

Abstract
FANCA is a key player in the canonical Fanconi anemia (FA) repair pathway. We have recently shown that FANCA also plays an important role in the single-strand annealing sub-pathway (SSA) of DNA double-strand break (DSB) repair by biochemically catalyzing single-strand annealing. Here, we report that a steroidal lactone withaferin A (WA) specifically impedes SSA repair by promoting FANCA downregulation at a sub-micromolar concentration range. We find that WA causes FANCA downregulation post-translationally in a proteasome-dependent manner. This WA-mediated downregulation is achieved through HSP90 inhibition and disruption of the FANCA-HSP90 interaction. WA-mediated FANCA degradation significantly reduces cellular SSA repair, abolishes FANCD2 monoubiquitination, elevates sensitivity to mitomycin C, and results in accumulation of DSBs. Importantly, the WA-induced defect in SSA repair is highly dependent on the absence of FANCA protein and overexpression of exogenous WT-FANCA protein in WA-treated cells significantly complements the repair defect.
1. Introduction
DSBs are highly cytotoxic DNA lesions, which can lead to cell death or mutagenic consequences that drive genome instability and tumorigenesis [1]. Indeed, disruption of many DNA DSB repair genes predispose to breast cancer, including mutations in BRCA1 and BRCA2. Depending on cell cycle phases and availability of sequence homology, DSBs are repaired predominantly by four distinct pathways: 1) Homologous recombination (HR), 2) Single strand annealing (SSA), 3) Microhomology-mediated end joining (MMEJ, alternative end-joining Alt-EJ), or 4) Non-homologous end joining (NHEJ). While HR is error free, SSA, MMEJ, and NHEJ are highly error-prone pathways that are responsible for genome instability in cells [2–9].
The Fanconi anemia (FA) pathway of DNA repair is specialized in repairing DNA interstrand crosslinks (ICLs). It is composed of at least 22 FANC proteins, of which deficiency in any causes hypersensitivity to crosslinking agents, chromosomal instability, and predisposition to cancer [10, 11]. FANCA is one of the FA core complex proteins [12, 13] and the most commonly affected complementation group in FA patients, accounting for ~64% of all mutations [14]. Outside of the canonical FA pathway, evidence has emerged that supports FA proteins’ role in repairing DSBs through the HR and SSA sub-pathways [15–17]. Our previous work showed that FANCA promotes the SSA sub-pathway of DNA DSB repair by biochemically catalyzing single-strand annealing [18].
Withaferin A (WA) is a steroidal lactone isolated from winter cherry (Withania somnifera) that possesses pharmacological activities for cancer treatments [19, 20]. Its biological activity has been studied in various cancer models where elevated ER stress, reactive oxygen species, cell cycle arrest, and apoptosis were observed (reviewed in [21]). These pleiotropic effects of WA may be attributed to its covalent interaction with chaperone protein HSP90 and Vimentin [22, 23]. HSP90 chaperones folding, maturation and stabilization of a number of client proteins primarily in cellular signal transduction pathways. A minor subset of HSP90 clients includes DNA damage response and DNA repair factors such as ATR, BRCA2, and FANCA [24, 25].
In this report, we evaluate the impact of withaferin A on DNA repair and demonstrate that withaferin A specifically impedes the SSA sub-pathway of DNA DSB repair through degrading FANCA.
2. Methods
2.1. Cell culture
The effects of WA were tested on several established breast cancer cell lines, including MDA-MB-231, SUM-149, MCF-7, and an immortalized breast epithelia cell line MCF-10A. The four U2OS cell lines carrying MMEJ, NHEJ, FIR, and SSA reporter constructs were generous gifts from Dr. Jeremy Stark at the City of Hope Medical Center [26]. SUM-149 cells were cultured in DMEM/F-12 medium supplemented with 10% FBS, 5 μg/mL insulin, 1 μg/mL hydrocortisone and L-glutamine. MCF-10A cells were maintained in DMEM/F-12 medium supplemented with 5% horse serum, 20 ng/mL EGF, 10 μg/mL insulin, 100 ng/mL cholera toxin, 0.5 μg/mL hydrocortisone and L-glutamine. All others were grown in DMEM medium supplemented with 10% FBS and L-glutamine with the exception of MCF-7, which additionally receives 10 ng/mL EGF, 10 μg/mL insulin and 0.5 pg/mL hydrocortisone. MDA-MB-231 cells stably expressing FANCA was generated by transfection of plvx-ires-FANCA [18] and maintained with 400 pg/mL neomycin. Proliferation analysis of cells in response to WA and mitomycin C or WA alone was carried out in 96-well plates with Alamar blue after 48 hours of drug exposure. Knockout of FANCA was carried out as previously described [18] and verified by Western blot.
2.2. Cell-based DSB repair assay
I-Scel based fluorescent reporter assay were carried out as previously described with some modifications [26]. U2OS cells were seeded in 6-well plates at a density of 200K/well. At the following day, cells were subjected to transfection of 1 μg per well of I-Scel plasmid by using lipofectamine 2000 to initiate double-strand break production. WA at indicated concentrations was added 3 hours after adding transfection mixture. 48 hours after transfection, cells were washed once with PBS, trypsinized with 300 μL trypsin, and neutralized with 400 μL media. Cell suspension was processed immediately for FACS analysis. PE channel was used to assist the exclusion of auto-fluorescence. Complementation of SSA repair defect induced by WA is done by co-transfection of 1 pg per well of plvx-ires-FANCA or its corresponding vector plvx-ires-neo.
