

Summary
Influenza viruses inhabit a wide range of host environments using a limited repertoire of protein components. Unlike viruses with stereotyped shapes, influenza produces virions with significant morphological variability even within clonal populations. Whether this tendency to form pleiomorphic virions is coupled to compositional heterogeneity and whether it affects replicative fitness remains unclear. Here we address these questions by developing a strain of influenza A virus amenable to rapid compositional characterization through quantitative, site-specific labeling of viral proteins. Using this strain, we find that influenza A produces virions with broad variations in size and composition from even single infected cells. This phenotypic variability contributes to virus survival during environmental challenges including temperature changes and exposure to antivirals. Complementing genetic adaptations that act over larger populations and longer times, this ‘low fidelity’ assembly of influenza A virus allows small populations to survive environments that fluctuate over individual replication cycles.
Graphical Abstract
In Brief
Live imaging of influenza A viral particles demonstrates that virion protein composition and morphology varies widely within the population produced by a single infected cell, suggesting that stochastic viral assembly may mediate phenotypic diversity and viral evolution.
Introduction
In order to replicate, viruses must navigate complex and unpredictable environments inside and outside the host. Genetic variability within virus populations is central to this, helping viruses evade host immunity (Dingens et al., 2017), withstand antiviral drugs (Belshe et al., 1988), expand host range (Ma et al., 2016), and adopt new routes of transmission (Imai et al., 2012). In RNA viruses, the basis for genetic variability is virus-encoded error-prone polymerases that enhance the rate of mutation several orders of magnitude above that of the host (Pauly et al., 2017; Sanjuan et al., 2010). Combined with the large number of progeny (~103–104) produced from an individual infected cell (Chen et al., 2007; Stray and Air, 2001), this allows viruses to sample a large genetic space more rapidly than their hosts, helping them to evolve around adaptive and therapeutic defenses, and to benefit from cooperative interactions among co-infecting quasispecies (Clavel and Hance, 2004; Vignuzzi et al., 2006). Recently, large pools of genetic diversity have also been shown to be instrumental in the evolution of drug resistance via epistasis, allowing permissive mutations to reside in a population that can stabilize subsequent mutations conferring resistance (Bloom et al., 2010; Pennings, 2012; Rong et al., 2010).
In addition to genetic variability, many RNA viruses share significant morphological variability, as revealed by electron microscopy (Battisti et al., 2012; Bharat et al., 2012; Calder et al., 2010). Influenza A virus (IAV), an enveloped respiratory virus with recurring pandemic potential, is found in a morphologically heterogeneous ‘filamentous’ form that predominates in human isolates, with virions having a uniform diameter of ~80–100 nm but lengths ranging in size from ~100nm to several microns (Badham and Rossman, 2016; Chu et al., 1949; Dadonaite et al., 2016). Although this morphological variability is known to be influenced by the sequence of the viral matrix protein (Burleigh et al., 2005; Elleman and Barclay, 2004), relatively little is known regarding how it may influence infectivity and transmission or whether virus composition is similarly variable. Unlike viruses with highly ordered capsids and envelopes, the glycoprotein spikes of IAV do not appear to be highly ordered (Harris et al., 2006; Wasilewski et al., 2012), suggesting that IAV may lack a specific mechanism of tightly regulating envelope composition in addition to particle size.
Heterogeneity in virus morphology and composition, which we refer to together as phenotypic variability, could influence IAV transmission in a number of ways. As the virus releases from an infected cell, transports within or between hosts, and binds to a naive target cell, it must form sufficiently stable attachments to enable infection, while still being able to break those attachments to enable transmission. To accomplish these competing tasks, IAV packages separate proteins in its envelope to bind host receptors (hemagglutinin, or HA, which binds to sialic acid) and destroy these same receptors (neuraminidase, or NA, which cleaves sialic acid). Consistent with their essential but competing activities, the functional balance between HA and NA has been found to drive compensatory evolutionary responses when this balance is perturbed (Wagner et al., 2002), and it serves as the target of the most widely used antiinfluenza interventions, neuraminidase inhibitors (Nicholson et al., 2000). If sialic acid binding and cleaving activities varied at the level of individual viruses due to differences in HA and NA packaging and virion size, the efficacy of treatments specifically targeting either protein could be reduced. However, little is known about how the abundance of HA and NA on individual virions varies within an IAV population.
We sought to determine whether the variable – or ‘low fidelity’ – influenza A virus assembly process that produces heterogeneity in virion morphology could also cause heterogeneity in HA and NA composition and, if so, whether this phenotypic variability aids in the transmission of progeny virions in unpredictable growth environments. Although the adaptive benefits of phenotypic variability have been documented in many other systems (Balaban, 2004; Baldwin, 1896; Rego et al., 20l7), measuring its effects in viruses has traditionally been challenging. This is largely due to a lack of available methods for measuring virus phenotype – in particular, size, shape, protein abundance, and protein spatial organization – at the level of individual virions.
Methods such as mass spectrometry (Hutchinson et al., 2012, 2014; Shaw et al., 2008) and electron microscopy (Calder et al., 2010; Harris et al., 2006; Noda et al., 2012) have been useful for investigating various aspects of IAV composition and morphology but are limited in their ability to link phenotype with replicative fitness. As a result, it remains challenging to measure protein abundance at the single virus level across an entire population. More challenging still is to do so in a way that does not compromise the function of the proteins being measured or the replicative fitness of the virus as a whole so that complete infection cycles can be studied. To address this challenge and to characterize the intrinsic heterogeneity in genetically uniform populations of IAV, we developed a strain of the virus harboring small (~10 amino acid) tags on each of the virus’s most abundant structural proteins – HA, NA, M1, M2, and NP. Coupling small molecule fluorophores to these tags provides a handle by which we can quantify the abundance of specific proteins inside live, infected cells, or in individual released virions.
Using these fluorescently-tagged strains, we find that IAV composition and morphology vary widely, with total abundance of HA and NA each covering a ~100-fold range and the length of released virions ranging up to 20 µm within a viral population. Remarkably, the same variation in HA and NA incorporation found in virions released from multicellular monolayers is also found in virions produced by a single infected cell, indicating that low fidelity in the assembly process itself generates compositional variability, rather than differences among infected cells. Our data also show that HA and NA abundance and virus length can change significantly and reversibly over the course of a single replication cycle, and that composition is coupled to morphology, with longer virions more enriched in HA relative to NA than shorter virions. Critically, we find that the observed phenotypic variability serves as a determinant of viral escape from the cell surface in response to temperature changes and exposure to neuraminidase inhibitors, underscoring the functional significance of morphological and compositional heterogeneity. The ability of IAV to produce virions with broad stoichiometric distributions of its two competing surface proteins, HA and NA - in the absence of genetic changes to either protein – results in functionally versatile virus populations that can enhance virus survival in complex and unpredictable environments.
Results
Creating fluorescently-tagged IAV strains
Fluorescence microscopy offers several attractive features that could address the challenge of capturing protein abundance of individual viruses across a population while maintaining infectivity. Labeling proteins with specific fluorophores, either genetically or chemically (Sletten and Bertozzi, 2009), provides high contrast and allows proteins to be quantified and localized with high precision in a biochemically complex background. Additionally, fluorescence measurements can be performed on non-fixed, live samples, enabling the observation of dynamic phenomena that are inaccessible using electron microscopy. These features come at the cost of engineering tags into the virus that can be disruptive to virus replication; to date, efforts to rescue viruses harboring fluorescent fusion proteins on viral proteins have been limited (Lakdawala et al., 2014), presumably due to the large size of the tags (~25 kDa). An alternative approach is to use site-specific labeling to insert small tags (~5–10 amino acids) into viral proteins which can then be enzymatically modified with (or tightly bound by) a fluorophore. The small size of these fluorophores and their associated tags (~1 kDa each) reduce the likelihood that they will inhibit protein function. Site-specific labeling of IAV proteins has recently been reported for HA, NA, and NS1 (Li et al., 2010; Popp et al., 2012); however, this approach has not yet been harnessed to simultaneously and orthogonally label multiple viral proteins in infectious strains with replicative fitness comparable to wild type laboratory strains.
We sought to construct an IAV strain that is amenable to simultaneous fluorescence imaging of multiple viral proteins with minimal disruption to protein function and virus replication (Figure 1A). Using site-specific labeling via a combination of enzymes and probes - SFP synthase (Yin et al., 2006), Sortase A (Theile et al., 2013), transglutaminase 2 (Lin and Ting, 2006), and microbial transglutaminase (Sugimura et al., 2008) - as well as the membrane-permeable dye FlAsH (Griffin, 1998), we were able to rescue multiple variants harboring three orthogonal tags, and one with four (Figure S1A, Supplemental Data 1, STAR Methods). Cells infected with these variants shed high titers of virus that can be collected, labeled, and immobilized on coverslips for imaging (Figure 1B), or the live, infected cells can be labeled and imaged (Figure S1B&C). Viruses prepared in this way show heterogeneity in size and composition (Figure 1C). Large morphological variability is a hallmark of clinical isolates of influenza A virus and is also seen in our engineered strain expressing M1 from A/Udorn/1972 in an A/WSN/1933 background, which produces particles ranging from sub-diffraction limited foci to filamentous virions up to ~20µm in length. Although larger viruses (>1µm) are a relatively minor percentage of the total population (~10%), they comprise roughly one third of the total viral material that buds from cells during infection (Figure 1D). Both the insertion of site-specific tags in the viral genome and the attachment of fluorophores to these tags are minimally disruptive; depending on the tags, viruses containing multiple sites for fluorescence modification replicate with kinetics comparable to or modestly lower than the parental strain (Figure 1E), and viruses labeled with fluorophores maintain high infectivity (>70% of an unlabeled control) (Figure 1F).
Figure 1: Multi-spectral strains of influenza A virus permit fluorescence-based quantification of virus composition and morphology.

(A) Schematic of the virus labeling strategy. Inserting small (~5–10aa) tags into the structural proteins of influenza A virus renders the virus amenable to labeling via the site-specific attachment of fluorophores. (B) Work flow for collecting and labeling IAV proteins on intact and infectious virions. (C) Viruses labeled and immobilized on a coverslip exhibit diverse morphology and composition. (D) Length distribution of virions containing M1 from A/Udorn/1972 showing significant size variation. Viruses >1µm in length comprise a minority of ~10% of the population, but represent ~34% of the viral material shed during infection (estimated as the product of measured particle size and frequency within the population). Plot shows mean and standard deviation from six biological replicates with N > 44000 viruses in each. (E) Viruses with up to four orthogonal tags show replication kinetics similar to (HA/NP/M2/NA + M1Ud) or modestly slower than (HA/M1/M2) the parental strain in MDCK cells (error bars represent S.D.). (F) Labeling HA and NA on the surface of the virus preserves ~85% infectivity relative to unlabeled tagged strains, while labeling HA, NA, and NP preserves ~75% infectivity. Quantification shows the mean +/− S.D. of labeled virus titers relative to unlabeled samples for three biological replicates. See also Figures S1–S4 and Supplemental Data 1.
At moderate multiplicity of infection (MOI ~ 0.1–1, based on measured PFU/ml), cells infected with tagged virus are distinguishable from surrounding uninfected cells by the presence of fluorescence associated with multiple tags, allowing us to calculate the frequency with which pairs of viral proteins are expressed in cells infected by relatively few viruses (Figure S2A). Consistent with previous reports, we find that expression of pairs of viral proteins can be positively or negatively linked (Brooke et al., 2013). For example, while cells labeling positive for non-vRNP proteins (HA, NA, or M2) are positive for NP >99% of the time, cells labeling positive for NP are positive for these other viral proteins only ~75–85% of the time. The lower frequency of expression of non-vRNP genes (whose secondary transcription requires at least 5 segments) relative to a vRNP gene (whose secondary transcription requires at least 4), suggests that the protein expression we observe is due to secondary transcription from newly-synthesized vRNP complexes, since primary transcription from virally-delivered vRNPs can proceed at low levels with as few as one segment (Russell et al., 2018), which would tend to level these probabilities. Importantly, these percentages we measure hold whether proteins are labeled enzymatically or detected via an antibody that is independent of the engineered tags (Figure S2B & C).
After measuring the efficiency of labeling for our typical enzymatic reactions (~50% for SrtA-labeled HA and ~100% for SFP-labeled NA, Figure S3) and verifying that non-specific labeling is low (Figure S4A&B), we sought to determine the average number of molecules of HA and NA on the viral surface (STAR Methods and Figure S4C–E). This analysis yields a median density of ~22800 labeled HA molecules and ~2090 labeled NA molecules per µm2 of virus area; for a spherical particle 120 nm in diameter, this corresponds to 1031 HAs (~340 trimers) and 95 NAs (~24 tetramers). These values are comparable to those previously reported by mass spectrometry (294/98 HA monomers/trimers, 23/6 NA monomers/tetramers (Hutchinson et al., 2014)) and by direct counting of proteins on two viruses imaged using cryoelectron tomography (870–903 HA monomers, 152–200 NA monomers (Harris et al., 2006)).
Low fidelity of influenza A virus assembly produces diverse and infectious progeny
To determine if protein composition is as heterogeneous as virus morphology, we measured the relative abundance of labeled HA, NA, or M2 in individual particles (Figure 2A). We find that although the relative means of the distributions are in general agreement with published values from population measurements (Hutchinson et al., 2014; Zebedee and Lamb, 1988), HA, NA, and M2 abundance in individual virions each span a ~100-fold range (with 50% of each population concentrated within a three- to five-fold range), reflecting significant compositional heterogeneity. This variation appears to be affected by morphology, as replacing M1 from A/Udorn/1972 with the sequence from A/WSN/1933 results in more monodisperse particles and a narrower distribution of HA abundance (Figure 2B). Comparing viruses with either M1 or NP labeled using FlAsH-EDT2 gives a median value for M1 that is roughly three-fold that of NP (Figure 2C), also consistent with previous measurements (Hutchinson et al., 2014; Oxford et al., 1981). As with the other viral proteins, there is considerable variation in M1 levels that could arise from differences in virus size, as well as particles lacking a complete matrix layer (Harris et al., 2006). The bimodal distribution of NP abundance we observe suggests that many of the particles released during infection (~33% in this case, based on fitting two log-normal distributions to the data) have missing or defective genomes. The percentage of NP-negative viruses is roughly twice as high in longer (>1µm) filamentous particles, suggesting a link between particle morphology and the presence of NP, as a proxy for genome packaging (Figure S5A).
