

Summary
Regulatory T (Treg) cells maintain immune tolerance through the master transcription factor, FOXP3, which is crucial for Treg cell function and homeostasis. We identified an Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) patient with a FOXP3 mutation in the domain swap interface of the protein. Recapitulation of this Foxp3 variant in mice led to an autoimmune syndrome consistent with an unrestrained T helper type 2 (Th2) immune response. Genomic analysis of Treg cells by RNA-seq, Foxp3 ChIP-seq and H3K27ac-HiChIP revealed a specific de-repression of the Th2 transcriptional program leading to the generation of Th2-like Treg cells that were unable to suppress extrinsic Th2 cells. Th2-like Treg cells showed increased intra-chromosomal interactions in the Th2 locus leading to type-2 cytokine production. These findings identify a direct role for Foxp3 in suppressing Th2-like Treg cells and implicate additional pathways that could be targeted to restrain Th2 trans-differentiated Treg cells.
Keywords: IPEX, Foxp3, regulatory T cells, GATA3, Th2-like Treg, autoimmunity
Graphical Abstract
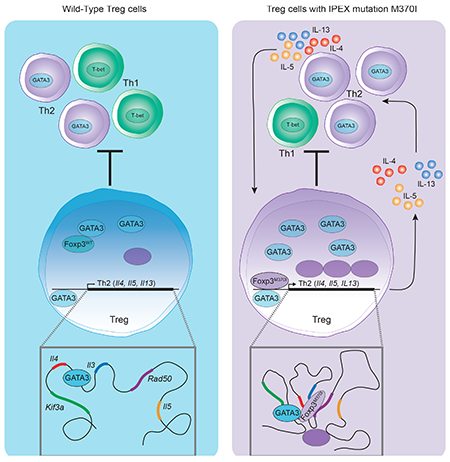
Introduction
IPEX syndrome is a recessive disease affecting exclusively males (d’Hennezel et al., 2012). This life-threatening disease is characterized by widespread multi-organ autoimmunity and most of the afflicted patients die before age 3 unless treated with a bone marrow transplant. In 2001, genome linkage analysis and positional cloning identified the forkhead/winged helix family transcription factor FOXP3 as the gene responsible for the IPEX phenotype in humans (Bennett et al., 2001; Wildin et al., 2001) and scurfy phenotype in mice (Brunkow et al., 2001). FOXP3 is the key transcription factor expressed predominantly in a small population of CD4+CD25hi T cells, termed regulatory T (Treg) cells, which are essential for maintenance of peripheral tolerance (Fontenot et al., 2003; Hori et al., 2003; Khattri et al., 2003)
To date there are over 75 different FOXP3 mutations described in IPEX patients (Bacchetta et al., 2016) which cluster in the N-terminal proline-rich domain, the central leucine zipper domain and the C-terminal FKH domain (Barzaghi et al., 2017). While a majority of IPEX patients do not have circulating Treg cells (Gavin et al., 2006; Moes et al., 2010), some mutations, located around the C-terminus of the protein, result in the generation of FOXP3+CD4+ lymphocytes at frequencies similar than those found in patients expressing wild type FOXP3 (d’Hennezel et al., 2009; Kinnunen et al., 2013). Nonetheless, Treg cells in these patients express reduced levels of FOXP3, CD25 and show a severe loss of suppressive function as compared to Treg cells isolated from healthy controls. These naturally occurring mutations have been invaluable to understand the relative importance of each structural domain of FOXP3 (Barzaghi et al., 2017; Bin Dhuban et al., 2017; Hayatsu et al., 2017).
As a dominant suppressive population, Treg cells can inhibit the development and differentiation of T effector memory (Tem) cells and reverse autoimmunity through a diverse functional profile, including suppressive cytokines production, cell contact-dependent regulation and metabolic control of the inflammatory environment (Lu et al., 2017). Molecular characterization of Treg cell subsets reveal populations defined based on co-expression of transcription factors specific to Th cell lineages (T-bet: Th1, IRF4: Th2, STAT3: Th17 and BCL6: Tfh) (Campbell and Koch, 2011). The co-expression of these selective lineage-determining transcription factors generates functionally distinct subsets that can localize to distinct sites of inflammation and acquire suppressive and tissue repair programs that effectively control ongoing tissue-specific immune responses. However, Treg plasticity and Foxp3 instability that can occur at these inflammatory sites can present a significant concern due to the loss of suppressive function and potentially by the acquisition of Tem functionality (DuPage and Bluestone, 2016). To execute its functional program, Foxp3 binds to over 1400 genes, acting as both a transcriptional repressor and activator, depending on the promoter to which it is bound (Arvey et al., 2014; Samstein et al., 2012). The complex regulatory function mediated by Foxp3 is achieved by interacting with a network of other transcription factors such as RUNX1, NFAT and GATA3 (Rudra et al., 2012). For example, GATA3 promotes Foxp3 expression and suppresses IL-17 cytokine production (Rudra et al., 2012; Wang et al., 2011; Wohlfert et al., 2011). Treg-specific deletion of GATA3 results in a spontaneous inflammatory disorder in mice, highlighting its crucial role in Treg cell homeostasis and function (Rudra et al., 2012; Wang et al., 2011). However, GATA3 is also the “master regulator” for Th2 cell differentiation and controls Th2 cytokine production (Yu et al., 2015). Thus, it is essential that Foxp3 mediates a transcriptional program that allows for GATA3 expression while repressing the rest of the Th2 signature to prevent Th2-mediated immune disorders. In this regard, it has been striking that Th2 differentiation is the default developmental and trans-differentiation program in both human and mouse Treg cells after downregulation of Foxp3 (Hansmann et al., 2012; Wan and Flavell, 2007) and that many IPEX patients present with typical Th2 pathology like atopy, eczema, eosinophilic inflammation, and hyper-IgE syndrome (Bacchetta et al., 2016).
Bandukwala and colleagues solved a partial crystal structure of the Forkhead DNA-binding domain (FKH domain), located at the C-terminal end of the Foxp3 protein, as part of the Foxp3:NFAT1:DNA complex (Bandukwala et al., 2011). The structure suggested that the FKH domain supports the formation of a dimer through a domain-swap interface. The Foxp3 FKH domain is optimized to form a domain-swapped dimer, due to the presence of tryptophan 348 and methionine 370 at the center of a network of hydrophobic and aromatic residues (Andersen et al., 2012; Bandukwala et al., 2011). The large number of IPEX patients with point mutations purported to destabilize the domain-swapped dimer of FOXP3 highlights the functional importance of this domain (Barzaghi et al., 2017).
In this report, we identified a FOXP3 gene mutation in a young male patient that manifested with severe IPEX syndrome and exhibited several signs of Th2-like disease. This mutation resulted in the substitution of methionine for isoleucine at amino acid 370 (M370I) located at the core of the dimerization motif in the FKH domain of FOXP3. We hypothesized that this mutations in the domain-swap interface altered FOXP3 function and its transcriptional control of the Treg cell program, potentially causing transdifferentiation into Th2 cells. In order to perform a detailed phenotypic, biochemical and functional analysis of this FOXP3 gene mutation, we generated a transgenic mouse that recapitulated the patient mutation. Introduction of the Foxp3 M370I gene mutation led to the formation of impaired CD4+Foxp3+ Treg cells with a highly plastic phenotype and acquisition of a Th2-like program, resulting in the development of a Th2-biased autoimmune disease. Molecular and functional analyses revealed chromatin remodeling at the Th2 locus with an increase in enhancer-promoter interactions, de novo binding of M370I Foxp3 to the Th2 cytokine locus, and dysregulation of the locus leading to Th2 cytokine production by the Tregs and resulting pathogenicity.
RESULTS
Mutation M370I in the FKH Domain-Swap interface of FOXP3 results in IPEX syndrome.
We identified a male infant exhibiting IPEX syndrome. Shortly after birth, the patient was diagnosed with insulin-dependent diabetes mellitus and within the first month of life developed thrombocytopenia, eczematous skin rash, elevated eosinophil count and high level of IgE. DNA sequencing revealed a point mutation of guanine to adenine at nucleotide position 1110 in the FOXP3 gene (exon 10), resulting in substitution of methionine for isoleucine at amino acid 370 (M370I, Figure 1A and supplemental Figure 1A), which is identical to that in a patient previously identified by An et al. (An et al., 2011).
Figure 1. M370I mutant mice recapitulate the phenotype of the IPEX patient.
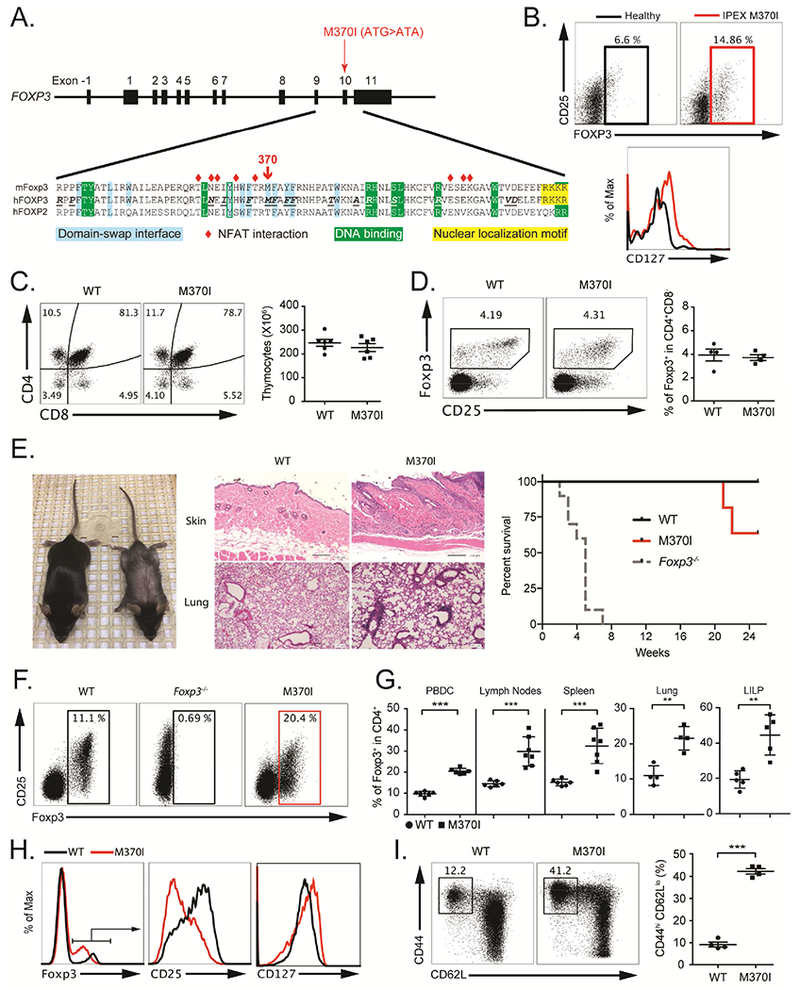
(A) Localization of IPEX M370I mutation: schematic representation of FOXP3 genomic DNA and sequences alignment of mouse and human FOXP3 with human FOXP2 FKH domain. Amino acids involved in NFAT interactions are labeled with red diamonds; residues at the domain swap interface are shaded in blue. The nuclear localization motif is highlighted in yellow and the residues interacting with DNA are shaded in green. Amino acids in bold and underline in the human FOXP3 sequence indicated positions of IPEX mutations. (B) Flow cytometric analysis of FOXP3, CD25 and CD127 gated on CD4+ cells from IPEX M370I patient and healthy control’s PBMCs. (C) Flow cytometric analysis (left panel) and quantification (right panel) of thymocytes in wild-type and M370I mice (3-4 weeks old). (D) Expression analysis (left panel) of Foxp3 and CD25 on Treg cells among CD4+CD8− thymocytes and quantification of Foxp3+ Treg cells among CD4+CD8− thymocytes (right panel). (E) Representative appearance (left panel) and histological analysis (middle panel) by hematoxylin and eosin staining of skin and lung samples of 12 weeks old wild-type and M370I mice. Kaplan-Meier survival curve (right panel) of wild-type (n=5), Foxp3−/− (n=5) and M370I (n=10) mice (F) Expression of CD25 and Foxp3 by CD4+ PBMCs from 3-4 week-old wild-type, Foxp3−/− and M370I littermates. (G) Quantification of Treg cells in PBMC, lymph nodes, spleens, lungs and large intestine lamina propria (LILP) from 3-4-weeks old mice for the PBMCs and 6-9-week-old wild-type and M370I littermates for other tissues. (H) Expression of Foxp3, CD25 and CD127 by CD4+ PBMCs from 6-9-week-old wild-type and M370I littermates. (I) Flow cytometric analysis (top panel) of CD44 and CD62L expression on PBMCs CD4+ T cells in 6-9-week-old wild-type mice and M370I littermates. Quantification (bottom panel) of activated-memory CD4+ T cells. All data points reflect individual biological replicates (error bars depict ±SD). See also Figures S1 and S2.
