

Abstract
Genomics approaches are increasingly utilized to probe host-viral interactions and identify mechanisms of viral pathogenesis and host-subversion. Here we review recent studies that utilize Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 screens, transcriptomics and epigenomics to gain insight into Dengue and Zika virus infections in humans. We discuss the benefits and limitations of recently utilized techniques that separate virally-infected cells from neighboring uninfected cells to identify the mechanisms by which these viruses regulate host responses. We conclude by discussing how these approaches can best advance our understanding of Dengue and Zika virus pathogenesis in humans.
Introduction:
Dengue virus (DENV) and Zika virus (ZIKV) pathogenesis
DENV causes dengue fever (DF) and dengue hemorrhagic fever/dengue shock syndrome (DHF/DSS), also known as Severe Dengue. It is the most prevalent viral illness transmitted by mosquitoes. Each year there are approximately 400 million DENV infections, of which 100 million are symptomatic [1]. There are over 2 million DHF/DSS cases and approximately 20,000 deaths each year [2]. DF is characterized by fever, arthralgia, myalgia, abdominal pain, and rash. In contrast, a hallmark of DHF/DSS is plasma leakage, which can lead to hemoconcentration, pleural effusion, ascites, and shock. Neurologic abnormalities, including encephalitis, are rare [3].
Similar to DENV, approximately 20% of ZIKV infections result in a DF-like, self-limited febrile disease. However, ZIKV infections in utero can cause Congenital Zika Syndrome (CZS), which includes microcephaly, cerebral malformations, ophthalmological and hearing defects, and arthrogryposis [4]. In a small number of adults, ZIKV may cause Guillain-Barre syndrome, an autoimmune peripheral neuropathy characterized by acute, symmetric limb weakness with decreased or absent deep-tendon reflexes [5–8].
DENV and ZIKV are transmitted to humans by the mosquitoes Aedes aegypti and Aedes albopictus, placing an estimated 3.6 billion people worldwide at risk for infection [1,9,10]. ZIKV can also be sexually transmitted [11,12]. Despite the significance of these pathogens, no specific therapies are available.
Genomic approaches to understanding DENV and ZIKV pathogenesis
All human pathogenic viruses must utilize host factors and disarm and/or avoid immune responses to survive. Therapeutics that inhibit viral utilization of essential host factors or viral suppression of critical immune responses could be effective anti-viral agents. Conventional approaches to studying antiviral responses have primarily focused on the function of a single or a limited subset of molecules. These studies have provided incredible insights into viral and host factors that influence virus replication and infection outcome. However, the response to viral infections is dynamic, complex and incompletely understood. Complementary, genome-wide methods, including CRISPR, transcriptomics and epigenomics, provide an unbiased and comprehensive way of expanding our understanding of host-viral interactions. However, bulk genomic approaches that examine mixed population of cells have important limitations that must be overcome, including the inability to differentiate responses originating from virally infected compared to bystander cells and distinguish cell-types to interogate cell-cell communication.
Main text of the review:
CRISPR-Cas9 loss of function screens
Viruses are obligate intracellular parasites that require host factors for survival. The CRISPR-Cas system is a prokaryotic immune defense engineered to serve as a genome editing tool [13–18]. Pooled CRISPR loss-of-function screens can identify host factors, practically genome-wide, required for viral infection that in turn could be targeted by therapeutics to prevent disease. Compared to RNAi, CRISPR–Cas appears to result in a greater signal-to-noise ratio and the identification of fewer false-positives, and is continuously being optimized to decrease its off-target effects [15,19–21].
As flaviviruses, which include DENV, ZIKV and West Nile virus (WNV), are cytolytic, infecting a pooled group of cells in which Cas9 and whole genome gRNAs have been introduced and cultured together allows selection of rare mutant cells that do not support viral entry, translation, replication or virus-induced cell death under highly stringent conditions. Independent genome-wide CRISPR–Cas9 loss of function screens using DENV [22,23], WNV [24,25] and ZIKV [26] have identified functionally related ER complexes that are required for flaviviral replication. A CRISPR screen in DENV2-infected Huh7.5.1 cells identified multiple subunits of the translocon-associated protein (TRAP), ER-associated protein degradation (ERAD), and oligosaccharyltransferases (OST) complexes [22]. The translocon and TRAP complexes transport secreted proteins across, or integrate membrane proteins into, the mammalian ER membrane [27,28]. The OST can associate with the translocon and N-glycosylate translocating proteins [29,30]. In the ER lumen, the ERAD pathway provides a protein quality control mechanism that targets incorrectly folded proteins for retrotranslocation into the cytosol for proteasomal degradation [31,32]. Remarkably, in an independent CRISPR screen that used the same cell line, Huh7.5.1, and virus, DENV2 strain 16681, 9 out of top 10 genes identified as essential for DENV infection were also in the top 25 scoring genes in the previous screen [22], and included members of the OST complex [23].
A genome-wide CRISPR/Cas9-based cell death screen in WNV-infected 293T cells identified seven human genes that were later validated to reduce WNV, DENV and ZIKV infection [25]. These genes are members of the translocon as well as the OST, signal peptidase (SPase), and endoplasmic reticulum membrane protein (EMC) complexes. The SPases typically cleave the signal peptide from translocating proteins and were shown to be required for proper cleavage of the flavivirus structural proteins (prM and E) and secretion of viral particles [25,29].
Loss of a SPase subunit, SPCS1, alone suppressed many members of Flaviviridae family, including WNV, DENV, ZIKV, yellow fever virus (YFV), Japanese encephalitis virus (JEV), and hepatitis C viruses (HCV), but had little impact on unrelated positive- or negative- sense RNA viruses [25]. An independent genome-wide CRISPR screen in WNV-infected 293T cells, identified mutations in the ERAD machinery that conferred strong protection against WNV induced cell death but did not appear to block viral replication [24].
The only ZIKV genome-wide CRISPR screen was performed in H1-HeLa cells infected with the African strain of ZIKV, MR766 [24]. In addition to identifying a putative ZIKV receptor (AXL), and genes involved in endocytosis, members of the TRAP, OST, and EMC complexes were identified as essential host genes for ZIKV replication.