Figure 2. Influenza A virus composition and morphology vary broadly at the single-virion level.

(A) Distributions of HA, NA, and M2 labeled via SFP synthase (NA and M2) and SrtA (HA). The relative means of abundance are in general agreement with prior measurements, but individual viruses vary considerably. (B) This variation diminishes by ~30% when M1WSN is used to produce viruses of more homogeneous size. (C) Similar distributions as in (A), but with NP and M1, quantified after labeling with FlAsH-EDT2. (D) Segmenting images of immobilized viruses and integrating the intensities of labeled HA and NA allows us to measure distributions of these proteins across a virus population, represented as a two-dimensional histogram showing the log-abundance of HA (horizontal axis) and NA (vertical axis) measured for 187309 viruses compiled from three independent experiments. Data from individual replicates is plotted in Figure S5B and tabulated in Supplemental Data 2. Contrast in the image to the left is enhanced to increase visibility of viruses over a wide range of intensities. (E) Smaller viruses (<~300nm) exhibit more variability in HA:NA ratio, which is bimodally distribution in viruses shorter than ~300nm in length (population labeled i) but has an increasingly narrow distribution as virus size increases (populations from ii and iii). (F) Distribution of infectious potential across the particle population. “NP+”: particles that label positive for NP; “semi-infectious”: particles that deliver at least one vRNA segment to an infected cell; “PFU”: particles capable of initiating multiple rounds of replication. Data is normalized within each condition either to the total particle count (grey bars) or to the number of NP+ particles (percentages above bars); all values are mean +/− S.D. Un-normalized data for the three individual replicates is given in Table S1.
We next characterized covariation in protein abundance in individual viral particles, focusing on HA and NA to determine if variation in their relative levels might influence their functional balance (Wagner et al., 2002). In contrast to the monomodal distributions of HA and NA measured individually, the ratio HA/NA is distributed bimodally in smaller (< ~300 nm) virions and gradually reduces to a monomodal distribution as virus size increases from sub-diffraction limited spots to particles approaching ~1µm in length (Figure 2D, Figure S5B). Although viruses higher in NA are more likely to possess high levels of NP (a necessary but not sufficient indicator that the viral genome is present), both subsets of the population contain a substantial fraction of particles that label positive for NP and thus are at least potentially infectious (64% out of 43518 higher-NA particles, versus 57% of 145704 lower-NA particles; Figure S5C).
The ratio of total particles to fully-infectious virions (PFU) for IAV has been reported to range from ~10–100 (Donald and Isaacs, 1954; Martin and Helenius, 1991; Noton et al., 2009; Schulze, 1972). In addition, IAV produces a high percentage of semi-infectious particles, which fail to deliver at least one segment of the viral genome to the host cell nucleus, and thus do not result in the production of infectious progeny. The ratio of semi-infectious to fully-infectious particles in a viral population has been estimated to range from 1:1 or 2:1 (35–50% of infected cells expressing all eight viral mRNAs) (Russell et al., 2018) up to ~7:1 (13% of infected cells expressing all viral proteins) (Brooke et al., 2013).
In order to place our work in this context, we measured the total number of particles in our samples (both with and without NP), along with the number that are semi- and fully-infectious, by quantifying plaque formation and viral protein expression in cells following treatment with a known number of particles (STAR Methods). This resulted in a particle:PFU ratio of 18.3 ± 7.1 and semi-infectious-to-fully-infectious ratio of 6.0 ± 2.0 (Figure 2F, Table S1, STAR Methods). The agreement between our measurements and published values for both ratios suggests that the samples we characterized reasonably represent the proportion of fully-infectious and semiinfectious particles present in laboratory preparations of IAV, despite their heterogeneity. Additionally, restricting our analysis to only those particles that include NP, we find that ~70% are at least semi-infectious while ~10% are fully-infectious (Figure 2F). Comparing this latter value to PFU:vRNA ratios suggests that our samples are comparable to the high-purity ones reported elsewhere (Russell et al., 2018). Altogether, this data supports the conclusion that, despite being compositionally heterogeneous and possessing filamentous morphology, the infectious potential of the particles characterized here is approaching the upper limit of what has been reported for influenza A virus.
Compositionally and morphologically variable viruses are produced by single cells during one replication cycle
We next investigated the source of the compositional and morphological variability we observed. We reasoned that phenotypic variability in a population of viruses could arise from three general sources that are not mutually exclusive: (i) genetic diversity in the virus population, (ii) heterogeneity within the population of infected cells, or (iii) variability that arises as the virus is assembling. If phenotypic variability within a virus population requires genetic diversity or heterogeneity among infected cells, then a single virus infecting a single cell should not be capable of producing progeny that reflect the full diversity in morphology and composition seen in virus released from a monolayer of cells. To test this, we developed experiments to characterize the composition and morphology of progeny virus produced by individual cells infected with individual infectious units (MOI ~ 0.01) over a single round of replication (Figure 3A, STAR Methods). Viruses budding from the apical surface of a cell in these experiments are trapped by antibody-functionalized coverslips after limited lateral diffusion, creating clusters of immobilized viruses that originated from individual cells (Figure 3B). When we measure the composition of viruses located within separate clusters, we observe similar distributions of HA-NA content and length as for virus raised in populations of cells (Figure 3C & D). This supports the hypothesis that the virus assembly process itself is ‘low fidelity’ and a primary contributor to phenotypic variability, and that neither genetic diversity in the virus population nor variability among infected cells is needed for influenza A virus progeny to access a broad phenotypic space.
Figure 3. Phenotypic variability is encoded in the influenza A virus assembly process.
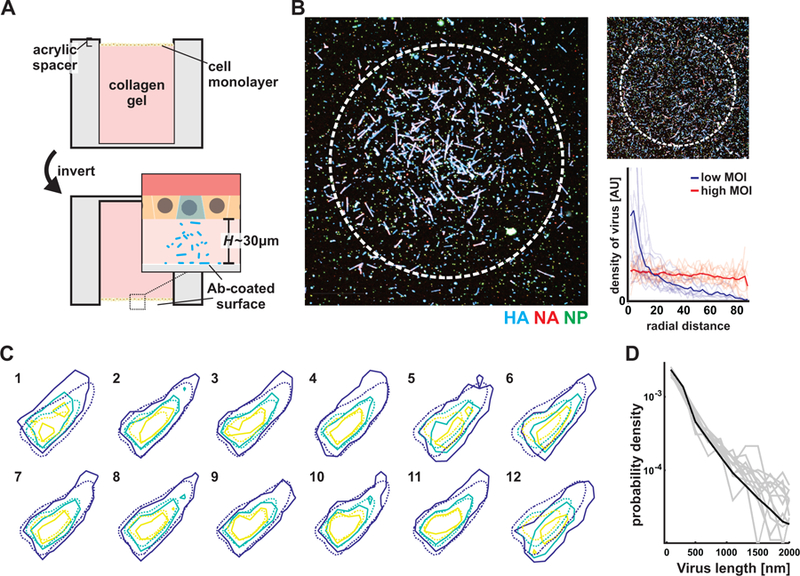
(A) Characterizing virus shed from individual infected cells by positioning the apical surface of an epithelial monolayer tens of microns from a functionalized coverslip and infecting at low (~0.01) MOI. (B) Measurements of virus density as a function of radial distance from the center of virus clusters. Clusters captured at low MOI decay to ~10% of their maximum value at a radial distance of ~50µm. Uniform densities of virus are collected when the same experiment is performed at high MOI. (C) Distributions of HA-NA content in virus shed from 12 separate cells (solid contours; average of 723 viruses per cluster) across two biological replicates, overlaid with the population-wide distribution (dashed contours). See also Supplemental Data 2. (D) Overlaid length distributions from the same 12 single-cell virus populations whose composition is plotted in C (gray lines), with the population-wide distribution plotted in black.
Phenotypic variability is altered by growth temperature and is broadly tolerated during internalization into cells
To test whether phenotypic variability is influenced by environmental conditions during assembly in the absence of genetic differences, we changed the growth temperature of cells infected with virus bearing tagged HA, NA, and NP, along with the V43I mutation to PB1, from 37°C to 33°C (temperatures roughly representative of the lower and upper airways), after infecting for one hour at 37°C to control for potential temperature dependences in virus entry. After collecting virus at 16 hpi and evaluating virion composition through fluorescent labeling and imaging (Figure 4A & B, Figure S7), we found that lower temperature shifts the virus population towards higher NA content within a single round of replication, and that this shift is reversible from one generation to the next (Figure 4B). This plasticity in viral composition does not extend to all viral proteins, as the percentage of particles containing NP is not significantly affected by temperature shifts within this range (Figure 4C). This data provides further evidence that virus heterogeneity is not inherently linked to viral genotype and that aspects of viral phenotype - in this case, packaging of HA and NA, but not NP - can reversibly shift in the absence of sufficient time for mutations to accumulate.
Figure 4. Compositional variability in individual viruses responds reversibly to changes in growth conditions.

(A) Compositional variability in viruses raised under different environmental conditions was investigated by infecting cells at 37°C for one hour and allowing infection to proceed at either 33°C or 37°C. Viruses grown at 33°C for one generation are then returned to 37°C to evaluate reversibility of observed viral phenotypes. (B) Virus phenotype is malleable to environmental conditions, shifting to higher NA when the temperature is decreased from 37°C to 33°C. These shifts are reversible from one generation to the next, suggesting that they originate from phenotypic variability rather than changes in genotype. Plots show combined data from three biological replicates, with N > 30000 viruses per condition in each replicate. Individual replicates are displayed in Figure S6A and tabulated in Supplemental Data 2. (C) In contrast to the relative abundance of HA and NA (top), the fraction of particles labeling positive for NP is not significantly affected by temperature shifts between 33°C and 37°C. Quantification in all cases shows the mean and standard deviation of population medians from three biological replicates, with N > 30000 viruses per condition in each replicate. Significance evaluated using a paired-sample T-test. (D) Comparing the total distribution of HA and NA in virus populations (as measured by immobilizing virus on coverslips using an HA-specific antibody, top row) to the distribution of HA and NA in viruses competent for binding to cells (middle row) and becoming internalized (bottom row). Under both 33°C and 37°C growth conditions, viruses capable of binding to cells and becoming internalized closely match the population as a whole, indicating that adhesion can occur for a broad range of HA and NA abundance. Regions marked by dashed lines indicate “higher NA” and “higher HA” subpopulations, with the percentage of particles in the “higher NA” group indicated in the upper left. Intensity values differ from those in (B) due to differences in the acquisition settings used for these three-dimensional samples (STAR Methods). Data is from four biological replicates, with individual replicates plotted in Figure S6B and tabulated in Supplemental Data 2. (E) Histograms of antibody binding discriminate between peripherally-bound particles (high labeling) and internalized particles (low labeling). Virus raised at 33°C show slightly higher tendency towards internalization than viruses raised at 37°C (data from individual replicates plotted in Figure S6B).
We next sought to determine if broad variation in HA and NA abundance results in subsets of the virus population that are incapable of binding to the cell surface and becoming internalized, due to an imbalance in the activity of these proteins. When we compared HA and NA abundance in viruses bound to coverslips to that of viruses both bound to and internalized by MDCK cells, we found that the distributions largely match for viruses raised at both 33°C and 37°C (Figure 4D), although the higher-NA viruses raised at 33°C showed a slightly higher tendency to become internalized (Figure 4E). This data shows that, in a permissive host such as MDCK cells, there are not stringent requirements for HA and NA abundance during binding and internalization, although particle composition may influence these processes.
A subset of IAV populations escape challenge with NA inhibitors
The phenotypic variability of IAV allows individual virions to sample from a broad range of possible morphologies and compositions, resulting in a population of viruses with differing propensities to bind and cleave host receptors, but which are nonetheless broadly capable of binding to and entering naive cells under permissive conditions. This diversity could have an adaptive advantage in situations where the density of sialic acid varies unpredictably, as would occur during challenge with neuraminidase inhibitors (NAIs). To test how virus populations are shaped over single replication cycles by such a challenge, we characterized how treatment with a commonly used NAI - oseltamivir - influenced viral phenotype in clonal, non-resistant populations (Figure 5A, STAR Methods). As NAI concentration was increased up to 1µM (an approximate upper bound for bioavailability in the sinuses (He et al., 1999)), released viruses become increasingly enriched in higher-NA virions and depleted in the higher-HA subset of the population (Figure 5B, Figure S7A). In contrast, viruses that remained bound to the cell surface (imaged after removing them with the soluble sialidase NanI from C. perfringens) exhibit higher – HA content and are more filamentous (Figure 5B, lower panels). These shifts are also apparent in the population’s HA/NA ratio, which is significantly decreased in viruses released from cells treated with 100nM NAI and significantly increased in viruses that remain bound (Figure 5C).
Figure 5: A compositional and morphological subpopulation of viruses challenged by neuraminidase inhibitors succeed in escaping from host cells.