To quantify and phenotype Treg cells, we performed flow cytometry analysis on the patient’s peripheral blood mononuclear cells (PBMCs). The percentage of FOXP3-expressing CD4+ Treg cells was similar to or even higher than in control PBMCs (Figure 1B and An et al.). However, mutant Treg cells exhibited increased expression of CD127 and a decreased CD25 surface expression (Figure 1B). Although mutant Treg cells expressed FOXP3, the functional activity appeared to be disrupted in vivo as the patient suffered from extensive autoimmune sequelae. Ultimately, the patient required a bone marrow transplant at 4.5 months of age to replace the dysfunctional Treg cells.
M370I mutant mice recapitulate the phenotype of the IPEX patient.
To understand the molecular and functional effects of the M370I mutation identified in the IPEX patient, we generated a transgenic (Tg) mouse carrying a Bacterial Artificial Chromosome (BAC) containing the mutant Foxp3 allele (Zhou et al., 2008) (Figure 1A and supplemental Figure 1A). BAC positive founder mice were bred with Foxp3-deficient animals (Foxp3−/−) (Lin et al., 2005) in order to generate a mouse expressing only mutant Foxp3 (Foxp3−/−M370I Tg, subsequently referred to as M370I Tg) and thereby, study the developmental and functional consequences of the M370I mutation on Treg cells.
Thymic development was not affected in M370I Tg mice. The absolute number of thymocytes and the frequency of CD4+Foxp3+ Treg cells were similar between wild-type and M370I Tg mice (Figures 1C and D), suggesting that thymic-derived Treg cells were generated despite this mutation within the domain-swap interface of Foxp3. However, the M370I Tg mice developed severe signs of skin and lung inflammation associated with an exacerbated Th2 immune response characterized by the presence of circulating Th2 cytokines (IL-4, IL-5, IL-13) and an overproduction of IgE without affecting total IgG concentration in the sera of M370I-Tg mice at 10-12 weeks of age (Figure S2A). The clinical symptoms of the disease required euthanizing the animals starting at approximately 20 weeks of age (Figure 1E). Consistent with the patient phenotype, the autoimmune manifestations were not due to the absence of Treg cells as more than 20% of CD4+ T cells present in PBMCs expressed Foxp3, a higher frequency than observed in wild type mice (Figures 1F and 1G).
We observed increased absolute numbers and percentages of M370I Treg cells in lymphoid (spleen, lymph nodes) and non-lymphoid tissues (lung, colon (LILP) and skin) (Figure 1G and Figure S2). However, the M370I Treg cells expressed lower levels of CD25 and higher levels of CD127 (Figure 1H), similar to that observed in the IPEX patient, confirming the biologic consequences of the M370I IPEX patient mutation. Moreover, phenotypic analysis revealed an increased number and frequency of activated CD4+ T cells (Figure 1I) suggesting severe disruption of immune homeostasis. Thus, the mouse model faithfully recapitulated aspects of the human disease and serves as an effective tool to dissect the in vivo effects of the Foxp3 mutation on Treg cell function and disease pathology.
M370I mutation maintains regulatory function of Treg cells.
The suppressive activity of M370I Treg cells was analyzed in vitro and in vivo using conventional Treg suppression assays. Treg cells isolated from lymph nodes and spleen were functionally suppressive in vitro; inhibiting the proliferation of naïve CD4+ T cells as efficiently as the wild-type Treg cells (Figure 2A). Both wild type and M370I Treg cells were able to suppress in vivo in a therapeutic mouse model of colitis induced by the transfer of CD4+CD45RBhigh T cells into immunodeficient Rag1−/− mice. Adoptive transfer of the naïve T cells resulted in weight loss that was ameliorated with bot wild-type and M370I mutant Treg cells (Figure 2B). Moreover, phenotypic analyses of splenic and lymph nodes Treg cells showed that mutant Foxp3+ cells expressed key Treg cell markers (Nrp-1, Helios, CTLA-4, CD39, GITR and IL-10 (Figures 2C and 2D)). Consistent with higher numbers and percentages of Treg cells in the M370I Tg animal, we observed an increased expression of the proliferation marker Ki67 on Foxp3 mutant cells (Figure 2E). This proliferation was associated with a Tem phenotype characterized by increased expression of CD44 and downregulation of CD62L and CCR7 (Figure 2E).
Figure 2. Phenotypic Characterization of mouse M370I Treg cells.
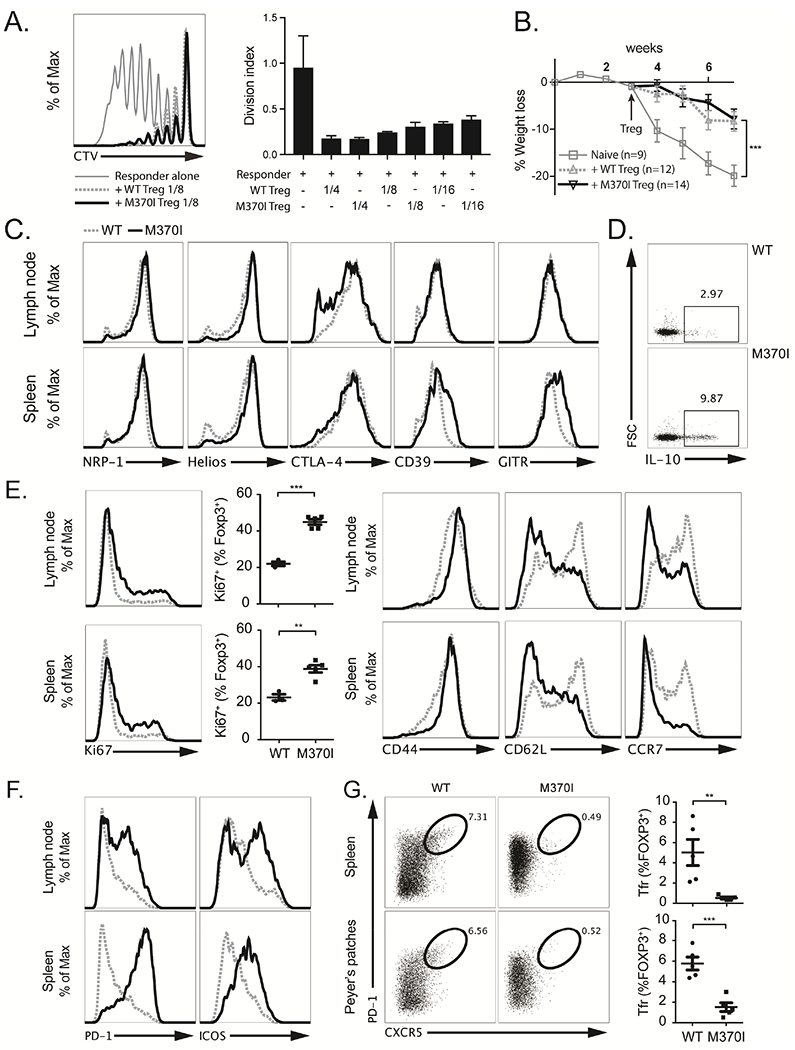
(A) Representative histograms of three independent experiments (left panel) depicting Celltrace Violet (CTV) dilution profile of responder CD4+ T cells cultured alone (thin grey line) or with either wild-type Treg cells (dashed grey line) or M370I Treg cells (black line) at a ratio of 1:8 (Treg cells:responders). Quantification (right panel) of the in vitro suppression assay by the division index at different Treg cell:responder ratios. (B) Weight loss (as a percentage of starting weight) over time after injection of naïve CD4+ T cells into immunodeficient Rag1−/− mice followed by WT or M370I Treg cells three weeks later. At the time of Treg injections, mice were randomly assigned to the three experimental groups. Error bars depict ±SEM, Two-way ANOVA test: naïve T cells/naiïve T cells + WT Tregs; week 7 (P < 0.001), naïve T cells/naiïve T cells + M370I Tregs; week 7 (P < 0.001). (C) Phenotypic analysis of Treg cells from LNs or spleen from wild-type (dashed grey line) and M370I littermates (black line) by staining for indicated proteins at 6-12 weeks of age. (D) Representative intracellular staining for IL-10 after PMA-ionomycin activation of splenic Treg cells from wild-type and M370I littermates at 6-12 weeks of age. (E) Flow cytometric analysis for proliferation (Ki67) and activation (CD44, CD62L and CCR7) markers on Treg cells from LNs and spleen from wild-type and M370I littermates. Expression and quantification from mice analyzed between 6 and 12 weeks of age. (F) Representative histograms for PD-1 and ICOS expression on Treg cells from LNs and spleen from wild-type and M370I littermates at 6-12 weeks of age (G) Representative flow cytometric analysis (left panel) and quantification (right panel) of Tfr cells in spleen and peyer’s patches from 6-12-week-old wild-type and M370I littermates. All data points reflect individual biological replicates (error bars depict ±SD, unless otherwise stated).
Programmed cell Death protein-1 (PD-1) and Inducible T-cell COStimulator (ICOS) were significantly upregulated in M370I Treg cells (Figure 2F). These two receptors have been used to identify follicular Treg cells (Tfr), so we analyzed the expression of the chemokine receptor CXCR5, one of the most common defining marker for Tfr cells (Vinuesa and Cyster, 2011). CXCR5 surface expression was profoundly affected on M370I Treg cells isolated from the spleen and Peyer’s patches, suggesting a defect in the recruitment and function of Tfr cells in the M370I Tg mice (Figure 2G). In summary, the M370I mutation resulted in the induction of highly proliferative population of effector-memory Treg cells expressing immunoregulatory molecules similar to those seen on wild-type Treg cells. Nonetheless, M370I mutation altered the ability of Treg cells to maintain immune homeostasis.
The M370I Foxp3 mutation drives the differentiation of Th2 Treg cells.
To analyze the molecular changes associated with the M370I mutation, we performed RNA sequencing (RNA-seq) on Treg cells isolated from secondary lymphoid organs of 3-weeks-old asymptomatic wild-type and M370I Tg mice. Among the top upregulated genes, (>5-fold change and adjusted p-value <0.05) we noted that Th2 cytokines such as Il4, Il5 and Il13 were highly expressed in M370I Treg cells compared to wild-type Treg cells (Figure 3A). Expression of Th2 cytokines was validated by qRT-PCR (Figure 3B), ELISA (Figure 3C) and intracellular flow cytometry (Figure 3D). This Th2 transcriptional program was associated with an upregulation of the Th2 master transcription factor GATA3 in M370I Treg cells. Up to 50% of M370I Foxp3+ cells co-expressed ICOS and GATA3 compared to only 10-20% of wild-type Treg cells isolated from lymph nodes (Figure 3E). The α-chain of the cytokine receptor IL-33R (St2 or IL1RL1) and the differentiation marker KLRG1, which are associated with GATA3 expression in Treg cell and Th2 cells (Delacher et al., 2017), were not upregulated in the M370I Treg cells as compared to wild type Tregs (Figure 3F). M370I Treg cells also expressed high levels of CD85k, also known as ILT3 (Figure 3G), a receptor discovered on a Treg subpopulation that promotes Th2 immune responses in vitro and in vivo by inducing the maturation of IRF4+PD-L2+ dendritic cells (DC) (Harakal et al., 2016; Ulges et al., 2015). Indeed, the M370I Tg mice presented an increased frequency of Th2-promoting IRF4+PD-L2+ cells among the CD11c+MHCII+ splenic DCs (Figure 3G). Finally, mutant Treg cells exhibited increased expression of PD-1 (Figure 2E), which has previously been shown to impair the ability of Treg cells to suppress Th2 cell differentiation (Yang et al., 2017). Taken together, these results suggest that M370I Treg cells could promote Th2 immune responses through multiple mechanisms, the production of Th2 cytokine, the expression of CD85k and finally, the upregulation of PD-1, which hinders the ability to control Th2 effector cells.
Figure 3. M370I Treg cells acquire Th2-like phenotype.