Together, these studies identified a critical role for several, often functionally related, ER complexes in flavivirus biology. These findings are consistent with the ER being the major site of flavivirus translation, polyprotein processing, replication, and virion assembly [33]. Importantly, the high reproducibility of genome-wide CRISPR screens contrasts with low overlap among results from siRNA-based screens for pro-viral factors. Thus at present, CRISPR–Cas9 represents an efficient, unbiased and reproducible way of identifying essential host factors in flavivirus infections. Alternatively, CRISPR screens performed in naturally resistant cells, or using CRISPR-mediated gene activation (CRISPRa) in susceptible cells, could identify key viral restriction factors important for flaviviral immunity [34–36].
Transcriptomic analyses to understand ZIKV infection in iPSC-derived or primary human cells
The neurodevelopmental dysfunctions of CZS have been associated with ZIKV infection of neural progenitor cells (NPCs) [37–46]. Transcriptional profiling demonstrated that ZIKV infection in human NPCs (hNPCs) leads to downregulation of genes involved in cell-cycle progression [45,47] and neurogenesis [47]. These findings are consistent with many studies showing ZIKV impairs NPC proliferation [37,40,43], differentiation [47], and survival [37–46], all of which may contribute to CZS. During pregnancy, ZIKV productively infects placental macrophages and cytotrophoblasts which may allow dissemination of the virus into the fetal circulation [48–53]. Weisblum et al. analyzed the response of human maternal-decidual tissues grown ex vivo as three-dimensional (3D) organ cultures during infection with ZIKV compared to human cytomegalovirus (HCMV), another major cause of perinatal infections. Despite more robust ZIKV replication in decidual tissue, the genome-wide expression analysis revealed that ZIKV induced far fewer genes involved in immune cell activation and trafficking compared to HCMV [54]. Furthermore, the chorionic villus responses to ZIKV were selectively enriched for apoptosis, cell death, and necrosis when compared to HCMV.
Although these gene expression studies have provided important insights into ZIKV pathogenesis in clinically relevant cell types, they have been limited to measurements from whole tissues or bulk cell populations that include multiple cell types and both infected and uninfected bystander cells (Figure 1A). This approach masks gene expression differences between infected and bystander cells, and obscures cell type-specific expression patterns that play important functions in cell-cell communication [55–57]. Conventional approaches to overcome these obstacles include the use of reporter viruses that can be used to identify or enrich for infected cells or the infection of highly susceptible cell lines. Both of these approaches can produce confounding results, since insertion of reporter proteins into a relatively small flaviviral genome may alter their virulence and prohibit characterization of low passage clinical isolates. Additionally, transformed cell lines that support robust viral replication often lack important anti-viral signaling responses and likely do not reflect the responses in clinically relevant, primary cells. New genome-wide approaches, such as single cell RNA-seq (scRNA-seq) [57] and fixed cell fluorescence activated cell sorting for next-generation sequencing (FACS-NGS) [56], that can compare virally infected and uninfected cells and cell-type specific responses are emerging to analyze host-flavivirus interactions and improve our understanding of human responses to flavivirus infection and viral subversion mechanisms (Figure 1B and1C).
Figure 1: Genome-wide approaches to understanding host responses to flaviviral infection.
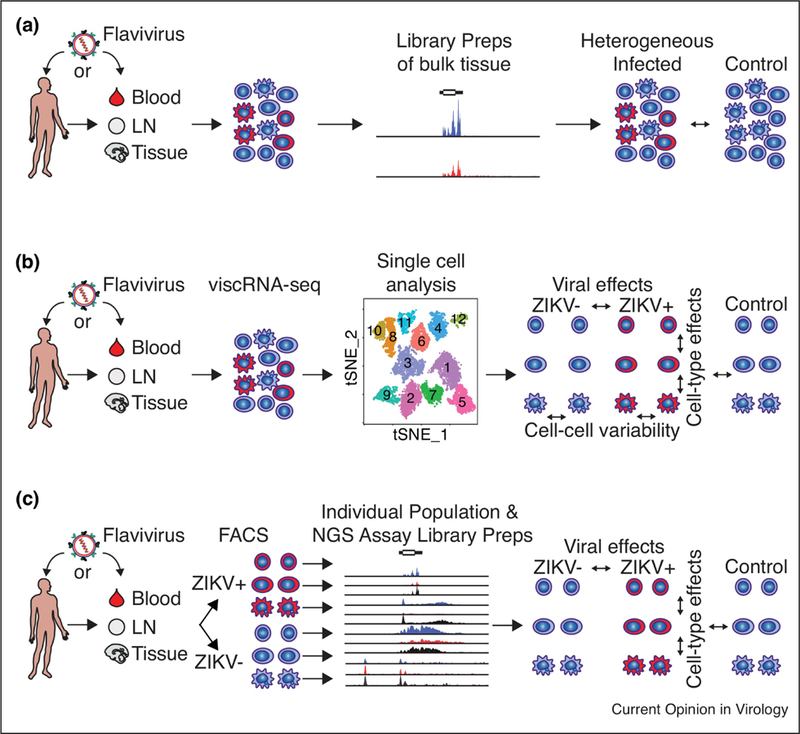
(A) Analysis comparing infected and uninfected tissue or bulk heterogeneous cell populations cannot differentiate signals from infected and uninfected bystander cells or cell type-specific expression patterns. (B) viscRNA-seq and (C) FACS-NGS approaches differentiate host responses in infected and bystander cells and between cell types providing insight into viral subversion of host responses and cell-specific reactions. Additionally, viscRNA-seq identifies transcriptional variability across single cells and measures viral abundance in individual cells while FACS-NGS is adaptable to many RNA and chromatin based NGS approaches. Abbreviations: ZIKV, Zika virus; viscRNA-seq, virus-inclusive single cell RNA-seq; LN, lymph node; tSNE, T-distributed Stochastic Neighbor Embedding; FACS, Fluorescence-activated cell sorting.