(A) Comparing the heterogeneity of populations of virus exposed to NAIs that are able to detach from infected cells (red dotted circle) to those that cannot (blue dotted circle). (B) Treatment with physiological doses of the NAI oseltamivir shifts the phenotype of released viruses towards lower HA and higher NA abundance (top row). Viruses that remain bound under these conditions have predominantly high-HA phenotypes (“+exo. NA”, bottom row). Percentages give the number of particles within the gated region defined by dashed lines. Data from individual replicates are plotted in Figure S7A and tabulated in Supplemental Data 2. (C) Comparison of HA-NA ratio in viruses released under different challenge (“NAI”) or permissive (“exo. NA”) conditions. Viruses that release from NAI-treated cells have significantly lower HA/NA ratios than those that release in the absence of inhibitor or in the presence of both inhibitor and exogenous NA. Bars represent mean +/− S.D. for population medians, with the number of replicates (n) given below each condition (p-values determined using a two-sample T-test). (D) Composition of filamentous viruses (L > 1µm) released under challenge and/or permissive conditions. While the density of HA on filamentous particles (total HA intensity divided by particle length) does not change, filamentous viruses shed from cells challenged with NAI show a slight but significant increase in NA density. Bars represent mean +/− S.D. for population medians measured in four independent experiments with between 499 and 2067 filamentous viruses each (p-values determined using a two-sample T-test). (E) Measuring the propensity of viruses to detach from sialic acid coated coverslips as a function of their shape and composition. (F) Viruses that detach within a 1 hr observation period have significantly lower HA/NA ratios and significantly shorter lengths, as shown in (G). Quantification is based on three independent experiments with N > 366 released viruses and N > 991 attached viruses recorded in each. Data from individual experiments are plotted in Figure S7B. P-values calculated using a two-sample K-S test (F) and a two-sample T-test (G).
Shifts in the abundance of HA and NA in individual viruses could result from changes in virus morphology (as smaller viruses will have fewer of both proteins) as well as from changes in protein density on the viral surface. Although the surface density of receptor-binding HA on filamentous viruses (length > 1µm) does not differ significantly with NAI challenge, the average density of receptor-cleaving NA increases ~30% after NAI challenge (Figure 5D), suggesting that packaging more NA confers an advantage in these conditions. This result indicates that HA/NA ratio contributes to the effective binding affinity of a virus. To test this idea more directly, we developed an in vitro virus detachment assay, in which we monitor the dissociation of viruses from sialic acid coated coverslips as a function of their morphology and composition (Figure 5E). For viruses bound to sialic acid at densities approximating the cell surface during NAI treatment (~1 molecule/nm2 (Rosenberg, 1972)), those that dissociate over the course of an hour have significantly lower HA/NA, consistent with the live cell experiments (Figure 5F, Figure S7B).
In addition to the importance of HA-NA compositional balance in virus dissociation, we find that viruses that dissociate from the surface in our in vitro assay are also significantly shorter than those that remain stably bound (Figure 5G). Consistent with this, we observe substantial changes in the size distributions of released viruses under NAI challenge, with virion lengths >1µm significantly depleted from the pool of released virus (Figure 6A, left column). Although filamentous particles are recovered from NAI-treated cells following the addition of the exogenous sialidase (Figure 6A, right column), the total population of viruses produced in the presence of NAI treatment (i.e. those released naturally as well as those detached by sialidase treatment) reveals an overall reduction in virus length (Figure 6B), suggesting that virus morphology is malleable under NAI treatment. We note that, for these conditions, the percentage of particles containing NP in the absence of NAI is statistically indistinguishable from the percentage containing NP in its presence (59±9% versus 62±8%; p=0.51 from a two-sample T-test). These data show that IAV can reliably escape from host cells treated with even high concentrations of NAI, but that the viruses released are only a subset of the phenotypically diverse population produced, with shorter virions having higher NA and lower HA better suited to escape NAI treatment. In comparison, the larger particles that remain sequestered on the cell surface (even in the absence of NAI challenge: Figure 6A, top-right panel) would be incapable of transmission over long distances, although they may nonetheless be able to infect through direct contact with neighboring cells. Altogether, the low fidelity assembly process of IAV that results in phenotypic heterogeneity appears to help virus subpopulations survive environmental fluctuations, including acute stresses, and quickly reestablish phenotypic diversity without the need for genetic changes (Figure 7).
Figure 6: Morphological variability of influenza A virus is reduced by neuraminidase inhibitors challenge.

(A) Treatment with NAI at approximate physiological concentrations shortens the typical length of released viral filaments (“Released” column), while longer filaments are preferentially sequestered on the cell surface (“Attached” column). (B) Frequency plots showing the distribution of particle lengths within released, attached, and total populations of viruses. Plots show the mean +/− S.D. for length distributions determined from four biological replicates. For released viruses, differences are significant for all lengths (P < 0.01; two-sample t-test); for attached viruses as well as the total population, differences are significant for lengths above ~2.5µm (P < 0.05; two-sample t-test).
Figure 7: Low fidelity assembly in IAV contributes to survival in fluctuating environments by creating broad and malleable phenotypic variability.
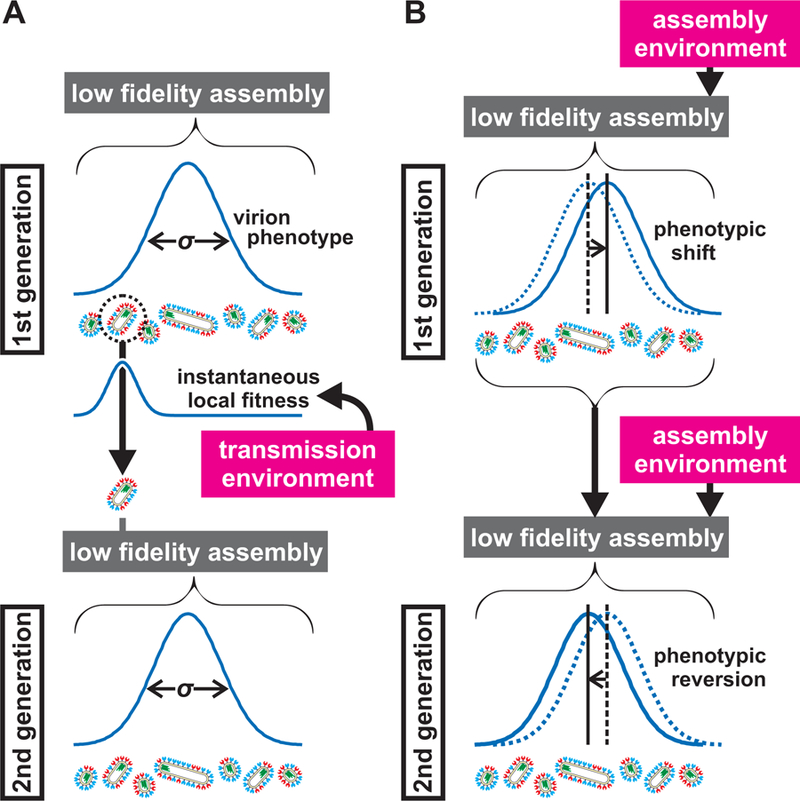
(A) Environmental inputs during transmission can restrict viral fitness, selecting phenotypic subsets (illustrated here by a single surviving virus) within the broader virus population (schematized by a distribution with variance σ). The low-fidelity assembly of IAV allows surviving subpopulations to reestablish phenotypic variability in the subsequent generation. (B) Environmental inputs also act during assembly, influencing virus phenotype through non-genetic shifts in virus composition and morphology that are reversible from one generation to the next.
Discussion
The infectiousness of a virus is a complex characteristic, arising from the specific activities of viral proteins and the multiple barriers presented by the host. Genetic mutations that alter (for example) receptor binding specificity or innate immune suppression can have dramatic effects on host range and virulence, ultimately driving viral evolution. However, even as evolutionary processes shape viral populations over multiple generations, individual viruses face immediate and local challenges to productive transmission and infection. The ability of an individual virion to circumvent these challenges depends both on the genetically-specified activity of its proteins as well as factors that may not be under direct genetic control: the shape of the virus and the abundance of viral and cellular proteins that are packaged during assembly. In the case of influenza A virus, these characteristics can vary considerably within a population. Because the contributions of these factors can be genetically silent, their significance has been difficult to determine and often overlooked in laboratory studies of infectivity. However, the long-standing observation that in vivo replication favors forms of influenza A virus that have more heterogeneous morphology – a characteristic that is genetically influenced by lAV’s matrix protein M1 – suggests that phenotypic variability may confer benefits to IAV under certain circumstances.
In the present work, we have focused on measuring variation in size and the abundance of HA and NA across populations of IAV. Our findings demonstrate that heterogeneity in these characteristics at the individual virus level changes the balance of intrinsic protein activity, creating virions with varying proclivity towards receptor binding or receptor destruction. Although more work is needed to determine how these results translate into replicative fitness, phenotypic variability in viral populations could be adaptive if it increased the probability of survival in an unpredictable environment (Kussell, 2005). Creating diverse progeny could contribute to the persistence of a virus population and allow sufficient time for an adaptive genetic response to evolve. This would be particularly relevant during a population bottleneck when genetic diversity is limited, such as during the initial infection of a new host (Poon et al., 2016; Varble et al., 2014).
Alternatively, phenotypic diversity could be detrimental if it increases the percentage of particles that are non-functional. The infectious potential of the heterogeneous population of particles characterized here appears to be broadly representative of IAV populations previously studied in vitro, with ~10% of NP-containing particles being fully-infectious. The nature of the remaining particles remains unclear; the percentage of semi-infectious particles in a population (~60% in our case) has been reported to be several times greater than the percentage of particles reported to package some but not all vRNAs (Chou et al., 2012; Nakatsu et al., 2016). Since these particles superficially appear to be genetically complete, it is not clear whether they are inherently defective due to their composition or if they simply fail to deliver a subset of vRNA segments at a post-fusion step owing to chance. In this view, all viral particles are subject to the probabilistic processes of binding, internalization, fusion, uncoating, nuclear import, and any other process necessary for replication. Although particle morphology or composition may be critical for any of these steps, our results suggest that wide ranges of HA and NA abundance on the incoming particle are not detrimental in permissive environments, at least for binding and internalization. In environments where the virus is not genetically well-adapted (as could occur during zoonotic infection), more work is needed to determine to what extent phenotypic outliers in the virus population may contribute to establishing infection.
The consequences of phenotypic variability discussed here would not be exclusive to IAV, raising the possibility that similar considerations may be relevant to other viruses as well. Interestingly, pleomorphism is common among enveloped viruses, with filamentous morphology common in the filoviridae, pneumoviridae, and paramyxoviridae families, in addition to the example of IAV within orthomyxoviridae (Compans et al., 1966; Roberts et al., 1995). Even within virus families where particle morphology is comparatively uniform (e.g. flaviviridae), other forms of functionally-relevant phenotypic variability may be present, such as in the balance between immature and mature glycoproteins on the viral surface (Pierson and Diamond, 2012). More generally, the size, composition, and epitopes displayed on the surface of many viruses influence how they gain entry into a cell and interact with the innate and adaptive immune response (Dejnirattisai et al., 2016; Geng et al., 2007; Rossman et al., 2012), suggesting that variability in these properties could influence replicative fitness or viral tropism. The tendency to produce diverse progeny rather than many copies of a stereotyped particle could contribute to viral persistence in a wide variety of viruses.
The underlying mechanisms responsible for IAV’s phenotypic variability remain an active topic of research (Chlanda et al., 2015; Rossman and Lamb, 2011). As demonstrated by in vitro reconstitution experiments and computational modeling, highly-ordered viral capsids can, under certain circumstances, successfully avoid kinetic trapping to reach a uniform, minimum energy state (Butler and Klug, 1971; Hagan and Chandler, 2006). In contrast, IAV particles could be kinetically trapped structures, with HA and NA content locked in during assembly through interactions with other structural proteins and unable to equilibrate before the reaction is terminated by membrane scission. In this view (which is consistent with the shifts in virus populations we see at different growth temperatures), stochastic incorporation of proteins (HA, NA, and host proteins) from the local plasma membrane into the viral envelope could account for compositional variability, while morphological variability could arise from stochasticity in the timing or mechanism of membrane scission.
Our findings provide a first assessment of phenotypic variability at the level of individual virions in influenza A virus populations. Although more work is needed to determine if this variability is adaptive overall, our results offer evidence that it can promote virus survival under some circumstances. These experiments focus on a single laboratory strain of IAV engineered to produce filamentous particles in vitro, and future work will determine the generality of these findings to different strains and subtypes, as well as to in vivo models of infection. The tools we describe here for minimally disruptive fluorescent labeling of influenza A virus provide a means of assessing this generality and a conceptual guide for asking similar questions about other filamentous viruses. The large variability that we measure in HA/NA ratio raises the possibility that other aspects of virus composition and organization may be similarly heterogeneous, with implications throughout the replication cycle. Since therapeutic interventions against IAV could alter viral phenotypes in genetically-independent ways, direct characterization of viral populations in the manner described here may help in evaluating the impact of emerging antiviral therapies (Furuta et al., 2013; Strauch et al., 2017). Overall, understanding the extent to which viral heterogeneity contributes to viral persistence could help to guide the development of a new class of therapies aimed at reducing natural variability, or applying combinatorial pressure that mitigates the survival benefits of phenotypic variability.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Daniel A. Fletcher (fletch@berkeley.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
MDCK-II cell line:
This female canine kidney epithelial cell line was maintained in DMEM with 10% fetal bovine serum (FBS, Life Technologies), and 1% Pen-Strep (Life Technologies), at 37C, 5% CO2. Cells were negative for mycoplasma as verified with Mycoalert mycoplasma detection kit (Lonza).
HEK293T cell line:
This human kidney epithelial cell line was maintained in DMEM with 10% fetal bovine serum (FBS, Life Technologies), and 1% Pen-Strep (Life Technologies), at 37C, 5% CO2. Cells were negative for mycoplasma as verified with Mycoalert mycoplasma detection kit (Lonza).