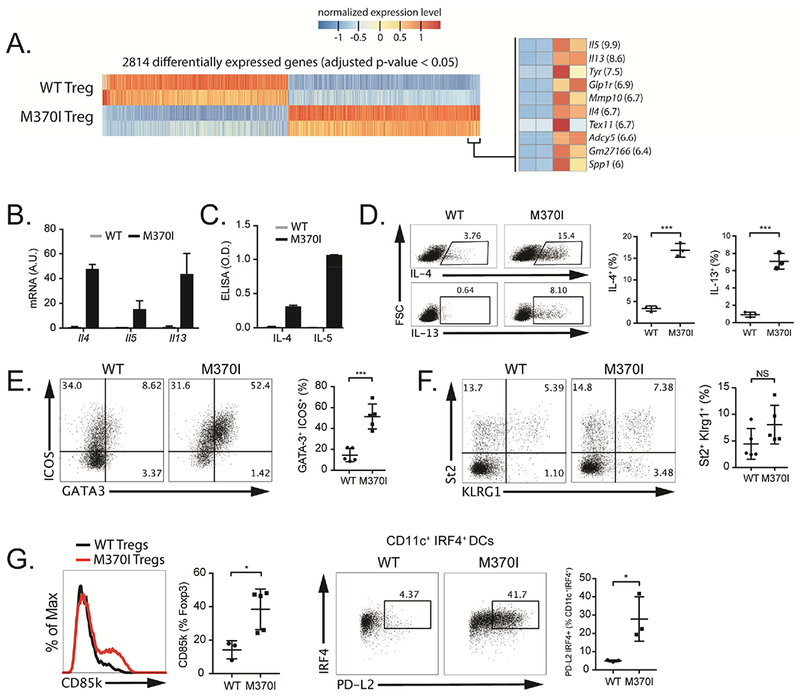
(A) RNA-seq analysis, heat map representation of differentially expressed genes between wild-type and M370I Treg cells from LNs and spleen from 3-week-old wild-type and M370I littermates mice. (left panel) most enriched DEGs in the M370I Treg (right panel, fold-change between WT and M370I Treg is shown in parenthesis). (B) Quantification by RT-qPCR of the expression of Il4, Il5 and Il13 in wild-type and M370I Treg cells. (C) Quantification by ELISA of IL-4 and IL-5 secreted by wild-type and M370I Tregs cells after overnight PMA-ionomycin activation. (D) Representative flow cytometric analysis (left panel) and quantification (right panel) of IL-4 and IL-13 producing Treg cells from PMA-ionomycin activated LNs from wild-type and M370I mice. (E) Representative flow cytometric analysis (left panel) and quantification (right panel) of Th2-like Treg cells (GATA3+ICOS+) present in the LNs from wild-type and M370I mice. (F) Characterization (left panel) and quantification (right panel) of St2 and KLRG1 expression on LNs Treg cells from wild-type and M370I mice. (G) Flow cytometric analysis and quantification (left panel) of CD85k expression in splenic Treg cells from wild-type and M370I mice. Representative flow cytometric analysis (right panel) and quantification of CD11c+MHCII+PD-L2+IRF4+ splenic DCs from wild type and M370I mice. Expression and quantification of results from mice analyzed between 6 and 12 weeks of age, unless otherwise stated. All data points reflect individual biological replicates (error bars depict ±SD) and representative of at least three independent experiments.
To test whether Treg cells can control effector differentiation programs, we activated wild-type naïve T cells with antigen presenting cells (APC) alone or co-cultured with wild-type or M370I Treg cells. The mutant Treg cells were significantly less efficient at inhibiting GATA3 expression induced during the activation of CD4+ T cells (Figure 4A) as compared to wild-type Treg cells. Nonetheless, M370I Treg cells were able to control proliferation (Figure 2A) and T-bet expression (Figure 4A). Even in the absence of proliferation, 40% of the effector T cells co-cultured with M370I Treg cells expressed GATA3. To determine if GATA3 upregulation was dependent on IL-4 produced by mutant Treg cells, we co-cultured IL-4 and IL-13 deficient naïve T cells with wild-type or M370I Treg cells and activated the cells in an APC free system. GATA3 was specifically induced in co-culture with M370I Treg cells but not with wild-type Treg cells (Figure 4B) In these co-cultures, M370I Treg cells were the only source of IL-4. In fact, when IL-4 activity was neutralized with an anti-IL-4 blocking antibody, the responder cells expressed the same level of GATA3 in the presence of wild-type or M370I Treg cells (Figure 4B). Interestingly, under these neutral culture conditions (no APCs and no cytokines added) the mutant Treg cells regulated T-bet expression in T effector cells to the same extent or better as the wild-type Treg cells (Figure S3).
Figure 4. IL-4 production by M370I Treg cells is cell intrinsic and drives Th2 differentiation.
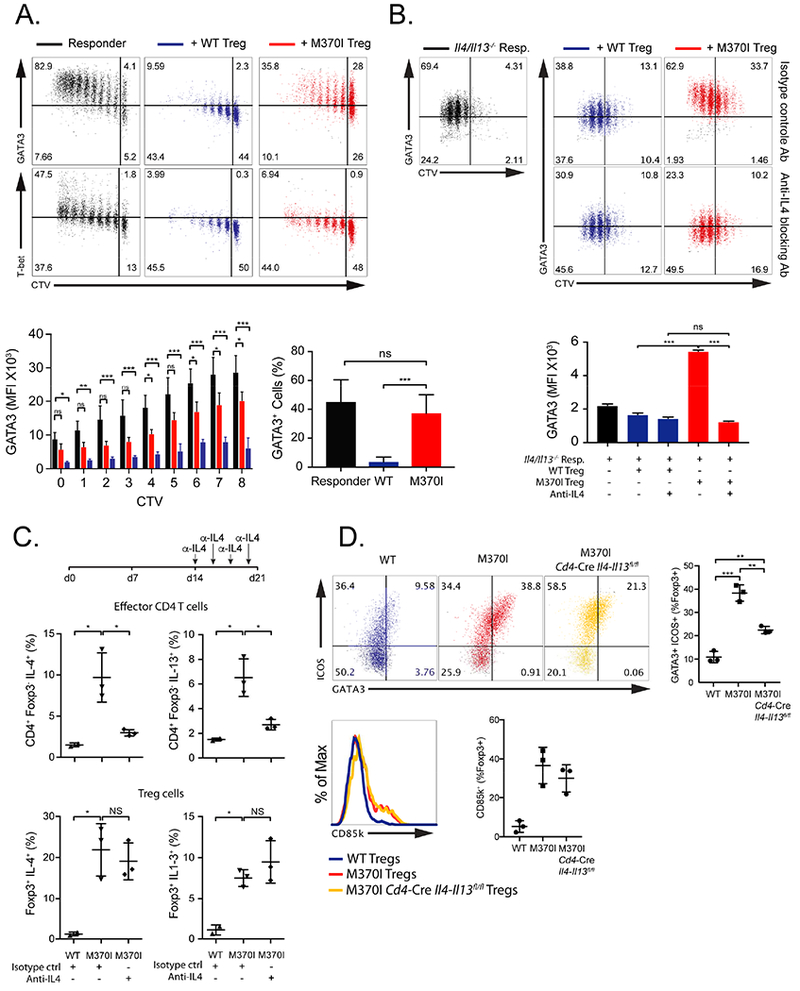
(A) Representative dot plot (Top panel) depicting CTV dilution profile and GATA3 (Top panel) or T-bet (middle panel) expression of responder CD4+ T cells stimulated for 72-96 hours with anti-CD3 and APCs alone (black dots) or with either wild-type Treg cells (blue dots) or M370I Treg cells (red dots) at a ratio of 1:10 (Treg cells:responders). Normalized geometric mean fluorescent intensity (MFI) values of GATA3 expression are summarized for each cell cycle division (lower left panel) and frequency of GATA3+ CD4+ responder T cells in undivided cells (lower right panel). Data are representative of three independent experiments (error bars depict ±SD). (B) Representative dot plot depicting CTV profile and GATA3 expression of II4/II13−/− responder CD4+ T cells cultured with plate bound CD3 and CD28 alone (black dots) or with either wild-type Treg cells (blue dots) or M370I Treg cells (red dots) at a ratio of 1:2 (Treg cells:responders) with isotype (Top panel) or anti-IL-4 blocking antibodies (middle panel). Values of GATA3 expression are summarized for each treatment (lower panel). Data are representative of two independent experiments (error bars depict ±SD). (C) Scheme of in vivo IL-4 blocking antibody treatment (upper panel). Quantification of IL-4 and IL-13 producing effector CD4+ T cells (middle panel) or Treg cells (lower panel) from PMA-ionomycin activated spleen cells from wild-type and M370I mice treated with isotype control or anti-IL4. Data are representative of two experiments with n=3. (D) Representative flow cytometric analysis and quantification (upper panel) of Th2-like Treg cells (GATA3+ICOS+) and CD85k expressing Treg cells (lower panel) present in the spleens from 3 weeks old wild-type, M370I mice or M370 T cell-specific II4-II13 deficient mice. Data are representative of three independent experiments with n=3. All the cells were sorted as describe in methods from wild-type and M370I littermates at 3-4 weeks of age, unless otherwise stated. See also Figure S3.
Next, we determined whether IL-4 produced by effector T cells contributed to the Th2 phenotype of mutant Treg cells in vivo. We injected 2 week-old Foxp3 M370I Tg mice with anti-IL-4 blocking antibody (Figure 4C top panel). The antibody treatment reduced the secretion of Th2 cytokines by the effector CD4+ T cells in the M370I Tg mice to the levels seen in wild-type mice (Figure 4C middle panel). However, mutant Treg cells continued to secrete IL-4 and IL-13 in the presence of the IL-4 blocking antibody (Figure 4C bottom panel). These results indicate that the dysregulated Th2 program, including Th2 cytokines production by mutant Treg cells, was a cell-intrinsic effect driven by M370I Foxp3 independent of an IL-4 feedback loop generated by the autoimmune disease. We also adopted a genetic strategy to confirm the key role of M370I Foxp3 mutation in the generation of the Th2-like phenotype in mutant Treg cells. T cell-specific IL-4 and IL-13 deficient mice were crossed with the M370I mutant animal. In the absence of IL-4 and IL-13 production by T cells, we observed an increased percentage of ICOS+GATA3+ and increased expression of CD85k (Figure 4D) on mutant Treg cells, although Th2 cytokines signaling was important for maximal GATA3 expression (Figure 4D). Together, these results confirm the critical role of IL-4 signaling in the activation and maintenance of GATA3 expression in CD4+ T cells. The data also substantiates the intrinsic role of M370I mutation in the generation of Th2-cell-like Treg cells, characterized by the expression of GATA3 and the secretion of the canonical Th2 effector cytokines. Overall, these data demonstrate that Treg cell dysregulation induced by the IPEX mutation is due to a cell-intrinsic Treg defect driven by M370I Foxp3 independent of IL-4 and IL-13 produced by T effector cells. Nonetheless, maximal expression of GATA3 in M370I Treg and presumably Th2 effector cells is controlled by a feed-forward loop that stabilizes and/or enhances the Th2-cell-like phenotype of the mutant Treg cells.
M370I mutation leads to increased enhancer-promoter (E-P) interaction at a limited number of loci.