Genome-wide expression analysis at the single cell level using viscRNA-seq
DENV and ZIKV are positive-sense, single-stranded RNA viruses with genomes approximately 10.7 kb in length. The 5’ end of the positive strand genomic RNA has a N7 methylated (me7)-guanosine cap, and the 3’ end is non-polyadenylated and terminates in a stable stem-loop structure [33,58,59]. Typical scRNA-seq approaches rely on oligo(dT) priming, thus they would not be expected to distinguish flavivirus-infected from uninfected cells. Zanini et al. developed viral inclusive scRNA-seq (viscRNA-seq) approach that uses both oligo(dT) and virus-specific primers to amplify the whole transcriptome of single cells together with the viral RNA (vRNA) from the same cell (Figure 1B and Table 1) [57]. They performed a time-series of DENV and ZIKV infections in the human hepatocellular carcinoma cell line huh-7. First they noted a large dynamic range for intracellular virus abundance. By correlating gene expression with DENV level in the same cell, they identified several cellular functions known to be involved in flavivirus replication, including ER translocation, N-linked glycosylation and intracellular membrane trafficking. Using loss-of-function and gain-of-function screens targeting highly correlated genes they identified novel proviral (RPL31, TRAM1, and TMED2) and antiviral (ID2, CTNNB1) factors involved in DENV infection [57]. In a subsequent study, the same group combined FACS with viscRNA-Seq using pan-DENV primers to profile the landscape of host transcripts and DENV RNA in thousands of single blood cells from 6 patients acutely infected with DENV [60]. This approach identified not only cell-type specific immune activation but also virus association with both naive B cells and monocytes. Although monocytes are well-accepted to be cellular hosts of DENV, the role of B cells in supporting productive DENV infection has been unclear [61]. Further studies using DENV patient samples and primary human cell culture models are now needed to validate B cell permissivity to DENV infection. Additionally, this study identified candidate predictive biomarkers of Severe Dengue that will also need to be validated in a larger cohort.
Table 1:
Benefits and limitations of recently described viscRNA-seq (Zanini F et al.) and FACS-NGS methods (Carlin AF et al.).
| viscRNA-seq | FACS-NGS | |
|---|---|---|
| Special Equipment | scRNA-seq platform with viral primers | FACS, antibodies |
| Sample and library preparation | Easy - hours | Medium - days |
| Compares single cells | Yes | No |
| Compatible NGS assays | RNA-seq | RNA-seq, ChIP-seq, ATAC- seq, Hi-C |
| RNA-seq library types | PolyA | PolyA, Ribo-depleted, miRNA, etc. |
| Viral abundance/cell | Yes | No |
Understanding transcription at the level of chromatin:
Epigenetics can help us understand why distinct cell types or environments lead to differential responses to the same viral infection, how the cellular responses during viral infections are coordinated by specific transcription factors (TFs) and chromatin states, and how human genetic variation can alter viral susceptibility. This knowledge is critical since viral responses are often cell-type and context-dependent. For example, ZIKV is highly cytotoxic to neural progenitors but glial cells can propagate ZIKV [62]. Additionally, ZIKV infection induces massive apoptosis of iPSC-derived NPCs but causes limited apoptosis and viral persistance in cells derived from primary neural progenitors or in NSCs within infected organotypic slice cultures [37,38,41,45,47,63]. These differential responses, with clear implications for ZIKV pathogenesis, could be driven by differences in gene expression determined by cell origin (iPSC-derived vs. human in vivo) and/or the environment (monoculture vs. organ culture), as both cell state and environmental stimuli can drive divergent gene expression programs by differentially activating a common enhancer repertoire [64–66].
The epigenetic landscape of each cell type is established by pioneer TFs or lineage-determining TFs (LDTF) that initiate enhancer selection [67–76]. This epigenomic landscape allows transcription of constitutively expressed genes and provides the geography (chromatin state) that directs binding of signal-dependent TFs (SDTFs), like NFkB or interferon regulatory factors (IRFs) [68]. This means that the epigenomic landscape controls cell-type-specific constitutive gene expression and the initial transcriptional response to stimuli, like DENV or ZIKV infections [67,68,72,76]. Viral infections can further change the epigenetic landscape and thus reprogram the host and viral transcriptional profile [77–80]. This epigenetic remodeling may involve activation or repression of enhancers, modifications of DNA or changes in chromatin architecture. Thus, simultaneous probing of epigenomic and transcriptomic responses can explain (1) cell-type-dependent and context-dependent (for example, tissue environment) cellular responses to ZIKV/DENV infection while identifying (2) how pro- and anti-viral cellular pathways are activated and suppressed in a genome-wide unbiased manner.
Genomics analyses in viral infected and bystander cells using FACS
We recently developed a FACS-based approach that distinguishes flavivirus-infected from uninfected cells for genome-wide studies (Figure 1C and Table 1) [56]. In this study, primary human macrophages from multiple donors were infected with two different strains of Asian ZIKV, including a low passage clinical isolate representing contemporaneous lineage. At different time points post-infection, the cells are fixed with formaldehyde and ZIKV-infected (ZIKV+) cells are separated from uninfected bystanders (ZIKV-) by intracellular antibody staining that labels the common flavivirus envelope antigen. High quality RNA or chromatin is recovered for genome-wide transcriptional and epigenetic studies. Using this method we observed strikingly divergent transcriptional and epigenetic responses between ZIKV-infected and uninfected bystander macrophages derived from human blood monocytes. If fact, we revealed that studying mixed populations could produce misleading results about how ZIKV modifies cellular responses. In contrast, direct comparison of ZIKV+ infected to ZIKV- bystander cells that are exposed to the same environmental stimuli demonstrated how ZIKV manipulates human transcriptional responses. In particular, ZIKV+ macrophages exhibited a delayed and attenuated transcriptional response with suppression of multiple inflammation and immune response-related categories including IFN signaling, cytokine signaling, antigen presentation and pattern recognition receptor (PRR) response pathways, whereas ZIKV- bystander cells rapidly mounted and sustained a robust interferon-stimulated gene (ISG) response. ZIKV degrades STAT2 in ZIKV+ but not in ZIKV- bystander macrophages consistent with previous studies [81–83]. Thus, bystander macrophages can upregulate ISG expression by responding to secreted IFN-β using the canonical type I IFN pathway. A benefit of our approach is that sorted cell populations can be processed for epigenetic analyses such as ChIP-seq, ATAC-seq or genome 3D-structural analyses such as Hi-C. Using ChIP-seq, we identified genomic regions exhibiting gain or loss of H3K27ac in ZIKV+ or