METHOD DETAILS
Designing tagged IAV strains
To construct strains of influenza A virus that would allow specific labeling of the virus’s most abundant structural proteins, we first sought to optimize the identity and location of tags in the hopes of recovering virus with unaltered replication kinetics. From a labeling perspective, lAV’s most abundant structural proteins can be broadly categorized according to whether they contain extracellular/extraviral domains (HA, NA, and M2), or if they are exclusively intracellular/intraviral (NP, M1). The exposed extracellular domains of HA, NA, and M2 make these proteins amenable to labeling via membrane-impermeable enzymes and dyes. For hemagglutinin, we reasoned that the head domain and N-terminal portions of HA1 could be attractive sites owing to the wide genetic diversity observed in these regions and their distance from portions of the protein involved in the conformational rearrangements accompanying fusion. Based on this, we designed two versions of tagged HA. The first consists of a 5*Gly sequence immediately following the HA signal sequence that, when cleaved by signal peptidase, exposes the N-terminal glycine recognized by SrtA for attachment of fluorescent peptide conjugates. This strategy for labeling HA via sortase A compliments a previously reported strategy (Popp et al., 2012), with the potential benefit of not requiring activation of HA for effective labeling. However, it requires that the N-terminus of the protein remain free from N-acetylation; although this modification has not been reported for HA (Hutchinson et al., 2012) and it does not appear to effect labeling in MDCK or HEK cells, this warrants attention if the labeling strategy reported here is to be extended to other systems. Nonetheless, enzymatic liberation of the N-terminus (via engineered TEV, trypsin, or thrombin cleavage sites) could restore efficient labeling if necessary. The second strategy for enzymatic modification of HA that we use leverages the specificity of tissue transglutaminase 2 from guinea pig liver (Sigma T5398) towards sequences containing consecutive glutamines. Based on its position at the apex of the molecule and relatively low conservation, we inserted a Q3-tag after G171 in the head domain of WSN HA (Figure S1A). Although both (Q3)HA and (G5)HA support robust virus replication and labeling, we use (G5)HA for the majority of experiments reported here.
For neuraminidase, we initially sought to incorporate an enzymatic tag in the stalk, focusing on motifs recognized by SFP synthase or microbial transglutaminase (mTG) that would provide orthogonality to the N-terminal sortase tag on HA. Although insertions of these motifs were well tolerated by the virus, they resulted in poor labeling efficiency (presumably due to poor accessibility of the enzymes and conformational constraints on the recognized sequence). As an alternative, we instead attached the 11 amino-acid ybbR tag (recognized by SFP synthase) to the protein’s C-terminus, followed by a stop codon and silent repetition of the native RNA sequence to allow efficient vRNA packaging (Figure S1A).
In order to perform site-specific labeling of M2, we again targeted the extracellular domain for insertion of motifs recognized by SFP or mTG. However, following the M1/M2 splice junction, 45 nucleotides are shared between M1 and M2, comprising the M1 C-terminus and a significant portion of the M2 ectodomain (residues 10–25). To minimize disruptions to the M1 C-terminus, we inserted M48 and ybbR tags into the M2 ectodomain following the M1 stop codon (Figure S1A). Disulfide bonds (via C17 and C19 in M2) and glycosylation (at N20, which matches the consensus sequence for N-linked glycosylation, NxS/T) upstream of the inserted tag dramatically reduce labeling efficiency, likely by blocking accessibility of the tag. To improve labeling, we introduce the mutations C17S, C19S, and C20S in to the M2 ectodomain and restore the disulfide bonds downstream of the tag. Although C17S and N20S mutations are silent in M1, C19S results in the mutation M248I in M1. However, this mutation does not influence viral fitness or morphology in vitro, and the mutations to M2 dramatically improved labeling. Because both M48 and ybbR tags were tolerated by the virus, we choose either as needed to assure orthogonal labeling relative to HA and/or NA.
Finally, for the intracellular / intraviral proteins M1 and NP, we used tetracysteine tags combined with the biarsenical dye FlAsH to perform site specific labeling. Based on the partial structure of M1 and models for its oligomerization, we identified eight sites as potential candidates for insertion of the tag, one of which (a loop between predicted helices 11 and 12) yielded viable virus. However, when we attempted to rescue virus using Udorn-M1 with the tag inserted in the same site, the resulting virus exhibited altered morphology from those seen with wildtype Udorn-M1. Thus, further work is needed to identify sites in M1 that preserve both viral fitness and the filamentous morphology. Finally, for engineering a tagged NP, a similar approach for choosing candidate sites led us to identify the C-terminus as the best location for the tag over any of the internal loops tested. Due to the proximity of this insertion to regions of the vRNA important for packaging, we repeated the native C-terminal sequence of NP following the stop codon (Figure S1A).
Cloning and culturing viruses
Genes coding for modified versions of HA, NA, M1, M2, and NP were cloned via PCR and Gibson assembly into a dual promotor vector based on pcDNA3.1+, with a CMV promoter driving mRNA transcription, and a human poll promoter driving transcription of negative-sense viral genomic RNA, (following the strategy of (Hoffmann et al., 2000)). These vectors are listed in the Key Resources table. For transfection, we use near-confluent co-cultures of HEK293T cells and MDCK cells, grown in 6-well plates. Approximately 24 hours after transfection (1µg of each of the eight IAV rescue plasmids per well, mixed with 25µl Mirus TransIT-293 and 250µl OptiMEM per the manufacturer’s instructions), we change media to virus growth media (MEM, 0.25% BSA, 1µg/ml TPCK-treated trypsin, and penicillin/streptomycin). After an additional 24–48 hours, we collect media from transfected cells and perform a plaque assay on MDCK monolayers to determine viral titer and isolate clones for further growth and characterization. To check for the preservation of fluorescent tags and (for viruses containing M1 from A/Udorn/72) a filamentous phenotype, we amplify each plaque and infect cells for labeling and imaging, to test for the brightness and frequency of labeling across the population, and for the presence of the filamentous phenotype.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Influenza A H1N1 HA antibody | Thermo Fisher | RRID: AB_2552278 |
| Influenza A H1N1 NA antibody | Thermo Fisher | RRID: AB_2549711 |
| Goat Anti-Rabbit IgG (Alexa Fluor 488) | Abcam | RRID: AB_2630356 |
| Bacterial and Virus Strains | ||
| Rosetta DE3 | EMD Millipore | Cat#70956 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Alexa Fluor 555 maleimide | Thermo Fisher | Cat#A20346 |
| Alexa Fluor 647 maleimide | Thermo Fisher | Cat#A20347 |
| FlAsH-EDT2 | Toronto Research Chemicals | Cat#F335200 |
| DBCO-Sulfo-Cy5 | Jena Bioscience | Cat#CLK-A130–1 |
| 3-azido-1-propanamine | Sigma | Cat#762016 |
| Oseltamivir carboxylate | Toronto Research Chemicals | Cat#O700980 |
| CLPETGG peptide | Genscript | N/A |
| Coenzyme A trilithium salt | Sigma | Cat#C3019 |
| BMPS | Thermo Fisher | Cat#22298 |
| NH2-PEG-OH | Rapp Polymere | Cat#122000–2 |
| NH2-PEG-NH2 | Rapp Polymere | Cat#112000–2 |
| NH2-PEG-Biotin | Rapp Polymere | Cat#133000–25-20 |
| Recombinant WSN HA | Thermo Fisher | Cat#11692V08H25 |
| Protein A | Abcam | Cat#ab155695 |
| Experimental Models: Cell Lines | ||
| Canine: MDCK-II | UC Berkeley Cell Culture Facility | https://bds.berkeley.edu/facilities/cell-culture |
| Human: HEK293T | UC Berkeley Cell Culture Facility | https://bds.berkeley.edu/facilities/cell-culture |
| Recombinant DNA | ||
| pcDNA3.1+ | Thermo Fisher | Cat#V79020 |
| pcDNA3.1+ vPA (WSN) | This paper | N/A |
| pcDNA3.1+ vPB1 (WSN) | This paper | N/A |
| pcDNA3.1+ vPB2 (WSN) | This paper | N/A |
| pcDNA3.1+ vN (WSN) | This paper | N/A |
| pcDNA3.1+ vNP (WSN) | This paper | N/A |
| pcDNA3.1+ vNP(FlAsH) (WSN) | This paper | N/A |
| pcDNA3.1+ vHA (WSN) | This paper | N/A |
| pcDNA3.1+ vHA(G5) (WSN) | This paper | N/A |
| pcDNA3.1+ vHA(Q3) (WSN) | This paper | N/A |
| pcDNA3.1+ vNA (WSN) | This paper | N/A |
| pcDNA3.1+ vNA(ybbR) (WSN) | This paper | N/A |
| pcDNA3.1+ vM (WSN) | This paper | N/A |
| pcDNA3.1+ vM (Ud M1) | This paper | N/A |
| pcDNA3.1+ vM(FlAsH / ybbR) (WSN) | This paper | N/A |
| pcDNA3.1+ vM(M48) (Ud M1) | This paper | N/A |
| pRSETa | Thermo Fisher | Cat#V35120 |
| pRSETa-mTG(His)6 | This paper | N/A |
| pET28b | EMD Millipore | Cat#69865 |
| pET28b-(His)6SrtA | This paper | N/A |
| pET28b-SFP(His)6 | This paper | N/A |
| Software and Algorithms | ||
| Matlab R2017b | Mathworks | RRID: SCR_001622 |
| Other | ||
| TransIT-293 | Mirus | MIR 2704 |
| TPCK-treated Trypsin | Thermo Fisher | Cat#20233 |
| Collagen | Corning | Cat#354249 |
| Fetuin from fetal bovine serum | Sigma | Cat#F3004 |
| Transglutaminase 2 from guinea pig liver | Sigma | Cat#T5398 |
| Neuraminidase from Clostridium perfringens | Sigma | Cat#N2876 |
To compare viruses with spherical and filamentous morphologies in an otherwise identical genetic background, we use A/WSN/33 as the parental strain and substitute M1 from A/Udorn/72 (containing the amino acid substitutions V41A, K95R, T167A, D204E, I205V, and T218A relative to WSN-M1) to generate the filamentous phenotype (Elleman and Barclay, 2004). For experiments evaluating phenotypic variability (Figures 3–6), we use tagged IAV strains containing the V43I mutation in the PB1 gene, which decreases the mutation rate of the viral polymerase by a few-fold, without affecting replication in vitro (Cheung et al., 2014).
To determine viral titers for growth curves and for evaluating the effects of fluorophores on infectivity (Figures 1E & F), we infect 6-well plates of confluent MDCK cells with serially-diluted viral stocks adjusted to a total volume of 400µl per well. After infecting for one hour at 37°C with periodic rocking, we overlay each well with 2ml 0.4% agarose in viral growth media. At ~48hpi, we remove the agarose overlay, stain with crystal violet, and count plaques to determine titer.
Purification of enzymes and preparation of probes for virus and cell labeling
Sortase A and SFP synthase were purified as recombinant, histidine-tagged enzymes from E. coli, following established protocols (Theile et al., 2013; Yin et al., 2006). Microbial transglutaminase (mTG) from Streptomyces mobaraensis was cloned into the bacterial expression vector pRSETa, with a c-terminal 6x-histidine tag and including the N-terminal prodomain of mTG. After harvesting and lysing cells expressing mTG overnight at 16°C, we activate the enzyme by removing the pro-domain with TPCK-treated trypsin added directly to the lysate to a final concentration of 50µg/ml. After incubating at room temperature for one hour, the lysate is bound to affinity resin at 4°C for two hours, washed in buffer containing 50mM Tris, pH 8.0, 300mM NaCl, and 10mM imidazole, and eluted in a linear gradient of the same buffer with increasing concentrations of imidazole up to 500mM. We pooled the eluted fractions and further purified via size exclusion chromatography on a HiLoad 16/600 Superdex 200pg equilibrated with 50mM Tris pH 8.0, 300mM NaCl, 10% glycerol, and 0.2mM TCEP. The enzyme ran as a single monodisperse peak, which we collected and concentrated to a final concentration of 500µM.
Probes for enzymatic labeling via Sortase A and SFP synthase were created in-house, using thiol-reactive dyes (Alexa-555 maleimide, Alexa-647 maleimide, DyLight 405 maleimide) and either Coenzyme A (Sigma C3019) or peptides with the sequence CLPETGG (Genscript custom peptide synthesis, >95% purity). After dissolving dyes in anhydrous DMF (20mM concentration) and peptides/CoA in PBS with 2mM EDTA (10mM concentration), we mix the two reactants to final concentrations of 5mM peptide/CoA and 7.5mM dye. Approximately 24 hours after starting the reaction, we quench any unreacted dye by adding 2-mercaptoethanol to a final concentration of 10mM. This yields a ~5mM stock of labeled product (verified by thin layer chromatography) which we use directly for subsequent virus and cell labeling.
Labeling live IAV-infected cells
Cells infected with engineered IAV strains were labeled at 13hpi following overnight growth at 33°C. One hour before labeling cell surface proteins (12hpi), we wash the infected cells with OptiMEM and replace virus growth media with OptiMEM supplemented with 1µM FlAsH EDT2 (Toronto Research Chemicals F335200) before returning cells to a 33°C incubator. After 30 minutes, we wash out media containing the dye and replace with media supplemented with 1mM EDT2 (10 minutes at 33°C), which forms a dark complex with unbound or non-specifically bound FlAsH-EDT2, reducing background. After washing out EDT2-containing media, if the cells to be labeled contain an M48 tag on M2 (recognized by microbial transglutaminase, mTG), we incubate cells at 33°C for 45 minutes in virus growth media supplemented with 10µM mTG and 1mM azidopropanamine (Sigma, 762016).
The infected cells at this point are ready for the surface attachment of fluorescent dyes. To minimize background caused by endocytosis of free dye, we incubate the cells on ice for 10 minutes prior to adding the labeling reaction. The labeling reaction consists of OptiMEM supplemented with the following: 0.25% BSA, 5mM CaCl2, 5mM MgCl2, 200µM SrtA, 5µM SFP synthase, 50µM CLPETGG probe, 2.5µM CoA probe, and 0.5µM DBCO (for click labeling of azide-modified M2, if used; Jena Bioscience, CLK-A130–1). After labeling on ice for one hour, cells are returned to room temperature and washed in virus growth media prior to immediate imaging.
Virus labeling and immobilization
Coverslips functionalized with Staphylococcus aureus Protein A (ab155695) were created as described previously (Fourniol et al., 2014), with small modifications. Briefly, coverslips coated with diamino PEG (Rapp Polymere 112000–2) were reacted with NHS-maleimide bifunctional crosslinker (BMPS, ThermoFisher Scientific 22298). After washing to remove excess crosslinker, custom-made wells of polydimethylsiloxane (PDMS) were placed on the surface of the coverslip and 50pl of recombinant Protein A with a C-terminal cysteine was added at a 32µM in 300mM NaCl, 20mM HEPEs pH 7.2, and 2mM EDTA. After incubating for two hours at room temperature, the coupling reaction was quenched though the addition of 2-ME to a final concentration of 10mM. Following thorough washing with PBS to remove uncoupled Protein A, wells were incubated overnight at 4°C with an anti-H1N1 HA polyclonal antibody (Thermo PA5–34929) diluted to 2µg/ml in PBS.
