

Complex allelic variation hampers the assembly of haplotype-resolved sequences from diploid genomes. Here we present trio binning, an approach that simplifies haplotype assembly by resolving allelic variation prior to assembly. In contrast to prior approaches, the effectiveness of our method improves with increasing heterozygosity. Trio binning uses short reads from two parental genomes to first partition long reads from an offspring into haplotype-specific sets. Each haplotype is then assembled independently, resulting in a complete diploid reconstruction. We use trio binning to recover both haplotypes of a diploid human genome and identify complex structural variants missed by alternative approaches. We sequence an F1 cross between cattle subspecies Bos taurus taurus and Bos taurus indicus, and completely assemble both parental haplotypes with NG50 haplotig sizes >20 Mbp and 99.998% accuracy, surpassing the quality of current cattle reference genomes. We suggest that trio binning improves diploid genome assembly and will facilitate new studies of haplotype variation and inheritance.
Genome sequences must be reconstructed from many shorter read sequences in a complex process known as assembly1. Repetitive sequences longer than the sequencing read lengths prevent a complete reconstruction of chromosomes, so assembly typically results in a collection of contiguous sequences (contigs) that are interrupted by repeats or gaps. The advent of long-read sequencing technologies has improved the quality of genome assemblies by resolving many such repeats 2. However, even these technologies have not overcome the challenge of completely assembling both haplotypes of a diploid genome. Instead, most genome assembly tools simply co-assemble the haplotypes into a mosaic consensus, resulting in an assembly that does not accurately represent either original haplotype. Collapsing haplotypes into a single consensus representation introduces false variants not present in either haplotype, leading to annotation and analysis errors 3. Ideally, a genome should be represented as a complete set of haplotypes rather than an artificial mixture.
A common approach to skirt the issue is to reduce the problem of haplotype variation by sequencing an inbred individual (e.g. fly 4, mouse 5). However, this is impractical for many species and, even when possible, can result in a genome that is not representative of variation found in the natural population. An alternative approach is to use haploid, clone-based genomic libraries, as was done for the human genome project 6. More recently, a diploid human assembly was constructed using tiled fosmids 7, but such cloning is often impractical. Alternatively, homozygous cell lines such as CHM1hTERT can be targeted 8–10, but such cell lines often develop unstable karyotypes in culture and are not always available. Other attempts have been made to separate haplotypes de novo from whole-genome sequencing. For example, the highly polymorphic sea squirt Ciona savignyi was first assembled using modifications to the Arachne assembler 11 designed to split haplotypes based on read overlap information 12. However, this was an extreme case as the reference individual had an estimated heterozygosity of 4.6%. Early attempts to assemble a diploid human genome, with heterozygosity of just 0.1%, first collapsed the haplotypes into a combined assembly and then phased alleles over a short range using pairs of heterozygous variants observed on a single read or read pair 13. Current phasing tools operate similarly, and map sequencing reads to a reference sequence to infer blocks of variants that originate from the same haplotype 14–16. More sophisticated library preparations such as chromosome sorting 17, Strand-seq 18, and Hi-C 19 can link variants over a much longer range, delivering chromosome-scale phase blocks. However, methods that rely on reference mapping typically fail in regions of high heterozygosity and/or significant structural variation between haplotypes, yielding a limited view of genetic diversity.
A more comprehensive solution to the diploid assembly problem is to integrate haplotype separation into the assembly process itself. However, this approach is limited by the fragment length of the sequencing process. Sequencing reads alone do not always contain enough information to link variants across longer regions of homozygosity, resulting in relatively short phase blocks. As a compromise, diploid assemblers such as FALCON-Unzip 20 and Supernova 21 output “pseudo-haplotypes” that represent a single allele at each position, but do not preserve phase across long homozygous alleles or assembly gaps. In addition, these assemblers can confuse repeats with diverged alleles, leading to artefactual duplications or deletions. One potential solution is to combine long-read sequencing with additional types of information such as linked reads and/or bacterial artificial chromosomes (BACs) 22, Strand-seq 23, or Hi-C 24. However, no de novo assemblers currently integrate these data types, so this can be a manual and expensive process.
We provide a simple solution to the diploid assembly problem that assembles accurate, genome-scale haplotypes de novo. Unlike other methods that are limited to phasing individual chromosomes, we produce two complete, haploid genomes — one for each parental haplotype. These complete haplotypes are versatile and can be analyzed individually or recombined via alignment into a diploid genome graph. If contiguity is paramount, a maximally contiguous pseudo-haplotype could be defined as a walk through this graph (e.g. a repeat might be resolved on one haplotype but not the other). Alternatively, to capture complex structural variation, multiple haplotypes from a population could be combined to create a pan-genome reference. These potential applications are simplified by the initial reconstruction of complete, linear haplotypes.
Key to our method is the separation of haplotypes prior to assembly using a father-mother-offspring trio. Each haplotype is then assembled separately without the interference of inter-haplotype variation. Trios have long been used in genomics to infer inheritance, including for the HapMap project 25, the 1000 Genomes Project 26, and the creation of “platinum” variant catalogs 27. Trios were also used by trio-sga to simplify heterozygous diploid genome assembly 28, but reliance on short-read sequencing limited the haplotype-specific contigs (haplotigs) to an average size of a few kilobases. In contrast, our long-read method enables the assembly of multi-megabase haplotigs and complete parental haplotypes.