ZIKV- cells. Motif analysis of these regions demonstrated a reduction of enrichment of IRF/ISRE motifs as well as motifs for PU.1 and C/EBP, two macrophage LDTFs in ZIKV+ cells. In contrast, NF-κB and STAT1 motifs were equally represented. This unbiased approach showed that ZIKV specifically suppresses ISRE-dependent signaling but does not suppress signaling via NF-κB and STAT1 in macrophages. We then performed ChIP-seq for RNA polymerase II (RNApol2) in order to identify genes that are being actively transcribed during ZIKV infection. We observed a striking reduction of RNApol2 DNA occupancy in ZIKV+ cells, and in line with this result, decreased protein levels of RPB1, the largest RNApol2 subunit that plays critical roles in both DNA binding and transcriptional elongation. The mechanism by which ZIKV reduces RBP1 levels and RNApol2 DNA occupancy is as yet to be identified. Multiple viruses, including Influenza, Chikungunya and herpes simplex virus type 1 (HSV-1) produce viral proteins that interact with RNApol2 and suppress its function by various mechanisms [84–86]. A ZIKV protein may activate the degradation of RPB1 directly or degrade/inactivate one of the many host proteins involved in forming the RNApol2 pre-initiation complex. In our study, the loss of RNApol2 was particularly notable at genes important for core macrophage functions, many of which are associated with super-enhancers (SEs), or stretch enhancers, in human macrophages. SEs are regions of disproportionately high densities of active chromatin regulatory marks and binding of TFs near genes that play essential roles in the identity and function of cell types [87–90]. Conceivably, loss of RNApol2 driven transcription of genes involved in core cellular identity could contribute to cell death and/or impaired differentiation of that particular cell type in various ZIKV-infected tissues, including NPCs in the brain. Importantly, changes in RNApol2 in ZIKV+ cells could not be identified using gene expression analyses alone, highlighting the need to integrate both transcriptomic and epigenetic information towards deciphering virus-host interactions. Future studies using simultaneous transcriptional and epigenetic analyses to study ZIKV-relevant primary human cells, including placental macrophages, trophoblasts and cells in the central nervous system, will help reveal how cell-type and context influence host responses to ZIKV.
Influence of human genetic variation on flavivirus responses
Host genetics can strongly influence human immunity and disease severity during viral infections [91–96]. Accordingly, ZIKV infection differentially affects the neuronal differentiation of human fetal brain NSCs derived from different donors, thereby suggesting a role for human individual differences in susceptibility to ZIKV-neuropathology [47]. Consistent with this idea, Caires-Junior LC et al. infected NPCs from three pairs of dizygotic twins in which one fetus was CZS-affected and the other was CZS-unaffected [97]. NPCs from CZS-affected individuals had significantly higher ZIKV replication and reduced cell growth compared to CZS-unaffected. RNAseq analysis prior to infection revealed baseline differences in gene expression in CZS-affected vs. CZS-unaffected NPCs. Whole-exome analysis of 18 affected CZS-affected babies compared to 5 unaffected twins and 609 controls did not suggest a monogenic model to explain CZS susceptibility. The authors suggested that the differences in gene expression and ZIKV susceptibility may therefore be related to oligogenic and/or epigenetic mechanisms. Thus, a potential benefit of epigenetic analyses of host-viral interactions is determination of the mechanisms by which human genetic variation alters viral susceptibility. The majority (~88%) of risk loci for common diseases in genome-wide association studies (GWASs) are outside of the protein-coding genome [98], and functional variants for GWAS loci can reside in signal-dependent enhancers [93,99–102]. Thus, a significant fraction of disease-causing mechanisms originate in context-dependent regulatory states. Mapping epigenetics, especially in the appropriate context (i.e. during flaviviral infections), will be beneficial because it can be used to overlap with expression quantitative trait loci (eQTLs) or GWAS-risk associated variants to help elucidate their potential epigenetic mechanism of action.
Influence of flaviviral genetic variation on human cell responses
Flaviviral genotypes can also affect human cell responses during infection, thereby modulating disease severity. The association of Asian DENV serotype 2 (DENV2) with severe disease in the Americas during 1980s and the increased virulence of the Asian strains relative to American DENV2 in human blood monocyte-derived dendritic cells and macrophages mapped to multiple changes in the viral genome, including substitutions at amino acid 390 of the envelope (E) protein and in the 5’ and 3’ untranslated regions (UTRs) [103][104,105]. Additionally, the epidemiological fitness of DENV2 clade PR-2B in 1994 to 1995 in Puerto Rico increased [106,107] due to substitutions in the 3’ UTR that enhanced production of subgenomic flavivirus RNA (sfRNA), which can bind tripartite motif 25 (TRIM25) and inhibit IFNB1 expression [108]. Similarly, sequence comparison of the 2015–2016 epidemic ZIKV isolates to pre-epidemic ZIKV strains have identified multiple mutations of possible signifiance. A single amino acid serine-to-asparagine mutation, S139N, in the prM protein was found to increase ZIKV infectivity of human NPCs and neurovirulence in neonatal mice [109]. Additionally, an alanine-to-valine amino acid substitution, A188V, in the NS1 protein increases NS1 secretion thereby potentiating ZIKV infection of Aedes aegypti mosquitos [110]. This same mutation also enables NS1 to bind TBK1 and reduce IFNB1 induction [111]. Thus, phylogenetic and molecular analyses of clinically important flaviviral strains can provide important insights into how viral mutations contribute to immune evasion, epidemic potential, and clinical outcomes.
Conclusions:
Genomic technologies are increasingly used to study host-viral interactions because they can rapidly identify key pro-viral and anti-viral pathways in an unbiased genome-wide manner. However, many challenges remain to using these technologies to best understand human responses to flavivirus infections.
The reported flavivirus CRISPR–Cas9 screens appear to be very efficient, unbiased and reproducible methods of identifying essential host factors. Viral host factor requirements may be viral strain- and cell-type specific. For example, African and Asian ZIKV isolates show phenotypic differences in vitro and in vivo with only recent Asian epidemic strains being associated with congenital abnormalities [112,113]. Additionally, specific flaviviral mutations can influence the antiviral response of the host cell. Ideally, CRISPR studies should use clinically and epidemiologically important viral isolates and confirm with multipe strains. Moreover, host-factors may be cell-type specific, especially in highly specialized cells such as neural stem cells during ZIKV infection. Cell-type specific factors that are important for understanding human pathology may only be revealed if CRISPR screens are performed in the appropriate cell contexts.
