper solution (Corning)
Exosome free media (See Note 1)
2.2. Density gradient centrifugation
OptiPrep density gradient medium (60% iodixanol) (Sigma) (See Note 2).
Homogenization buffer: 0.25 M sucrose, 1 mM EDTA, 10 mM Tris-HCl, pH 7.4
50% Iodixanol (See Note 3)
Beckman floor-mode ultracentrifuge Optima XPN 90
Beckman SW 28 Ti rotor
Beckman SW 40 Ti rotor (See Note 4)
Beckman ultracentrifuge tubes
10 mL BD disposable syringe (BD)
50 mL Steriflip-GP filter unit (Millipore)
Metal hub blunt point needles, 4-inch (Hamilton)
2.3. Fraction analysis
Qubit protein assay Kit (Thermo Fisher Scientific)
Qubit 3.0 fluorometer (Thermo Fisher Scientific)
Nanosight LM14 (Malvern Instruments)
2.4. SDS-PAGE and western blotting
ImageQuant LAS 4000
Anti-TSG101 antibody (Santa Cruz Biotechnology, sc-7964)
Anti-Alix antibody (Cell Signaling Technology, 2171)
IRDye 680RD goat anti-mouse IgG (LI-COR)
IRDye 800CW goat anti-rabbit IgG (LI-COR)
Odyssey infrared imaging system
2.5. Flow cytometry
FITC anti-mouse CD81 antibody (Thermo Fisher Scientific, HMCD8101)
Alexa Fluor 647 anti-mouse CD9 antibody (BioLegend, 124810)
Sulfate latex beads, 4% w/v, 5 μm (Thermo Fisher Scientific)
Glycine 1 M solution (Sigma)
Bovine serum albumin (BSA), 7.5% w/v (Sigma)
2.6. Exosome RNA analysis
mirVana™ PARIS™ RNA and Native Protein Purification Kit (Thermo Fisher Scientific)
Agilent 2100 bioanalyzer instrument
Agilent RNA 6000 pico kit
Agilent small RNA kit
3. Methods
3.1. Preparation of macrophage-conditioned media
Prepare single cell suspension from mouse bone marrow and pass cells through 70μm cell strainer.
Centrifuge at 300 × g for 10 minutes at 4 °C and resuspend cells in 1 mL of ACK buffer to lyse red blood cells.
Incubate for 5 minutes at room temperature.
Add 10 mL D-PBS and centrifuge at 300 × g for 10 minutes. Decant the supernatant and Add 25 mL complete DMEM media supplemented with M-CSF (40 ng/mL). Culture the cells in a 150-mm cell culture dish in a 37 °C incubator with 5% CO2.
After 7 days of culture, decant the supernatant; add 3 mL Cellstripper to the cells.
Incubate for 5 minutes in 37 °C incubator. Gently tap the dishes to detach the cells.
Add 20 mL D-PBS, and transfer the cells to 50 mL tube and centrifuge at 300 × g for 10 mins. Decant the supernatant and resuspend the cells in 25 mL serum-free DMEM. Count the cells and place 1.5 × 107 cells into a fresh 150-mm cell culture dish.
After 2 hours, decant the supernatant and wash the cells three times with D-PBS, add 20 mL exosome-free media.
After 24~48 hours, collect conditioned media into 50- mL conical tubes.
3.2. Nanoparticle concentration on 60% iodixanol cushion
Combine the conditioned media from two 150-mm cell culture dishes and centrifuge the 40 mL media at 300 × g for 10 minutes at 4 °C. Aspirate and transfer 38 mL media from the top to a fresh 50-mL conical tube; be careful NOT to disturb the pellet.
Centrifuge at 2000 × g for 10 minutes at 4 °C to remove large cellular debris. Aspirate and transfer 36 mL media from the top to a fresh 50- mL conical tube; be careful NOT to disturb the pellet.
Filter the supernatant by passing through a 50 mL Steriflip-GP filter unit (0.22 μm) to remove remaining cell debris and large vesicles (See Note 5).
Add the 36 mL pre-cleared media into ultracentrifuge tube (See Note 6); carefully underlay the media with 2 mL of 60% iodixanol using Hamilton blunt point 4-inch needles. (See Note 7)
After balancing the tubes within 0.1 g of each other, centrifuge in a SW 28 Ti rotor at 100,000 × g for 2~4 hr in Optima XPN 90 ultracentrifuge at 4 °C (See Note 8).
Carefully use blunt end needles to collect a volume of 3 mL from the bottom of the ultracentrifuge tubes that consists of 2 mL iodixanol cushion and 1 mL media, resulting in a mixture of 40% iodixanol. Mix thoroughly.
3.3. Exosome isolation by density gradient ultracentrifugation
Prepare 5%, 10%, and 20% iodixanol by mixing defined volumes of 50% Iodixanol with homogenization buffer according to Table 1, and allow 30 min mixing time for each solution.
Create a discontinuous gradient by first placing 3 mL of the 5% iodixanol solution to the bottom of the centrifuge tube with a long blunt end needle. Subsequently, underlay 3 mL of the 10% iodixanol, followed by 3 mL of the 20% iodixanol (See Note 9).
Place 3 mL of the 40% iodixanol solution containing nanoparticles from Section 3.2 Step 6 at the bottom of the discontinuous gradient.
After balancing the tubes within 0.1 g of each other, centrifuge in a SW 40 Ti rotor at 100,000 × g for 18 hrs in Optima XPN 90 ultracentrifuge.
After centrifugation, gently collect 12 individual 1 mL fractions from the top of the tubes.
Table 1.
Iodixanol gradient solutions for exosome isolation
| Iodixanol solutions, w/v (%) |
50% Iodixanol, w/v (mL) |
Homogenization buffer (mL) |
Total (mL) |
Density (g/mL) |
|---|---|---|---|---|
| 5 | 2 | 18 | 20 | 1.054 |
| 10 | 4 | 16 | 20 | 1.079 |
| 20 | 8 | 12 | 20 | 1.127 |
3.4. Fraction density determination (See Note 10)
Add 100 μL of the 5%, 10% and 20% iodixanol standards along with each of the fractions into 100 μL of water in a 96-well plate. Subject the samples to thorough mixing by pipetting up and down three times. Perform each measurement in triplicate.
Measure the absorbance of the individual solutions in each well using a plate reader containing a 340 nm filter.
Create a standard curve by plotting the densities of the 5%, 10% and 20% iodixanol solutions (table 1) against the mean absorbance.
Use the mean absorbance of each fraction to calculate the density of each fraction from the standard curve.
3.5. Protein quantification by Qubit protein assay
Label 3 tubes for the standards and 1 tube for each fraction
Prepare Qubit Working Solution by diluting the Qubit Protein Reagent 1:200 in Qubit Protein Buffer.
Add 190 μL Qubit working solution and 10 μL standards or samples into the corresponding assay tube.
Vortex standards and samples for 2–3 seconds and incubate at room temperature for 15 minutes.
Select Protein Assay on the Qubit 3.0 Fluorometer to calibrate with standards and read the samples.
3.6. Protein characterization by SDS-PAGE and immunoblotting
Mix 90 μL of each fraction with 10 μL of 10X RIPA buffer, PMSF, and Laemmli buffer. Boil the mixed samples at 95 °C for 5 minutes.
Prepare 10% polyacrylamide gels containing SDS as a denaturant.
Load the gels with a molecular-weight marker spanning 10 kDa to 250 kDa along with the denatured samples and resolve the proteins by electrophoresis.
Visualization of resolved proteins in the PAGE is achieved by incubating the gel with Coomassie stain for 2 hrs.
Subsequently, the gels are de-stained in 5% methanol and 7.5% glacial acetic acid solution and images are captured using an ImageQuant LAS 4000.
For western blot detection, transfer proteins resolved in the PAGE onto nitrocellulose membranes using a standard western blot transfer protocol.
Block the unbound sites within the membrane with 5% non-fat milk for 1 hour at room temperature.
Immunostain proteins with anti-TSG101 antibody (1:200) and anti-Alix antibody (1:1000) diluted in 5% non-fat milk and agitate membranes overnight at 4 °C.
Wash membranes for 5 minutes 4 times at room temperature with 1X PBST.
Incubate membranes with IRDye 680RD Goat anti-Mouse IgG (1:15,000) and IRDye 800CW Goat anti-Rabbit IgG (1:15,000) in 5% non-fat milk for 1 hour at room temperature. Protect membranes from light starting at this step.
Wash membranes for 5 minutes 4 times at room temperature with 1X PBST in the dark. Rinse membranes twice with D-PBS.
Scan membranes with Odyssey Infrared Imaging System at 700 and 800 nm.
Determine the presence of exosome markers in the iodixanol gradient fractions.
3.7. Exosome characterization by flow cytometry
Incubate 10 μg purified exosomes with 10 μL latex beads for 15 min at room temperature in a 1.5-mL Eppendorf tube with continuous rotation.
Add 1 mL D-PBS and incubate 4°C overnight with continuous rotation.
Add 110 μL of 1 M glycine (i.e., 100 mM final), mix gently and incubate at room temperature for 30 min.
Pellet the beads at 4000 rpm for 3 min at room temperature, remove the supernatant and discard.
Resuspend the pellet in 1 mL D-PBS/0.5% BSA, and re-pellet the beads at 4000 rpm for 3 min, remove the supernatant and discard.
Resuspend beads in 0.5 mL D-PBS /0.5% BSA.
Incubate 10 μL coated beads with 10 μL anti-exosomal protein antibody diluted in D-PBS/0.5% BSA 30 min at 4°C.
Wash twice with 1 mL D-PBS/0.5% BSA.
Add 200 μL of D-PBS /0.5% BSA and analyze the beads by flow cytometry.
3.8. Exosome enumeration by nanoparticle tracking analysis
Identify the optimal region in LM14 Nanosight for video capture; flush the chamber with 2 mL pure water (See Note 11).
Dilute the exosome samples with pure water to create 1 mL of solution at an optimal concentration (See Note 12).
Aspirate the exosome sample into a 1 mL syringe and infuse 0.5 mL into the sample chamber, taking care to ensure that no air bubbles are introduced to the system.
Fine tune to refocus the particles and set the infusion rate at 40.
Set appropriate camera level and screen gain, record three videos, 1 min each.
Set appropriate detection level and analyze the captured video files.
3.9. Exosomal RNA extraction
Add 400 μL exosome sample in 2 mL tubes
Add 400 μL pre-warmed 2X Denaturing Solution. Immediately mix thoroughly.
Add 800 μL of the bottom phase of Acid-Phenol:Chloroform.
Vortex for 30–60 sec to mix.
Centrifuge for 5 min at 14,000 × g at room temp to separate the mixture into aqueous and organic phases.
Carefully remove the aqueous (upper) phase without disturbing the lower phase or the interphase, and transfer it to a fresh tube.
Add 1.25 volumes of 100% ethanol to the aqueous phase and mix thoroughly.
For each sample, place a filter cartridge into one of the collection tubes.
Pipet the lysate/ethanol mixture onto the filter cartridge. Apply a maximum of 700 μL to a filter cartridge at a time.
Centrifuge at 10,000 × g for ~30 sec.
Discard the flow-through, and repeat until all of the lysate/ethanol mixture is through the filter. Save the collection tube for the washing steps.
Apply 700 μL miRNA Wash Solution 1 to the Filter Cartridge and centrifuge at 10,000 × g for ~15 sec, discard the flow-through from the collection tube, and replace the filter cartridge into the same collection tube.
Apply 500 μL Wash Solution 2/3 and draw it through the Filter Cartridge as in the previous step.
Repeat with a second 500 μL of Wash Solution 2/3.
After discarding the flow-through from the last wash, replace the filter cartridge in the same collection tube and spin the assembly for 1 min at to remove residual fluid from the filter.
Transfer the filter cartridge into a fresh collection tube. Apply 50 μL of preheated (95°C) nuclease-free water to the center of the filter, and close the cap. Centrifuge for ~30 sec to recover the RNA.
Collect the eluate and store it at –80°C.
3.10. Exosomal total RNA characterization
Using an Agilent 2100 Bioanalyzer system, prepare a Gel-Dye mix by mixing 1 μL RNA dye concentrate with 65 μL filtered gel, vortex solution well and spin the tube at 13000 × g for 10 min at room temperature.
Load 9 μL Gel-Dye mix to appropriate well and prime the RNA Pico chip
Load 9 μL Gel-Dye mix and the RNA conditioning solution to appropriate well
Pipette 5 μL of RNA marker in the well for ladder and all 11 sample wells.
Pipette 1 μL of the heat denatured ladder into the ladder well;
Pipette 1 μL of sample in each of the 11 sample wells
Vortex for 1 min at 2400 rpm in the IKA station;
Run the chip in the Agilent 2100 Bioanalyzer within 5 min.
3.11. Exosomal small RNA characterization
Prepare the Gel-Dye mixture by combining the RNA dye concentrate with filtered gel, vortex the solution well and spin the tube at 13000 × g for 10 min at room temperature.
Load 9 μL Gel-Dye mix to appropriate well and prime the small RNA chip
Load 9 μL Gel-Dye mix and the RNA conditioning solution to appropriate wells
Pipette 5 μL of RNA marker in the well for ladder and all 11 sample wells.
Pipette 1 μL of the heat denatured ladder into the ladder well;
Pipette 1 μL of sample in each of the 11 sample wells
Vortex for 1 min at 2400 rpm in the IKA station;
Run the chip in the Agilent 2100 Bioanalyzer within 5 min.
4. Notes
Standard Fetal Bovine Serum (FBS) contains very high levels of bovine extracellular vesicles and lipoproteins which will complicate downstream analyses if not thoroughly removed prior to use for cell culture. It is therefore important to eliminate these native nanoparticles by an overnight centrifugation at 100,000 × g.
The use of Iodixanol offers several advantages over sucrose. First, solutions of iodixanol display lower viscosity than sucrose solutions, making it easier to handle. Second, iodixanol is metabolically inert and non-toxic to cells, allowing subsequent functional assays to be performed without the need for its removal. Lastly, Iodixanol solutions also display lower osmolality than sucrose solutions at similar density and can be made iso-osmotic at high densities, allowing exosomes to be purified under iso-osmotic conditions and thus better preserve their integrity and functionality.
It is recommended to prepare a 50% iodixanol stock solution, which makes it easier than 60% iodixanol to create 5%, 10% and 20% iodixanol solutions. To prepare a 50% iodixanol stock, mix 5 volumes of OptiPrep with 1 volume of 1.5 M sucrose, 6 mM EDTA, 60 mM Tris-HCl, pH 7.4. On standing, iodixanol tends to settle; always shake the bottle of OptiPrep to obtain a homogeneous solution before use.
Swinging bucket rotors are advantageous over fixed-angle rotors for gradient work, since the gradient always maintains the same orientation with the axis of the tube and the longer path length permits better separation.
Cells, cellular debris, and other large microparticles are first removed from the conditioned medium by differential centrifugation and filtration. Omission of these clarification steps may lead to a loss of exosomes due to their entrapment into aggregates that rapidly form and sediment as large particles at high centrifugal force.
The yields of exosomes vary considerably depending on cell type and culture condition. A larger volume of conditioned media may be required for some downstream experiments, such as western blot protein detection and RNA sequencing. Ultrafiltration devices, e.g. Amicon Ultra-15 centrifugal filters, can be used for concentrating exosomes from conditioned media before ultracentrifugation.
Iodixanol solutions tend to adhere to the walls of a pipette, making it difficult to accurately transfer with pipetting. The use of syringes is therefore recommended for accurate dispensing. It is best to take up more of iodixanol medium than is required as it is more accurate to empty the syringe to a graduation mark than to empty it completely.
At a given centrifugal g-force, the time required for complete sedimentation of vesicles largely depends on the path length of the rotor. As swinging-bucket rotors have a greater path length than fixed angle rotors, the run time for swinging-bucket rotors will be longer for efficient sedimentation.
A convenient way to create discontinuous gradients is by under-laying successively denser solutions beneath lighter ones. A syringe fitted with a metal filling cannula is an ideal tool for this procedure. Add 10% or 20% solutions very slowly to avoid mixing with 5% or 10% solutions, respectively. It is also important to avoid air bubble formation during dispensing, as it will disturb the lower density layers.
Density analysis can be simply performed by measuring the absorbance at 340 nm on gradient fractions using multi-well plate readers. However, because there is a linear relationship between refractive index and density, the solutions density can also be determined by measuring the refractive index of the gradient fraction, if a wide-range Refractometer is available.