Prior to collecting virions for imaging, PDMS wells were thoroughly washed to remove unbound antibody and exchange to NTC buffer (100nM NaCl, 20mM Tris pH 7.6, 5mM CaCl2). Freshly harvested virions were collected, spun at 2000g for 5 minutes, and treated with exogenous sialidase (C. Perfringens, Sigma N2876) at 37°C for one hour. Treatment with sialidase prior to immobilization helped reduce virion aggregation, especially viruses raised in the presence of neuraminidase inhibitor. To release virions tethered to cells (as in Figures 5 & 6), media containing released virions was replaced with media supplemented with exogenous sialidase and incubated at 37C for one hour. Following sial idase treatment, all virions were spun once more at 2000g for 5 min (stronger centrifugation lead to the loss of larger filamentous particles), and added to the antibody-coated wells at a total titer of ~105 PFU.
After allowing virus to bind for 2 hours at room temperature, unbound virus and media was washed out in preparation for labeling. Tetracysteine-tagged proteins (NP, M1) were labeled using FIAsH-EDT2 at 1µM (30 min, RT). Membrane proteins were subsequently labeled in parallel using Sortase A (180pM enzyme, 50pM CLPETGG peptide) and SFP synthase (5pM enzyme, 5pM CoA probe) in NTC buffer supplemented with 5mM MgCl2. Enzymatic labeling was allowed to proceed for 90 minutes, exchanging a freshly-prepared labeling solution after the first 45 minutes. Afterwards, immobilized virions were washed to remove excess labeling solution and treated with 1pg/ml TPCK-trypsin (15min, RT) to activate HA and remove minor amounts of background fluorescence.
For labeling viruses in solution (performed for virus dissociation assays and SDS-PAGE analysis), the same enzyme and probe concentrations are used, as well as the same incubation times. Following labeling, Capto Core 700 beads (GE Healthcare; ~1:1 resin volume to sample volume) are used to remove residual dyes and enzymes from the solution of labeled virus.
SDS-PAGE analysis of labeled virus
Viruses labeled under the same conditions as those used to prepare intact virions for fluorescence imaging were subjected to analysis by SDS-PAGE by concentrating virus samples by centrifugation and lysing in tris-glycine sample buffer with 20mM TCEP (for viruses with FlAsH tag-containing proteins) or 350mM β-mercaptoethanol (for viruses with labeled M2, which requires more stringent conditions to reduce disulfide-linked tetramers). Samples were then run on 4–20% gels and imaged using a Typhoon Trio imager.
Fluorescence imaging of viruses on coverslips
For confocal imaging of virus samples and cells, we use a Nikon Eclipse Ti microscope body, equipped with a Yokogawa CSU-X spinning disk, Andor Zyla sCMOS camera, 60X and 100X APO TIRF objectives (1.49 NA), and a Spectral Applied Research ILE-3003 laser launch for illumination via laser lines at 405nm, 488nm, 561nm, and 640nm. Because the vast majority of viruses immobilized on coverslips lie in-plane in our samples with a thickness less than our depth of focus, we are able to image the entire particle with a single z-slice. For data from different conditions within replicates, we imaged samples in immediate succession to avoid any effect from drift in the optical system over time; similarly, we acquired data from replicates to be combined typically within ~1 month of each other. For TIRF imaging, we use the same microscope body, objectives, and laser lines, but an Andor iXon Ultra EMCCD camera in place of the sCMOS camera for the increased sensitivity necessary for calibrating counts of molecules on the surface of labeled viruses (see “Quantifying HA and NA abundance”).
Measuring virus length and composition from fluorescence images
To determine virus length and composition from fluorescence images, we image 10–20 fields of view containing ~1000–3000 particles per field of view for each condition in an experiment. For each field of view, we create a binary image for both the HA and NA channels using as a threshold value the median pixel value plus one standard deviation. After eliminating spurious pixels through successive erosion and dilation, we use the combined HA and NA binarized images to segment the field of view into individual particles, from which we extract pixel values for each of the channels imaged, along with the major and minor axes of the binarized mask. Unless stated otherwise, the intensity values reported throughout this study correspond to the sum of the intensities of masked pixels, with the background intensity of the field of view (calculated as the median dark level) subtracted from each pixel value before summation.
To estimate virus lengths, we subtract the minor axis length from the major axis length determined from segmented particles, multiple by the number of nanometers per pixel, and add 80 nm. This value corresponds to the approximate width of a filamentous particle; adding it back to the length accounts for the earlier subtraction of the minor axis length. For a significant portion of the particles we image, the length is comparable to or below the limit of diffraction. As a result, length measurements on smaller particles (less than ~500nm) are likely to be less accurate than measurements of longer ones.
Characterizing virus released from individual infected cells
As illustrated in Figure 3A, we culture MDCK cells in custom-built chambers made using 4.5mm-thick cast acrylic, cut into hollow 5mm-diameter cylinders using a VersaLASER laser cutter with a 30W CO2 laser and sealed on the bottom by cementing a 200µm-thick sheet of cast acrylic cut to the same diameter. Following UV sterilization, we fill each chamber with a type I-A collagen and allow it to gel at 37°C for 30 minutes, creating a smooth surface for cell attachment that is offset by ~100µm from the rim of the acrylic chamber. After culturing cells to confluence on these collagen gels with a droplet of media on top, we infect at MOI ~ 0.01 for one hour and replace media with virus growth media supplemented with 100mU/ml exogenous sialidase (Sigma N2876) to prevent progeny virus from attaching to neighboring, uninfected cells. Finally, we invert the cylindrical culture chambers so that the apical surface of the monolayer is positioned directly over an antibody-functionalized, PEGylated surface for growth overnight and virus binding. After allowing 16 hours for virus growth and release, we remove the cell culture chamber and proceed to label and image immobilized virus as described above.
Counting total, semi-infectious, and fully-infectious particles
To obtain particle counts, we serially diluted labeled virus stocks and bound them to antibody-functionalized coverslips overnight with gentle rocking. This allows us to approximate the total number of particles present by counting the number of particles immobilized after an extensive incubation from a known volume of virus. We then measure the number of particles per unit area on the coverslip (using fluorescence imaging of ~40 fields of view), multiply this by the total area of the sample, and divide this by the volume of virus added. Note that because this incubation period exceeds the one-hour incubation we use when measuring PFU in cells, it may underestimate the total percentage of particles within a population that are fully-infectious.
In parallel with obtaining particle counts in this manner, we performed plaque-forming assays and fluorescence imaging of NP on the same viral stocks. To determine PFU/ml, we dilute 0.004 µl of virus into 400 µl of media and incubate with confluent cells in a 6-well plate for one hour. To determine NP-expressing cells, we dilute 0.4 µl of virus into 400 µl of media in a separate well of the same 6-well plate used to count PFU; this assures that virus is given equal opportunity to bind and infect in our counts of both PFU and semi-infectious-particles. We count plaques as described above in “Cloning and culturing virus”. To count NP-expressing cells, we label cells challenged with virus at ~8 hpi using FlAsH-EDT2 as described in “Labeling live IAV infected cells”. NP-expressing cells are then counted based on images of ~40 fields of view, and the number of total NP-expressing cells is determined by extrapolating from this data across the area of the well.
To determine the number of semi-infected cells, we make the simplifying assumption that the probability of an infected cell expressing any particular vRNA segment is equal to p, such that the probability of an infected cell expressing all 8 segments (i.e. a fully-infected cell) is p8, and the probability of an infected cell expressing the four segments necessary for secondary transcription (PA, PB1, PB2, and NP) is p4. In our analysis of viral protein expression in infected cells, we find that the expression of non-vRNP genes is typically ~75–85% as frequent as the expression of the vRNP gene NP (Figure S2C). This is consistent with the expression of non-vRNP genes (HA, NA, or M2 in this case) being dependent on the packaging of at least five segments (PB1, PB2, PA, NP, plus the segment of interest) and the expression of NP being dependent on only four. This leads us to conclude that the protein expression we observe is restricted to cells undergoing secondary transcription of viral genes, and thus our count of NP-expressing cells underestimates the total number of semi-infected cells. To correct this estimate, we calculate the number of semi-infectious particles (SI) - those that deliver at least one vRNA to the cell - using:
Comparing HA-NA distributions of virus immobilized on coverslips with virus bound to or internalized by cells
We compared distributions of HA and NA abundance in virus capable of binding and entering cells to the population as a whole by preparing viruses with labeled HA and NA and incubating these either with antibody-functionalized coverslips (to measure the population as a whole) or with cells for one hour at 33°C. This lower incubation temperature allowed cells to internalize virus while minimizing the dispersal or degradation of viral components following membrane fusion. Virus internalization precludes us from directly measuring particle size, as live imaging of fluorescent virus has revealed that filaments become severely distorted as they are taken in. However, total HA and NA abundance correlates with size (Figure 2D & E) and serves as a reasonable proxy. Following incubation with labeled virus, we wash cells twice with PBS and fix without permeabilization. We then probe with anti-HA and fluorescent secondary antibodies to discriminate between virus attached to the cell surface (positive for antibody labeling), and those that are internalized (negative for antibody labeling). Because these samples are not confined to a single plane, we collect z-stacks at 0.5µm intervals and analyze virus composition based on three-dimensional segmented stacks, changing the typical intensities compared to analysis based on a single z-section. To ensure accurate comparison to virus bound on coverslips, these samples are also imaged as stacks and analyzed in the same manner.
NAI challenge assay
For challenge experiments with the neuraminidase inhibitor oseltamivir, we infect a polarized monolayer of MDCK cells grown on a collagen gel at an MOI of 1–3. This relatively high MOI limits virus replication to a single round; combined with the use of virus containing the V43I mutation in PB1, this minimizes the probability of genetic adaptation to NAI treatment occurring over the course of the experiment. After incubating cells with virus for one hour at 37°C, we wash to remove excess virus, replacing media with virus growth media supplemented with or without a specified concentration of oseltamivir carboxylate (Toronto Research Chemicals O700980), but without TPCK-treated trypsin. At 16 hours post infection, we remove the virus containing media for labeling and imaging, and replace with media supplemented with 1U/ml NanI from Clostridium perfringens (Sigma N2876). After treating with this exogenous sialidase for one hour at 37°C, we again collect cell culture media for virus labeling and imaging.
In vitro virus detachment assay
Functionalized coverslips were prepared as described under “Virus labeling and immobilization”, except that NH2-PEG-OH (Rapp Polymere, 122000–2) was used in place of diamino PEG, with the addition of 2.5 mole-percent NH2-PEG-Biotin (Rapp Polymere, 133000–25-20) for subsequent attachment of sialylated proteins. Following PEGylation, coverslips were incubated for 10 minutes at RT with streptavidin at 50µg/ml in 150 mM NaCl, 25mM HEPES, pH 7.2, and washed 5X in the same buffer. Fetuin (Sigma F3004) labeled with NHS-biotin was then added at 100nM and incubated ~30 minutes at RT. Coverslips were then washed 5X in NTC buffer and equilibrated to 4°C in preparation for virus binding. Viruses with HA and NA enzymatically labeled as described previously were bound to coverslips equilibrated to 4°C for one hour on ice. Immediately before imaging, excess virus was washed with pre-chilled NTC, and the sample was mounted on the microscope stage. For imaging virus detachment, we use TIRF microscopy, which allows us to minimize sample bleaching over extended acquisitions. After allowing the chamber to equilibrate to room temperature, the buffer in the chamber was exchanged to NA buffer (100mM NaCl, 50mM MES pH 6.5, 5mM CaCl2), and images were acquired at 30 second intervals for one hour. We analyze the resulting image series similar to our analysis of confocal microscopy images, with an additional step to track particle trajectories between frames to identify viruses that detach over the course of the acquisition. Measuring the HA and NA intensity or morphological features of particles that do or do not detach over the course of imaging yields the plots shown in Figure 5E–G.
Measuring labeling efficiencies
We used Western blot analysis to determine the labeling efficiency of our typical reactions, comparing enzymatically-labeled HA and NA to recombinant protein labeled with an NHS dye. The results of this analysis are shown in Figure S3. Figure S3A shows the same blot imaged at two wavelengths (top: 785nm, bottom: 685nm) in a LiCor Odyssey CLx. Following the ladder in the leftmost lane, Lanes 1 through 6 contain 0.4 pmol each of recombinant soluble WSN HA (Thermo Fisher, 11692V08H25) labeled with Alexa Fluor 647 NHS ester at different degrees of labeling (Lane 1 = 100%, Lane 2 = 50%, Lane 3 = 25%, Lane 4 = 12.5%, Lane 5 = 6.25%, Lane 6 = 0%). Lanes 7 and 8 contain equal amounts of IAV (500pl culture), grown at 33°C and labeled with SrtA (HA) and SFP (NA), respectively, using Alexa Flour 647 probes. The blots are probed simultaneously with rabbit anti-HA and rabbit anti-NA antibodies, which are both detected by anti-rabbit antibody conjugated to IRDye 800CW. The relative intensities of the AF647 dye and the secondary antibodies allows us to compare IAV samples to recombinant samples at known labeling ratios, and thus to estimate the labeling efficiency of our typical reactions. Since the anti-HA antibody we use recognizes a C-terminal antigen within the extracellular portion of HA2, we focus on the HA0 band in the IAV samples, which contains signal from both the antibody as well as the N-terminally modified glycine. This analysis suggests that ~50% of the HA in our samples has been labeled with AF647 dye (Figure S3B, top).