2.3. FANCA protein and in vitro biochemical assay
cDNAs for FANCA were obtained from Dr. Weidong Wang at the National Institute on Aging, NIH. The FANCA gene was cloned into pFastBac1 vectors and subsequently sequenced. Suspected mutations were screened against the human single nucleotide polymorphism (SNP) collection at NCBI (http://www.ncbi.nlm.nih.gov/sites/entrez). True mutations were corrected by PCR-mediated site-specific mutagenesis and verified by resequencing. Baculoviruses were subsequently prepared according to the manufacturer’s protocol (Invitrogen). Purification of FANCA was carried out as described previously [27]. In brief, upon expression of the recombinant FANCA proteins in insect cells, the cells were homogenized using a Dounce homogenizer to prepare extracts. FANCA were purified by using HiTrap Q Sepharose Fast Flow, 5-mL HiTrap Blue, Mono S, Mono Q, and/or Superdex 200 gel filtration columns (GE Flealthcare, Piscataway, NJ), and/or a 2-mL high-resolution hydroxylapatite column (Calbiochem, La Jolla, CA) and by tracing FANCA protein through SDS-PAGE and Western blot.
DNA binding EMSA analysis was performed as described previously [27] in a 10 μl reaction containing 25 mM Tris-HCI pH 7.5, 100 mM NaCI, 5 mM EDTA,1 mM DTT, 6% glycerol, 1 nM 5’-32P-labeled oligonucleotide substrate A1, 260 ng FANCA protein and indicated amount of WA. The reactions were incubated at room temperature for 45 min, followed by the addition of 4 μl of 50% (w/v) sucrose buffered by 10 mM Tris-HCI pH 7.5. The reaction mixtures were resolved by electrophoresis through a 4% non-denaturing polyacrylamide gel in 40 mM Tris acetate (pH 7.6) and 10 mM EDTA with 6% glycerol at 100 V (~1.5 watts/gel) for 40 min. DNA substrates and shifted bands were visualized by autoradiography.
Assessment of strand annealing activities was carried out as previously described [18]. In brief, a total of 0.5 nM 5’-32P-labeled DNA substrate (annealed A1/A2) and 260 ng FANCA protein were incubated in a 10 μl reaction of 25 mM Tris-HCI pH8.0, 100 mM NaCI, 1 mM EDTA with presence of indicated amount of WA. The reaction mixture was incubated at room temperature for 40 min and stopped with 1 μl of 10x stop solution (200 mM EDTA, 32% Glycerol, 1% SDS, 0.024% Bromophenol Blue), 3 pg proteinase K, and 10 min incubation at room temperature. Products were separated on a 6% native PAGE gel at 100V for 1.5 hr. Substrate and product bands were visualized by autoradiography.
2.4. Immunoblot, immunoprecipitation, and immunofluorescence staining
Primary antibodies used in this study include: FANCA (Bethyl, A301-980A), RAD52 (Novus, NBP1-19429), FAAP20 (Invitrogen, PA5-58555), Actin (Santa Cruz, sc-47778), HSP90 (Santa Cruz, Sc-515081), FANCD2 (kind gift from Dr. Weidong Wang, NIH), GAPDH (Cell signaling, 8884), and γH2AX (Cell signaling, 9718).
Cell lysis buffer is comprised of 50 mM Potassium Phosphate buffer PH 8.0, 15% glycerol, 500 mM NaCI, 5 mM β-Mercaptoethanol, 0.5% NP-40, 2mM DTT, and supplemented with benzonase, protease inhibitors and orthovanadate. Protein concentration was measured with Coomassie Plus reagent (Pierce, 23236). For western blots, protein samples were resolved in 7% (for visualizing FANCD2 monoubiquitination) or 10% SDS-PAGE and transferred onto 0.45 μm nitrocellulose membrane. Antibodies were diluted in 5% milk supplemented 1X PBST (0.1% tween-20) with working concentration determined according to manufacturers’ recommendation for western blots. For immunoprecipitation, 1 mL of cells lysate in mild lysis buffer (20 mM Tris, 137 mM NaCI, 5 mM EDTA, 1% Triton X-100, 10% glycerol, protease inhibitors and orthovanadate) with total protein concentration of 1 mg/mL was incubated with 30 μL protein A/G agarose resin and 3 μL of Hsp90 antibody in RT for 2 hours. Ice-cold lysis buffer supplemented with PMSF was used three times for washes. At last, resin was boiled in 35 μL of 1X SDS loading buffer and loaded onto SDS-PAGE for immunoblotting.
For immunofluorescence staining, cells were grown on 8-well culture slide (Corning, 354656) and received WA of indicated concentration for 48 hours. Cells were fixed in 4% buffered formaldehyde for 15 minutes at room temperature, permeabilized for 5 min in PBS supplemented with 0.2% Triton X-100, and blocked in DMEM containing 10% FBS. FANCA and yH2AX antibodies were used at dilutions of 1:100 and 1:150 in DMEM containing 10% FBS. After 2 hours of primary antibody incubation, cells were washed three times with PBST (PBS containing 0.2% Tween). Fluorescence-conjugated secondary antibody incubations were performed at room temperature for 1 hour. Cells received two PBST washes, PBS containing DAPI (0.5 μg/mL) for 10 minutes, and one final wash with PBS. After removal of chamber module, slides were applied with proLong gold antifade mountant (Life technologies, P36934), and covered with cover glasses for confocal imaging.