We lack mechanistic understanding of how human responses, especially responses in heterogenous tissues, to DENV and ZIKV contribute to morbidity and mortality. Here we describe two approaches, viscRNA-seq (Figure 1B) and FACS-NGS (Figure 1C) that overcome these limitations to provide deeper insight into host-DENV and -ZIKV interactions. Some of the benefits and limitations of each method as currently described are presented in Table 1. Both viscRNA-seq and FACS-NGS techniques could be applied to better understand how cell-cell communication between infected and bystander cells as well as between different cell types in heterogenous tissues contributes to the observed phenotypes. By correlating intracellular virus abundance with gene expression in individual cells, viscRNA-seq can identify gene expression that is strongly correlated (positive or negative) with viral replication. In addition to assessing transcriptional responses, FACS-NGS is adaptable to many epigenetic analyses. This allows simultaneous comparison of the epigenetic landscape and gene expression profiles to determine how cell origin, differentiation method and tissue microenvironments establish context-dependent patterns of gene expression that drive differential outcomes to the same viral infection.
Direct application of the recently utilized techniques, viscRNA-seq and FACS-NGS, to study tissue responses during in vivo human infections is an important next step to understand human disease. Development of new genomics approaches, including those capturing 3D genome structures and requiring small cell numbers, and rigorous testing of both existing and new techniques will be needed to identify the best methods for sample isolation and preservation that most reliably capture in vivo human responses and can also be performed in DENV- and ZIKV-endemic countries with limited resources. Additionally, improved computational methods for integrating large, diverse data-sets that extract maximal understanding are required.
Box 1: Outstanding Questions.
How does cell-type- and environment-specific enhancers control flavivirus responses and infection outcome?
Does human genetic and epigenetic variation control DENV and ZIKV susceptibility and if so where are these mutations located?
What are the best methods to isolate and preserve in vivo responses to viral infections for downstream genomics approaches?
What are the optimal computational methods for integrating multiple types of genomics data, including trascriptomic, epigenomic, and 3D organization of chromatin, towards identifying the dominant pro-viral and anti-viral pathways?
Acknowledgements
The authors would like to thank Dr. Christopher Benner for his helpful suggestions.
Funding: This research was supported by NIAID/NIH grants R01 AI116813, R21 NS100477, R21AI127988 and Interactive Fund grant from Kyowa Kirin Pharmaceutical Research to SS, and KL2 Scholars: 1KL2TR001444 and a Career Award for Medical Scientists from the Burroughs Wellcome Fund to AFC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, et al. : The global distribution and burden of dengue. Nature 2013, 496:504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gubler DJ: The economic burden of dengue. Am J Trop Med Hyg 2012, 86:743–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Solomon T, Dung NM, Vaughn DW, Kneen R, Thao LT, Raengsakulrach B, Loan HT, Day NP, Farrar J, Myint KS, et al. : Neurological manifestations of dengue infection. Lancet 2000, 355:1053–1059. [DOI] [PubMed] [Google Scholar]
- 4.Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR: Zika Virus and Birth Defects--Reviewing the Evidence for Causality. N Engl J Med 2016, 374:1981–1987. [DOI] [PubMed] [Google Scholar]
- 5.Cao-Lormeau VM, Blake A, Mons S, Lastere S, Roche C, Vanhomwegen J, Dub T, Baudouin L, Teissier A, Larre P, et al. : Guillain-Barre Syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. Lancet 2016, 387:1531–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malkki H: CNS infections: Zika virus infection could trigger Guillain-Barre syndrome. Nat Rev Neurol 2016, 12:187. [DOI] [PubMed] [Google Scholar]
- 7.Parra B, Lizarazo J, Jimenez-Arango JA, Zea-Vera AF, Gonzalez-Manrique G, Vargas J, Angarita JA, Zuniga G, Lopez-Gonzalez R, Beltran CL, et al. : Guillain-Barre Syndrome Associated with Zika Virus Infection in Colombia. N Engl J Med 2016, 375:1513–1523. [DOI] [PubMed] [Google Scholar]
- 8.Watrin L, Ghawche F, Larre P, Neau JP, Mathis S, Fournier E: Guillain-Barre Syndrome (42 Cases) Occurring During a Zika Virus Outbreak in French Polynesia. Medicine (Baltimore) 2016, 95:e3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bogoch II, Brady OJ, Kraemer MUG, German M, Creatore MI, Kulkarni MA, Brownstein JS, Mekaru SR, Hay SI, Groot E, et al. : Anticipating the international spread of Zika virus from Brazil. Lancet 2016, 387:335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gubler DJ: Dengue, Urbanization and Globalization: The Unholy Trinity of the 21(st) Century. Trop Med Health 2011, 39:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Ortenzio E, Matheron S, Yazdanpanah Y, de Lamballerie X, Hubert B, Piorkowski G, Maquart M, Descamps D, Damond F, Leparc-Goffart I: Evidence of Sexual Transmission of Zika Virus. N Engl J Med 2016, 374:2195–2198. [DOI] [PubMed] [Google Scholar]
- 12.Moreira J, Peixoto TM, Siqueira AM, Lamas CC: Sexually acquired Zika virus: a systematic review. Clin Microbiol Infect 2017, 23:296–305. [DOI] [PubMed] [Google Scholar]
- 13.Barrangou R, Horvath P: A decade of discovery: CRISPR functions and applications. Nat Microbiol 2017, 2:17092. [DOI] [PubMed] [Google Scholar]
- 14.Doudna JA, Charpentier E: Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346:1258096. [DOI] [PubMed] [Google Scholar]
- 15.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et al. : Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shalem O, Sanjana NE, Zhang F: High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet 2015, 16:299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang T, Wei JJ, Sabatini DM, Lander ES: Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343:80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y, Wei W: High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 2014, 509:487–491. [DOI] [PubMed] [Google Scholar]
- 19.DeJesus R, Moretti F, McAllister G, Wang Z, Bergman P, Liu S, Frias E, Alford J, Reece-Hoyes JS, Lindeman A, et al. : Functional CRISPR screening identifies the ufmylation pathway as a regulator of SQSTM1/p62. Elife 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evers B, Jastrzebski K, Heijmans JP, Grernrum W, Beijersbergen RL, Bernards R: CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat Biotechnol 2016, 34:631–633. [DOI] [PubMed] [Google Scholar]
- 21.Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. : High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163:1515–1526. [DOI] [PubMed] [Google Scholar]
- 22.Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, Swaminathan K, Mata MA, Elias JE, Sarnow P, et al. : Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 2016, 535:159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin DL, Cherepanova NA, Bozzacco L, MacDonald MR, Gilmore R, Tai AW: Dengue Virus Hijacks a Noncanonical Oxidoreductase Function of a Cellular Oligosaccharyltransferase Complex. MBio 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma H, Dang Y, Wu Y, Jia G, Anaya E, Zhang J, Abraham S, Choi JG, Shi G, Qi L, et al. : A CRISPR-Based Screen Identifies Genes Essential for West-Nile-Virus-Induced Cell Death. Cell Rep 2015, 12:673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang R, Miner JJ, Gorman MJ, Rausch K, Ramage H, White JP, Zuiani A, Zhang P, Fernandez E, Zhang Q, et al. : A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 2016, 535:164–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Savidis G, McDougall WM, Meraner P, Perreira JM, Portmann JM, Trincucci G, John SP, Aker AM, Renzette N, Robbins DR, et al. : Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell Rep 2016, 16:232–246. [DOI] [PubMed] [Google Scholar]
- 27.Johnson AE, van Waes MA: The translocon: a dynamic gateway at the ER membrane. Annu Rev Cell Dev Biol 1999, 15:799–842. [DOI] [PubMed] [Google Scholar]
- 28.Fons RD, Bogert BA, Hegde RS: Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J Cell Biol 2003, 160:529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nyathi Y, Wilkinson BM, Pool MR: Co-translational targeting and translocation of proteins to the endoplasmic reticulum. Biochim Biophys Acta 2013, 1833:2392–2402. [DOI] [PubMed] [Google Scholar]
- 30.Cherepanova N, Shrimal S, Gilmore R: N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr Opin Cell Biol 2016, 41:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Harper JW, Kopito RR: Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol 2011, 14:93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wideman JG: The ubiquitous and ancient ER membrane protein complex (EMC): tether or not? F1000Res 2015, 4:624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fields BN, Knipe DM, Howley PM: Fields virology edn 5th Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2007. [Google Scholar]
- 34.Dominguez AA, Lim WA, Qi LS: Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol 2016, 17:5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gebre M, Nomburg JL, Gewurz BE: CRISPR-Cas9 Genetic Analysis of Virus-Host Interactions. Viruses 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heaton BE, Kennedy EM, Dumm RE, Harding AT, Sacco MT, Sachs D, Heaton NS: A CRISPR Activation Screen Identifies a Pan-avian Influenza Virus Inhibitory Host Factor. Cell Rep 2017, 20:1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, et al. : Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165:1238–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JL, Guimaraes KP, Benazzato C, Almeida N, Pignatari GC, Romero S, et al. : The Brazilian Zika virus strain causes birth defects in experimental models. Nature 2016, 534:267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dang J, Tiwari SK, Lichinchi G, Qin Y, Patil Veena S, Eroshkin Alexey M, Rana Tariq M: Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids through Activation of the Innate Immune Receptor TLR3. Cell Stem Cell 2016, 19:258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcez PP, Loiola EC, Madeiro da Costa R, Higa LM, Trindade P, Delvecchio R, Nascimento JM, Brindeiro R, Tanuri A, Rehen SK: Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352:816–818. [DOI] [PubMed] [Google Scholar]
- 41.Hanners NW, Eitson JL, Usui N, Richardson RB, Wexler EM, Konopka G, Schoggins JW: Western Zika Virus in Human Fetal Neural Progenitors Persists Long Term with Partial Cytopathic and Limited Immunogenic Effects. Cell Rep 2016, 15:2315–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS: A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19:720–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li C, Xu D, Ye Q, Hong S, Jiang Y, Liu X, Zhang N, Shi L, Qin CF, Xu Z: Zika Virus Disrupts Neural Progenitor Development and Leads to Microcephaly in Mice. Cell Stem Cell 2016, 19:120–126. [DOI] [PubMed] [Google Scholar]
- 44.Miner JJ, Cao B, Govero J, Smith AM, Fernandez E, Cabrera OH, Garber C, Noll M, Klein RS, Noguchi KK, et al. : Zika Virus Infection during Pregnancy in Mice Causes Placental Damage and Fetal Demise. Cell 2016, 165:1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, Yao B, Shin J, Zhang F, Lee EM, et al. : Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 2016, 18:587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu KY, Zuo GL, Li XF, Ye Q, Deng YQ, Huang XY, Cao WC, Qin CF, Luo ZG: Vertical transmission of Zika virus targeting the radial glial cells affects cortex development of offspring mice. Cell Res 2016, 26:645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGrath EL, Rossi SL, Gao J, Widen SG, Grant AC, Dunn TJ, Azar SR, Roundy CM, Xiong Y, Prusak DJ, et al. : Differential Responses of Human Fetal Brain Neural Stem Cells to Zika Virus Infection. Stem Cell Reports 2017, 8:715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jurado KA, Simoni MK, Tang Z, Uraki R, Hwang J, Householder S, Wu M, Lindenbach BD, Abrahams VM, Guller S, et al. : Zika virus productively infects primary human placenta-specific macrophages. JCI Insight 2016, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Noronha L, Zanluca C, Azevedo ML, Luz KG, Santos CN: Zika virus damages the human placental barrier and presents marked fetal neurotropism. Mem Inst Oswaldo Cruz 2016, 111:287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petitt M, Tabata T, Puerta-Guardo H, Harris E, Pereira L: Zika virus infection of first-trimester human placentas: utility of an explant model of replication to evaluate correlates of immune protection ex vivo. Curr Opin Virol 2017, 27:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quicke KM, Bowen JR, Johnson EL, McDonald CE, Ma H, O’Neal JT, Rajakumar A, Wrammert J, Rimawi BH, Pulendran B, et al. : Zika Virus Infects Human Placental Macrophages. Cell Host Microbe 2016, 20:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwartz DA: Viral infection, proliferation, and hyperplasia of Hofbauer cells and absence of inflammation characterize the placental pathology of fetuses with congenital Zika virus infection. Arch Gynecol Obstet 2017, 295:1361–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tabata T, Petitt M, Puerta-Guardo H, Michlmayr D, Wang C, Fang-Hoover J, Harris E, Pereira L: Zika Virus Targets Different Primary Human Placental Cells, Suggesting Two Routes for Vertical Transmission. Cell Host Microbe 2016, 20:155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weisblum Y, Oiknine-Djian E, Vorontsov OM, Haimov-Kochman R, Zakay-Rones Z, Meir K, Shveiky D, Elgavish S, Nevo Y, Roseman M, et al. : Zika Virus Infects Early- and Midgestation Human Maternal Decidual Tissues, Inducing Distinct Innate Tissue Responses in the Maternal-Fetal Interface. J Virol 2017, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bowen JR, Ferris MT, Suthar MS: Systems biology: A tool for charting the antiviral landscape. Virus Res 2016, 218:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carlin AF, Vizcarra EA, Branche E, Viramontes KM, Suarez-Amaran L, Ley K, Heinz S, Benner C, Shresta S, Glass CK: Deconvolution of pro- and antiviral genomic responses in Zika virus-infected and bystander macrophages. Proc Natl Acad Sci U S A 2018. [DOI] [PMC free article] [PubMed]
- 57.Zanini F, Pu SY, Bekerman E, Einav S, Quake SR: Single-cell transcriptional dynamics of flavivirus infection. Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cleaves GR, Dubin DT: Methylation status of intracellular dengue type 2 40 S RNA. Virology 1979, 96:159–165. [DOI] [PubMed] [Google Scholar]
- 59.Wengler G, Wengler G, Gross HJ: Studies on virus-specific nucleic acids synthesized in vertebrate and mosquito cells infected with flaviviruses. Virology 1978, 89:423–437. [DOI] [PubMed] [Google Scholar]
- 60.Zanini FRM; Croote D; Sahoo MK; Sanz AM; Ortiz-Lasso E; Albornoz LL; Suarez FR, Montoya JG, Pinsky BA; Quake S; Einav S: Virus-inclusive single cell RNA sequencing reveals molecular signature predictive of progression to severe dengue infection. bioRxiv 2018, 388181. [DOI] [PMC free article] [PubMed]
- 61.Lin YW, Wang KJ, Lei HY, Lin YS, Yeh TM, Liu HS, Liu CC, Chen SH: Virus replication and cytokine production in dengue virus-infected human B lymphocytes. J Virol 2002, 76:12242–12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Muffat J, Li Y, Omer A, Durbin A, Bosch I, Bakiasi G, Richards E, Meyer A, Gehrke L, Jaenisch R: Human induced pluripotent stem cell-derived glial cells and neural progenitors display divergent responses to Zika and dengue infections. Proc Natl Acad Sci U S A 2018, 115:7117–7122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Retallack H, Di Lullo E, Arias C, Knopp KA, Laurie MT, Sandoval-Espinosa C, Mancia Leon WR, Krencik R, Ullian EM, Spatazza J, et al. : Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc Natl Acad Sci U S A 2016, 113:14408–14413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, et al. : Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014, 159:1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP, et al. : An environment-dependent transcriptional network specifies human microglia identity. Science 2017, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I: Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014, 159:1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Banerji J, Rusconi S, Schaffner W: Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 1981, 27:299–308. [DOI] [PubMed] [Google Scholar]
- 68.Heinz S, Romanoski CE, Benner C, Glass CK: The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol 2015, 16:144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shlyueva D, Stampfel G, Stark A: Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet 2014, 15:272–286. [DOI] [PubMed] [Google Scholar]
- 70.Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. : An atlas of active enhancers across human cell types and tissues. Nature 2014, 507:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Consortium EP: An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. : Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. : Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 2007, 39:311–318. [DOI] [PubMed] [Google Scholar]
- 74.Levine M: Transcriptional enhancers in animal development and evolution. Curr Biol 2010, 20:R754–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. : Integrative analysis of 111 reference human epigenomes. Nature 2015, 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Romanoski CE, Link VM, Heinz S, Glass CK: Exploiting genomics and natural genetic variation to decode macrophage enhancers. Trends Immunol 2015, 36:507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gross AM, Jaeger PA, Kreisberg JF, Licon K, Jepsen KL, Khosroheidari M, Morsey BM, Swindells S, Shen H, Ng CT, et al. : Methylome-wide Analysis of Chronic HIV Infection Reveals Five-Year Increase in Biological Age and Epigenetic Targeting of HLA. Mol Cell 2016, 62:157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Heinz S, Texari L, Hayes MGB, Urbanowski M, Chang MW, Givarkes N, Rialdi A, White KM, Albrecht RA, Pache L, et al. : Transcription Elongation Can Affect Genome 3D Structure. Cell 2018, 174:1522–1536 e1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hammerschmidt W: The Epigenetic Life Cycle of Epstein–Barr Virus. In Epstein Barr Virus Volume 1 Edited by Münz C: Springer; 2015. vol 390.] [DOI] [PubMed] [Google Scholar]
- 80.Vedham V, Verma M: Cancer-associated infectious agents and epigenetic regulation. Methods Mol Biol 2015, 1238:333–354. [DOI] [PubMed] [Google Scholar]
- 81.Bowen JR, Quicke KM, Maddur MS, O’Neal JT, McDonald CE, Fedorova NB, Puri V, Shabman RS, Pulendran B, Suthar MS: Zika Virus Antagonizes Type I Interferon Responses during Infection of Human Dendritic Cells. PLoS Pathog 2017, 13:e1006164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sanchez-Seco MP, Evans MJ, Best SM, et al. : Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19:882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kumar A, Hou S, Airo AM, Limonta D, Mancinelli V, Branton W, Power C, Hobman TC: Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep 2016, 17:1766–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Akhrymuk I, Kulemzin SV, Frolova EI: Evasion of the innate immune response: the Old World alphavirus nsP2 protein induces rapid degradation of Rpb1, a catalytic subunit of RNA polymerase II. J Virol 2012, 86:7180–7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fraser KA, Rice SA: Herpes simplex virus immediate-early protein ICP22 triggers loss of serine 2-phosphorylated RNA polymerase II. J Virol 2007, 81:5091–5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vreede FT, Fodor E: The role of the influenza virus RNA polymerase in host shut-off. Virulence 2010, 1:436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dowen JM, Fan ZP, Hnisz D, Ren G, Abraham BJ, Zhang LN, Weintraub AS, Schujiers J, Lee TI, Zhao K, et al. : Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 2014, 159:374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, Young RA: Super-enhancers in the control of cell identity and disease. Cell 2013, 155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Parker SC, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, van Bueren KL, Chines PS, Narisu N, Program NCS, et al. : Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A 2013, 110:17921–17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA: Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bigham AW, Buckingham KJ, Husain S, Emond MJ, Bofferding KM, Gildersleeve H, Rutherford A, Astakhova NM, Perelygin AA, Busch MP, et al. : Host genetic risk factors for West Nile virus infection and disease progression. PLoS One 2011, 6:e24745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Glass WG, McDermott DH, Lim JK, Lekhong S, Yu SF, Frank WA, Pape J, Cheshier RC, Murphy PM: CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med 2006, 203:35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee MN, Ye C, Villani AC, Raj T, Li W, Eisenhaure TM, Imboywa SH, Chipendo PI, Ran FA, Slowikowski K, et al. : Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 2014, 343:1246980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Quach H, Rotival M, Pothlichet J, Loh YE, Dannemann M, Zidane N, Laval G, Patin E, Harmant C, Lopez M, et al. : Genetic Adaptation and Neandertal Admixture Shaped the Immune System of Human Populations. Cell 2016, 167:643–656 e617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vannberg FO, Chapman SJ, Hill AV: Human genetic susceptibility to intracellular pathogens. Immunol Rev 2011, 240:105–116. [DOI] [PubMed] [Google Scholar]
- 96.Venegas M, Brahm J, Villanueva RA: Genomic determinants of hepatitis C virus antiviral therapy outcomes: toward individualized treatment. Ann Hepatol 2012, 11:827–837. [PubMed] [Google Scholar]
- 97.Caires-Júnior LC, Goulart E, Melo US, Araujo BHS, Alvizi L, Soares-Schanoski A, de Oliveira DF, Kobayashi GS, Griesi-Oliveira K, Musso CM, et al. : Discordant congenital Zika syndrome twins show differential in vitro viral susceptibility of neural progenitor cells. Nature Communications 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA: Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 2009, 106:9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJ, Shishkin AA, et al. : Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518:337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Heintzman N, Ren B, Fu XD, Topol EJ, Rosenfeld MG, et al. : 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature 2011, 470:264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Raj T, Rothamel K, Mostafavi S, Ye C, Lee MN, Replogle JM, Feng T, Lee M, Asinovski N, Frohlich I, et al. : Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science 2014, 344:519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ye CJ, Feng T, Kwon HK, Raj T, Wilson MT, Asinovski N, McCabe C, Lee MH, Frohlich I, Paik HI, et al. : Intersection of population variation and autoimmunity genetics in human T cell activation. Science 2014, 345:1254665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Leitmeyer KC, Vaughn DW, Watts DM, Salas R, Villalobos I, de C, Ramos C, Rico-Hesse R: Dengue virus structural differences that correlate with pathogenesis. J Virol 1999, 73:4738–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cologna R, Rico-Hesse R: American genotype structures decrease dengue virus output from human monocytes and dendritic cells. J Virol 2003, 77:3929–3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pryor MJ, Carr JM, Hocking H, Davidson AD, Li P, Wright PJ: Replication of dengue virus type 2 in human monocyte-derived macrophages: comparisons of isolates and recombinant viruses with substitutions at amino acid 390 in the envelope glycoprotein. Am J Trop Med Hyg 2001, 65:427–434. [DOI] [PubMed] [Google Scholar]
- 106.Bennett SN, Holmes EC, Chirivella M, Rodriguez DM, Beltran M, Vorndam V, Gubler DJ, McMillan WO: Molecular evolution of dengue 2 virus in Puerto Rico: positive selection in the viral envelope accompanies clade reintroduction. J Gen Virol 2006, 87:885–893. [DOI] [PubMed] [Google Scholar]
- 107.Rigau-Perez JG, Vorndam AV, Clark GG: The dengue and dengue hemorrhagic fever epidemic in Puerto Rico, 1994–1995. Am J Trop Med Hyg 2001, 64:67–74. [DOI] [PubMed] [Google Scholar]
- 108.Manokaran G, Finol E, Wang C, Gunaratne J, Bahl J, Ong EZ, Tan HC, Sessions OM, Ward AM, Gubler DJ, et al. : Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 2015, 350:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yuan L, Huang XY, Liu ZY, Zhang F, Zhu XL, Yu JY, Ji X, Xu YP, Li G, Li C, et al. : A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 2017, 358:933–936. [DOI] [PubMed] [Google Scholar]
- 110.Liu Y, Liu J, Du S, Shan C, Nie K, Zhang R, Li XF, Zhang R, Wang T, Qin CF, et al. : Evolutionary enhancement of Zika virus infectivity in Aedes aegypti mosquitoes. Nature 2017, 545:482–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xia H, Luo H, Shan C, Muruato AE, Nunes BTD, Medeiros DBA, Zou J, Xie X, Giraldo MI, Vasconcelos PFC, et al. : An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat Commun 2018, 9:414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Simonin Y, Loustalot F, Desmetz C, Foulongne V, Constant O, Fournier-Wirth C, Leon F, Moles JP, Goubaud A, Lemaitre JM, et al. : Zika Virus Strains Potentially Display Different Infectious Profiles in Human Neural Cells. EBioMedicine 2016, 12:161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Smith DR, Sprague TR, Hollidge BS, Valdez SM, Padilla SL, Bellanca SA, Golden JW, Coyne SR, Kulesh DA, Miller LJ, et al. : African and Asian Zika Virus Isolates Display Phenotypic Differences Both In Vitro and In Vivo. Am J Trop Med Hyg 2018, 98:432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]