Although we were unable to obtain pure, recombinant, WSN NA to perform an analogous test of NA labeling via SFP synthase, we can nonetheless estimate NA labeling through comparisons to HA in the two virus samples. Combining the signal from the HA0 and HA1 bands in Lane 7 results in an estimate of ~1.7 pmol of labeled HA per 500ul of virus culture, while the signal from labeled NA in Lane 8 suggests ~1.1 pmol from an equivalent volume (Figure S3B, bottom). The corresponding HA:NA ratio of 1.54 is lower than published values, suggesting that the labeling efficiency of NA is higher than that of HA. (Note that we cannot infer HA:NA ratio from antibody staining intensity due to unknown differences in affinity between the two antibodies used and their epitopes). This lower than expected HA:NA ratio is likely also influenced by having grown these IAV samples at 33°C, a condition which increases NA content (Figure 4). Labeling efficiencies >50% for NA are also consistent with the shift we observe in the anti-NA bands in Lanes 7 and 8 (shown magnified and as line scans in Figure S3C), where attachment of the dye has caused a visible shift towards higher molecular weight.
An important caveat when performing enzymatic labeling is that the identity of the dye molecule can influence the efficiency of labeling. We found this to be the case for our samples; a similar analysis using IRDye 680RD in place of AF647 resulted in an estimate of ~90% HA labeling and a labeled HA:NA ratio of 4.4 (0.105 pmol labeled HA to 0.024 pmol labeled NA from 150ul of virus culture), suggesting that NA labeling with this dye may be less efficient than with AF647. This thus presents an important consideration in developing optimal labeling strategies.
Measuring non-specific labeling
To determine the importance of non-specific labeling due to interactions between dye and untagged virus, we have imaged untagged viruses alongside their tagged counterparts after treating both with enzymes and fluorescent probes under identical conditions. This allows us to quantify non-specific labeling in a manner that is directly comparable to our experimental data. The results of these experiments are summarized in Figure S4. To determine background for sortase A and its Alexa 647 probe (used to label HA throughout this work), we compared virus containing the N-terminal pentaglycine on HA with wildtype virus lacking this tag. To detect virus on the coverslip, we labeled all virus with an anti-HA antibody and detected it with a fluorescent secondary (Figure S4B, left panel). Because these fluorescent secondaries will cross-react with antibodies on the coverslip used to immobilize viruses in most of our experiments, we switched our immobilization strategy for this test, using coverslips functionalized with the heavily sialylated protein fetuin. This analysis demonstrates that virus containing the pentaglycine tag are, on average, more than 500 times brighter than the untagged counterpart. However, we observe a minor portion of particles lacking the pentaglycine tag that label ~10-fold lower than the tagged virus. These may represent wildtype virus on which the native N-terminal glycine of HA2 has become labeled. However, since they comprise <~5% of the particles measured and contribute relatively little signal, they would not significantly alter our analysis or conclusions.
We performed a similar analysis for NA and NP, using SFP synthase (with Alexa 555 CoA, the dye used for NA labeling throughout our manuscript) and FlAsH-EDT2 to label tagged and untagged viruses (Figure S4B, middle and right panels). Background in both cases is modest relative to signal (about 40-fold lower than signal for labeling with SFP and about 30-fold lower than signal for FlAsH), with 27% (SFP) and 24% (FlAsH) of untagged particles having slight negative values for intensity after our routine background correction. For FlAsH labeling of NP, it is worth noting that the level of background signal in untagged particles matches that of particles that we consider to be “lacking a genome”. Thus the signal we see in these particles is not necessarily attributable to the incorporation of small amounts of NP, but may instead come from background.
This analysis supports the conclusion that non-specific labeling is not likely to influence our results. However, it does potentially constrain the applications for which this approach would be useful. For example, we have deliberately avoided attempting detailed counts of the amount of NP or the low-abundance M2 present in viruses due to these technical limitations. Additionally, non-specific labeling is an important consideration when selecting dyes (Hughes et al., 2014), a challenge that we have dealt with by choosing the most soluble dyes available at given wavelengths.
Quantifying HA and NA abundance
We estimated the labeling efficiency of different tagged proteins by recording images of labeled viruses using TIRF microscopy and comparing the intensity of viruses labeled in this way to the measured intensities of individual dye molecules. Since images that yield good signal-to-noise ratio for an individual dye molecule will yield saturated images of entire virions, it was necessary to calibrate dye intensities under two different imaging conditions. First, we non-specifically adsorb dye molecules onto a clean coverslip. This produces fields of view comprised of mostly single dye molecules with a lower abundance of small dye aggregates. We image multiple such fields of view using TIRF microscopy, first using a laser power that matches that used for imaging viruses, and second under a higher laser power, where single molecules can be imaged continuously and identified through their bleaching characteristics. These pairs of images for each field of view allows us to use the intensity of small dye aggregates (visible without saturation under both settings) to determine the intensity of single dye molecule under the imaging settings appropriate for characterizing viruses. Dividing the total intensity of a virus particle by the total intensity of a fluorophore determined in this way provides a first estimate for the number of fluorophores conjugated to that virus (Figure S4C).
Using this approach to analyze particles whose lengths are easily resolvable (typically ~1–10µm in length), we can estimate the density of labeled molecules on the viral surface. Dividing the number of fluorophores by the surface area of the virus (modeled as a cylinder of a length which we measure and an assumed radius of 40nm (Calder et al., 2010; Harris et al., 2006), capped by two hemispheres) gives the number of fluorophores per unit area: ~6000 molecules of HA and ~1100 molecules of NA per µm2 for N = 26 filamentous viruses with an average length of 3µm. At this density, a spherical virus 120nm in diameter would have ~271 HAs and ~50 NAs. Since our labeling of HA with SrtA is ~50% efficient and our labeling of NA with SFP is ~100% efficient, correcting these estimates results in ~542 HAs and ~50 NAs
The brightness of a fluorophore on the surface of a virus will depend on its position within the evanescent field, and thus its height above the coverslip. This distance will vary from ~20nm to ~100nm, given the diameter of a filamentous virus (~80nm) and our immobilization strategy (PEG2K-biotin to streptavidin to fetuin; Figure S4C). In contrast, the fluorophores that we use for calibration are in direct contact with the coverslip. Since the rate of photon emission should depend linearly on the excitation intensity (Dempsey et al., 2011), correcting for this requires weighting the contribution from fluorophores a distance z from the coverslip by the intensity of the evanescent field at this height, and integrating over the height of the virus. The resulting intensity, relative to the intensity that would result if all fluorophores were located on the coverslip (z = 0) is given by:
Evaluating this ratio with z0 = 20nm, a = 40nm, and d = 80nm gives ~0.5 for a wide range of virus lengths, L. Applying this correction increases the estimate for labeled HA and NA for the smallest virus particles to ~1031 HAs and ~95 NAs. These are the numbers we report in the manuscript.
An additional factor that influences the intensity of fluorophores on the virus is interactions between dye molecules. This could occur through photophysical effects (e.g. resonance energy transfer between spectrally overlapping dyes, or self-quenching between molecules of the same dye in close proximity), or through steric repulsion of new dye molecules when the degree of labeling is high. The dense packing of HA trimers and NA tetramers on the virus surface could position fluorophores sufficiently close (~5nm) to exhibit either of these effects. This is particularly relevant for NA, where the C-termini of monomers within the tetramer are positioned within a few nanometers of each other, making them susceptible to self-quenching. To determine the significance of self-quenching in multiply-labeled NA tetramers, we prepared virus in which NA labeling was performed with a mixture of AlexaFluor 555 - conjugated CoA, and dark CoA with a capped thiol. By varying the relative amounts of fluorescent CoA from 12.5% (1/8th) to 100% and quantifying virus intensity, we can detect self-quenching through deviations from linearity, provided that neither substrate (dark or fluorescent) is strongly preferred by their respective enzymes. As expected, intensity is roughly linear at ratios of AF555:dark CoA of 25% or less; however, when NA is labeled with 50% or 100% CoA-555, intensity is diminished relative to expectations by ~40%, suggesting self-quenching of the dye (Figure S4D). Importantly, however, if this self-quenching is attributable to dye molecules within (and not between) NA tetramers, it should apply uniformly across a population of viruses, regardless of their NA content. Although we observe a similar saturation effect for HA labeled with Alexafluor 647 (Figure S4D), in all cases brightest labeling is achieved by using 100% labeled conjugates.
Although these additional considerations are important, we choose not to include them in our reported values of HA and NA densities because we do not yet understand their origins, and thus we do not know precisely how they should be accounted. For example, if the reason that HA labeling saturates is because conjugated dye molecules sterically or electrostatically exclude further labeling, then this effect would be captured by the 50% labeling ratio. Alternatively, if the effect were photophysical, this would not be the case. Since labeling of NA with SFP has higher efficiency, we believe that saturation in this case may be due to self-quenching, though this is speculation.
Finally, we note that for viruses labeled with combinations of fluorophores, there are also potential interactions between spectrally distinct dyes; in particular, AF555 and AF647 serve as a donor-acceptor pair for FRET. For viruses labeled with these fluorophores, the apparent intensity of AF555-labeled NA could be diminished through resonance energy transfer to AF647-labeled HA. To quantify the importance of this transfer, we measured fluorescence dequenching of NA by bleaching AF647-labeled HA and comparing the original and dequenched NA intensity (Figure S4E). This gives an increase in median NA intensity of ~12% upon dequenching, a modest effect compared to other contributions discussed here.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was calculated for using Matlab R2017b and the Statistics and Machine Learning Toolbox. The number of biological replicates (defined as experiments performed on separate cell cultures using separate virus stocks), the precision and dispersion measures used, the statistical significance, and the statistical test used to determine the significance are indicated in each figure or figure legend where quantification is reported.
Supplementary Material
(A) All sequences are cloned into the pcDNA3.1+ backbone, with mRNA expression driven by a CMV promoter, and negative-sense vRNA expression driven by the human polI promoter. Numbers below sequences denote residue position in the respective segment for the WSN genome. The NA(ybbR) vector is cloned by inserting the 11a.a. ybbR tag with a 5a.a. linker at the C-terminus of NA, followed by the non-coding repetition of 156 nucleotides from the 5’ end of the viral RNA (inserted in the positive sense) to improve segment packaging. HA amenable to N-terminal labeling by SrtA is created by inserting five consecutive glycines (“5xGly”) following the site of signal peptidase cleavage. HA amenable to labeling by transglutaminase 2 contains a Q3-tag inserted within a variable region located at the top of the molecule. NP(FlAsH) consists of a tetracysteine tag attached directly to the C-terminus of NP, followed by 144 nucleotides from the 5’ end of the viral RNA repeated (in the positive-sense orientation) following the stop codon as in NA(ybbR) to improve segment packaging. To construct M1 (FlAsH), we insert a tetracysteine motif plus an additional glycine following Gly194 between two predicted helices in M1. For both M2 (M48) (labeled by microbial transglutaminase) and M2 (ybbR) (labeled by SFP synthase), we insert the respective tags downstream of the M1 stop codon. Within the M1 coding region, we introduce three mutations (highlighted in yellow) which are silent in M1 except for a non-deleterious M248I mutation. These mutations remove disulfide bonds and a glycosylation site upstream of the M2 tags that disrupt labeling. Coloring scheme: green - amino acids separate from the tag but modified to improve labeling; blue - nucleotides / amino acids comprising labeling tags; orange - nucleotides / amino acids inserted as linkers between native sequences and tags. See also Supplemental Data 1 for complete sequences of modified segments. (B) Field of view showing a monolayer of MDCK cells with ~50% of cells infected with an engineered strain of IAV harboring tagged HA (DyLight 405), NA (Alexa 555), M2 (Sulfo-Cy5), and NP (FlAsH) (scale bar = 20µm). Cells were grown at 33°C overnight following infection and labeled at ~12 hpi. Images show maximum intensity projections. (C) Detail of a single infected cell, showing filamentous virions emerging from the apical surface. Budding viruses are enriched in HA and NA, while M2 mostly remains on the cell body. Newly-exported vRNP complexes are visible as punctate structures emanating from the perinuclear region (scale bar = 10µm).
Supplemental Data 1. Sequences of IAV modified genome segments used in this work, Related to Figure 1.
The following data from this paper is organized on separate tabs in this file. Figure 2D & E: Data from the four replicates giving HA, NA, and NP intensities along with particle lengths; Figure 3C: HA and NA intensity data for 12 virus clusters; Figure 4B: HA, NA, and NP intensity values for each of three replicates; Figure 4D: HA, NA, and antibody intensity values for each of the four replicates for virus raised at 33°C and 37°C; Figure 5B: HA, NA, and NP intensity values from three replicates for each of the six conditions plotted in Figure 5B and Figure S7; Figure 5C: Population medians and sizes plotted in Figure 5C. Columns represent paired replicates. Figure 5D: Population medians and sizes plotted in Figure 5D.
(A) Monolayers of MDCK cells infected at MOI ~ 0.1–1 with different engineered IAV strains and labeled at 13hpi following growth at 33°C. Cells show a mosaic labeling pattern, indicating a mixture of infected and uninfected cells. By manually segmenting cells, we integrate the intensity in each labeled channel as a measure of the abundance of proteins available for virus production (note that membrane protein labeling is specific to only the cell surface population, leaving the intracellular pool unlabeled). Images show maximum intensity projections. (B) Two-dimensional histograms showing the covariance in labeling for pairs of proteins, with each point representing a single cell, and with infected cells occupying the upper right quadrant, and uninfected cells occupying the lower left quadrant. Upper left and lower right quadrants, which correspond to cells expressing one tag but not the other, are sparsely occupied. One exception to this are cells which label positive for NP, but not for other viral proteins (NA, M2, or HA, with the latter labeled either enzymatically by SrtA or using an anti-HA antibody). These cells also do not show signs of vRNP export visible in other members of the population, suggesting that they lack multiple viral proteins and represent viruses which failed to deliver all eight genomic segments to the infected cell, although they likely contain the four segments necessary for expression of NP: NP, PB1, PB2, and PA. (C) We calculate the percentage of cells expressing a particular tag by defining a threshold, which we then use to calculate two conditional probabilities: (i) the probability of expressing X given Y and (ii) the probability of expressing Y given X. The conditional probabilities for the nine conditions we measure are shown in the bar plots. All probabilities are >90%, except for the aforementioned population of cells in which we detect NP but no other tagged proteins (HA, NA, M2).
(A) Western blots comparing recombinant HA standards (left 6 lanes, not including the ladder) and IAV with either labeled HA or NA (2 rightmost lanes, besides the ladder). The blot is probed with both anti-HA and anti-NA antibodies (top), or imaged using fluorescence from directly-labeled proteins (bottom). (B) Comparing the AF647:aHA signal in the IAV sample to the recombinant standard results in an estimate of 48% labeling efficiency for HA on the surface of intact viruses. (C) Comparing the total AF647 fluorescence from HA and NA relative to the recombinant standard results in an estimate of 0.17pmol labeled HA and 0.11pmol labeled NA. The higher than expected NA:HA ratio suggests that NA labeling is more efficient than HA labeling. (C) Consistent with >50% efficient labeling of NA, we observe a mobility shift in NA towards higher molecular weight after modification with AF647.
(A) Tagged influenza A virus strains were labeled using either SFP + Alexa 555 (NA), SrtA + Alexa 647 (HA), and FlAsH (NP) (sample i); gpTgase2 + Alexa 647 (HA) (sample ii); or FlAsH (M1) and SFP + Alexa 647 (M2) (sample iii). After labeling, viruses were subjected to SDS-PAGE analysis, using sample buffer containing 20mM TCEP (samples I and ii) or 350mM beta-mercaptoethanol (sample iii) and lightly boiled. High concentrations of β-ME disrupt binding of the biarsenical dye FlAsH, but are necessary to fully reduce disulfide-linked M2 tetramers. Molecular weights agree with values predicted from protein sequences, allowing for tags and fluorophores, as well as glycosylation (for HA and NA). Under conditions with low amounts of TPCK-treated trypsin, HA is present in both cleaved (HA1) and uncleaved (HA0) forms, with fluorophores attached via both SrtA and TGase2 running with HA1 in the cleaved form. (B) Comparison of HA labeled with SrtA (left), NA labeled with SFP (middle), and NP labeled with FlAsH (right). Virus lacking the pentaglycine-, ybbR-, or tetracysteine-tags have a median labeling intensity ~500-fold, ~40-fold, or ~30-fold lower than virus with the respective tags. (C) To obtain approximate counts of labeling densities on the surface of a virus, we immobilize viruses on a PEG support and image using TIRF microscopy. Comparing the intensity of individual fluorophores to the integrated intensity of the virus provides an initial estimate of the number of fluorophores per virus (see supplemental text). (D) Testing for fluorophore interactions in multiply-labeled NA tetramers and HA trimers. As the percentage of CoA-555 or CLPETGG-647 increases relative to dark CoA or CLPETGG peptide (capped with NEM) in a mixture with the CoA/peptide-conjugate concentration held constant, the apparent density of fluorophores varies sub-linearly with the percentage of the fluorescent species, suggesting interactions (physical or photophysical) between dye molecules. (E) FRET between Alexa555-labeled NA and Alexa647-labeled HA accounts for an additional underestimation of NA density by ~12%.
(A) In longer (>1µm) filamentous viruses, NP incorporation (a necessary but insufficient requirement for genome packaging, quantified through total NP intensity per particle and restricted to particles with NP foci at one end, to eliminate the possibility of smaller viruses bound along the filament length that contain NP) is less efficient than in the overall viral population (37% efficient versus 67% efficient, as in Figure 2C). (B) Data from Figure 2D, plotted as individual replicates. See also Supplemental Data 2. (C) The efficiency of NP incorporation (a necessary but insufficient requirement for genome packaging) in particles with relatively more NA (64%) is comparable to that measured in particles with relatively less NA (57%). HA-NA abundance in particles containing NP resembles that of the population as a whole (bottom panel).
(A)Separate replicates from temperature shift experiments (Figure 4B). (B) Separate replicates from binding and internalization experiments (Figure 4D & E). See also Supplemental Data 2.
(A) Separate replicates of particle distributions following treatment with oseltamivir (Figure 5B). (B) Separate replicates of in vitro virus detachment (Figure 5F). See also Supplemental Data 2.
Tabulation of results from three biological replicates: “particles (total)” refers to the total number of particles pulled down onto anti-HA coated coverslips after a 16 hour incubation with periodic mixing; “particles (NP+)” refers to the number of immobilized particles that contain fluorescently-labeled NP above a threshold level; “semi-infectious” refers to the total number of particles that deliver at least one vRNA segment to an infected cell, and is calculated based on the segment probability (p) and the measured number of fully-infectious particles (PFU) as PFU/p8; “vRNP-infectious” refers to particles which deliver at least the four segments necessary for secondary transcription (PA, PB1, PB2, and NP); “fully-infectious” refers to particles capable of initiating multiple rounds of replication (PFUs). Finally, “segment probability” is the probability (expressed as a fraction) of any particular vRNA segment being expressed by an infected cell.
Highlights.
Influenza A virus forms morphologically and compositionally heterogeneous progeny
Heterogeneity is encoded in assembly and can depend on the growth environment
Progeny of individual virions are as variable as the progeny of large populations
Phenotypic variability helps subsets of the population to escape NA inhibitors
Acknowledgements
The authors would like to acknowledge members of the Fletcher Lab for feedback and technical consultation. This work was supported by the NIH R01 GM114671 (DAF), the Immunotherapeutics and Vaccine Research Initiative at UC Berkeley (DAF), and the Chan Zuckerberg Biohub (DAF). M.D.V. was funded by a Burroughs Wellcome Fund CASI Fellowship. D.A.F. is a Chan Zuckerberg Biohub investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Badham MD, and Rossman JS (2016). Filamentous Influenza Viruses. Curr. Clin. Microbiol. Rep 3, 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban NQ (2004). Bacterial Persistence as a Phenotypic Switch. Science 305, 1622–1625. [DOI] [PubMed] [Google Scholar]
- Baldwin JM (1896). A New Factor in Evolution. Am. Nat 30, 441–451. [Google Scholar]
- Battisti AJ, Meng G, Winkler DC, McGinnes LW, Plevka P, Steven AC, Morrison TG, and Rossmann MG (2012). Structure and assembly of a paramyxovirus matrix protein. Proc. Natl. Acad. Sci 109, 13996–14000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshe RB, Smith MH, Hall CB, Betts R, and Hay AJ (1988). Genetic basis of resistance to rimantadine emerging during treatment of influenza virus infection. J. Virol 62, 1508–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharat TAM, Noda T, Riches JD, Kraehling V, Kolesnikova L, Becker S, Kawaoka Y, and Briggs JAG (2012). Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc. Natl. Acad. Sci 109, 4275–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom JD, Gong LI, and Baltimore D (2010). Permissive Secondary Mutations Enable the Evolution of Influenza Oseltamivir Resistance. Science 328, 1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooke CB, Ince WL, Wrammert J, Ahmed R, Wilson PC, Bennink JR, and Yewdell JW (2013). Most Influenza A Virions Fail To Express at Least One Essential Viral Protein. J. Virol 87, 3155–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burleigh LM, Calder LJ, Skehel JJ, and Steinhauer DA (2005). Influenza A Viruses with Mutations in the M1 Helix Six Domain Display a Wide Variety of Morphological Phenotypes. J. Virol 79, 1262–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler PJG, and Klug A (1971). Assembly of the Particle of Tobacco Mosaic Virus from RNA and Disks of Protein. Nature. New Biol 229, 47–50. [DOI] [PubMed] [Google Scholar]
- Calder LJ, Wasilewski S, Berriman JA, and Rosenthal PB (2010). Structural organization of a filamentous influenza A virus. Proc. Natl. Acad. Sci 107, 10685–10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HY, Di Mascio M, Perelson AS, Ho DD, and Zhang L (2007). Determination of virus burst size in vivo using a single-cycle SIV in rhesus macaques. Proc. Natl. Acad. Sci 104, 19079–19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlanda P, Schraidt O, Kummer S, Riches J, Oberwinkler H, Prinz S, Krausslich H-G, and Briggs JAG (2015). Structural Analysis of the Roles of Influenza A Virus Membrane-Associated Proteins in Assembly and Morphology. J. Virol 89, 8957–8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou Y. -y., Vafabakhsh R, Doganay S, Gao Q, Ha T, and Palese P (2012). One influenza virus particle packages eight unique viral RNAs as shown by FISH analysis. Proc. Natl. Acad. Sci 109, 9101–9106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CM, Dawson IM, and Elford WJ (1949). Filamentous forms associated with newly isolated influenza virus. The Lancet 253, 602–603. [DOI] [PubMed] [Google Scholar]
- Clavel F, and Hance AJ (2004). HIV Drug Resistance. N. Engl. J. Med 350, 1023–1035. [DOI] [PubMed] [Google Scholar]
- Compans RW, Holmes KV, Dales S, and Choppin PW (1966). An electron microscopic study of moderate and virulent virus-cell interactions of the parainfluenza virus SV5. Virology 30, 411–426. [DOI] [PubMed] [Google Scholar]
- Dadonaite B, Vijayakrishnan S, Fodor E, Bhella D, and Hutchinson EC (2016). Filamentous influenza viruses. J. Gen. Virol 97, 1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejnirattisai W, Supasa P, Wongwiwat W, Rouvinski A, Barba-Spaeth G, Duangchinda T, Sakuntabhai A, Cao-Lormeau V-M, Malasit P, Rey FA, et al. (2016). Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with zika virus. Nat. Immunol 17, 1102–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingens AS, Haddox HK, Overbaugh J, and Bloom JD (2017). Comprehensive Mapping of HIV-1 Escape from a Broadly Neutralizing Antibody . Cell Host Microbe 21, 777–787.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald HB, and Isaacs A (1954). Counts of Influenza Virus Particles. J. Gen. Microbiol 10, 457–464. [DOI] [PubMed] [Google Scholar]
- Elleman CJ, and Barclay WS (2004). The M1 matrix protein controls the filamentous phenotype of influenza A virus. Virology 321, 144–153. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Gowen BB, Takahashi K, Shiraki K, Smee DF, and Barnard DL (2013). Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Res 100, 446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, and Discher DE (2007). Shape effects of filaments versus spherical particles in flow and drug delivery. Nat. Nanotechnol 2, 249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin BA (1998). Specific Covalent Labeling of Recombinant Protein Molecules Inside Live Cells. Science 281, 269–272. [DOI] [PubMed] [Google Scholar]
- Hagan MF, and Chandler D (2006). Dynamic Pathways for Viral Capsid Assembly. Biophys. J 91, 42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A, Cardone G, Winkler DC, Heymann JB, Brecher M, White JM, and Steven AC (2006). Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl. Acad. Sci 103, 19123–19127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Massarella J, and Ward P (1999). Clinical Pharmacokinetics of the Prodrug Oseltamivir and its Active Metabolite Ro 64–0802: Clin. Pharmacokinet 37, 471–484. [DOI] [PubMed] [Google Scholar]
- Hutchinson EC, Denham EM, Thomas B, Trudgian DC, Hester SS, Ridlova G, York A, Turrell L, and Fodor E (2012). Mapping the Phosphoproteome of Influenza A and B Viruses by Mass Spectrometry. PLoS Pathog 8, e1002993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson EC, Charles PD, Hester SS, Thomas B, Trudgian D, Martfnez-Alonso M, and Fodor E (2014). Conserved and host-specific features of influenza virion architecture. Nat. Commun 5, 4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai M, Watanabe T, Hatta M, Das SC, Ozawa M, Shinya K, Zhong G, Hanson A, Katsura H, Watanabe S, et al. (2012). Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature [DOI] [PMC free article] [PubMed]
- Kussell E (2005). Phenotypic Diversity, Population Growth, and Information in Fluctuating Environments. Science 309, 2075–2078. [DOI] [PubMed] [Google Scholar]
- Lakdawala SS, Wu Y, Wawrzusin P, Kabat J, Broadbent AJ, Lamirande EW, Fodor E, Altan-Bonnet N, Shroff H, and Subbarao K (2014). Influenza A Virus Assembly Intermediates Fuse in the Cytoplasm. PLoS Pathog 10, e1003971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lu X, Li J, Berube N, Giest K-L, Liu Q, Anderson DH, and Zhou Y (2010). Genetically Engineered, Biarsenically Labeled Influenza Virus Allows Visualization of Viral NS1 Protein in Living Cells. J. Virol 84, 7204–7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C-W, and Ting AY (2006). Transglutaminase-Catalyzed Site-Specific Conjugation of Small-Molecule Probes to Proteins in Vitro and on the Surface of Living Cells. J. Am. Chem. Soc 128, 4542–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma EJ, Hill NJ, Zabilansky J, Yuan K, and Runstadler JA (2016). Reticulate evolution is favored in influenza niche switching. Proc. Natl. Acad. Sci 113, 5335–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K, and Helenius A (1991). Transport of incoming influenza virus nucleocapsids into the nucleus. J. Virol 65, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu S, Sagara H, Sakai-Tagawa Y, Sugaya N, Noda T, and Kawaoka Y (2016). Complete and Incomplete Genome Packaging of Influenza A and B Viruses. MBio 7, e01248–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson K, Aoki F, Osterhaus A, Trottier S, Carewicz O, Mercier C, Rode A, Kinnersley N, and Ward P (2000). Efficacy and safety of oseltamivir in treatment of acute influenza: a randomised controlled trial. The Lancet 355, 1845–1850. [DOI] [PubMed] [Google Scholar]
- Noda T, Sugita Y, Aoyama K, Hirase A, Kawakami E, Miyazawa A, Sagara H, and Kawaoka Y (2012). Three-dimensional analysis of ribonucleoprotein complexes in influenza A virus. Nat. Commun 3, 639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noton SL, Simpson-Holley M, Medcalf E, Wise HM, Hutchinson EC, McCauley JW, and Digard P (2009). Studies of an Influenza A Virus Temperature-Sensitive Mutant Identify a Late Role for NP in the Formation of Infectious Virions. J. Virol 83, 562–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxford JS, Corcoran T, and Hugentobler AL (1981). Quantitative analysis of the protein composition of influenza A and B viruses using high resolution SDS polyacrylamide gels. J. Biol. Stand 9, 483–491. [DOI] [PubMed] [Google Scholar]