2.5. RT-PCR
To investigate transcriptional changes of FANCA levels, MDA-MB-231 was first treated with 1.6 μM WA for indicated times. Five million cells were used for total RNA preps with the RNeasy mini kit from Qiagen (74104). RNA prep quality was examined on an agarose gel. cDNA synthesis was carried out by using reverse transcription kit from Promega (A3500) following manufacturer’s protocol. Sybr green (applied Biosystems cybrgreen 4367659) mixes of cDNA and primers were subject to quantitative PCR analysis in a Bio-Rad CFX system. The 2_ΔΔα method was employed to calculate relative fold changes of FANCA in WA treated cells. Primers used include FANCA-fwd 5’-GCGTGTACCATTCTTGTCAAC, FANCA-rev 5’-GCTACCATCTCCTGCAATCTG, 18sRNA-fwd 5’-GTAACCCGTTGAACCCCATT, and 18sRNA-rev 5’-CCATCCAATCGGTAGTAGCG.
2.6. Quantification and statistical analysis
ImageJ was used for quantification for Western blot. FlowJo was used for flow cytometry analysis. Microsoft excel software was used to perform all statistical analyses. Statistical differences between groups were determined by two-tailed Student t-test based on at least 3 repeats. Significance is denoted as * for p<0.05, and ** for p<0.01.
3. Results
3.1. WA impedes the SSA sub-pathway of DNA DSB repair in cells
HSP90 regulates DSB repair proteins [24, 25] and WA exerts its effect through HSP90 [22]. However, it is not established whether WA directly affects DSB repair in cells. To assess the impact of WA on DSB repair, we employed the l-Scel based GFP reporter assay in U2OS cells, which can monitor repair products from all four unique DSB repair sub-pathways [26]. As shown in Fig. 1A and 1B, WA treatment of U2OS cells causes significant reduction only in SSA repair, but not the other three sub-pathways. At 0.3 μM, WA reduces SSA repair by 40% (Fig. 1A). This result resembles our early findings with FANCA depleted cells [18]. As WA may cause changes in cell proliferation [21]. which in turn would affect the choice of repair pathways, we determined the cell proliferation profile and showed that the specified WA concentrations and treatment conditions do not alter cell proliferation or cause apparent cytotoxicity (Fig. 1C). Thus, the specific reduction of SSA repair is not due to altered U2OS cell proliferation and cytotoxicity caused by WA treatment.
Figure 1. WA specifically inhibits the SSA sub-pathway of DNA DSB repair.
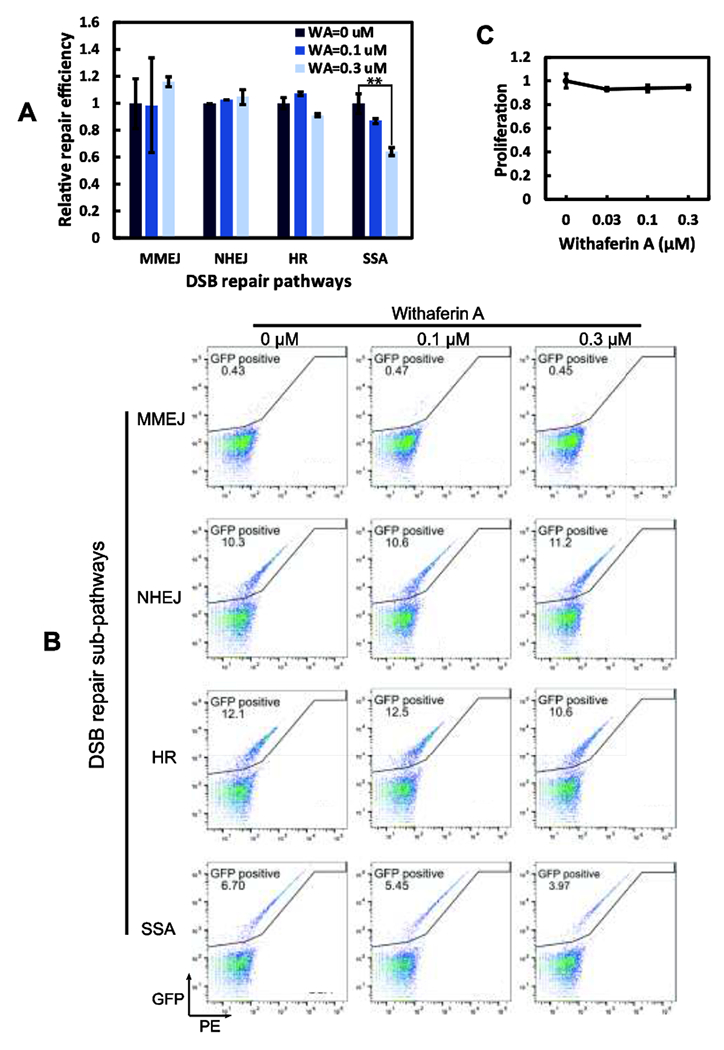
(A) I-Scel based GFP reporter assay was employed to evaluate the impact of WA on DSB repair. 0.3 μM WA significantly reduces SSA efficiency but not the other three pathways. p<0.01. (B) Representative FACS illustrations for WA’s effects on MMEJ, NHEJ, FIR, and SSA. Percentage of cells with successfully SSA repair (GFP positive) drops with WA administration. (C) U2OS maintains normal proliferation under the same repair conditions.
3.2. Reduction of FANCA protein is responsible for the WA-induced SSA repair defect
RAD52 and FANCA are the major catalytic factors in the SSA sub-pathway of DNA DSB repair [18, 28]. To test whether the WA-mediated SSA defect is due to changes in RAD52 and/or FANCA, we examined the protein levels of RAD52 and FANCA under the same low WA treatment conditions in the same U2OS cells (Fig. 2A). Intriguingly, 0.3 μM of WA treatment for 48h, which causes a defect in SSA repair (Fig. 1A), dramatically reduces the protein level of FANCA, but not that of RAD52 (Fig. 2A). Importantly, the reduction of FANCA protein is not due to changes in FAAP20 protein (Fig. 2A), a well-established factor that stabilizes FANCA [29, 30].
Figure 2. WA downregulates FANCA protein and causes defective FANCD2 monoubiquitination and elevated sensitivity to mitomycin C.

(A) Chronic treatments of U2OS cells with 0.1 and 0.3 μM WA downregulate FANCA but not RAD52 level. (B) WA treatment at a higher concentration (1.6 μM) causes quick reduction of FANCA protein. (C) WA treatments have no further impact on the SSA sub-pathway when FANCA is absent in a FANCA knockout line of U2OS. (D) Complementation of WA-treated U2OS cells with FANCA-WT (FANCA cmp) rescues SSA repair. Top panel, quantitative SSA repair, **p<0.01. Bottom panel, Western blot of FANCA protein levels. (E) WA-induced FANCA downregulation abrogates both spontaneous and MMC induced FANCD2 monoubiquitination. U2OS cells were treated with 1 μM of WA for 24 hours and receive 1.5 μM of MMC at the later 8 hours course before harvest for western blots. (F) WA treatment (0.3 μM) elevates cellular sensitivity to a DNA crosslinking agent mitomycin C, indicating that WA causes reduction of ICL repair.
When cells were challenged with a moderately higher concentration of WA (1.6 μM) within a short time course, FANCA protein was again dramatically downregulated, whereas no impact was observed for RAD52 (Fig. 2B, lanes 4-5). These results indicate that the reduction of FANCA protein levels is likely responsible for the WA-mediated SSA repair defect. To further test whether the WA-induced reduction of SSA repair is caused by factors other than FANCA, we carried out a WA-SSA repair assay using FANCA knockout U2OS cells that harbor the SSA-GFP reporter [18]. As shown in Fig. 2C, 0.1-0.3 μM WA treatment of FANCA knockout cells does not cause any additional defect in SSA repair, strongly suggesting that degradation of FANCA, but not other factors, is responsible for the WA-mediated SSA repair defect. Furthermore, complementation of WA-treated U2OS cells with an ectopically overexpressed FANCA protein significantly rescues the SSA repair defect (Fig. 2D, compare lane 2 with 4).
3.3. WA-mediated FANCA protein reduction compromises FANCD2 monoubiquitination, elevates sensitivity to ICL, and results in accumulation of DSBs
Besides its catalytic role in SSA, FANCA also participates in the canonical FA pathway of crosslink repair and in the SSA repair mediated by the FA pathway [15–17]. Therefore, it is likely that reduction of FANCA protein compromises FANCD2 monoubiquitination and ICL repair. Indeed, a WA treatment condition that depletes FANCA from U2OS cells abolishes the monoubiquitination of FANCD2 in the presence or absence of a DNA crosslinking agent (MMC) (Fig. 2E, compare lanes 2 and 4 with lanes 1 and 3 respectively). Consistent with compromised FANCD2 monoubiquitination, U2OS cells show elevated cellular sensitivity to a DNA crosslinking agent, mitomycin C (Fig. 2F). Thus, WA-induced reduction of FANCA downregulates SSA repair (Fig. 1) and inactivates the canonical FA pathway. Consequently, 0.3-μM WA treatment of U2OS cells causes dramatic increase of yFI2AX, an indicator of cellular DSBs [31, 32], when assessed by Western blot (Fig. 3A). We next evaluated U2OS cells exposed to 0.3 μM WA with immunofluorescent staining. Consistent with our model of FANCA-mediated activity of WA, elevated accumulation of μH2AX was observed in 0.3 μM WA treated cells while FANCA levels decreased (Fig. 3B). Quantification of fluorescence intensity suggests that this elevation is statistically significant (Fig. 3C).
Figure 3. WA treatment results in accumulation of DSBs.
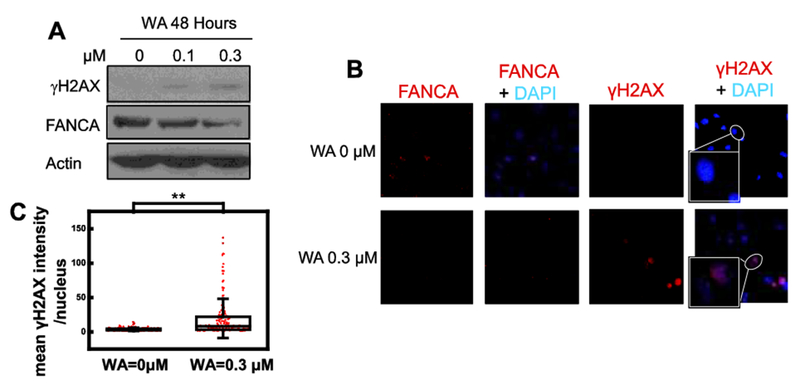
(A) DSB marker γH2AX increases along with WA treatments. FANCA is a replica of the corresponding gel in Fig. 2A. (B and C) Confocal microscopy analysis of immunofluorescence staining reveals elevated γH2AX in 0.3 μM WA treated nucleus (B) with statistical significance (C, ** p<0.01). Intensity of γH2AX fluorescence is quantified by using imageJ with DAPI masks.