Here we introduce trio binning and demonstrate that it reconstructs accurate and complete parental haplotypes for a wide range of zygosity and genome sizes. We first report results for benchmark datasets with both high (Arabidopsis) and low (human) levels of heterozygosity, and illustrate that prior methods do not completely recover both haplotypes of a diploid genome. We then report a complete diploid assembly of an F1 hybrid between Bos taurus taurus and Bos taurus indicus, and demonstrate that the quality of each haplotype exceeds that of even the best livestock reference genomes. The results demonstrate that trio binning of outbred genomes is an easy, accurate, and superior method for assembling diploid reference genomes.
Results
Complete haplotype assembly with TrioCanu
We have implemented trio binning and haplotype assembly as the TrioCanu module of the Canu assembler 29. The method requires moderate coverage of short, high-quality sequencing reads (e.g. 30× Illumina) from two parental genomes to identify short, length k subsequences (k-mers) that are specific to each parent. These k-mers are presumed to be specific to the corresponding haplotypes of the offspring. Next, long reads are collected from the offspring to sufficiently cover both haplotypes (e.g. 40× PacBio per haplotype). Long reads from the offspring are then binned into paternal and maternal groups based on the presence of the haplotype-specific k-mers, and assembled separately (Figure 1, Online Methods).
Figure 1. Outline of trio binning and haplotype assembly.
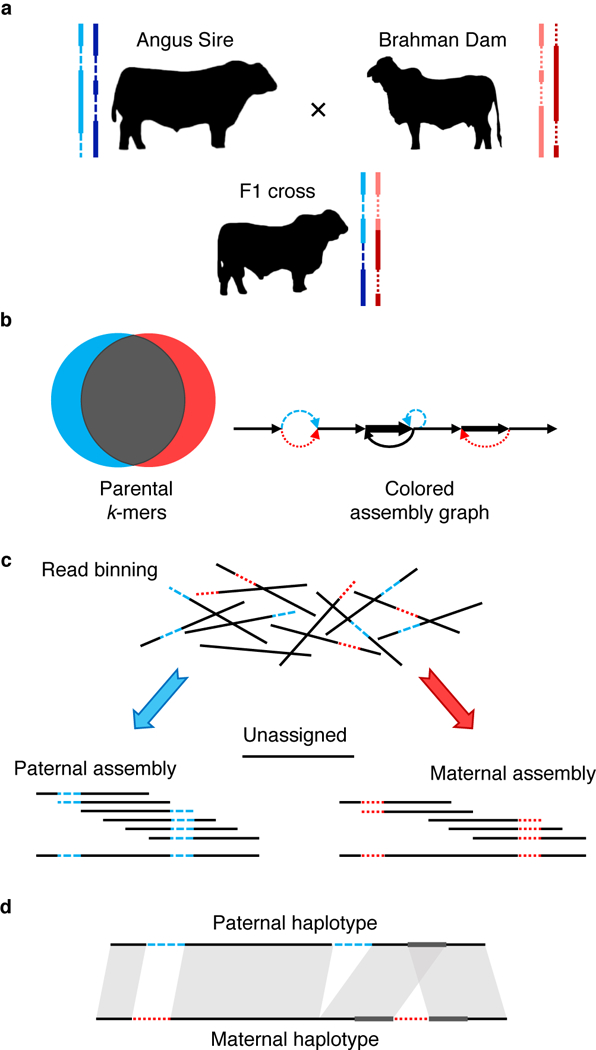
a) Two parents constitute four haplotypes including shared sequence in both parents (solid lines) and sequence unique to one parent (dashed lines). The offspring inherits a recombined haplotype from each parent (blue, paternal; red, maternal). b) Short-read sequencing of the parents identifies unique length-k subsequences (k-mers), which can be used to infer the origin of heterozygous alleles in the offspring’s diploid genome. c) Trio binning simplifies assembly by first partitioning long reads from the offspring into paternal and maternal sets based on these k-mers. Each haplotype is then assembled separately without the interference of heterozygous variants. Unassignable reads are homozygous and can be assigned to both sets or assembled separately. d) The resulting assemblies represent genome-scale haplotypes, and accurately recover both point and structural variation.
Trio binning performs best for a uniformly heterozygous offspring, which maximizes the probability that any given read will contain at least one haplotype-specific k-mer. Each heterozygous single-nucleotide variant is expected to induce 2k haplotype-specific k-mers. As a result, the fraction of haplotype-specific k-mers is greater than the single-nucleotide heterozygosity. In human, for example, where single-nucleotide heterozygosity is estimated to be only ~0.1%, nearly 2% of the 21-mers are haplotype specific (Table 1 and Supplementary Table 1). Thus, k-mers are powerful haplotype markers that can also capture complex insertions, deletions, and fusion events.
Table 1.
Trio heterozygosity, binning, and assembly statistics.
| Species | Total cov. | Haplotype | Haplotype k-mers |
Assigned bases | Haplotype coverage | No. haplotigs | Haplotig NG50 (Mbp) | Assembly size (Mbp) |
|---|---|---|---|---|---|---|---|---|
| A. thaliana | 180.6× | