

Abstract
Heme degradation through the action of heme oxygenase (HO) is unusual in that it utilizes heme as both a substrate and cofactor for its own degradation. HO catalyzes the oxygen-dependent degradation of heme to biliverdin with the release of CO and “free” iron. The characterization of HO enzymes from humans to bacteria reveals a similar overall structural fold that contributes to the unique reaction manifold. The heme oxygenases share a similar heme-dependent activation of O2 to the ferric hydroperoxide as that of the cytochrome P450s and peroxidases. However, whereas the P450s promote cleavage of the ferric hydroperoxide O—O bond to the oxoferryl species the HOs stabilize the ferric hydroperoxide promoting hydroxylation at the heme edge. The alternate reaction pathway in HO is achieved through the conformational flexibility and extensive hydrogen bond network within the heme binding site priming the heme for hydroxylation. Until recently it was believed that all heme degrading enzymes converted heme to biliverdin and iron, with the release of carbon monoxide (CO). However, the recent discovery of the bacterial IsdG-like heme degrading proteins of Staphylococcus aureus, Bacillus anthracis and Mycobacterium tuberculosis has expanded the reaction manifold of heme oxidation. Characterization of the heme degradation products in the IsdG-like reaction suggests a mechanism distinct from the classical HOs. In the following review we will discuss the structure–function of the canonical HOs as it relates to the emerging alternate reaction manifold of the IsdG-like proteins.
Keywords: Heme degradation, Heme oxygenase, Biliverdin, Staphylobilin, Mycobilin, Coupled oxidation, Verdoheme
Introduction
The biological degradation of heme (iron-protoporphyrin IX) is catalyzed by a family of enzymes termed heme oxygenases that convert heme to biliverdin, with the release of carbon monoxide (CO) and iron (Fig. 1A) [1–4]. Although first described in mammals as a “housekeeping” enzyme for its role in the recycling of iron, HO1 is now recognized for its role in antioxidant defense, cellular signaling and the biosynthesis of light-sensing bilins [5–10]. Furthermore, pathogenic bacteria utilize HOs as a means of acquiring iron that is essential for virulence and survival [11–15]. The HO-dependent conversion of heme to biliverdin consumes three molecules of oxygen and seven reducing equivalents. In mammals the reducing equivalents for the oxidation of heme are supplied by NADPH-cyto-chrome P450 reductase and the resulting biliverdin IXα (biliverdin BVIXα) is converted to bilirubin by the action of biliverdin reductase. In plants, algae and cyanobacteria a ferredoxin-dependent heme oxygenase generates biliverdin IXα as a precursor for the synthesis of light-harvesting pigments [16, 17]. In the case of the canonical bacterial HOs no physiological electron donors have been identified [18–20].
Fig. 1.
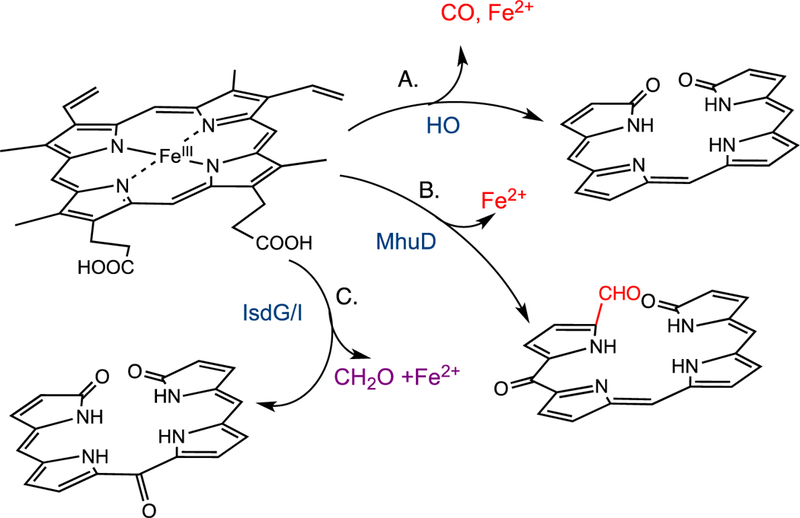
Reaction products of the canonical HO and non-canonical IsdG/I-like heme oxygenases.
The recent discovery of the Staphylococcus aureus and Bacillus anthracis IsdG-like proteins encoded within a cluster of genes termed iron-regulated surface determinants (isd) has expanded the reaction manifold of heme cleavage [21–23]. Although initially reported to convert heme to biliverdin [22, 23] recent studies have shown the mechanism and reaction products of the IsdG-like proteins to be distinct from those of the canonical HOs [24, 25]. The S. aureus IsdG reaction converts heme to a mixture of 5-oxo-bilirubin and 15-oxo-bilirubin, collectively termed “staphylobilins” (Fig. 1C) [25]. Moreover, recent studies have shown that oxidative cleavage of heme by IsdG leads to the release of the α-meso carbon as formaldehyde, precluding verdoheme as an intermediate [24]. Similarly, the structurally related Mycobacterium tuberculosis MhuD protein catalyzes oxidative ring cleavage with retention of the meso-carbon as an aldehyde suggesting the MhuD and IsdG-like proteins lie along the same reaction pathway (Fig. 1B) [26]. In contrast to the canonical HOs a physiological electron donor to the IsdG/I enzymes of S. aureus has recently been identified [27].
Although the canonical HOs and non-canonical IsdG-like proteins are mechanistically distinct from each other they belong to a unique class of heme enzymes that channel the activated Fe(III)-OOH toward heme hydroxylation through modulation of the heme electronic configuration. This reaction is distinct from that of other monoxygenases such as the cytochrome P450s that diverge through an alternate pathway that promotes cleavage of the activated Fe(III)–OOH to the oxoferryl Fe(IV)=O species.
Structural diversity of heme oxidation
The canonical HO enzymes from bacteria to mammals have a similar overall α-helical structural fold (Fig. 2A) [28–30]. Heme is held between the proximal and distal helices and anchored in the pocket through interactions of the propionates with surface exposed Lys residues. Interestingly, the iron-regulated HemO from Pseudomonas aeruginosa gives rise to an altered regioselectivity from that of all other HO’s as a consequence of an alternate seating of the heme within the active site. The in-plane rotation of the heme is a consequence of alternate propionate interactions with the protein scaffold [29]. In addition to the conserved proximal His ligand all canonical HO’s retain an ordered hydrogen-bonding network required for proton delivery to the coordinated Fe(II)—O2 to form activated Fe(III)–OOH (Fig. 3A).
Fig. 2.
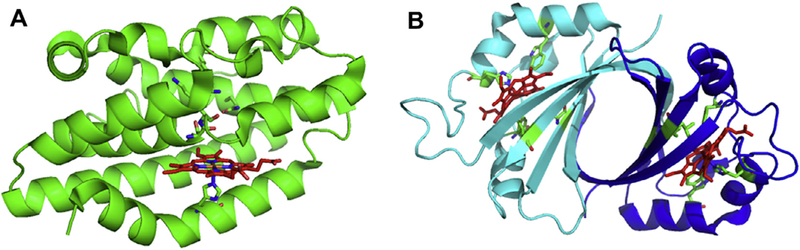
Comparison of the oxygen activation pathways of HO and cytochrome P450.
Fig. 3.
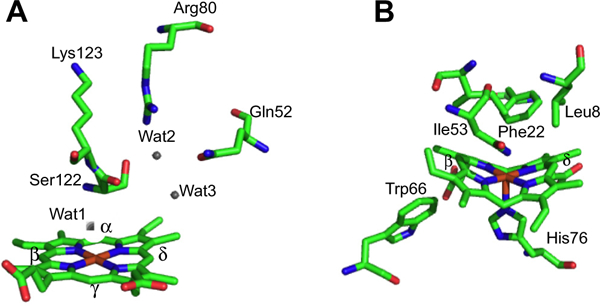
Overall fold of the canonical HemO of P. aeruginosa and IsdI of S. aureus. (A) HemO shown with heme in red and active site hydrogen bond contributing residues shown in stick form. (B) IsdG shown with heme in red and active site residues in stick form. Protein Data Bank (PDB) codes 1SK7 (HemO) and 3LGM (IsdI).
In contrast the recently identified non-canonical IsdG/I and MhuD heme degrading enzymes of S. aureus and M. tuberculosis, respectively, have an overall structural fold distinct from that of the classical HOs (Fig. 2B) [31,32]. The Isd protein family are homodimers, with each monomer adopting a ferredoxin-like α/β-sand-wich fold that comes together to form a β-barrel at the dimer interface. Each monomer binds one heme in a hydrophobic cleft on either side of the β-barrel with the coordinating His residue provided by the peripheral helix/loops surrounding the β-barrel. In the CoPPIX–IsdI complex the porphyrin is ligated through His-76 with the propionates being anchored in the back of the pocket through interactions with Arg-21 and Arg-25. Interestingly, the ordered hydrogen-bonding network found in the canonical HO enzymes is absent in the IsdG-like proteins (Fig. 3B). Moreover, the resting state heme in the Isd proteins undergoes significant distortion from planarity induced through steric interaction of the heme macrocycle with several residues within the heme pocket (Fig. 3B) [32]. The structural aspects of the respective proteins required for heme reactivity will be discussed in the following sections.
Steric versus electronic contributions to heme hydroxylation
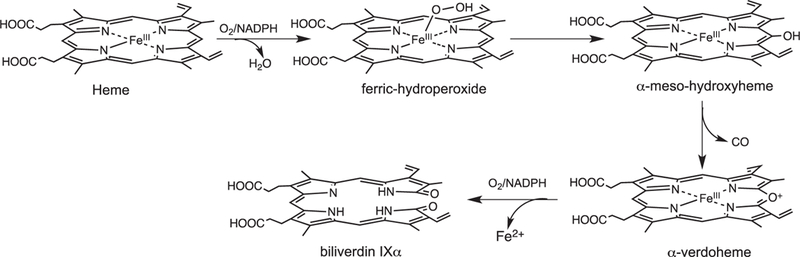
The initial step in heme oxidation involves the reduction of the Fe(III)–heme complex to the Fe(II)–O2 complex. Reduction of the Fe(II)–O2 complex to the activated Fe(III)–peroxo species leads to the formation of α-meso-hydroxyheme, which in the presence of oxygen is rapidly converted to verdoheme (Fig. 4) [33]. Early studies by Ortiz de Montellano and Wilks further showed a molar equivalent of H2O2 could substitute for O2 and reducing equivalents in the conversion of heme to α-meso-hydroxyheme [34]. In contrast reaction with most alkyl or acyl hydroperoxides does not lead to heme hydroxylation but rather to the formation of a ferryl complex as a result of O—O bond cleavage. Based on the fact H2O2 can substitute for O2 and reducing equivalents in the initial meso-hydroxylation of heme it was proposed that heme hydroxylation occurred by one of two mechanisms: (a) electrophilic addition of the terminal oxygen of the ferric peroxo species (Fe(III)–OOH) to the α-meso-carbon, or (b) nucleophilic addition of the terminal oxygen of the protonated (Fe(III)–OO−) species (Fig. 5). In these initial studies a radical mechanism was ruled out based on the indiscriminate nature of such a reactive species (Fig. 5c). Subsequently, it was shown that ethyl hydroperoxide reacted to yield α-meso-ethoxyheme seemingly eliminating nucleophilic addition as a viable mechanism [35]. Evidence of the Fe(III)–OOH species was obtained by cryogenic ENDOR and EPR spectroscopy following one-electron reduction of the Fe(II)–O2 complex [36]. Temperature annealing experiments identified the Fe(III)–OOH intermediate as the immediate precursor of α-meso-hydroxyheme (Fig. 5a). The activation of Fe(II)–O2 to Fe(III)–OOH is a common step in heme containing monoxygenases and heme oxygenase.[37] However, the subsequent steps diverge significantly where, in contrast to the self-hydroxylation by heme oxygenase, the Fe(III)–OOH intermediate in the P450-type monooxygenases decays to a ferryl species (Fe(IV)=O) (Fig. 5d).
Fig. 4.
Active site structure of the canonical HemO and non-canonical IsdI. (A) Hydrogen bond network of the heme-HemO distal pocket. (B) Active site of heme-IsdG with the active site residues as labeled. Protein Data Bank (PDB) codes 1SK7 (HemO) 3LGM (IsdI).
Fig. 5.
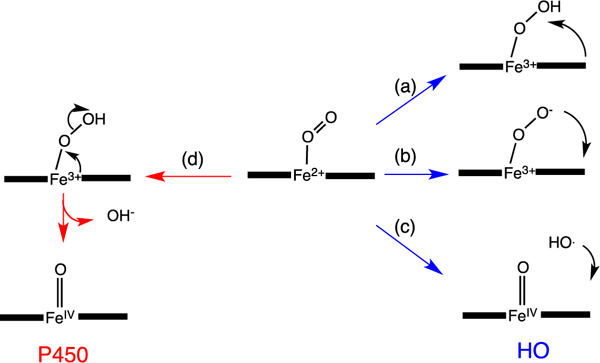
Reaction intermediates in the heme oxygenase catalyzed degradation of heme to biliverdin.
A significant distinction between the canonical HO’s and the P450/peroxidase monoxygenases is the absence of a residue providing the polar side chain that stabilizes the bound O2. The stabilization of the O2-ligand in HO is a combination of the distal helix conserved Gly–Gly motif and the extensive hydrogen bond network coordinated through bridging H2O molecules (Fig. 3B) [28–30,38,39]. Furthermore, the absorption spectrum of the Fe(II)–O2 heme HO-1 complex is similar to that of oxmyoglobin [33] and has a unique oxygen-isotope shift in the resonance Raman spectrum suggesting the bound O2 is highly bent [40]. The structure of the Corynebacterium diphtheriae Fe(II)–O2 heme-HmuO complex confirmed the highly bent O—O bond angle, a consequence of interactions with the distal helix Gly–Gly motif and the hydrogen bond network [39]. Indeed disruption of the hydrogen bond network in the mammalian HO-1 enzymes on mutation of Asp-140 resulted in destabilization of the Fe–OOH intermediate and accelerated decay to the Fe(IV)=O species [41,42]. This was supported by EPR and ENDOR experiments that confirmed Asp-140 hydrogen bonds to the ordered water molecule that stabilizes the Fe(III)–O–O− species. NMR studies further confirmed the hydrogen bond network is critical in stabilizing the ordered water molecule required for the donation of a proton to the coordinated Fe(II)–O2 [43,44]. In contrast mutation of the conserved Asp-136 in the C. diphtheriae HmuO did not lead to loss of enzymatic activity highlighting the importance of the nearby water that hydrogen bonds to the distal oxygen of the coordinated Fe(II)–O2 [45]. Furthermore the bacterial HOs from Neisseria meningtidis and P. aeruginosa despite retaining a rigid hydrogen bond network within the pocket lack the conserved Asp residue found in the eukaryotic and C. diphtheria enzymes [29, 38]. The lack of conservation of the conserved Asp in the bacterial HOs confirmed the critical role of the nearby water molecule in providing a hydrogen bond to the distal oxygen of the Fe(II)–O2, and as a conduit for proton transfer in activation of the Fe(III)–OOH. Ikeda-Saito and coworkers proposed a water-driven oxygen activation where the steric constraints of the highly bent Fe(III)–OOH, with its slightly longer O—O bond length, places the terminal oxygen in direct contact with the meso-carbon [45].
In contrast to the sterically constrained water-mediated activation, Rivera and coworkers proposed the initial hydroxylation is facilitated by the active participation of the heme [46,47]. This hypothesis was based on the unusual EPR fingerprint of the Fe(III)–OOH HO-1 complex with the sum of the g values (∑g2) ~14 being indicative of a novel electronic structure (dxz,dyz)4(dxy)1 where the unpaired electron resides in the dxy orbital. The (dxy)1 electronic configuration is proposed to place a large amount of unpaired electron density on the meso carbons resulting in significant ruffling of the heme [48]. Furthermore, density functional calculations suggest such an electronic structure places significant π-radical character on the meso carbons consistent with self catalyzed heme hydroxylation [49].
The unique electronic configuration of the Fe(III)–OOH intermediate leading to heme hydroxylation was proposed to be a combination of flexibility in the heme binding site and a stronger hydrogen bond network than that observed for the globins [46]. Thus the HO distal site drives the heme by providing a hydrogen bond to the coordinated Fe(III)–OOH and lowering the sigma donating capability that subsequently leads to the characteristic non-planar deformations of the porphyrin required for heme hydroxylation. In earlier mechanistic studies O—O bond homolysis and addition of hydroxyl radical (·OH) to the meso-carbon was ruled out based on the indiscriminate nature of the hydroxyl radical (Fig. 5c) [34]. However, the (dxz,dyz)4(dxy)1 configuration and subsequent ruffling of the heme further advanced support for a radical mechanism [47].
However, recent NMR studies by La Mar and coworkers on the Fe(III)–CN and Fe–N3 heme-HO-1 complex have raised questions as to the relationship between electronic structure and HO catalysis [50]. Prior NMR studies reported that in contrast to the globins, where the Fe(III)–N3 and Fe(III)–CN complexes have similar contact shift patterns and ground states, the P. aeruginosa and N. meningitides HemO Fe(III)–N3 and Fe(III)–CN complexes exhibited different orbital ground states [51]. The larger magnetic moment of the Fe(III)–N3 when compared to the Fe(III)–CN was interpreted to suggest a novel S = 3/2, (dyz)2(dxy)1(dxz)1 ground state rather than the S =1/2, (dxy)2(dyz)2(dxz)1 for the Fe(III)–CN complex. The novel ground state was attributed to a weaker axial field strength resulting from the stronger hydrogen-bond ligand than that observed for the ferric globins. This data was interpreted as further evidence for the modulation of the heme electronics as a diagnostic of catalytic competence. In contrast La Mar and coworkers reported that the switch in the heme contact shift pattern for the Fe(III)–CN and Fe(III)–N3 complexes is a general HO characteristic but concluded that ground state for the Fe(III)–N3 complex is S =1/2 as for the Fe(III)–CN complex. This was based on the observation that the azide orientation in the distal pocket increases its ligand field strength. The authors concluded that in contrast to the globins the HO active site hydrogen bond donor is weaker consistent with the more planar (dxy)2(dyz)2(dxz)2 electronic configuration. Furthermore, retention of a similar ground state in the Asp-140 Ala mutant indicates that the unique electronic properties of the heme are not correlated with catalytic activity. Although contradictory to the hypothesis that the weaker axial ligand field strength stabilizes the (dxy)1 configuration and subsequent heme ruffling required for catalysis, the data is consistent with the sterically constrained water-driven oxygen activation previously proposed by Ikeda-Sai-to [45]. Recent theoretical QMM studies suggested the energy barrier for a concerted mechanism involving O—O bond cleavage and O—Cmeso bond formation is energetically unfavorable [52]. The more favorable reaction pathway indicates a non-synchronous effectively concerted mechanism involving O—O bond homolysis and attack of an extremely “short-lived” ·OH at the meso carbon. The authors further suggest this mechanism is consistent with the water cluster controlling O—O cleavage along with the required steric constraints, while providing a strong hydrogen bond to the leaving ·OH in directing attack at the meso-carbon.
As previously mentioned heme oxygenase lies along a reaction manifold where the “default” Fe(III)–OOH heme hydroxylation reaction outweighs constraints required for the alternate pathway to the Fe(IV)=O species. Interestingly, the critical hydrogen bond network responsible for stabilizing the Fe(III)–OOH intermediate and activating the heme for hydroxylation in the canonical HOs, is absent in the structures of IsdG/I and MhuD [31,32]. However, the heme in the resting state ferric heme–IsdI complex is extremely ruffled through direct contact of the heme γ-meso carbon with Phe-22 (Fig. 3B). A recent 1H NMR study of the Fe(III)–CN− complex of IsdI revealed large shifts for the meso-H and much smaller shifts for the heme methyl groups suggesting the heme exhibits predominantly the (dxz,dyz)4(dxy)1 electronic configuration [53]. This is also consistent with the extreme distortion of the heme visible in the structure of the Fe(III)–CN− IsdI complex [53]. Furthermore, the (dxz,dyz)4(dxy)1 configuration in the alkaline Fe(III)–OH of IsdI represents a much greater population than the ~2% observed for the less distorted Fe(III)–OH heme-HemO complex [46]. The authors proposed that the increased heme distortion and stabilization of the (dxz,dyz)4(dxy)1 configuration in the Isd enzymes facilitates self-hydroxylation of the heme in the absence of the hydrogen bond network. However, at the present time the nature of the hydroxylating species in the IsdG-catalyzed reaction is not known. It is also unclear if as in the canonical HOs H2O2 can substitute for oxygen and reducing equivalents in the initial heme hydroxylation. A detailed spectroscopic analysis similar to those performed on the HO enzymes will no doubt aid in identifying the hydroxylating species in the IsdG catalyzed reaction.
Oxidative cleavage of meso-hydroxyheme: a new paradigm?
Beyond the initial hydroxylation of the IsdG/MhuD-heme the remaining steps in the release of iron diverge from that of the canonical HOs. In studies of the canonical HOs the Fe(III)–meso-hydroxyheme decays to Fe(III)–verdoheme in the presence of oxygen [35,54–56]. EPR and resonance Raman spectroscopy suggest meso-hydroxyheme following deprotonation to the oxophlorin, is in equilibrium between the ferric non-radical and ferrous radical state (Fig. 6) [54,57]. In aerobic conditions the free radical state binds oxygen to give a ferrous peroxy radical intermediate where either the peroxy radical interacts with the ferrous iron or internal electron transfer gives rise to the Fe(III)–OOH, that then leads to an unstable ferryl alkoxy radical. The unstable ferryl alkoxy radical triggers the release of the meso- carbon as CO (Fig. 6). The resulting meso-carbon radical is oxidized on electron transfer to give the Fe(IV)=O intermediate and the resulting carbocation is trapped as Fe(III)-verdoheme. In contrast to meso-hydroxyheme interacting with O2 prior addition of a reducing equivalent [54,58] it was previously proposed that reduction of meso-hydroxyheme to Fe(II)-verdoheme was required prior to O2 binding [57]. However, the reaction of Fe(III)-meso-hydroxyheme with O2 is significantly faster than reduction of the iron, suggesting under physiological conditions the formation of Fe(III)-verdoheme precedes the injection of a reducing equivalent to yield Fe(II)-verdoheme [59].
Fig. 6.
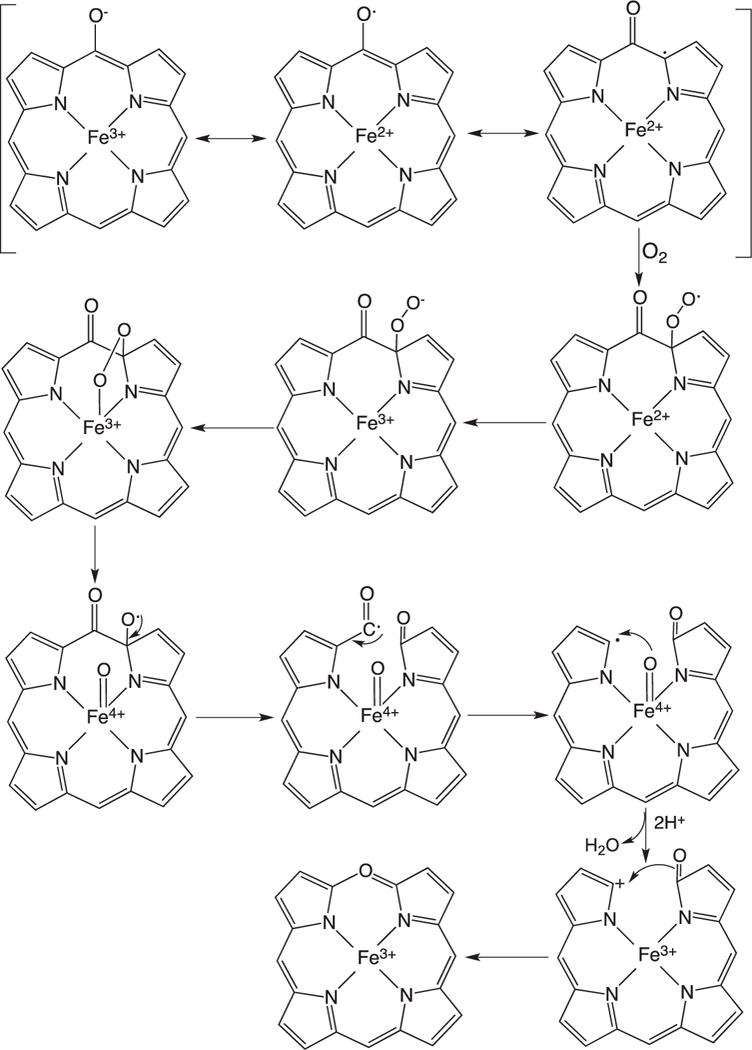
Proposed mechanism for the auto-oxidation of meso-hydroxyheme to verdoheme.
Skaar and coworkers invoked a similar mechanism for the IsdG/I catalyzed conversion of heme to staphylobilin that proceeds through either Fe(III)-dihydroxyheme or Fe(III)-meso-hydroxyverdoheme (Fig. 7) [21]. In the first pathway (Fig. 7A) a two-electron reduction and formation of the activated Fe(III)–OOH intermediate leads to hydroxylation and formation of Fe(III)-β/δ-dihydroxyheme. A second one-electron reduction to Fe(II)-hydroxyverdoheme in the presence of oxygen leads to ring opening to hydroxyverdoheme. In the alternate pathway (Fig. 6B) the reaction proceeds as for the canonical HO’s with conversion to Fe(III)-verdoheme. On further reduction and oxygen binding a second hydroxylation step at either the β- or δ-meso-carbon yields β- or δ-Fe(III)-hydroxyverdoheme, respectively. The common intermediate in both pathways Fe(III)- hydroxyverdoheme requires four electrons and one molecule of oxygen to yield either the ring-opened 5-oxo-bilirubin (ring cleavage at the β-meso-carbon) and 15-oxo-bilirubin (ring cleavage at the δ-meso-carbon). However, there exists no spectroscopic evidence of either the Fe(III)-verdoheme or Fe(III)-hydroxyverdoheme as intermediates in the IsdG catalyzed reaction.
Fig. 7.
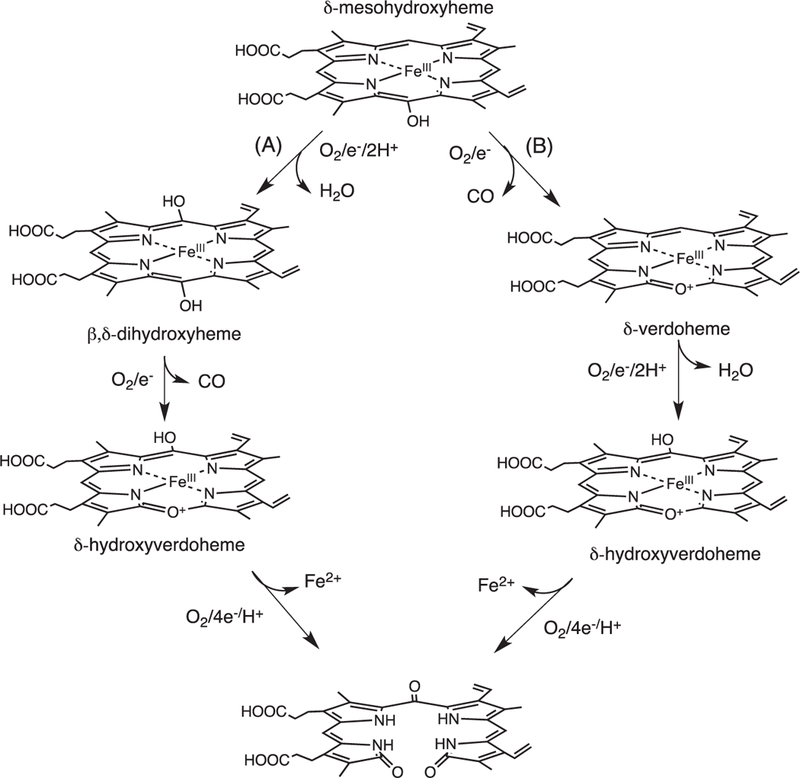
Proposed mechanism for the conversion of heme to staphylobilin via a “heme oxygenase” like reaction. Adapted from [Ref. 15].
In contrast recent studies by Ikeda-Saito and colleagues have shown oxidative cleavage of heme by the MhuD enzyme does not give rise to CO as a by-product precluding verdoheme as an intermediate [26]. The product of the MhuD catalyzed reaction cleaves the porphyrin ring with retention of the α-meso carbon as an aldehyde and further modification of the adjacent meso-carbon to a carbonyl at either the β- or δ-meso (mycobilin-b) carbon (Fig. 1C) [26]. Based on the structural homology between MhuD and IsdG it was hypothesized the reaction mechanism may be similar to that of MhuD. In a similar manner heme conversion to staphylobilin leads to the release of the meso-carbon as formaldehyde and not CO as previously proposed [21,24]. Thus the IsdG-like proteins present a paradigm shift in bacterial heme degradation the physiological consequences of which will be discussed below.
Although the detailed mechanism of IsdG-like catalyzed degradation of heme have yet to be elucidated the unique protein fold and heme electronic structure are thought to play a significant role. The radical character of the ferrous oxophlorin intermediate generated on deprotonation of meso-hydroxyheme in the canonical HOs rapidly autoxidizes to verdoheme. In contrast the IsdG-like protein scaffold has evolved to suppress the autoxidation reaction by altering the reactivity of meso-hydroxyheme. This suppression of the autoxidation step is most likely due to the extreme heme ruffling and altered electronic configuration of the meso-hydroxyheme or equivalent intermediate [53].
Despite the similarity between MhuD and IsdG/I in ring opening to an aldehyde intermediate, the enzymes have distinct regioselectivities. The diagonal modification at the β- and δ-meso carbons in the IsdG protein is due solely to the extreme heme ruffling placing the terminal oxygen of the putative Fe(III)–OOH in proximity to both the β and δ-meso-carbon. In contrast MhuD cleaves the heme at the α-meso carbon with carbonyl modification of either the adjacent β- or δ-meso carbon. The cleavage at the α-meso-carbon is most likely the result of an in-plane rotation of the heme in MhuD compared to IsdG placing the α-meso carbon of the heme at the site of activation. The alternate heme orientation is supported in the di-heme-MhuD structure where heme I is rotated 90o from that of the heme in Isdl or heme II in MhuD [31]. The carbonyl modification at either the β or δ-meso carbon most likely results from orientational disorder around the α/γ-axis.
It is unclear at the present time if the alternate regioselectivy in the MhuD and IsdG proteins plays a role in determining if the aldehyde is retained or released, respectively. Furthermore, it is not known if the initial meso-carbon hydroxylation leads to ring opening prior to the carbonyl modification or vice versa. This and many other mechanistic questions will require a more detailed analysis and characterization of intermediates along the reaction pathway. The overall stoichiometry of the reaction and the origin of the oxygen atoms in the final reaction product will provide the foundation for future kinetic and spectroscopic studies. The freeze-quench spectroscopic techniques employed in characterizing reactive intermediates in the canonical HO catalyzed reaction will be invaluable in determining the sequence of intermediates in the IsdG-like reactions.
It is important to consider the biological significance of the alternate heme degradation products in the context of bacterial pathogenesis. The products of the HO reaction, namely biliverdin and CO, have long been reported to have important antioxidant and anti-proliferative/anti-inflammatory properties in mammals [60–63]. Moreover, it is interesting to speculate that CO produced as a by-product of heme utilization may provide an advantage for some pathogens in establishing chronic infection by aiding in suppressing inflammation. It has been shown that cystic fibrosis patients with chronic infection due to P. aeruginosa exhale higher levels of CO [64]. In many of the gastro-enteric pathogens that cause acute invasive disease heme utilization may involve mechanism similar to that of S. aureus where CO as a by-product would not be beneficial. Induction of colonic HO-1 or exposure to CO in animal models of colitis increases bactericidal activity of macrophages and thus reduces intracellular invasion and bacterial load [65, 66].
The absence of CO production in the non-canonical HO’s of M. tuberculosis and S. aureus would indicate strategies for infection that do not favor CO production. In M. tuberculosis it has been shown CO can activate the dormancy regulon through the heme-dependent two-component sensor kinases DosS and DosT [67]. Thus avoiding activation of the dormancy genes in an immune-competent state is important for long-term survival of the organism. In contrast CO-releasing molecules have been shown to produce bactericidal effects in S. aureus although the effects may be related to activation of reactive oxygen species [68, 69]. Alternatively, the unique linear tetrapyrroles and/or production of formaldehyde may have as yet unidentified physiological functions in the host-pathogen interaction.
Ring opening and the conversion of verdoheme to biliverdin
The HO dependent conversion of Fe(II)-verdoheme to biliverdin until recently was the least understood step in the reaction. It was initially thought the conversion of verdoheme to biliverdin proceeded through a hydrolytic reaction with insertion of an oxygen molecule from water [70], a mechanism at odds with early 18O2/16 O2-labelling studies identifying the terminal lactam oxygens as being derived from separate O2-molecules [71–73]. However, Sano and coworkers in a series of studies showed redox dependent conversion of verdoheme to biliverdin by O2 and H2O2 [74, 75].
The ring-opening of verdoheme requires four electrons and O2: however, the mechanisms and site of O2-binding have remained elusive. Under physiological conditions where product release is not rate limiting the ring-opening of Fe(II)-verdoheme is the rate-determining step [55]. Pioneering studies by Ortiz de Montellano suggested that the resonance structure of Fe(II)-verdoheme (Fig. 8) would allow for a mechanism where O2 binding could occur through the iron or on the α-pyrrole carbon (Fig. 9a and b) [2]. In the initial structural elucidation of the Fe(II)-verdoheme hHO-1 the distal water network is absent and the verdoheme is five-coordinate [76]. Furthermore, in the Fe(II)–NO verdoheme complex the NO is bent toward the α-meso oxygen that together with the lack of a distal water network was thought to favor nucleophilic attack through a bridged oxygen (Fig. 9b) [76]. This scenario is consistent with attack of the oxygen via direct addition to the α-pyrrole carbon or through binding to the iron (Fig. 9a and b).
Fig. 8.
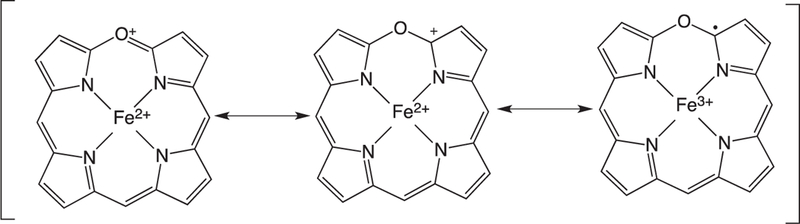
Resonance structures of Fe(II)-verdoheme.
Fig. 9.
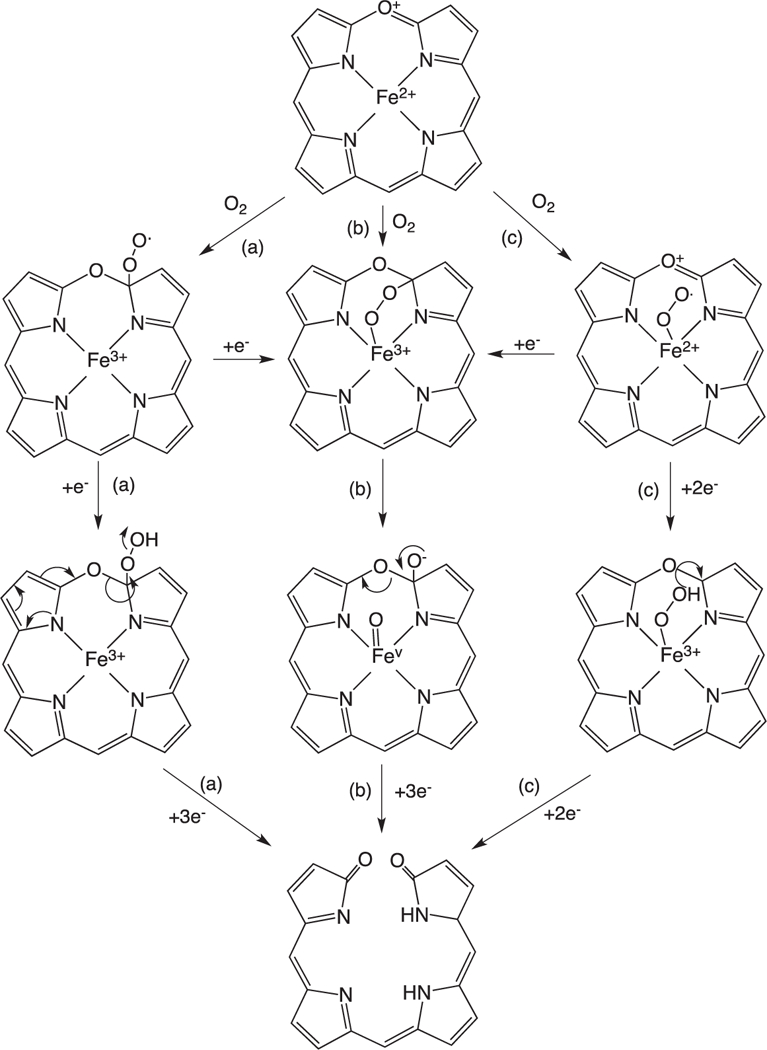
Proposed mechanisms for the conversion of Fe(II)-verdoheme to biliverdin.
In contrast Ikeda-Saito and colleagues proposed that similar to the initial heme hydroxylation step the conversion of Fe(II)-verdo-heme to biliverdin proceeds via electrophilic addition of the Fe(III)–OOH intermediate (Fig. 8c). This hypothesis is based on observation that H2O2 and alkyl-peroxides can substitute for O2 and two reducing equivalents in converting Fe(II)-verdoheme to biliverdin and alkoxy-biliverdin, respectively [77,78]. A mechanism involving nucleophilic attack through the bridged intermediate cannot be ruled out for the reaction with H2O2 as unlike the alkyl peroxides H2O2 can be deprotonated. Furthermore, it is difficult to reconcile the proposed electrophilic addition with the structure of the Fe(II)- and Fe(II)–NO verdoheme hHO-1 complexes where the distal ordered water network required for protonation of the Fe(III)–OOH intermediate is absent [76]. Further complicating the picture a 2.2 Å structure of the Fe(II)–N3 verdoheme rHO-1 complex although not seen to have azide (N3) bound revealed the retention of the ordered distal water network observed in the heme-HO complexes [79,80]. Moreover, an electron density peak above the iron was tentatively interpreted to be a water molecule or hydroxide ion. Further insight into the nature of the Fe(II)-verdoheme binding site was revealed in the 1.7 structure of the Fe(II)–N3 verdoheme HmuO complex indicating a highly bent Fe–N–N(–N) with an angle of ~110° similar to that observed for the oxy–heme HmuO complex [39,81]. The distal N atom is directed toward the α-meso position through a hydrogen-bond interaction with the distal water, similar to that of the Fe(II)–O2 heme complex. The authors suggested that similar to the initial hydroxylation step the structural data is consistent with electrophilic addition through a Fe(III)–OOH species. Quantum mechanical/ molecular mechanical (QM/MM) calculations on the Fe(II)–N3 verdoheme HmuO structure support a mechanism that proceeds through a Fe(II)–OOH with O—O bond cleavage being coupled to electron transfer from the verdoheme to the departing OH− and proton transfer to and from the water cluster, thus enabling a nucleophilic attack of OH− on the α-pyrrole carbon [81]. Furthermore, mutation of the distal Asp-140 in rHO-1 decreases the formation of verdoheme indicating that the ordered hydrogen bond network, as in the initial hydroxylation step, plays an important role in the conversion of verdoheme to biliverdin [77]. The conflicting reports surrounding the structure of the Fe(II)-verdoheme HO distal pocket are most likely due to the unstable nature of the Fe(II)-verdoheme intermediate. Although Fe(II)-verdoheme was reported to be five coordinate in the initial crystal structure previous early and recent spectroscopic studies suggest Fe(II)-verdoheme is six coordinate and low spin [59,82,83]. Although challenging many of the remaining questions regarding the mechanism of Fe(II)-verdoheme conversion to biliverdin will be answered through detailed spectroscopic analysis of the electronic structure of Fe(II)-verdoheme and its O2-reacted intermediates.
The similarities between the mechanistic aspects of the initial heme hydroxylation and the redox dependent conversion of Fe(II)-verdoheme to biliverdin represent a conservation of protein structure and function. Ikeda-Saito and colleagues have further proposed that this pathway, in contrast to the less energy consuming hydrolytic conversion of Fe(II)-verdoheme to biliverdin, may be advantageous in the physiological role of HO in O2-sensing and protection from oxidative stress [1,77]. Specifically they suggest that increased H2O2 levels during oxidative stress may be ameliorated by the action of HO producing biliverdin and subsequently bilirubin, both of which have antioxidant properties [9,84].
Concluding remarks
For decades the biological degradation of heme was thought to be restricted to oxidative cleavage of heme to biliverdin with the release of CO. The discovery and characterization of the non- canonical IsdG-like proteins is significant on many levels. The monooxygenases, peroxidases and heme oxygenases all share a similar mechanism in the activation of O2 to the Fe(III)–OOH but then diverge through an Fe(IV)=O species or reaction of the ferric hydroperoxide with the heme meso-carbon, respectively. In the canonical HOs heme hydroxylation is a combination of the decreased tendency to undergo O—O bond cleavage while simultaneously activating the heme. This is achieved by the conformational flexibilty of the heme binding site and the extensive hydrogen bond network. Where the IsdG-like proteins lie along this activation pathway is of great interest and will provide further insight into the role of structural distortion in heme reactivity. Finally, the evolution of heme oxygenation offers some intriguing possibilities for the biological significance of the HO and IsdG-like catalyzed reactions within their environmental and physiological niches.
Acknowledgments
AW would like to thank Paul R. Ortiz de Montellano for the opportunity to be there in the “early days” of the HO story and Thomas L. Poulos for his help and guidance over the years.
1. Abbreviations used:
- CoPPIX
cobalt protoporphyrin
- HO
heme oxygenase
- hHO-1
human heme oxygenase
- rHO-1
rat heme oxygenase
- HmuO
Corynebacterium diphtheriae heme oxygenase
- HemO
Pseudomonas heme oxygenase
- Isd
iron surface determinant
- Mhu
Mycobacterium heme utilization
- EPR
electron paramagnetic resonance
- NMR
nuclear magnetic resonance
- ENDOR
electron-nuclear double resonance
References
- [1].Matsui T, Unno M, Ikeda-Saito M, Acc. Chem. Res. 43 (2010) 240–247. [DOI] [PubMed] [Google Scholar]
- [2].Ortiz de Montellano PR, Curr. Opin. Chem. Biol. 4 (2000) 221–227. [DOI] [PubMed] [Google Scholar]
- [3].Tenhunen R, Marver H, Pimstone NR, Trager WF, Cooper DY, Schmid R, Biochemistry 11 (1972) 1716–1720. [DOI] [PubMed] [Google Scholar]
- [4].Yoshida T, Kikuchi G, J. Biol. Chem. 253 (1978) 4230–4236. [PubMed] [Google Scholar]
- [5].Bhoo SH, Davis SJ, Walker J, Karniol B, Vierstra RD, Nature 414 (2001) 776–779. [DOI] [PubMed] [Google Scholar]
- [6].Davis SJ, Vener AV, Vierstra RD, Science 286 (1999) 2517–2520. [DOI] [PubMed] [Google Scholar]
- [7].Frankenberg-Dinkel N, Antioxid. Redox Signal. 6 (2004) 825–834. [DOI] [PubMed] [Google Scholar]
- [8].Otterbein LE, Soares MP, Yamashita K, Bach FH, Trends Immunol. 24 (2003) 449–455. [DOI] [PubMed] [Google Scholar]
- [9].Poss KD, Tonegawa S, Proc. Natl. Acad. Sci. USA 94 (1997) 10925–10930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sedlak TW, Saleh M, Higginson DS, Paul BD, Juluri KR, Snyder SH, Proc. Natl. Acad. Sci. USA 106 (2009) 5171–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brickman TJ, Vanderpool CK, Armstrong SK, Infect. Immun. 74 (2006) 1741–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Skaar EP, Humayun M, Bae T, DeBord KL, Schneewind O, Science 305 (2004) 1626–1628. [DOI] [PubMed] [Google Scholar]
- [13].Wandersman C, Delepelaire P, Annu. Rev. Microbiol. 58 (2004) 611–647. [DOI] [PubMed] [Google Scholar]
- [14].Wilks A, Barker KD, in: Kadish KM, Smith KM, Guilard R (Eds.), Handbook of Porphyrin Science, World Scientific, Singapore, 2011, pp. 357–398. [Google Scholar]
- [15].Wilks A, Burkhard KA, Nat. Prod. Rep. 24 (2007) 511–522. [DOI] [PubMed] [Google Scholar]
- [16].Dammeyer T, Frankenberg-Dinkel N, Photochem. Photobiol. Sci. 7 (2008) 1121–1130. [DOI] [PubMed] [Google Scholar]
- [17].Montgomery BL, Lagarias JC, Trends Plant Sci. 7 (2002) 357–366. [DOI] [PubMed] [Google Scholar]
- [18].Ratliff M, Zhu W, Deshmukh R, Wilks A, Stojiljkovic I,J. Bacteriol. 183 (2001) 6394–6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wilks A, Schmitt MP, J. Biol. Chem. 273 (1998) 837–841. [DOI] [PubMed] [Google Scholar]
- [20].Zhu W, Wilks A, Stojiljkovic I, J. Bacteriol. 182 (2000) 6783–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Reniere ML, Ukpabi GN, Harry SR, Stec DF, Krull R, Wright DW, Bachmann BO, Murphy ME, Skaar EP, Mol. Microbiol. 75 (2010) 1529–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Skaar EP, Gaspar AH, Schneewind O, J. Biol. Chem. 279 (2004) 436–443. [DOI] [PubMed] [Google Scholar]
- [23].Skaar EP, Gaspar AH, Schneewind O, J. Bacteriol. 188 (2006) 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Matsui T, Nambu S, Ono Y, Goulding CW, Tsumoto K, Ikeda-Saito M, Biochemistry 52 (2013) 3025–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Reniere ML, Haley KP, Skaar EP, Biochemistry 50 (2011) 6730–6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nambu S, Matsui T, Goulding CW, Takahashi S, Ikeda-Saito M, J. Biol. Chem. 288 (2013) 10101–10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Loutet SA, Kobylarz MJ, Chau CH, Murphy ME, J. Biol. Chem. 288 (2013) 25749–25759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hirotsu S, Chu GC, Unno M, Lee DS, Yoshida T, Park SY, Shiro Y, Ikeda- Saito M, J. Biol. Chem. 279 (2004) (1947) 11937–11947. [DOI] [PubMed] [Google Scholar]
- [29].Friedman J, Lad L, Li H, Wilks A, Poulos TL, Biochemistry 43 (2004) 5239–5245. [DOI] [PubMed] [Google Scholar]
- [30].Schuller DJ, Wilks A, Ortiz de Montellano PR, Poulos TL, Nat. Struct. Biol. 6 (1999) 860–867. [DOI] [PubMed] [Google Scholar]
- [31].Chim N, Iniguez A, Nguyen TQ, Goulding CW, J. Mol. Biol. 395 (2010) 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee WC, Reniere ML, Skaar EP, Murphy ME, J. Biol. Chem. 283 (2008) 30957–30963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yoshida T, Noguchi M, Kikuchi G, J. Biol. Chem. 255 (1980) 4418–4420. [PubMed] [Google Scholar]
- [34].Wilks A, Ortiz de Montellano PR, J. Biol. Chem. 268 (1993) 22357–22362. [PubMed] [Google Scholar]
- [35].Wilks A, Torpey J, Ortiz de Montellano PR, J. Biol. Chem. 269 (1994) 29553–29556. [PubMed] [Google Scholar]
- [36].Davydov RM, Yoshida T, Ikeda-Saito M, Hoffman BM, J. Am. Chem. Soc. 121 (1999) 10656–10657. [Google Scholar]
- [37].Garcia-Serres R, Davydov RM, Matsui T, Ikeda-Saito M, Hoffman BM, Huynh BH, J. Am. Chem. Soc. 129 (2007) 1402–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Schuller DJ, Zhu W, Stojiljkovic I, Wilks A, Poulos TL, Biochemistry 40 (2001)11552–11558. [DOI] [PubMed] [Google Scholar]
- [39].Unno M, Matsui T, Chu GC, Couture M, Yoshida T, Rousseau DL, Olson JS, Ikeda-Saito M, J. Biol. Chem. 279 (2004) 21055–21061. [DOI] [PubMed] [Google Scholar]
- [40].Takahashi S, Ishikawa K, Takeuchi E, Ikeda-Saito M, Yoshida T, Rousseau DL, J. Am. Chem. Soc. 117 (1995) 6002–6006. [Google Scholar]
- [41].Fujii H, Zhang X, Tomita T, Ikeda-Saito M, Yoshida T, J. Am. Chem. Soc. 123 (2001)6475–6484. [DOI] [PubMed] [Google Scholar]
- [42].Lightning LK, Huang H, Moenne-Loccoz P, Loehr TM, Schuller DJ, Poulos TL, de Montellano PR, J. Biol. Chem. 276 (2001) 10612–10619. [DOI] [PubMed] [Google Scholar]
- [43].Li Y, Syvitski RT, Auclair K, Ortiz de Montellano P, La Mar GN,J. Am. Chem. Soc. 125 (2003) 13392–13403. [DOI] [PubMed] [Google Scholar]
- [44].Syvitski RT, Li Y, Auclair K, Ortiz De Montellano PR, La Mar GN, J. Am. Chem. Soc. 124 (2002) 14296–14297. [DOI] [PubMed] [Google Scholar]
- [45].Matsui T, Furukawa M, Unno M, Tomita T, Ikeda-Saito M, J. Biol. Chem. 280 (2004) 2981–2989. [DOI] [PubMed] [Google Scholar]
- [46].Caignan GA, Deshmukh R, Zeng Y, Wilks A, Bunce RA, Rivera M, J. Am. Chem. Soc. 125 (2003) (1852) 11842–11852. [DOI] [PubMed] [Google Scholar]
- [47].Rivera M, Caignan GA, Astashkin AV, Raitsimring AM, Shokhireva T, Walker FA, J. Am. Chem. Soc. 124 (2002) 6077–6089. [DOI] [PubMed] [Google Scholar]
- [48].Walker F, Coord. Chem. Rev. 185–186 (1999) 471–534. [Google Scholar]
- [49].Ghosh A, Gonzalez E, Vangberg T, J. Am. Chem. Soc. 103 (1999) 1363–1367. [Google Scholar]
- [50].Ogura H, Evans JP, Peng D, Satterlee JD, Ortiz de Montellano PR, La Mar GN, Biochemistry 48 (2009) 3127–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zeng Y, Caignan GA, Bunce RA, Rodriguez JC, Wilks A, Rivera M, J. Am. Chem. Soc. 127 (2005) 9794–9807. [DOI] [PubMed] [Google Scholar]
- [52].Chen H, Moreau Y, Derat E, Shaik S, J. Am. Chem. Soc. 130 (2008) 1953–1965. [DOI] [PubMed] [Google Scholar]
- [53].Takayama SJ, Ukpabi G, Murphy ME, Mauk AG, Proc. Natl. Acad. Sci. USA 108 (2011) 13071–13076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Liu Y, Moenne-Loccoz P, Loehr TM, Ortiz de Montellano PR, J. Biol. Chem. 272 (1997)6909–6917. [DOI] [PubMed] [Google Scholar]
- [55].Liu Y, Ortiz de Montellano PR, J. Biol. Chem. 275 (2000) 5297–5307. [DOI] [PubMed] [Google Scholar]
- [56].Ortiz de Montellano PR, Wilks A, Adv. Inorg. Chem. 51 (2000) 359–402. [Google Scholar]
- [57].Matera KM, Takahashi S, Fujii H, Zhou H, Ishikawa K, Yoshimura T, Rousseau DL, Yoshida T, Ikeda-Saito M, J. Biol. Chem. 271 (1996) 6618–6624. [DOI] [PubMed] [Google Scholar]
- [58].Migita CT, Fujii H, Mansfield Matera K, Takahashi S, zhou H, Yoshida T, Biochim. Biophys. Acta 1432 (1999) 203–213. [DOI] [PubMed] [Google Scholar]
- [59].Sakamoto H, Takahashi K, Higashimoto Y, Harada S, Palmer G, Noguchi M, Biochem. Biophys. Res. Commun. 338 (2005) 578–583. [DOI] [PubMed] [Google Scholar]
- [60].Kim HP, Ryter SW, Choi AM, Annu. Rev. Pharmacol. Toxicol. 46 (2006) 411–449. [DOI] [PubMed] [Google Scholar]
- [61].Rochette L, Cottin Y, Zeller M, Vergely C, Pharmacol. Therapeutics 137 (2013) 133–152. [DOI] [PubMed] [Google Scholar]
- [62].Snyder SH, Baranano DE, Neuropsychopharmacology 25 (2001) 294–298. [DOI] [PubMed] [Google Scholar]
- [63].Wegiel B, Hanto DW, Otterbein LE, Trends Mol. Med. 19 (2013) 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Antuni JD, Kharitonov SA, Hughes D, Hodson ME, Barnes PJ, Thorax 55 (2000)138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Naito Y, Uchiyama K, Takagi T, Yoshikawa T, Curr. Med. Chem. 19 (2012) 70–76. [DOI] [PubMed] [Google Scholar]
- [66].Onyiah JC, Sheikh SZ, Maharshak N, Steinbach EC, Russo SM, Kobayashi T, Mackey LC, Hansen JJ, Moeser AJ, Rawls JF, Borst LB, Otterbein LE, Plevy SE, Gastroenterology 144 (2013) 789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kumar A, Deshane JS, Crossman DK, Bolisetty S, Yan BS, Kramnik I, Agarwal A, Steyn AJ, J. Biol. Chem. 283 (2008) 18032–18039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Tavares AF, Nobre LS, Saraiva LM, FEMS Microbiol. Lett. 336 (2012) 1–10. [DOI] [PubMed] [Google Scholar]
- [69].Nobre LS, Seixas JD, Romao CC, Saraiva LM, Antimicrob. Agents Chemotherap. 51 (2007) 4303–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Jackson AH, Iron in Biochemistry and Medicine, Academic, New York, 1974. [Google Scholar]
- [71].Docherty JC, Firneisz GD, Schacter BA, Arch. Biochem. Biophys. 235 (1984) 657–664. [DOI] [PubMed] [Google Scholar]
- [72].Docherty JC, Schacter BA, Firneisz GD, Brown SB, J. Biol. Chem. 259 (1984) 13066–13069. [PubMed] [Google Scholar]
- [73].King RF, Brown SB, Biochem. J. 174 (1978) 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Saito S, Itano HA, Proc. Natl. Acad. Sci. USA 79 (1982) 1393–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Sano S, Sano T, Morishima I, Shiro Y, Maeda Y, Proc. Natl. Acad. Sci. USA 83 (1986) 531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Lad L, Ortiz de Montellano PR, Poulos TL, J. Inorg. Biochem. 98 (2004) 1686–1695. [DOI] [PubMed] [Google Scholar]
- [77].Matsui T, Nakajima A, Fujii H, Matera KM, Migita CT, Yoshida T, Ikeda- Saito M, J. Biol. Chem. 280 (2005) 36833–36840. [DOI] [PubMed] [Google Scholar]
- [78].Matsui T, Omori K, Jin H, Ikeda-Saito M, J. Am. Chem. Soc. 130 (2008) 4220–4221. [DOI] [PubMed] [Google Scholar]
- [79].Sato H, Sugishima M, Sakamoto H, Higashimoto Y, Shimokawa C, Fukuyama K, Palmer G, Noguchi M, Biochem. J. 419 (2009) 339–345. [DOI] [PubMed] [Google Scholar]
- [80].Unno M, Matsui T, Ikeda-Saito M, J. Inorg. Biochem. 113 (2012) 102–109. [DOI] [PubMed] [Google Scholar]
- [81].Lai W, Chen H, Matsui T, Omori K, Unno M, Ikeda-Saito M, Shaik S, J. Am. Chem. Soc. 132 (2010) 12960–12970. [DOI] [PubMed] [Google Scholar]
- [82].Damaso CO, Bunce RA, Barybin MV, Wilks A, Rivera M, J. Am. Chem. Soc. 127 (2005) 17582–17583. [DOI] [PubMed] [Google Scholar]
- [83].Takahashi S, Matera KM, Fujii H, Zhou H, Ishikawa K, Yoshida T, Ikeda- Saito M, Rousseau DL, Biochemistry 36 (1997) 1402–1410. [DOI] [PubMed] [Google Scholar]
- [84].Baranano DE, Rao M, Ferris CD, Snyder SH, Proc. Natl. Acad. Sci. USA 99 (2002) 16093–16098. [DOI] [PMC free article] [PubMed] [Google Scholar]