3.4. WA-mediated reduction of FANCA is a general mechanism across different cell lines
To test whether WA induces reduction of FANCA protein in other cells, we treated MDA-MB-231, SUM-149, MCF-7, MCF-10A, and U2OS cells with WA (Fig. 4A). MDA-MB-231 and SUM-149 are triple-negative breast cancer (TNBC) lines; MCF-7 is an ER- and PR-positive breast cancer cell line; MCF-10A is a non-tumorigenic epithelial cell line; and U2OS is an osteosarcoma cell line that is the platform of our DSB repair reporter assays. As shown in Fig. 4A, the WA-mediated reduction of FANCA protein is conserved in all cells in an acute response to WA concentration, suggesting a general mechanism. Intriguingly, the basal level of FANCA protein varies in different cell lines. MDA-MB-231 has the highest and MCF-10A has the lowest amount of FANCA protein.
Figure 4. WA-mediated reduction of FANCA is a general mechanism across different cell lines.
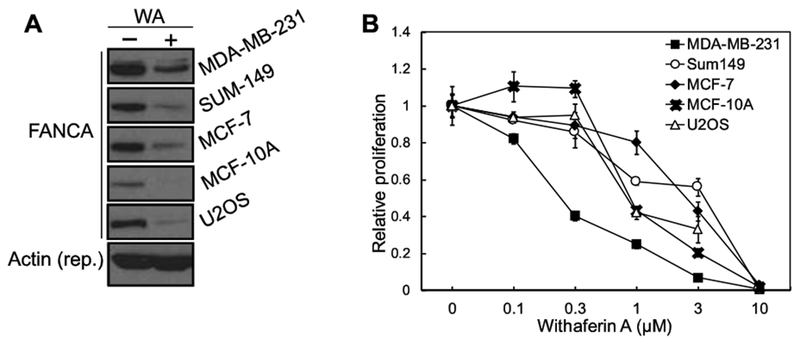
(A) Multiple cell lines with various levels of FANCA protein show downregulated FANCA level in response to WA treatment. All panels were from the same gel with same loading and exposure settings. (B) The TNBC line MDA-MB-231 with highest FANCA expression exhibits higher sensitivity among all tested cell lines to WA treatment.
A comparison of cell proliferation in response to WA treatment indicates that MDA-MB-231 , which has the highest amount of WT-FANCA protein expression (Fig. 4A), is the most sensitive cell line (Fig. 4B). At 0.37 μM, WA inhibits proliferation of MDA-MB-231 by ~60%. However, other cell lines are relatively refractory to this concentration of WA treatment (Fig. 4B). Intriguingly, overexpression of FANCA, similar to the naturally high FANCA expression in cells, also sensitizes MDA-MB-231 cells to WA treatment (Supplementary Fig. 1A). These data suggest that FANCA degradation mediated by WA is a general mechanism in cells and cellular FANCA protein level is likely associated with sensitivity to WA.
3.5. WA-mediated FANCA protein reduction is dependent on HSP90 and the proteasome mechanism
Reduction of protein levels in cells can be caused by downregulated gene expression and/or upregulated protein degradation. A qPCR test indicates that, instead of decreased expression, transcription of FANCA is upregulated in response to WA treatment (Fig. 5A), strongly suggesting that WA-mediated reduction of FANCA is not due to suppressed gene expression. A cycloheximide chase experiment reveals that FANCA has a long half-life of beyond 4 hours in MDA-MB-231 cells (Fig. 5B), suggesting that the decrease in FANCA level (Figs. 2 and 4) is likely caused by upregulated degradation. Indeed, temporary disturbance of proteasome function by MG132 is able to rescue WA-induced reduction of FANCA protein level (Fig. 5C), suggesting that the proteasome pathway is responsible for FANCA clearance.
Figure 5. WA downregulates FANCA post-translationally through HSP90 inhibition and in a proteasome-dependent manner.

(A) RT-PCR reveals that transcription of FANCA is elevated with 1.6 μM of WA treatment. (B) Normal half-life of FANCA with 5 μg/mL cycloheximide (CHX). (C) WA-mediated FANCA destabilization (at the indicated concentration for 4 hours) is blocked with proteasomal inhibition by MG132 (10 μM, starts 4 hours prior to WA addition). (D) WA impedes the interaction between FANCA and FISP90. Another FISP90 inhibitor AUY922 (at 0.1 μM) also disrupts the interaction in a time-dependent manner. (E) Combinatorial use of WA (1.6 μM) and AUY922 (0.1 μM) for 1.5 hour results in additive destabilization.
As FANCA has been identified as a client of FISP90 [24], and WA impacts FISP90 activity through covalent modification [22], we reasoned that WA may destabilize FANCA through HSP90 targeting. In a co-immunoprecipitation assay using anti-HSP90 antibody, FANCA physically interacts with HSP90 (Fig. 5D, lane 2). Acute treatments of MDA-MB-231 cells with WA disrupts the FANCA-HSP90 interaction (Fig. 5D, compare FANCA in lanes 3-4 with that in lane 2). AUY922, a well-established HSP90 inhibitor, also causes dramatic reduction of FANCA-HSP90 interaction (Fig. 5D, compare FANCA in lanes 5-6 with that in lane 2). The combination of WA and AUY922 leads to additive reduction of FANCA when compared with single treatments (Fig. 5E).
It has been known that WA affects functions of Vimentin, C/EBPβ, and C377 through direct modification [33–36]. To test whether WA directly affects FANCA functions, we measured the DNA binding and single-strand annealing activities of purified human FANCA protein in vitro, in the presence or absence of WA [18, 27]. The results indicate that WA does not directly affect the biochemical activities of FANCA (Supplementary Figs. 1B and 1C).
4. Discussion
Recently, we reported that, outside of its canonical role in the FA pathway of DNA crosslink repair, FANCA promotes SSA repair of DSBs by annealing single-stranded DNA [18]. While WA can impact cells on many levels [22, 23], our study demonstrates that WA, within the tested concentration range, directly affects the choice of DSB repair pathways through the depletion of FANCA. With our cellular reporter assay for DSB repair in U2OS cells, WA reduces the SSA repair efficiency at a concentration of 0.3 μM (Fig. 1A) while cellular proliferation is not significantly impacted (Fig. 1C). However, at the same concentration, WA impacts defective proliferation of MDA-MB-231 (Fig. 4B), which has higher constitutive FANCA protein levels (Fig. 4A), supporting the FANCA dependency as one of the modes of WA action.
The ability to survive toxic genomic lesions, such as DSBs and ICLs, is a necessary trait for both normal and cancerous cells. The DNA damage load that is continuously placed on cells has served as a selective pressure for the evolution of a robust repair network that consists of multiple sub-pathways. To promote survival and prevent genomic instability, the Homologous Recombination (HR) pathway is highly active during DNA replication and restores the proper DNA sequence at a DSB break site through use of the sister chromatid as a repair template. Therefore, cancers bearing an HR deficient phenotype must become reliant on alternative repair pathways during S/G2 cell cycle phases in order to survive and continuously divide [37]. These alternative repair pathways can also serve as a mechanism for chemo/radiotherapy resistance by increasing the DNA damage load tolerated by cancer cells. The Single Strand Annealing (SSA) pathway is a mutagenic, alternative DSB repair pathway that is able to compensate for HR during S/G2 phases [38]. SSA functions through an extensive end-resection step that reveals homologous sequences on each strand [39]. The central step of SSA repair occurs through the catalytic annealing of these homologous sequences, making the success of this pathway highly contingent on the functions of the annealing proteins RAD52 or FANCA [18, 40]. Therefore, targeting factors that promote cancer survival and chemotherapy resistance through the SSA pathway could serve as a powerful approach for sensitizing tumors to DNA-damaging agents while reducing the collateral damage to non-cancerous tissue. The therapeutic potential of weakening SSA repair has been previously recognized, and has led to the development of RAD52 inhibitors [41 ]. However, no current strategy exists to block the participation of FANCA in SSA, which we have previously shown to be responsible for residual levels of SSA when RAD52 is depleted [18]. Consequently, all potential mediators of this pathway must be suppressed in order to ensure effective killing of SSA-dependent cancer cells.
High FANCA expression might be developed by cancer cells to survive excessive DNA damage with a price of elevated genomic instability through error-prone SSA repair. Importantly, genomic instability is not only a hallmark of cancer cells, but also an enabling factor for cancer development [42]. However, adaptations leading to a dependency on this repair pathway may grant cancer cells vulnerability to interventions by suppressing SSA. This study provides further evidence that targeting FANCA for cancer treatment is likely helpful and warrants additional studies. In addition, this downregulation is proteasome dependent and executed through HSP90 targeting, a consequence presumably conserved for most cell types (Fig. 4). While targeted therapies that aim to inhibit the repair activities of PARP1 [43–45], RAD52 [46, 47], and Polθ [48, 49] has been proposed and tested, our study implies an alternative approach with similar consequences. Through intervention of the chaperone complex that is important for the DNA repair network [24], perturbation of activity and stability can be imposed to a defined set of HSP90 clients. Given differential utilization of DNA repair pathways throughout cell cycles and in various cell types, extended efforts may be required to characterize the cell type-specific and cell cycle-specific response to HSP90 interference.
Supplementary Material
Highlights:
Withaferin A (WA) specifically impedes SSA repair of DNA DSB repair pathways at low concentrations.
WA promotes FANCA downregulation post-translationally in a proteasome-dependent manner.
WA-mediated FANCA degradation is achieved through HSP90 inhibition and disruption of the FANCA-HSP90 interaction
WA-mediated FANCA degradation abolishes FANCD2 monoubiquitination and results in accumulation of DSBs.
Acknowledgements
This work was supported by National Institutes of Health Grants HL131013 and ES027058 and a Flight Attendant Medical Research Institute grant to Yanbin Zhang. Thanks to Drs. Joyce Slingerland and Xin-Hai Pei at the Sylvester Comprehensive Cancer Center, University of Miami for MCF-10A, MCF-7, SUM-149, and MDA-MB-231 cells. Cells and constructs for DSB repair reporter assays were generously provided by Jeremy Stark at the City of Hope National Medical Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors do not have conflicts of interests.
References
- [1].Chapman JR, Taylor MR, Boulton SJ, Playing the end game: DNA double-strand break repair pathway choice, Mol Cell, 47 (2012) 497–510. [DOI] [PubMed] [Google Scholar]
- [2].Ceccaldi R, Rondinelli B, D’Andrea AD, Repair Pathway Choices and Consequences at the Double-Strand Break, Trends Cell Biol, 26 (2016) 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Singleton MR, Wentzell LM, Liu Y, West SC, Wigley DB, Structure of the single-strand annealing domain of human RAD52 protein, Proceedings of the National Academy of Sciences of the United States of America, 99 (2002) 13492–13497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jackson SP, Bartek J, The DNA-damage response in human biology and disease, Nature, 461 (2009) 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Goldstein M, Kastan MB, The DNA damage response: implications for tumor responses to radiation and chemotherapy, Annu Rev Med, 66 (2015) 129–143. [DOI] [PubMed] [Google Scholar]
- [6].Zeman MK, Cimprich KA, Causes and consequences of replication stress, Nat Cell Biol, 16 (2014) 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ciccia A, Elledge SJ, The DNA damage response: making it safe to play with knives, Mol Cell, 40 (2010) 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Burgess RC, Misteli T, Not All DDRs Are Created Equal: Non-Canonical DNA Damage Responses, Cell, 162 (2015) 944–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lieber MR, The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway, Annual review of biochemistry, 79 (2010) 181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Palovcak A, Liu W, Yuan F, Zhang Y, Maintenance of genome stability by Fanconi anemia proteins, Cell Biosci, 7 (2017) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Moldovan GL, D’Andrea AD, How the fanconi anem ia pathway guards the genome, Annu Rev Genet, 43 (2009) 223–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ceccaldi R, Sarangi P, D’Andrea AD, The Fanconi anaemia pathway: new players and new functions, Nature reviews. Molecular cell biology, 17 (2016) 337–349. [DOI] [PubMed] [Google Scholar]
- [13].Huang Y, Leung JW, Lowery M, Matsushita N, Wang Y, Shen X, Huong D, Takata M, Chen J, Li L, Modularized functions of the Fanconi anemia core complex, Cell Rep, 7 (2014) 1849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang AT, Smogorzewska A, SnapShot: Fanconi anemia and associated proteins, Cell, 160 (2015) 354–354 e351. [DOI] [PubMed] [Google Scholar]
- [15].Howard SM, Yanez DA, Stark JM, DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining, PLoS Genet, 11 (2015) e1004943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D’Andrea AD, Wang ZQ, Jasin M, Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair, Proc Natl Acad Sci U S A, 102 (2005) 1110–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yang YG, Herceg Z, Nakanishi K, Demuth I, Piccoli C, Michelon J, Hildebrand G, Jasin M, Digweed M, Wang ZQ, The Fanconi anemia group A protein modulates homologous repair of DNA double-strand breaks in mammalian cells, Carcinogenesis, 26 (2005) 1731–1740. [DOI] [PubMed] [Google Scholar]
- [18].Benitez A, Liu W, Palovcak A, Wang G, Moon J, An K, Kim A, Zheng K, Zhang Y, Bai F, Mazin AV, Pei XH, Yuan F, Zhang Y, FANCA Promotes DNA Double-Strand Break Repair by Catalyzing Single-Strand Annealing and Strand Exchange, Mol Cell, 71 (2018) 621–628 e624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stan SD, Hahm ER, Warin R, Singh SV, Withaferin A causes FOXO3a- and Bim-dependent apoptosis and inhibits growth of human breast cancer cells in vivo, Cancer Res, 68 (2008) 7661–7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nagalingam A, Kuppusamy P, Singh SV, Sharma D, Saxena NK, Mechanistic elucidation of the antitumor properties of withaferin a in breast cancer, Cancer Res, 74 (2014) 2617–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wu J, Liu T, Rios Z, Mei Q, Lin X, Cao S, Heat Shock Proteins and Cancer, Trends Pharmacol Sci, 38 (2017) 226–256. [DOI] [PubMed] [Google Scholar]
- [22].Yu Y, Hamza A, Zhang T, Gu M, Zou P, Newman B, Li Y, Gunatilaka AA, Zhan CG, Sun D, Withaferin A targets heat shock protein 90 in pancreatic cancer cells, Biochemical pharmacology, 79 (2010) 542–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bargagna-Mohan P, Paranthan RR, Hamza A, Dimova N, Trucchi B, Srinivasan C, Elliott GI, Zhan CG, Lau DL, Zhu H, Kasahara K, Inagaki M, Cambi F, Mohan R, Withaferin A targets intermediate filaments glial fibrillary acidic protein and vimentin in a model of retinal gliosis, J Biol Chem, 285 (2010) 7657–7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Oda T, Hayano T, Miyaso H, Takahashi N, Yamashita T, Hsp90 regulates the Fanconi anemia dNa damage response pathway, Blood, 109 (2007) 5016–5026. [DOI] [PubMed] [Google Scholar]
- [25].Ha K, Fiskus W, Rao R, Balusu R, Venkannagari S, Nalabothula NR, Bhalla KN, Hsp90 inhibitor-mediated disruption of chaperone association of ATR with hsp90 sensitizes cancer cells to DNA damage, Mol Cancer Ther, 10 (2011) 1194–1206. [DOI] [PubMed] [Google Scholar]
- [26].Gunn A, Stark JM, I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks, Methods Mol Biol, 920 (2012) 379–391. [DOI] [PubMed] [Google Scholar]