Foxp3 acts mainly as a repressor of gene transcription by binding to distal enhancers (Arvey et al., 2014). Thus, we examined the E-P looping interactions as a read out for molecular consequences of the M370I Foxp3 mutation and its effects on gene expression. In a recent paper, Mumbach et al showed that immunoprecipitation for histone modification H3K27ac, which correlates with active enhancers and promoters, could be coupled with a ligation-reaction in HiChIP to identify E-P chromatin interactions across the genome in primary cells (Mumbach et al., 2017). We performed H3K27ac-HiChIP analysis to create high-resolution contact maps of active enhancers in wild-type and M370I Treg cells (Figure S4). Comparison of the H3K27ac-loops between Foxp3 wild-type and Foxp3-M370I cells revealed a limited number of significant changes. Of a total of 9,895 loops, only 281 loops (2.8%), were differentially identified between wild-type and M370I samples (Figure 5A and Tables S1 and S2). Fifty percent of these loops showed an increased chromatin interaction in the M370I mutant cells (Figure 5A). These M370I-biased loops were shown to be Treg-specific as only 5% of the loops overlapped with loops enriched in the effector T cells isolated from the same Tg mice (Table S2 (loops highlighted in red)). The Treg-specific chromatin loops were compared to Foxp3 ChIPseq data (Arvey et al., 2014; Samstein et al., 2012) to identify the loops with Foxp3-binding sites. Of the Foxp3 mutant-biased loops, 26.62% contained Foxp3 ChIP peaks at their anchors (Table S2, loops highlighted in green) while only 6.34% of WT-biased loops had Foxp3 ChIP peaks (Table S2, loops highlighted in green). This is consistent with a predominant role of Foxp3 as a repressor and suggests that M370I mutation affects Foxp3 suppressive function and its capacity to inhibit E-P interactions by altering the chromatin architecture (3D interactions) of Foxp3 target genes. Gata3 gene was among the most enriched M370I Foxp3 mutant-biased loops, showing a significant increase in E-P interactions (Figures 5A and 5B). This up-regulation of specific peaks in the H3K27ac HiChIP signal was due to a direct loss of repressive activity by M370I Foxp3 rather than the mutation impairing DNA binding of Foxp3. Indeed, analysis of genome-wide Foxp3 DNA-binding sites in both wild-type and M370I Treg cells by using chromatin immunoprecipitation followed by high-throughput DNA sequencing (ChIP-Seq) revealed an overall increased DNA-binding of the M370I Foxp3 (Figure 5C). Over 2372 unique peaks were identified by Foxp3 ChIP-seq (Table S3) and the majority (70%) were shared between wild-type and M370I Foxp3. Four percent were enriched in the WT sample and 25% of peaks were enriched in the M370I samples (Figure 5C). Thus, the M370I mutation did not abrogate Foxp3 binding to DNA. To validate the robustness of our Foxp3 ChIP-seq, we compared our data set with previously published ChIP-seq data using the same anti-Foxp3 antibody (Samstein et al., 2012). There was a strong correlation between the two wild-type ChIP-seq data sets (Pearson correlation coefficient = 0.87) at the genome-wide level, suggesting good reproducibility. In addition, analysis of Cd28, Ctla4 and IL2ra, three well-defined Foxp3 target genes, demonstrated unchanged chromatin architecture and a similar binding pattern of Foxp3 between all the samples (Figures 5D and S5). In addition, a similar or even higher Foxp3 ChIP signal for the M370I mutant protein was observed when focusing on the E-P loops with highest H3K27ac HiChIP signals (Figure 5E). The deregulation of Gata3 was associated with increased looping at multiple Th2 signature genes such as Maf, Tnfsf8, Il10 and Il4, suggesting a selective loss of repressive activity for a subset of genes controlling Th2 effector function (Figures 5A and S5). In summary, the M370I mutation inhibited the repressive function of Foxp3 and led to increased interactions between enhancers and promoters at a small number of key genes involved in Th2 effector function. These results supported a cell-intrinsic defect of M370I Foxp3 in mutant Treg cells that leads to changes in Gata3, and other Th2-related gene expression and the development of dysfunctional Treg cells.
Figure 5. M370I Foxp3 mutation increased E-P interactions at Foxp3 target genes.
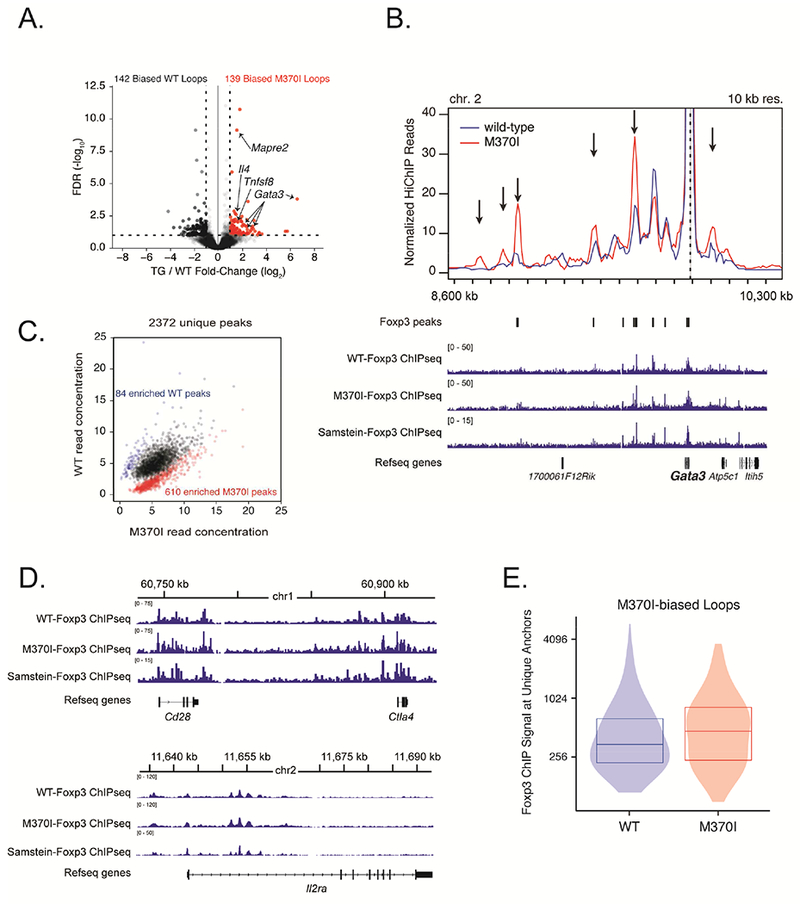
(A) Volcano plot of H3K27ac-HiChIP loops differentially expressed between wild-type and M370I Treg cells; M370I-biased loops are labelled in red (log2 (fold change) >1 and false discovery rate (FDR) <0.05). (B) Virtual 4C interaction profile at the Gata3 promoter in wild-type (blue line) and M370I (red line) Treg cells. Representative plots of Foxp3 ChIP-seq reads from wild-type and M370I Treg cells aligned to the genomic region containing Gata3 gene (bottom panel). Arrows indicated M370I-biased loops. Wild-type and M370I-Foxp3 ChIP-seq peaks are localize on the plot. (C) Scatterplot of normalized read for each peak region identified by Foxp3 ChIP-seq in either WT or M370I Treg cells. Each point represents a Foxp3 binding site; differentially bound sites are labelled in blue for the wild-type enriched sites (84 peaks) and in red for the M370I enriched sites (610 peaks). (D) Representative plots of Foxp3 ChIP-seq reads from wild-type and M370I Treg cells aligned to the genomic region containing Cd28 and Ctla4 genes (top panel) or Il2ra gene (bottom panel). (E) Quantification of Foxp3 ChIP-seq signal at unique M370I-biased loops anchors in wild-type and M370I Treg cells. See also Figures S5, S6, Tables S1, S2 and S3.
M370I mutation leads to binding of Foxp3 to the Th2 locus.
Integration of genome-wide RNA-seq, Foxp3 ChIP-seq and H3K27ac HiChIP from M370I mutant Treg cells enable the identification of Foxp3 target gene(s) that may contribute to the disease-associated phenotype. We focused on genes marked by H3K27ac loops that were bind by mutant M370I Foxp3. Indeed, the majority of the Foxp3-specific loops had increased E-P interactions with M370I mutation (Table S2, loops highlighted in green). In total, 698 genes were upregulated, 610 Foxp3-ChIP peaks were enriched and 139 H3K27ac HiChIP loops were identified in M370I mutant Treg cells. When combined in a Venn diagram (Figure 6A), only 7 genes were identified in all three data sets. Among these 7 genes, 5 were associated with Th2 immune responses (Cxcr6, Il4, Il10, Mapre2, Tnfsf8). Although the majority of these genes are known to be Foxp3 target genes, the II4 gene had not previously been identified as a Foxp3 target. Detailed analysis of the Th2 locus, which included Il4, Il5, Il13, Rad50 and Kif3a, revealed enriched binding of M370I Foxp3 to multiple sites along this locus encoding for Th2 cytokines (Figure 6B). These binding sites were unique to M370I mutant Foxp3 as no statistically significant bindings were identified in wild-type samples or in the published Foxp3 ChIP-seq data sets (Figure 6B, Samstein et al., 2012 and Kitagawa et al., 2017). Taken together, in depth genomic analysis of mutant Treg cells identify de novo Foxp3 binding sites in the Th2 locus, which were dependent on the Foxp3 M370I mutation.
Figure 6. M370I Foxp3 mutation induced Th2 effector programme by increasing chromatin interaction at the Th2 cytokine locus.
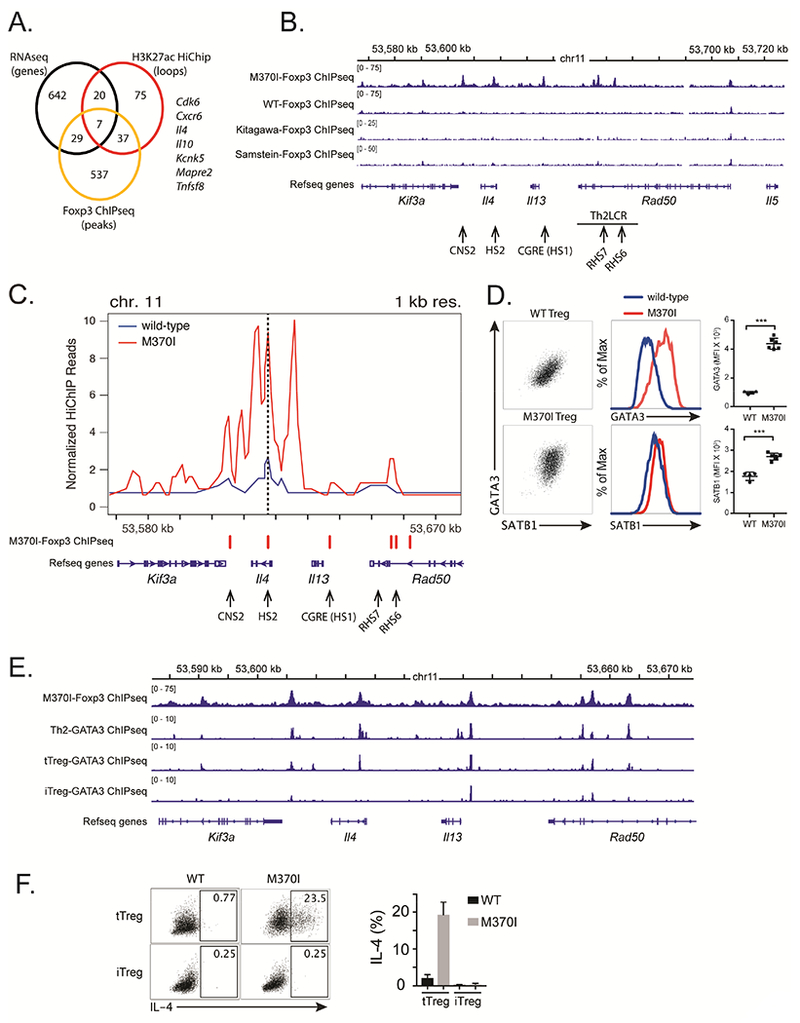
(A) Venn diagram showing the integration of genome-wide RNA-seq (log2 (fold change) of >1 and adjusted p-value <0.05), Foxp3 ChIP-seq and H3K27ac HiChIP. Pooling these three complementary approaches has generated a list of seven genes. (B) Representative plots of Foxp3 ChIP-seq reads from wild-type and M370I Treg cells aligned to the genomic region containing the Th2 locus. The arrows localize positions of cis-regulatory elements controlling the chromatin architecture of the Th2 locus. (C) Virtual 4C interaction profile at the Il4 promoter in wild-type (blue line) and M370I (red line) Treg cells. M370I-Foxp3 Chip-seq peaks and Th2 cis-regulatory elements are localize on the plot. (D) Representative flow cytometric analysis and quantification of GATA3 and SATB1 expression on activated Treg cells sorted from wild-type and M370I mice (Data are representative of 4 independent experiments). (E) Binding of Foxp3 and GATA3 at the Th2 cytokine locus. (F) Representative intracellular staining (left panel) and quantification (right panel) for IL-4 after activation of tTreg and iTreg cells from wild-type and M370I littermates at 6-12 weeks of age (Data are representative of 3 independent experiments).
M370I mutation leads to deregulation of the Th2 locus.
It is well established that the coordinated expression of Th2 cytokines is controlled by chromatin conformation at the locus encoding these genes (Lee, 2014). This spatial organization is generated by cis-regulatory elements and specific transcription factors that mediate the formation of long-range chromosomal interactions (Spilianakis and Flavell, 2004). Alignment of the Foxp3 ChIP peaks revealed that the M370I mutant Foxp3 binding sites were localized to c/s-acting conserved noncoding sequence 2 (CNS2) regulatory elements, DNase I-hypersensitive site 2 (HS2, the Il4 intronic enhancer), CGRE (Il13 distal promoter), and Rad50 DNase-I-hypersensitive sites, RHS6 and RHS7 (Figures 6B and 6C). Of interest, M370I mutant Foxp3 bound the Th2 locus control region (LCR), which controls the expression of all three Th2 cytokines and mediates intra-chromosomal interactions (Lee et al., 2003). In order to determine if the binding of M370I mutant Foxp3 affects the Th2 locus conformation, high-resolution contact maps were generated from the H3K27ac HiChIP data using the Il4 promoter as the anchor point (dot line, Figure 6C). The analysis enabled the visualization of virtual 4C plots for the Th2 locus (Figure 6C) and demonstrated a greater intra-chromosomal interaction in M370I Foxp3 mutant Treg cells. This pattern of DNA interaction between Il4 and the cis-regulatory element is unique to the mutant Treg cells as there were no similar loops observed in the wild-type Treg cells. These long-range chromosomal interactions are mediated by transcription factors, like GATA3 (Spilianakis and Flavell, 2004), and special AT-rich sequence-binding protein-1 (SATB1) (Cai et al., 2006). As predicted by the spatial reorganization of the Th2 locus, coordinated up-regulation of GATA3 and SATB1 was observed by flow cytometry in M370I Foxp3+ activated Treg cells (Figure 6D). GATA3 and Foxp3 have been shown to cooperate and occupy the same binding sites. Moreover, GATA3 directly binds to multiple sites within the Th2 locus. Therefore, we analyzed previously published GATA3 ChIP-seq data sets (Wei et al., 2011) from Treg and Th2 cells to determine whether GATA3 and M370I Foxp3 could co-occupy the same sites within the Th2 locus. By combining GATA3 and Foxp3 ChlP-seq data sets, we observed that M370I Foxp3 binding sites co-localized with GATA3 peaks within the Th2 locus in both Treg and Th2 cells (Figure 6E). These results suggested that GATA3 might act as “pioneer factor” to allow M370I Foxp3 to bind DNA and de-repress the Th2 locus in mutant Treg cells. GATA3 bound the Th2 locus only in in vivo generated Treg cells (most likely tTregs, Foxp3+Nrp1+Helios+), whereas GATA3 did not interact strongly with the Th2 locus in TGF-β induced Treg cells (iTregs) (Wei et al., 2011). This observation was consistent with experiments revealing that Th2 cytokines were not produced by iTreg cells expressing the mutant Foxp3 (Figure 6F), suggesting that M370I Foxp3 and GATA3 have to bind the Th2 cis-acting regulatory elements together to induce expression of Il4, Il5 and Il13 in Treg cells. To determine the molecular cooperation between GATA3 and M370I Foxp3, CRISPR-Cas9 gene editing system was used to knock out Gata3 in Treg cells from wild-type and M370I mutant mice. Rosa26Cas9 mice were crossed with the M370I mutant mice and a retroviral approach was used to introduce the sgRNA targeting Gata3. The majority (>80%) of the cells infected with virus expressing Gata3-specific sgRNAs down regulated GATA3 protein expression (Figure 7A). In the absence of GATA3, Th2 cytokine production by M370I Treg cells was reduced (Figures 7B and C) and the percentage of Treg cells producing cytokines were reduced to the level of the wild-type Treg cells (Figure 7C). These observations demonstrate a unique cooperation between M370I Foxp3 and GATA3, which led to the coordinated production of the canonical Th2 effector cytokines and drives a Th2-like phenotype in Treg cells.
Figure 7. Th2 effector cytokines production by M370I Treg cells requires GATA3.
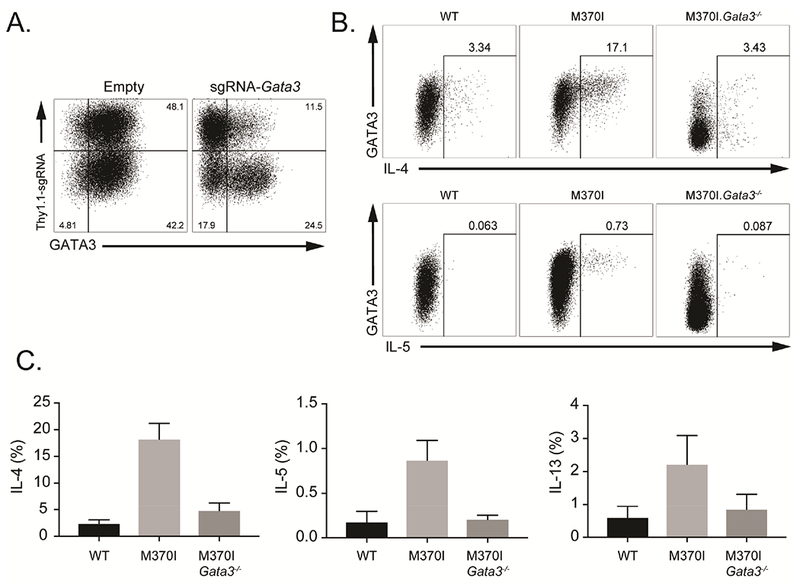
Representative flow cytometric analysis of expanded Treg cells infected with retrovirus expressing Thy1.1 or Thy1.1 and single guide RNA (sgRNA) targeting Gata3. (B) Representative flow cytometric analysis of IL-4 (bottom panel) and IL-5 (top panel) producing Treg cells after activation. Expanded Treg cells from Rosa26Cas9 (WT) or Rosa26Cas9 M370I Tg mouse were infected with retrovirus expressing Thy1.1 (M370I) or Thy1.1 and sgRNA targeting Gata3 (M370I Gata3−/−). (C) Quantification of cytokine producing cells. Data are mean ± SD and representative of at least three independent experiments.
Discussion
Naturally occurring IPEX mutations provide unique insight into FOXP3 biology that can be utilized to identify new pathways regulating Treg cell homeostasis and function (Bin Dhuban et al., 2017; Hayatsu et al., 2017). In this study, we identified an IPEX patient with a M370I mutation in FOXP3 who developed a number of autoimmune manifestations including several pathologies consistent with Th2 cell-mediated disease. We generated a transgenic mouse model in which the wild-type Foxp3 gene was disrupted and replaced with a mutant allele of Foxp3 that mimicked the mutation identified in the patient. This mutation led to the development of a disease similar to the one observed with the IPEX patient, namely, a Th2-like autoimmune syndrome.
A paradigm in T cell biology is the existence of different effector T cell subsets (Th1, Th2, Th17 and Tfh) defined by the expression of master transcription factors, T-bet, GATA3, RORγt and Bcl-6, respectively (Campbell and Koch, 2011). These key transcription factors are induced during the initiation of the immune response and control T cell differentiation, function and migration. Moreover, activated Treg cells can co-opt these transcriptional programs to tailor their phenotypic and functional characteristics to those of the CD4+ effector T cells they restrain. However, there is a risk that this plasticity can lead to loss of Foxp3 repressive functions and enable these Th-like Treg subsets to devolve into Tem cells that can generate a potent pathogenic response. Thus, the Foxp3 transcription factor has evolved to allow certain aspects of Th gene expression to develop, such as chemokine receptor expression, while continuing to repress cytokine production and Teff functions (DuPage and Bluestone, 2016). In this study, we show that a single point mutation in the FKH domain of Foxp3 disrupts the balance between the regulatory function of Foxp3 and the Th2 effector program.
Understanding the mechanisms that control the Th2 effector program in Treg cells is important because Th2-like Treg cells have been identified in multiple clinically relevant settings. Human and mouse Th2-like Treg cells have been identified in models of allergic diseases (Noval Rivas et al., 2015; Reubsaet et al., 2013), in tumor environments (Halim et al., 2017) and in the context of IL-2 therapy (Birjandi et al., 2016). Genome-wide integrative analysis of M370I mutant Treg cells by RNA-seq, ChIP-seq and H3K27ac HiChIP revealed de novo binding of M370I Foxp3 to Il4 and Il13 regulatory regions and the reorganization of the chromatin architecture at the Th2 locus, leading to a de-repression of the Th2 transcriptional program and the generation of Th2-like Treg cells. This study demonstrated a direct and active role for mutant Foxp3 protein in the generation of the Th2-like phenotype in Treg cells from IPEX samples. These observations are supported by previous studies suggesting a Th2 reprogramming of Treg cells by Foxp3 mutant proteins. Indeed, recently, Kwon and colleagues published an in vitro alanine-scan library of mouse Foxp3 (Kwon et al., 2018). Paradoxically, mutations of Foxp3 affecting DNA-binding and mutations localized in the FKH domain result in expression of unexpected target genes like Il4 and Il5. In addition, Hayatsu et al. describe a dysregulation of the Th2 immune response by one of the most common IPEX mutations, A384T (Hayatsu et al., 2017). A384T mutant Treg cells produce Th2 cytokines and our analysis of the Foxp3 ChIP-seq data set from the Hayatsu study revealed that A384T mutant Foxp3 also bound to the Th2 locus. The combined observations strongly suggest a role of Foxp3 in controlling Th2 programming in Treg cells.
Because neonatal immunity shows an inherent polarization of CD4+ T cells toward Th2 immunity (Restori et al., 2018) and because the Th2 arm of the immune response is the most sensitive to Treg dysfunction (Tian et al., 2011), it was crucial to separate a direct effect of M370I Foxp3 driving the Th2 transcriptional program in the mutant Treg cells versus a secondary effect due to an indirect response to a dominant Th2 environment. The systemic inhibition of IL-4 by neutralizing antibodies or the T cell-specific genetic ablation of Il4 and Il13 provided evidence for a direct effect of mutant Foxp3 on the Th2-like Treg phenotype. In addition, the expression of IL4ra by Treg cells enables a feed-forward loop to drive and stabilize the Th2-cell-like phenotype by maintaining optimal GATA3 expression (Noval Rivas et al., 2015) and amplifying Th2 reprogramming by both Foxp3 mutant-dependent and independent mechanisms.
In conclusion, IPEX mutations provide unique opportunities to leverage clinical observations to increase our understanding of fundamental Treg biology. Analysis of Foxp3 mutation M370I showed a direct role of a mutant Foxp3 protein in the Th2-like phenotype observed during Treg dysfunction and revealed the complex interplay between the regulatory function of Foxp3 and the Th2 effector program. A better understanding of the mechanisms controlling the differentiation of Th2-like Treg cells will inform novel therapeutic strategies for diseases like allergies, asthma and cancer.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeffrey A. Bluestone (jeff.bluestone@ucsf.edu).
Experimental Model and Subject Details
Human samples
PBMCs were collected from 4 months old male patient diagnosed with IPEX mutation M370I and from a healthy 7 years old male child. Cells were freshly isolated and evaluated for expression of CD4, CD25, CD127 and FOXP3. Intracellular staining was performed with FOXP3 staining kit according to the manufacturer’s instructions (BioLegend). Informed written consents were obtained in accordance with the reviewed and approved policies and procedures at the University of California, San Francisco (UCSF). Ethics approval was granted by the UCSF Institutional Review Board.
Mouse models
The single nucleotide variant c.1110G>A identified in IPEX patients was inserted via standard recombineering (Lee et al., 2001) into a BAC clone from the Children’s Hospital of Oakland Research Institute (CHORI) BACPAC Resource Center, clone 143d8 from the RPCI 23 Mouse BAC Library, which contained the Foxp3 gene and approximately 186Kb of flanking sequence carrying all regulatory regions required for Treg specific Foxp3 gene expression (Figure S1). The modified BAC was purified using cesium chloride gradient ultracentrifugation and microinjected by the UCSF Transgenic/Targeted Mutagenesis Core Facility into the pronuclei of C57BL/6 mouse embryos to generate Foxp3 M370I BAC Tg mice. Mice carrying the M370I transgene were breed with Foxp3−/− mice to analyze the function of IPEX mutation M370I. In order to facilitate the isolation of pure (>98%) populations of Foxp3+ Treg cells used for the H3K27ac HiChIP assays, the Foxp3-GFP-hcre transgene has been introduced to M370I Tg mice. For CRISPR/Cas9 genome editing, M370I-Tg mice have been cross with mice Rosa26cas9, expressing constitutively Cas9 endonuclease under the R26 locus. Mice with selective deletion of Il4-Il13 in T cells (Cd4-Cre crossed with IL4-IL13fl/fl) were kindly provided by Dr. R. M. Locksley. Age matched male mice were used within experiments, for sample size and age see corresponding figure legends. All mice were housed and bred under specific pathogen-free conditions at the UCSF Animal Barrier Facility. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of UCSF.