- Pauly MD, Procario MC, and Lauring AS (2017). A novel twelve class fluctuation test reveals higher than expected mutation rates for influenza A viruses. ELife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennings PS (2012). Standing Genetic Variation and the Evolution of Drug Resistance in HIV. PLoS Comput. Biol 8, e1002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson TC, and Diamond MS (2012). Degrees of maturity: the complex structure and biology of flaviviruses. Curr. Opin. Virol 2, 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon LLM, Song T, Rosenfeld R, Lin X, Rogers MB, Zhou B, Sebra R, Halpin RA, Guan Y, Twaddle A, et al. (2016). Quantifying influenza virus diversity and transmission in humans. Nat. Genet 48, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp MW-L, Karssemeijer RA, and Ploegh HL (2012). Chemoenzymatic Site-Specific Labeling of Influenza Glycoproteins as a Tool to Observe Virus Budding in Real Time. PLoS Pathog 8, e1002604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego EH, Audette RE, and Rubin EJ (2017). Deletion of a mycobacterial divisome factor collapses single-cell phenotypic heterogeneity. Nature 546, 153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SR, Compans RW, and Wertz GW (1995). Respiratory syncytial virus matures at the apical surfaces of polarized epithelial cells. J. Virol 69, 2667–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong L, Dahari H, Ribeiro RM, and Perelson AS (2010). Rapid Emergence of Protease Inhibitor Resistance in Hepatitis C Virus. Sci. Transl. Med 2, 30ra32–30ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA (1972). SIALIC ACIDS ON THE PLASMA MEMBRANE OF CULTURED HUMAN LYMPHOID CELLS: Chemical Aspects and Biosynthesis. J. Cell Biol 53, 466–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman JS, and Lamb RA (2011). Influenza virus assembly and budding. Virology 411, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman JS, Leser GP, and Lamb RA (2012). Filamentous Influenza Virus Enters Cells via Macropinocytosis. J. Virol 86, 10950–10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AB, Trapnell C, and Bloom JD (2018). Extreme heterogeneity of influenza virus infection in single cells. ELife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan R, Nebot MR, Chirico N, Mansky LM, and Belshaw R (2010). Viral Mutation Rates. J. Virol 84, 9733–9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze IT (1972). The structure of influenza virus. Virology 47, 181–196. [DOI] [PubMed] [Google Scholar]
- Shaw ML, Stone KL, Colangelo CM, Gulcicek EE, and Palese P (2008). Cellular Proteins in Influenza Virus Particles. PLoS Pathog 4, e1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten EM, and Bertozzi CR (2009). Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem. Int. Ed 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauch E-M, Bernard SM, La D, Bohn AJ, Lee PS, Anderson CE, Nieusma T, Holstein CA, Garcia NK, Hooper KA, et al. (2017). Computational design of trimeric influenza-neutralizing proteins targeting the hemagglutinin receptor binding site. Nat. Biotechnol 35, 667–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stray SJ, and Air GM (2001). Apoptosis by influenza viruses correlates with efficiency of viral mRNA synthesis. Virus Res 77, 3–17. [DOI] [PubMed] [Google Scholar]
- Sugimura Y, Yokoyama K, Nio N, Maki M, and Hitomi K (2008). Identification of preferred substrate sequences of microbial transglutaminase from Streptomyces mobaraensis using a phage-displayed peptide library. Arch. Biochem. Biophys 477, 379–383. [DOI] [PubMed] [Google Scholar]
- Theile CS, Witte MD, Blom AEM, Kundrat L, Ploegh HL, and Guimaraes CP (2013). Site-specific N-terminal labeling of proteins using sortase-mediated reactions. Nat. Protoc 8, 1800–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varble A, Albrecht RA, Backes S, Crumiller M, Bouvier NM, Sachs D, Garcfa-Sastre A, and tenOever BR (2014). Influenza A Virus Transmission Bottlenecks Are Defined by Infection Route and Recipient Host. Cell Host Microbe 16, 691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, and Andino R (2006). Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439, 344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner R, Matrosovich M, and Klenk H-D (2002). Functional balance between haemagglutinin and neuraminidase in influenza virus infections. Rev. Med. Virol 12, 159–166. [DOI] [PubMed] [Google Scholar]
- Wasilewski S, Calder LJ, Grant T, and Rosenthal PB (2012). Distribution of surface glycoproteins on influenza A virus determined by electron cryotomography. Vaccine 30, 7368–7373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Lin AJ, Golan DE, and Walsh CT (2006). Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nat. Protoc 1, 280–285. [DOI] [PubMed] [Google Scholar]
- Zebedee SL, and Lamb RA (1988). Influenza A virus M2 protein: monoclonal antibody restriction of virus growth and detection of M2 in virions. J. Virol 62, 2762–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) All sequences are cloned into the pcDNA3.1+ backbone, with mRNA expression driven by a CMV promoter, and negative-sense vRNA expression driven by the human polI promoter. Numbers below sequences denote residue position in the respective segment for the WSN genome. The NA(ybbR) vector is cloned by inserting the 11a.a. ybbR tag with a 5a.a. linker at the C-terminus of NA, followed by the non-coding repetition of 156 nucleotides from the 5’ end of the viral RNA (inserted in the positive sense) to improve segment packaging. HA amenable to N-terminal labeling by SrtA is created by inserting five consecutive glycines (“5xGly”) following the site of signal peptidase cleavage. HA amenable to labeling by transglutaminase 2 contains a Q3-tag inserted within a variable region located at the top of the molecule. NP(FlAsH) consists of a tetracysteine tag attached directly to the C-terminus of NP, followed by 144 nucleotides from the 5’ end of the viral RNA repeated (in the positive-sense orientation) following the stop codon as in NA(ybbR) to improve segment packaging. To construct M1 (FlAsH), we insert a tetracysteine motif plus an additional glycine following Gly194 between two predicted helices in M1. For both M2 (M48) (labeled by microbial transglutaminase) and M2 (ybbR) (labeled by SFP synthase), we insert the respective tags downstream of the M1 stop codon. Within the M1 coding region, we introduce three mutations (highlighted in yellow) which are silent in M1 except for a non-deleterious M248I mutation. These mutations remove disulfide bonds and a glycosylation site upstream of the M2 tags that disrupt labeling. Coloring scheme: green - amino acids separate from the tag but modified to improve labeling; blue - nucleotides / amino acids comprising labeling tags; orange - nucleotides / amino acids inserted as linkers between native sequences and tags. See also Supplemental Data 1 for complete sequences of modified segments. (B) Field of view showing a monolayer of MDCK cells with ~50% of cells infected with an engineered strain of IAV harboring tagged HA (DyLight 405), NA (Alexa 555), M2 (Sulfo-Cy5), and NP (FlAsH) (scale bar = 20µm). Cells were grown at 33°C overnight following infection and labeled at ~12 hpi. Images show maximum intensity projections. (C) Detail of a single infected cell, showing filamentous virions emerging from the apical surface. Budding viruses are enriched in HA and NA, while M2 mostly remains on the cell body. Newly-exported vRNP complexes are visible as punctate structures emanating from the perinuclear region (scale bar = 10µm).
Supplemental Data 1. Sequences of IAV modified genome segments used in this work, Related to Figure 1.
The following data from this paper is organized on separate tabs in this file. Figure 2D & E: Data from the four replicates giving HA, NA, and NP intensities along with particle lengths; Figure 3C: HA and NA intensity data for 12 virus clusters; Figure 4B: HA, NA, and NP intensity values for each of three replicates; Figure 4D: HA, NA, and antibody intensity values for each of the four replicates for virus raised at 33°C and 37°C; Figure 5B: HA, NA, and NP intensity values from three replicates for each of the six conditions plotted in Figure 5B and Figure S7; Figure 5C: Population medians and sizes plotted in Figure 5C. Columns represent paired replicates. Figure 5D: Population medians and sizes plotted in Figure 5D.
(A) Monolayers of MDCK cells infected at MOI ~ 0.1–1 with different engineered IAV strains and labeled at 13hpi following growth at 33°C. Cells show a mosaic labeling pattern, indicating a mixture of infected and uninfected cells. By manually segmenting cells, we integrate the intensity in each labeled channel as a measure of the abundance of proteins available for virus production (note that membrane protein labeling is specific to only the cell surface population, leaving the intracellular pool unlabeled). Images show maximum intensity projections. (B) Two-dimensional histograms showing the covariance in labeling for pairs of proteins, with each point representing a single cell, and with infected cells occupying the upper right quadrant, and uninfected cells occupying the lower left quadrant. Upper left and lower right quadrants, which correspond to cells expressing one tag but not the other, are sparsely occupied. One exception to this are cells which label positive for NP, but not for other viral proteins (NA, M2, or HA, with the latter labeled either enzymatically by SrtA or using an anti-HA antibody). These cells also do not show signs of vRNP export visible in other members of the population, suggesting that they lack multiple viral proteins and represent viruses which failed to deliver all eight genomic segments to the infected cell, although they likely contain the four segments necessary for expression of NP: NP, PB1, PB2, and PA. (C) We calculate the percentage of cells expressing a particular tag by defining a threshold, which we then use to calculate two conditional probabilities: (i) the probability of expressing X given Y and (ii) the probability of expressing Y given X. The conditional probabilities for the nine conditions we measure are shown in the bar plots. All probabilities are >90%, except for the aforementioned population of cells in which we detect NP but no other tagged proteins (HA, NA, M2).
(A) Western blots comparing recombinant HA standards (left 6 lanes, not including the ladder) and IAV with either labeled HA or NA (2 rightmost lanes, besides the ladder). The blot is probed with both anti-HA and anti-NA antibodies (top), or imaged using fluorescence from directly-labeled proteins (bottom). (B) Comparing the AF647:aHA signal in the IAV sample to the recombinant standard results in an estimate of 48% labeling efficiency for HA on the surface of intact viruses. (C) Comparing the total AF647 fluorescence from HA and NA relative to the recombinant standard results in an estimate of 0.17pmol labeled HA and 0.11pmol labeled NA. The higher than expected NA:HA ratio suggests that NA labeling is more efficient than HA labeling. (C) Consistent with >50% efficient labeling of NA, we observe a mobility shift in NA towards higher molecular weight after modification with AF647.
(A) Tagged influenza A virus strains were labeled using either SFP + Alexa 555 (NA), SrtA + Alexa 647 (HA), and FlAsH (NP) (sample i); gpTgase2 + Alexa 647 (HA) (sample ii); or FlAsH (M1) and SFP + Alexa 647 (M2) (sample iii). After labeling, viruses were subjected to SDS-PAGE analysis, using sample buffer containing 20mM TCEP (samples I and ii) or 350mM beta-mercaptoethanol (sample iii) and lightly boiled. High concentrations of β-ME disrupt binding of the biarsenical dye FlAsH, but are necessary to fully reduce disulfide-linked M2 tetramers. Molecular weights agree with values predicted from protein sequences, allowing for tags and fluorophores, as well as glycosylation (for HA and NA). Under conditions with low amounts of TPCK-treated trypsin, HA is present in both cleaved (HA1) and uncleaved (HA0) forms, with fluorophores attached via both SrtA and TGase2 running with HA1 in the cleaved form. (B) Comparison of HA labeled with SrtA (left), NA labeled with SFP (middle), and NP labeled with FlAsH (right). Virus lacking the pentaglycine-, ybbR-, or tetracysteine-tags have a median labeling intensity ~500-fold, ~40-fold, or ~30-fold lower than virus with the respective tags. (C) To obtain approximate counts of labeling densities on the surface of a virus, we immobilize viruses on a PEG support and image using TIRF microscopy. Comparing the intensity of individual fluorophores to the integrated intensity of the virus provides an initial estimate of the number of fluorophores per virus (see supplemental text). (D) Testing for fluorophore interactions in multiply-labeled NA tetramers and HA trimers. As the percentage of CoA-555 or CLPETGG-647 increases relative to dark CoA or CLPETGG peptide (capped with NEM) in a mixture with the CoA/peptide-conjugate concentration held constant, the apparent density of fluorophores varies sub-linearly with the percentage of the fluorescent species, suggesting interactions (physical or photophysical) between dye molecules. (E) FRET between Alexa555-labeled NA and Alexa647-labeled HA accounts for an additional underestimation of NA density by ~12%.
(A) In longer (>1µm) filamentous viruses, NP incorporation (a necessary but insufficient requirement for genome packaging, quantified through total NP intensity per particle and restricted to particles with NP foci at one end, to eliminate the possibility of smaller viruses bound along the filament length that contain NP) is less efficient than in the overall viral population (37% efficient versus 67% efficient, as in Figure 2C). (B) Data from Figure 2D, plotted as individual replicates. See also Supplemental Data 2. (C) The efficiency of NP incorporation (a necessary but insufficient requirement for genome packaging) in particles with relatively more NA (64%) is comparable to that measured in particles with relatively less NA (57%). HA-NA abundance in particles containing NP resembles that of the population as a whole (bottom panel).
(A)Separate replicates from temperature shift experiments (Figure 4B). (B) Separate replicates from binding and internalization experiments (Figure 4D & E). See also Supplemental Data 2.
(A) Separate replicates of particle distributions following treatment with oseltamivir (Figure 5B). (B) Separate replicates of in vitro virus detachment (Figure 5F). See also Supplemental Data 2.
Tabulation of results from three biological replicates: “particles (total)” refers to the total number of particles pulled down onto anti-HA coated coverslips after a 16 hour incubation with periodic mixing; “particles (NP+)” refers to the number of immobilized particles that contain fluorescently-labeled NP above a threshold level; “semi-infectious” refers to the total number of particles that deliver at least one vRNA segment to an infected cell, and is calculated based on the segment probability (p) and the measured number of fully-infectious particles (PFU) as PFU/p8; “vRNP-infectious” refers to particles which deliver at least the four segments necessary for secondary transcription (PA, PB1, PB2, and NP); “fully-infectious” refers to particles capable of initiating multiple rounds of replication (PFUs). Finally, “segment probability” is the probability (expressed as a fraction) of any particular vRNA segment being expressed by an infected cell.