Nanoparticles are present in many commercially available buffers. It is important to test if the buffer is free of particles prior to its use.
Accurate quantification of exosomes and other nanoparticles using NTA works best when sample concentrations are between 108 and 109 particles per mL of solution. An optimum dilution would present around 100 particles in the field of view when inserted into the sample chamber and visualized.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health; (5U19CA179512) Extracellular RNA Communication Consortium Common Fund, and HL133575 to (RLR) which was administered by the Northern California Institute for Research and Education. The work was performed at the Veterans Affairs Medical Center, San Francisco, California.
5. References
- 1.Colombo M, Raposo G, and Thery C 2014. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30:255–289. [DOI] [PubMed] [Google Scholar]
- 2.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, and Lotvall JO 2007. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9:654–659. [DOI] [PubMed] [Google Scholar]
- 3.Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, Buzas K, Casal E, Cappello F, Carvalho J, et al. 2015. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 4:27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gyorgy B, Hung ME, Breakefield XO, and Leonard JN 2015. Therapeutic applications of extracellular vesicles: clinical promise and open questions. Annu Rev Pharmacol Toxicol 55:439–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kourembanas S 2015. Exosomes: vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu Rev Physiol 77:13–27. [DOI] [PubMed] [Google Scholar]
- 6.Johnstone RM, Adam M, Hammond JR, Orr L, and Turbide C 1987. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J Biol Chem 262:9412–9420. [PubMed] [Google Scholar]
- 7.Thery C, Amigorena S, Raposo G, and Clayton A 2006. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol Chapter 3:Unit 3 22. [DOI] [PubMed] [Google Scholar]
- 8.Linares R, Tan S, Gounou C, Arraud N, and Brisson AR 2015. High-speed centrifugation induces aggregation of extracellular vesicles. J Extracell Vesicles 4:29509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Momen-Heravi F, Balaj L, Alian S, Mantel PY, Halleck AE, Trachtenberg AJ, Soria CE, Oquin S, Bonebreak CM, Saracoglu E, et al. 2013. Current methods for the isolation of extracellular vesicles. Biol Chem 394:1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nordin JZ, Lee Y, Vader P, Mager I, Johansson HJ, Heusermann W, Wiklander OP, Hallbrink M, Seow Y, Bultema JJ, et al. 2015. Ultrafiltration with size-exclusion liquid chromatography for high yield isolation of extracellular vesicles preserving intact biophysical and functional properties. Nanomedicine 11:879–883. [DOI] [PubMed] [Google Scholar]
- 11.Abramowicz A, Widlak P, and Pietrowska M 2016. Proteomic analysis of exosomal cargo: the challenge of high purity vesicle isolation. Mol Biosyst 12:1407–1419. [DOI] [PubMed] [Google Scholar]
- 12.Van Deun J, Mestdagh P, Agostinis P, Akay O, Anand S, Anckaert J, Martinez ZA, Baetens T, Beghein E, Bertier L, et al. 2017. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods 14:228–232. [DOI] [PubMed] [Google Scholar]
- 13.Elmowalid GA, Qiao M, Jeong SH, Borg BB, Baumert TF, Sapp RK, Hu Z, Murthy K, and Liang TJ 2007. Immunization with hepatitis C virus-like particles results in control of hepatitis C virus infection in chimpanzees. Proc Natl Acad Sci U S A 104:8427–8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greening DW, Xu R, Ji H, Tauro BJ, and Simpson RJ 2015. A protocol for exosome isolation and characterization: evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. Methods Mol Biol 1295:179–209. [DOI] [PubMed] [Google Scholar]
- 15.Lamparski HG, Metha-Damani A, Yao JY, Patel S, Hsu DH, Ruegg C, and Le Pecq JB 2002. Production and characterization of clinical grade exosomes derived from dendritic cells. J Immunol Methods 270:211–226. [DOI] [PubMed] [Google Scholar]
- 16.Lobb RJ, Becker M, Wen SW, Wong CS, Wiegmans AP, Leimgruber A, and Moller A 2015. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J Extracell Vesicles 4:27031. [DOI] [PMC free article] [PubMed] [Google Scholar]