Method Details
Induction of colitis
Experimental colitis is induced by transfer of 5 105 Sorted naïve CD4+ CD45RBhigh T cells into immunodeficient Rag1−/− mice, which were weighed weekly to detect signs of wasting disease. After 3 weeks (at the onset of disease symptoms), 7 104 freshly isolated Tregs (CD4+CD25+CD73+GITR+ cells) were injected i.p. into the same recipients. Percent weight change is calculated by comparing the current weight to the initial weight at day 0 as follows: percent weight change = ((weight at day 0 − current weight)/weight at day 0) × 100 × −1.0. Mice were euthanized 4 weeks after Treg transfer.
Lymphocyte isolation, Cell Sorting, and Flow Cytometry
Single-cell suspensions were prepared from lymph nodes (cervical, mediastinal, axillary, Inguinal), spleen and thymus of the indicated mice. We used DMEM-glutamax medium (Thermo Fisher Scientific) supplemented with 10% heat-inactivated fetal calf serum, 100 U/ml of penicillin, 100 U/ml of streptomycin, nonessential amino acids, 10 mM HEPES buffer and 50 μM β-mercaptoethanol (all from Thermo Fisher Scientific) for cell culture. Purified populations of Treg cells (confirmed ≥95%) were obtained by sorting single cells with FACSAria (BD Biosciences) machines. Treg cell are identified as CD4+CD8− CD25+GITR+FR4+Foxp3+, for H3K27ac HiChIP, BAC expressing Foxp3-GFP was used to identify Treg cell as GFP+CD4+CD8−CD25+ cells. For flow analysis, single-cell suspensions were used for flow cytometry analysis with the indicated antibodies. Cells were stain with a fixable live/dead stain (Invitrogen) and fixed with BD Cytofix/Cytoperm (BD Biosciences) for cytokines detection or eBioscience Foxp3 staining buffer set (ThermoFisher). Flow cytometry was performed on LSRII flow cytometer (BD Biosciences) and analyzed with FlowJo software and compiled using Prism.
Retroviral production.
The sgRNA against mouse gata3 (TTCCGTAGTAGGACGGGACG) was design with the sgRNA designing software at Benchling (https://benchling.com) and cloned under a U6 promoter into MSCV-IRES-Thy1.1. Retroviral Plat-E packaging cells were transfected with the indicated plasmids using Fugene and the retroviral supernatant was used fresh.
In vitro expansion Treg cells and retroviral transduction.
Sorted Treg cells were stimulated with anti-CD3 and anti-CD28 Dynabeads at a 1:3 cell:bead ratio, supplemented with 2,000 IU/ml rhIL-2 (Chiron Corp) in complete medium. The cultures were monitored daily and maintained at 0.7–1 × 106cells/ml by diluting with IL-2-supplemented complete medium. Twenty-four hours after activation, Treg cells were centrifuged (90 minutes, 6000 rcf at 25°C) with viral supernatant supplemented with 10 μg/ml polybrene and after 3 or 4 days cells were harvested, rested overnight (anti-CD3 and anti-CD28 beads were removed with magnetic rack) and stimulated with 0.5 μM ionomycin, 10 ng/ml phorbol myristate acetate (PMA) and 3 μg/ml of Brefeldin A at 37Ό for 3–4 hours before be ing fixed and permeabilized (Cytofix/Cytoperm) and stained for intracellular proteins.
Cytokine production
Lymphocyte cells were activated at 10×106 cells/ml with 0.5 μM ionomycin, 10 ng/ml PMA and 3 μg/ml of Brefeldin A at 37°C for 3–4 hour before labeling with LIVE/DEAD fixable dead stain, and staining for CD4, CD8, Foxp3, IL-10, IL-4, IL-5 and IL-13 using Cytofix/Cytoperm.
In vivo IL-4 neutralization
2 weeks old M370I mutant mice were treated every other day for one week with 0.5mg of IL-4 neutralizing antibody (11B11) or rat IgG1 isotype control. Spleens were collected at 3 weeks of age and CD4+ T cells were analyzed for IL-4 and IL-13 cytokines production after PMA-ionomycin activation.
In Vitro stimulation and Suppression Assays.
Graded numbers of fresh sorted Treg cells were added to 50,000 CD4+ T cells stimulated with 50,000 irradiated splenic APC (spleen of Rag1−/− mice, red blood cell lysis and γ-irradiation (2,000 rad)) and 1 μg/ml anti-CD3 (clone 145-2C11) in a U-bottomed 96-well plate. Responder CD4+ T cells were labeled with Cell Trace Violet (CTV) before the assay, and the level of proliferation was assessed by determining the dilution of CTV using flow cytometry 72-96 h after the initiation of the culture. Alternatively, Treg cells were co-cultured with CTV labeled responder CD4+ T cells in an APC free system in the presence of plate bound anti-CD3 (clone 145-2C11) and anti-CD28 (clone PV-1) mAbs (0.5 μg/ml each). After 4 days cells were harvested and labeled with LIVE/DEAD fixable dead stain before being fixed and permeabilized (Foxp3 Staining Kit, Thermo Fisher Scientific) for intracellular staining.
iTreg Induction
Naïve CD44lowCD62LhiCD25−CD4+ T cells were sorted from pooled LN and spleen, activated with plate bound anti-CD3 (clone 145-2C11) and anti-CD28 (clone PV-1) mAbs (0.5 μg/ml each) with 100U/ml of recombinant human IL-2 (Proleukin) and 20 ng/ml human recombinant TGF-β (Humanzyme). After 3 or 4 days cells were harvested, rest overnight and stimulated with 0.5 μM ionomycin, 10 ng/ml PMA and 3 μg/ml of Brefeldin A at 37°C for 3–4 hours before being fixed and perm eabilized (Cytofix/Cytoperm) and stained for intracellular proteins.
Cytokine and Ig ELISA
Cytokine concentrations were measured by commercial ELISA kits according to the manufacturer’s recommendations (Thermo Fisher Scientific). Cell cultures supernatant were obtained from triplicate cultures after overnight stimulation with PMA-ionomycin of Treg cells sorted from wild-type or M370I mice.
Histology
Lungs and skin were isolated from M370I and wild-type littermates, fixed in 10% formalin overnight, preserved in 70% EtOH, and embedded in paraffin. Samples were cut and sections stained with hematoxylin and eosin (H&E). Infiltrate identification was performed by blinded assessment of at least three sections of each tissue per mouse. Chromogenic multiplex immunohistochemistry was performed with anti-CD4 (EPR19514) at 1:1000 dilution, anti-CD8a (4SM15) at 1:50 dilution, and anti-Foxp3 (FJK-16 s) at 1:50 dilution.
RNA Isolation and RT q-PCR analysis
RNA was extracted using Qiagen’s miRNeasy kit or TRIzol homogenization and extraction (Thermo Fisher Scientific). RNA was converted to cDNA using Maxima First Strand cDNA synthesis Kit for RT-qPCR, and gene expression was measured in real time using Taqman probe for Il4, Il5, Il13 with Taqman Fast Universal mix. Samples were run in triplicate on the Applied Biosystems 7500/7900 Fast Real-Time PCR System and normalized to Hprt or 18S RNA.
RNA-seq
RNA-seq experiments were performed at the Sandler Asthma Basic Research Center Functional Genomics Core using the Illumina HiSeq Se 50pb platform with total RNA isolated from FACS-sorted Treg cells (CD4+CD8−CD25+GITR+FR4+Foxp3+, confirmed ≥95%) from wild-type and M370I mice. Quality of raw FASTQ sequences was assessed using FASTQC. To process RNA-Seq libraries, adaptor sequences were trimmed using Cutadapt version 1.14 (requiring a length greater than 10 nt after trimming) and quality-filtered by requiring all bases to have a minimum score of 20 (-m 20 -q 20). Only reads that passed the quality or length threshold on both strands were considered for mapping. Reads were aligned to the GRCm38/mm10 mouse genome reference with the STAR Aligner (version 020201). Ensembl reference annotation version 89 was used to define gene models for mapping quantification. Uniquely mapped reads for each gene model were produced using STAR parameter “—quantMode GeneCounts”. Signal files for IGV visualization were generated from STAR – outWigType bedGraph. Differential expression analysis was performed in R using DESeq2 (v.1.16.0) with the default parameters, including the Cook’s distance treatment to remove outliers.
Foxp3 ChIP-seq
Foxp3 ChIP-seq was performed as previously described (Zheng et al., 2007). Briefly, 3× 106 sorted Tregs were fixed with 1% formaldehyde for 10 min. The cells were washed twice with cold PBS and flash frozen at −80C. The chromatin was sonicated to yield fragments of 200-500bp in length, followed by overnight incubation with polyclonal rabbit Foxp3 antibody. Precipitated chromatin was washed, de-crosslinked and DNA was isolated using phenol-chloroform extraction method. ChIP DNA was prepared for high throughput sequencing library using Accel-NGS 2S Plus DNA library kit (Swift Biosciences) according to manufacturer’s protocol. ChIP DNA libraries were sequenced on an Illumina Hiseq4000 with a single-end 50bp run. Quality of raw FASTQ sequences was assessed using FASTQC. Since over 90% of the reads had sequence quality scores over Q33 and the lowest mean quality score per sample (per base) was 27, reads were not trimmed based on quality and the raw sequence data was used for further analysis. Reads were mapped to the mouse mm10 genome sequence (GRCm38) using BWA [Li H]. Duplicates removal was performed using PICARD MarkDuplicates tool (version 2.17.10) (http://picard.sourceforge.net), by only keeping uniquely mapped reads. ChIP-seq peaks were called using MACS2 (version 2.1.1.20160309). Differential binding events between conditions were calculated using the “bdgdiff” module in MACS2. BWA-aligned peaks and MACS2-determined peak positions were visualized using IGV.
H3K27ac HiChIP
BAC expressing Foxp3-GFP was used to identify Treg cell as GFP+CD4+CD8”CD25+ (confirmed ≥98%). HiChIP was performed as previously described (Mumbach et al 2017) using the H3K27ac antibody (Abcam ab4729). HiChIP paired-end reads were aligned to the mm9 genome using the HiC-Pro pipeline (Servant et al., 2015). Default settings were used to remove duplicate reads, assign reads to MboI restriction fragments, filter for valid interactions, and generate binned interaction matrices. HiC-Pro filtered reads were then processed into a .hic file using the hicpro2juicebox function (Servant et al., 2015). The Juicer pipeline HiCCUPS tool was used to identify high confidence loops using the same parameters as for the GM12878 in situ Hi-C map: hiccups -m 500 -r 5000,10000 -f 0.1,0.1 -p 4,2 -i 7,5 -d 20000,20000 .hic_input HiCCUPS_output (Durand et al., 2016a; Durand et al., 2016b; Rao et al., 2014). Virtual 4C plots were generated from dumped matrices generated with Juicebox. The Juicebox tools dump command was used to extract the chromosome of interest from the .hic file (Durand et al., 2016a; Durand et al., 2016b; Rao et al., 2014). The interaction profile of a specific 5 kb or 10 kb bin containing the anchor was then plotted in R. For 1 kb resolution profiles, matrices were obtained from HiC-Pro output (Servant et al., 2015). Virtual 4C profiles of different HiChIP samples were normalized by total cis interactions for each replicate. A counts matrix was constructed on the union HiCCUPS loop calls. For the wild-type and M370I pairwise comparison edgeR’s exact test (exactTest) was used and differential loops were called. Foxp3 ChIP signal at unique differential loop anchors was calculated using bedtools intersect.
Experimental Design and Statistical Analysis
Statistical tests used for bioinformatics analysis were described in methods or legends. p values from unpaired two-tailed Student’s t tests (calculated with Prism software) were used for all other statistical comparisons between two groups and data were displayed as the mean ± SE unless otherwise stated. p values are denoted in figures by: *, p < 0.05; **, p < 0.01; ***, p < 0.001. All data points reflect individual biological replicates.
Supplementary Material
Table S1: HiCCUPS differential loops in Treg and Teff cells from wild-type and M370I mice. Related to Figure 5A.
Table S2: HiCCUPS differential EIS in wild-type and M370 Treg cells. 281 loops, of a total of 9,895 loops (2.8%), were identified differentially between wild-type and M370I samples based on a log2 (fold change) >1 and false discovery rate <0.1. Sheet 1 listed all the M370I-biased loops (139) and sheet 2 listed all the WT-biased loops (142). The loops highlight in green have Foxp3 ChIP at the anchor, loops highlight in red overlap with the Teff H3K27ac HiChIP loops. Related to Figure 5A.
Table S3: Foxp3 ChIP peaks from the Foxp3 ChIP-seq analysis of wild-type and M370I Treg cells. Related to Figure 5C.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD4, PerCP (clone SK3) | BD Biosciences | Cat# 347324 |
| Anti-human CD127, PE (clone hIL-7R-M21) | BD Biosciences | Cat# 557938 |
| Anti-human CD25, APC (clone 2A3) | BD Biosciences | Cat# 340939 |
| Anti-human FOXP3, AF488 (clone 206D) | Biolegend | Cat# 320112 |
| Anti-mouse CD4, PerP (clone RM4-5) | BioLegend | Cat# 100538 |
| Anti-mouse CD8a, P. Orange (clone 5H10) | Invitrogen | Cat# MCD0830 |
| Anti-mouse CD25, Pe-Cy7 (clone PC61.5) | Thermo Fisher Scientific | Cat# 25-0251-82 |
| Anti-mouse CD39, PE (clone, 24DMS1) | Thermo Fisher Scientific | Cat# 12-0391-80 |
| Anti-mouse CD44, PE (clone, IM7) | BD Biosciences | Cat# 553134 |
| Anti-mouse CD45, BUV395 (clone, 30-F11) | BD Biosciences | Cat# 564279 |
| Anti-mouse CD62-L, APC (clone MEL-14) | Thermo Fisher Scientific | Cat# 17-0621-82 |
| Anti-mouse CD127, Pe-Cy7 (clone A7R34) | Thermo Fisher Scientific | Cat# 25-1271-82 |
| Anti-mouse CTLA4/CD152, PE (UC10-4F10) | BD Biosciences | Cat# 553720 |
| Anti-mouse FR4, Pe-Cy7 (clone eBio12A5) | Thermo Fisher Scientific | Cat# 25-5445-82 |
| Anti-mouse ICOS, PerCP/Cy5.5 (clone C398.4A) | BioLegend | Cat# 313518 |
| Anti-mouse KLRG1, PerCP/Cy5.5 (clone 2F1) | BioLegend | Cat# 138418 |
| Anti-mouse CXCR5, Biotin (clone 2G8) | BD Biosciences | Cat# 551960 |
| Anti-mouse CCR7/CD197, Biotin (clone 4B12) | Thermo Fisher Scientific | Cat# 13-1971-82 |
| Anti-mouse GITR (clone DTA-1) | Thermo Fisher Scientific | Cat# 17-5874-81 |
| Anti-mouse PD-1, Pe-Cy7 (RPM1-30) | BioLegend | Cat# 109110 |
| Anti-mouse PD-L2, PE (122) | Thermo Fisher Scientific | Cat# 12-9972-81 |
| Anti-mouse CD85k, AF647 (H1.1) | BioLegend | Cat# 144905 |
| Anti-mouse SATB1, AF647 (clone 14/SATB1) | BD Biosciences | Cat# 562378 |
| Anti-mouse IRF4, eF660 (clone 3E4) | Thermo Fisher Scientific | Cat# 50-9858-80 |
| Anti-mouse ST2, Biotin (clone DJ8) | MD Bioproducts | Cat# 101001 |
| Anti-mouse Ki67, PE (clone B56) | BD Biosciences | Cat# 556027 |
| Anti-mouse CD304 (Nrp1) | R&D systems | Cat# FAB566A |
| StreptAvidin APC | BioLegend | Cat# 405207 |
| StreptAvidin PE | BioLegend | Cat# 405204 |
| Anti-mouse Foxp3, FITC (clone FKJ-16s) | Thermo Fisher Scientific | Cat# 11-5773-82 |
| Anti-mouse GATA3, PE (clone TWAJ) | Thermo Fisher Scientific | Cat# 12-9966-41 |
| Anti-mouse T-bet, PE (clone eBio4B10) | Thermo Fisher Scientific | Cat# 12-5825-80 |
| Anti-mouse Helios, PE (clone 22F6) | BioLegend | Cat# 137216 |
| Anti-mouse IL-4, PE (clone 11B11) | BD Biosciences | Cat# 554435 |
| Anti-mouse IL-5, PE (clone TRFK5) | Thermo Fisher Scientific | Cat# 12-7052-81 |
| Anti-mouse IL-13, PE-Cy7 (clone eBio13A) | Thermo Fisher Scientific | Cat# 12-7133-81 |
| Anti-mouse IL-17A, APC (clone eBio17B7) | Thermo Fisher Scientific | Cat# 17-7177-81 |
| Anti-mouse IL-10, APC (clone JES5-16E3) | Thermo Fisher Scientific | Cat# 17-7101-81 |
| Anti-mouse IFNg, eF450 (clone XMG1.2) | Thermo Fisher Scientific | Cat# 48-7311-82 |
| anti-mouse CD8 (clone 4SM15) for Histology use | Thermo Fisher Scientific | Cat# 14-0808-80 |
| anti-mouse CD4 (clone EPR19514) for Histology use | Abcam | Cat# ab183685 |
| H3K27ac antibody (Rabbit polyclonal) | Abcam | Cat# ab4729 |
| Rabbit polyclonal anti-mouse Foxp3 peptide | Rudensky Lab | NA |
| Anti-CD3 and anti-CD28 Dynabeads | Thermo Fisher Scientific | Cat# 11456D |
| Anti-mouse CD3 (clone 145-2C11) for in vitro use | In house | NA |
| Anti-mouse CD28 (clone PV-1) for in vitro use | In house | NA |
| Anti-mouse IL-4 (clone 11B11) for in vitro use | Tonbo | Cat# 70-7041 |
| Anti-mouse IL-4 (clone 11B11) for in vivo use | BioXCell | Cat# BE0045 |
| Anti-trinitrophenol, Rat IgG1 Isotype control | BioXCell | Cat# BE0290 |
| Biological Samples | ||
| PBMC from 4 months old M370I IPEX male infant | UCSF children’s hospital | N/A |
| PBMC from 7 years old Healthy male child | UCSF children’s hospital | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| FuGENE HD Transfection Reagent | Promega | Cat# E2311 |
| Trizol Reagent | Thermo Fisher Scientific | Cat# 1559626 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma-Aldrich | Cat# P1585 |
| Ionomycin | Sigma-Aldrich | Cat# I0634 |
| Brefeldin A | Thermo Fisher Scientific | Cat# 00-4506-51 |
| LIVE/DEAD fixable dead stain | Thermo Fisher Scientific | Cat# L23105 |
| CellTrace Violet Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C34557 |
| Human recombinant TGF-β | Humanzyme | Cat# HZ-1011 |
| Human recombinant IL2 | Chiron | NA |
| Critical Commercial Assays | ||
| BD Cytofix/Cytoperm | BD Biosciences | Cat# 554714 |
| Foxp3/Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat# 00-5523-00 |
| FOXP3 Fix/Perm Buffer Set | BioLegend | Cat# 421403 |
| Mouse IFNg ELISA Ready-SET-Go | Thermo Fisher Scientific | Cat# 50-173-19 |
| Mouse IL-4 ELISA Ready-SET-Go | Thermo Fisher Scientific | Cat# 50-171-98 |
| Mouse IL-5 ELISA Ready-SET-Go | Thermo Fisher Scientific | Cat# 50-172-09 |
| Mouse IL-13 ELISA Ready-SET-Go | Thermo Fisher Scientific | Cat# 50-172-61 |
| Mouse IgG (Total) ELISA | Thermo Fisher Scientific | Cat# 88-50400-22 |
| Mouse IgG1 ELISA | Thermo Fisher Scientific | Cat# 88-50410-22 |
| Maxima First Strand cDNA Synthesis Kit for RT-qPCR | Thermo Fisher Scientific | Cat# K1671 |
| miRNeasy Mini Kit | Qiagen | Cat# 217004 |
| TaqMan Fast Universal PCR Master Mix | Thermo Fisher Scientific | Cat# 4352042 |
| Deposited Data | ||
| RNAseq , ChIP-seq and HiChIP files | This study | GEO: GSE112176 |
| Experimental Models: Cell Lines | ||
| Human: Plat-E cells | Cell Biolabs | Cat# RV-101 |
| Experimental Models: Organisms/Strains | ||
| Mouse: M370I Tg: B6.Tg(Foxp3-M370I)1aJbs/J | This study | N/A |
| C57BL/6J | Jackson laboratory | Stock# 000664 |
| Mouse: Rag1−/−: B6.129S7-Rag1tm1Mom/J | Jackson laboratory | Stock# 002216 |
| Mouse: Cd4-Cre: B6.Tg(Cd4-cre)1Cwi/BfluJ | Jackson laboratory | Stock# 017336 |
| Mouse: Foxp3−/−: B6.Cg-Foxp3tm1Tch/J | Jackson laboratory | Stock# 019933 |
| Mouse: Foxp3-GFP-hCre: B6J.129S-Tg(Foxp3-EGFP/icre)1aJbs/J | Jackson laboratory | Stock# 023161 |
| Mouse: Rosa26Cas9, B6J.129(Cg)-Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J | Jackson laboratory | Stock# 026179 |
| Mouse: Il4-Il13fl/fl: B6.129P2(Cg)-Il4/Il13tm1.1Lky/J | Dr. R.M. Locksley | N/A |
| Oligonucleotides | ||
| GATA3 sgRNA: TTCCGTAGTAGGACGGGACG | This study | N/A |
| TaqMan primer probes IL4 (Mm00445259_m1) | Thermo Fisher Scientific | Cat# 4331182 |
| TaqMan primer probes IL5 (Mm00439646_m1) | Thermo Fisher Scientific | Cat# 4331182 |
| TaqMan primer probes IL13 (Mm00434204_m1) | Thermo Fisher Scientific | Cat# 4331182 |
| Eukaryotic 18S rRNA Endogenous Control | Thermo Fisher Scientific | Cat# 4333760F |
| TaqMan primer probes HPRT (Mm00446968_m1) | Thermo Fisher Scientific | Cat# 4331182 |
| Recombinant DNA | ||
| pMSCV-U6-sgRNA-IRES-Thy1.1 | This study | N/A |
| Mouse Foxp3 BAC, M370I modified clone RP23-143D8 | This study | N/A |
| Software and Algorithms | ||
| FlowJo Software | Tree Star | N/A |
| GraphPad Prism | GraphPad Software, Inc. | N/A |
| Integrative Genomics Viewer (IGV) |
http://software. broadinstitute.org/ software/igv/ |
N/A |
ACKNOWLEDGMENTS
We deeply appreciate the support, cooperation, and trust of the patient family. In addition, this work wouldn’t be possible without the help of the clinical teams treating the patient. The authors thank Linda Vo and Jessica Barragan for helpful comments on the manuscript, A. Arvey, H. Bandukwala, A. Rao, R. Bacchetta and MG. Roncarolo for discussions and research insights. We thank D. Erle, A. Barczak, R. Barbeau, and W. Eckalbar of the UCSF Sandler Center Functional Genomics Core for assistance with RNAseq data; Jennifer Bolen and the UCSF immunohistochemistry core facility, M. Lee, V. Nguyen, and UCSF Flow Core for assisting with flow cytometry; D. Simeonov for help with sgRNA design and Yanli Wang for animal care. This work was supported by NIH P50-HG007735 (H.Y.C.) and the Sean N. Parker Autoimmune Research Laboratory (J.A.B.). A.M. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund, has received support for the Innovative Genomics Institute (IGI), and is an investigator at the Chan Zuckerberg Biohub. J.A.B. is the A.W. and Mary Margaret Clausen Distinguished Professor in Metabolism and Endocrinology. A.Y.R and H.Y.C. are Investigators of the Howard Hughes Medical Institute.
DECLARATION OF INTERESTS
J.A.B. is a consultant for Juno, a Celgene company; a stock holder and member of the Board of Directors on Rheos Medicines, a stock holder and member of the Scientific Advisory Boards of Pfizer Center for Therapeutic Innovation, Vir Therapeutics, Arcus Biotherapeutics, Quentis Therapeutics, Solid Biosciences, and Celsius Therapeutics. J.A.B. owns stock in MacroGenics Inc., Vir Therapeutics, Arcus Biotherapeutics, Quentis Therapeutics, Solid Biosciences, Celsius Therapeutics and Kadmon Holdings. A.M. is a co-founder of Spotlight Therapeutics. A.M. has served as an advisor to Juno Therapeutics and is a member of the Scientific Advisory Board at PACT Pharma. The Marson laboratory has received sponsored research support from Juno Therapeutics, Epinomics, Sanofi and a gift from Gilead. H.Y.C. is a co-founder of Accent Therapeutics and advisor to 10× Genomics and Spring Discovery.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession Numbers
The GEO accession number for the RNA-seq, Foxp3 CIP-seq and H3K27ac HiChIP data reported in this paper is GSE112176.
SUPPLEMENTAL INFORMATION
Supplemental Information includes five Figures and three Tables
REFERENCES
- An YF, Xu F, Wang M, Zhang ZY, and Zhao XD (2011). Clinical and molecular characteristics of immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome in China. Scandinavian journal of immunology 74, 304–309. [DOI] [PubMed] [Google Scholar]
- Andersen KG, Nissen JK, and Betz AG (2012). Comparative Genomics Reveals Key Gain-of-Function Events in Foxp3 during Regulatory T Cell Evolution. Frontiers in immunology 3, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvey A, van der Veeken J, Samstein RM, Feng Y, Stamatoyannopoulos JA, and Rudensky AY (2014). Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nature immunology 15, 580–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacchetta R, Barzaghi F, and Roncarolo MG (2016). From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Annals of the New York Academy of Sciences. [DOI] [PubMed] [Google Scholar]
- Bandukwala HS, Wu Y, Feuerer M, Chen Y, Barboza B, Ghosh S, Stroud JC, Benoist C, Mathis D, Rao A, and Chen L (2011). Structure of a domain-swapped FOXP3 dimer on DNA and its function in regulatory T cells. Immunity 34, 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, Nademi Z, Slatter MA, Ulloa ER, Shcherbina A, et al. (2017). Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: An international multicenter retrospective study. The Journal of allergy and clinical immunology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, and Ochs HD (2001). The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nature genetics 27, 20–21. [DOI] [PubMed] [Google Scholar]
- Bin Dhuban K, d’Hennezel E, Nagai Y, Xiao Y, Shao S, Istomine R, Alvarez F, Ben-Shoshan M, Ochs H, Mazer B, et al. (2017). Suppression by human FOXP3(+) regulatory T cells requires FOXP3-TIP60 interactions. Science immunology 2. [DOI] [PubMed] [Google Scholar]
- Birjandi SZ, Palchevskiy V, Xue YY, Nunez S, Kern R, Weigt SS, Lynch JP 3rd, Chatila TA, and Belperio JA (2016). CD4(+)CD25(hi)Foxp3(+) Cells Exacerbate Bleomycin-Induced Pulmonary Fibrosis. The American journal of pathology 186, 2008–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, and Ramsdell F (2001). Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nature genetics 27, 68–73. [DOI] [PubMed] [Google Scholar]
- Cai S, Lee CC, and Kohwi-Shigematsu T (2006). SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nature genetics 38, 1278–1288. [DOI] [PubMed] [Google Scholar]
- Campbell DJ, and Koch MA (2011). Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nature reviews. Immunology 11, 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Hennezel E, Ben-Shoshan M, Ochs HD, Torgerson TR, Russell LJ, Lejtenyi C, Noya FJ, Jabado N, Mazer B, and Piccirillo CA (2009). FOXP3 forkhead domain mutation and regulatory T cells in the IPEX syndrome. The New England journal of medicine 361, 1710–1713. [DOI] [PubMed] [Google Scholar]
- d’Hennezel E, Bin Dhuban K, Torgerson T, and Piccirillo CA (2012). The immunogenetics of immune dysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. Journal of medical genetics 49, 291–302. [DOI] [PubMed] [Google Scholar]
- Delacher M, Imbusch CD, Weichenhan D, Breiling A, Hotz-Wagenblatt A, Trager U, Hofer AC, Kagebein D, Wang Q, Frauhammer F, et al. (2017). Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nature immunology 18, 1160–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, and Bluestone JA (2016). Harnessing the plasticity of CD4(+) T cells to treat immunemediated disease. Nature reviews. Immunology 16, 149–163. [DOI] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, and Aiden EL (2016a). Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell systems 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, and Aiden EL (2016b). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell systems 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, and Rudensky AY (2003). Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nature immunology 4, 330–336. [DOI] [PubMed] [Google Scholar]
- Gavin MA, Torgerson TR, Houston E, DeRoos P, Ho WY, Stray-Pedersen A, Ocheltree EL, Greenberg PD, Ochs HD, and Rudensky AY (2006). Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proceedings of the National Academy of Sciences of the United States of America 103, 6659–6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim L, Romano M, McGregor R, Correa I, Pavlidis P, Grageda N, Hoong SJ, Yuksel M, Jassem W, Hannen RF, et al. (2017). An Atlas of Human Regulatory T Helper-like Cells Reveals Features of Th2-like Tregs that Support a Tumorigenic Environment. Cell reports 20, 757–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansmann L, Schmidl C, Kett J, Steger L, Andreesen R, Hoffmann P, Rehli M, and Edinger M. (2012). Dominant Th2 differentiation of human regulatory T cells upon loss of FOXP3 expression. Journal of immunology 188, 1275–1282. [DOI] [PubMed] [Google Scholar]
- Harakal J, Rival C, Qiao H, and Tung KS (2016). Regulatory T Cells Control Th2-Dominant Murine Autoimmune Gastritis. Journal of immunology 197, 27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayatsu N, Miyao T, Tachibana M, Murakami R, Kimura A, Kato T, Kawakami E, Endo TA, Setoguchi R, Watarai H, et al. (2017). Analyses of a Mutant Foxp3 Allele Reveal BATF as a Critical Transcription Factor in the Differentiation and Accumulation of Tissue Regulatory T Cells. Immunity 47, 268–283 e269. [DOI] [PubMed] [Google Scholar]
- Hori S, Nomura T, and Sakaguchi S (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061. [DOI] [PubMed] [Google Scholar]
- Khattri R, Cox T, Yasayko SA, and Ramsdell F (2003). An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nature immunology 4, 337–342. [DOI] [PubMed] [Google Scholar]
- Kinnunen T, Chamberlain N, Morbach H, Choi J, Kim S, Craft J, Mayer L, Cancrini C, Passerini L, Bacchetta R, et al. (2013). Accumulation of peripheral autoreactive B cells in the absence of functional human regulatory T cells. Blood 121, 1595–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HK, Chen HM, Mathis D, and Benoist C (2018). FoxP3 scanning mutagenesis reveals functional variegation and mild mutations with atypical autoimmune phenotypes. Proceedings of the National Academy of Sciences of the United States of America 115, E253–E262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, and Copeland NG (2001). A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73, 56–65. [DOI] [PubMed] [Google Scholar]
- Lee GR (2014). Transcriptional regulation of T helper type 2 differentiation. Immunology 141, 498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GR, Fields PE, Griffin TJ, and Flavell RA (2003). Regulation of the Th2 cytokine locus by a locus control region. Immunity 19, 145–153. [DOI] [PubMed] [Google Scholar]
- Lin W, Truong N, Grossman WJ, Haribhai D, Williams CB, Wang J, Martin MG, and Chatila TA (2005). Allergic dysregulation and hyperimmunoglobulinemia E in Foxp3 mutant mice. The Journal of allergy and clinical immunology 116, 1106–1115. [DOI] [PubMed] [Google Scholar]
- Lu L, Barbi J, and Pan F (2017). The regulation of immune tolerance by FOXP3. Nature reviews. Immunology 17, 703–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moes N, Rieux-Laucat F, Begue B, Verdier J, Neven B, Patey N, Torgerson TT, Picard C, Stolzenberg MC, Ruemmele C, et al. (2010). Reduced expression of FOXP3 and regulatory T-cell function in severe forms of early-onset autoimmune enteropathy. Gastroenterology 139, 770–778. [DOI] [PubMed] [Google Scholar]
- Mumbach MR, Satpathy AT, Boyle EA, Dai C, Gowen BG, Cho SW, Nguyen ML, Rubin AJ, Granja JM, Kazane KR, et al. (2017). Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nature genetics 49, 1602–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noval Rivas M, Burton OT, Wise P, Charbonnier LM, Georgiev P, Oettgen HC, Rachid R, and Chatila TA (2015). Regulatory T cell reprogramming toward a Th2-cell-like lineage impairs oral tolerance and promotes food allergy. Immunity 42, 512–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restori KH, Srinivasa BT, Ward BJ, and Fixman ED (2018). Neonatal Immunity, Respiratory Virus Infections, and the Development of Asthma. Frontiers in immunology 9, 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reubsaet L, Meerding J, Giezeman R, de Kleer I, Arets B, Prakken B, Beekman J, and van Wijk F (2013). Der p 1-induced CD4(+)FOXP3(+)GATA3(+) T cells have suppressive properties and contribute to the polarization of the TH2-associated response. The Journal of allergy and clinical immunology 132, 1440–1444. [DOI] [PubMed] [Google Scholar]
- Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, Leslie C, Shaffer SA, Goodlett DR, and Rudensky AY (2012). Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nature immunology 13, 1010–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, Neph S, Sabo P, Kim JM, Liao W, et al. (2012). Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 151, 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J, and Barillot E (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome biology 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilianakis CG, and Flavell RA (2004). Long-range intrachromosomal interactions in the T helper type 2 cytokine locus. Nature immunology 5, 1017–1027. [DOI] [PubMed] [Google Scholar]
- Tian L, Altin JA, Makaroff LE, Franckaert D, Cook MC, Goodnow CC, Dooley J, and Liston A (2011). Foxp3(+) regulatory T cells exert asymmetric control over murine helper responses by inducing Th2 cell apoptosis. Blood 118, 1845–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulges A, Klein M, Reuter S, Gerlitzki B, Hoffmann M, Grebe N, Staudt V, Stergiou N, Bohn T, Bruhl TJ, et al. (2015). Protein kinase CK2 enables regulatory T cells to suppress excessive TH2 responses in vivo. Nature immunology 16, 267–275. [DOI] [PubMed] [Google Scholar]
- Vinuesa CG, and Cyster JG (2011). How T cells earn the follicular rite of passage. Immunity 35, 671–680. [DOI] [PubMed] [Google Scholar]
- Wan YY, and Flavell RA (2007). Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature 445, 766–770. [DOI] [PubMed] [Google Scholar]
- Wang Y, Su MA, and Wan YY (2011). An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity 35, 337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G, Abraham BJ, Yagi R, Jothi R, Cui K, Sharma S, Narlikar L, Northrup DL, Tang Q, Paul WE, et al. (2011). Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity 35, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, et al. (2001). X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nature genetics 27, 18–20. [DOI] [PubMed] [Google Scholar]
- Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, Hall JA, Yagi R, Naik S, Bhairavabhotla R, et al. (2011). GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. The Journal of clinical investigation 121, 4503–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Blanco DB, Neale G, Vogel P, Avila J, Clish CB, Wu C, Shrestha S, Rankin S, Long L, et al. (2017). Homeostatic control of metabolic and functional fitness of Treg cells by LKB1 signalling. Nature 548, 602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Sharma S, Edwards J, Feigenbaum L, and Zhu J (2015). Dynamic expression of transcription factors T-bet and GATA-3 by regulatory T cells maintains immunotolerance. Nature immunology 16, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, and Rudensky AY (2007). Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature 445, 936–940. [DOI] [PubMed] [Google Scholar]
- Zhou X, Jeker LT, Fife BT, Zhu S, Anderson MS, McManus MT, and Bluestone JA (2008). Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. The Journal of experimental medicine 205, 1983–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: HiCCUPS differential loops in Treg and Teff cells from wild-type and M370I mice. Related to Figure 5A.
Table S2: HiCCUPS differential EIS in wild-type and M370 Treg cells. 281 loops, of a total of 9,895 loops (2.8%), were identified differentially between wild-type and M370I samples based on a log2 (fold change) >1 and false discovery rate <0.1. Sheet 1 listed all the M370I-biased loops (139) and sheet 2 listed all the WT-biased loops (142). The loops highlight in green have Foxp3 ChIP at the anchor, loops highlight in red overlap with the Teff H3K27ac HiChIP loops. Related to Figure 5A.
Table S3: Foxp3 ChIP peaks from the Foxp3 ChIP-seq analysis of wild-type and M370I Treg cells. Related to Figure 5C.