- [27].Yuan F, Qian L, Zhao X, Liu JY, Song L, D’Urso G, Jain C, Zhang Y, Fanconi anemia complementation group A (FANCA) protein has intrinsic affinity for nucleic acids with preference for single-stranded forms, The Journal of biological chemistry, 287 (2012) 4800–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Grimme JM, Honda M, Wright R, Okuno Y, Rothenberg E, Mazin AV, Ha T, Spies M, Human Rad52 binds and wraps single-stranded DNA and mediates annealing via two hRad52-ssDNA complexes, Nucleic Acids Res, 38 (2010) 2917–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Leung JW, Wang Y, Fong KW, Huen MS, Li L, Chen J, Fanconi anemia (FA) binding protein FAAP20 stabilizes FA complementation group A (FANCA) and participates in interstrand cross-link repair, Proceedings of the National Academy of Sciences of the United States of America, 109 (2012) 4491–4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yan Z, Guo R, Paramasivam M, Shen W, Ling C, Fox D 3rd, Wang Y, Oostra AB, Kuehl J, Lee DY, Takata M, Hoatlin ME, Schindler D, Joenje H, de Winter JP, Li L, Seidman MM, Wang W, A ubiquitin-binding protein, FAAP20, links RNF8-mediated ubiquitination to the Fanconi anemia DNA repair network, Mol Cell, 47 (2012) 61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hahm ER, Moura MB, Kelley EE, Van Houten B, Shiva S, Singh SV, Withaferin A-induced apoptosis in human breast cancer cells is mediated by reactive oxygen species, PloS one, 6 (2011) e23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chang HW, Li RN, Wang HR, Liu JR, Tang JY, Huang HW, Chan YH, Yen CY, Withaferin A Induces Oxidative Stress-Mediated Apoptosis and DNA Damage in Oral Cancer Cells, Front Physiol, 8 (2017) 634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bargagna-Mohan P, Hamza A, Kim YE, Khuan Abby Ho Y, Mor-Vaknin N, Wendschlag N, Liu J, Evans RM, Markovitz DM, Zhan CG, Kim KB, Mohan R, The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin, Chemistry & biology, 14 (2007) 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Thaiparambil JT, Bender L, Ganesh T, Kline E, Patel P, Liu Y, Tighiouart M, Vertino PM, Harvey RD, Garcia A, Marcus AI, Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation, International journal of cancer, 129 (2011) 2744–2755. [DOI] [PubMed] [Google Scholar]
- [35].Falkenberg KD, Jakobs A, Matern JC, Dorner W, Uttarkar S, Trentmann A, Steinmann S, Coulibaly A, Schomburg C, Mootz HD, Schmidt TJ, Klempnauer KH, Withaferin A, a natural compound with anti-tumor activity, is a potent inhibitor of transcription factor C/EBPbeta, Biochimica et biophysica acta, 1864 (2017) 1349–1358. [DOI] [PubMed] [Google Scholar]
- [36].Grossman EA, Ward CC, Spradlin JN, Bateman LA, Huffman TR, Miyamoto DK, Kleinman JI, Nomura DK, Covalent Ligand Discovery against Druggable Hotspots Targeted by Anti-cancer Natural Products, Cell chemical biology, 24 (2017) 1368–1376 e1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Anantha RW, Simhadri S, Foo TK, Miao S, Liu J, Shen Z, Ganesan S, Xia B, Functional and mutational landscapes of BRCA1 for homology-directed repair and therapy resistance, eLife, 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Her J, Bunting SF, How cells ensure correct repair of DNA double-strand breaks, The Journal of biological chemistry, 293 (2018) 10502–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mimitou EP, Symington LS, DNA end resection: many nucleases make light work, DNA Repair (Amst), 8 (2009) 983–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bhargava R, Onyango DO, Stark JM, Regulation of Single-Strand Annealing and its Role in Genome Maintenance, Trends Genet, 32 (2016) 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Huang F, Goyal N, Sullivan K, Hanamshet K, Patel M, Mazina OM, Wang CX, An WF, Spoonamore J, Metkar S, Emmitte KA, Cocklin S, Skorski T, Mazin AV, Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors, Nucleic acids research, 44 (2016) 4189–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, Cell, 144 (2011) 646–674. [DOI] [PubMed] [Google Scholar]
- [43].Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T, Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase, Nature, 434 (2005) 913–917. [DOI] [PubMed] [Google Scholar]
- [44].Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A, Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy, Nature, 434 (2005) 917–921. [DOI] [PubMed] [Google Scholar]
- [45].Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira-Frommer R, Safra T, Matei D, Fielding A, Spencer S, Dougherty B, Orr M, Hodgson D, Barrett JC, Matulonis U, Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial, The Lancet. Oncology, 15 (2014) 852–861. [DOI] [PubMed] [Google Scholar]
- [46].Sullivan K, Cramer-Morales K, McElroy DL, Ostrov DA, Haas K, Childers W, Hromas R, Skorski T, Identification of a Small Molecule Inhibitor of RAD52 by Structure-Based Selection, PloS one, 11 (2016) e0147230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chandramouly G, McDevitt S, Sullivan K, Kent T, Luz A, Glickman JF, Andrake M, Skorski T, Pomerantz RT, Small-Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA-Deficient Cancers, Chemistry & biology, 22 (2015) 1491–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, O’Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, Yusufzai T, D’Andrea AD, Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair, Nature, 518 (2015) 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, Sfeir A, Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination, Nature, 518 (2015) 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.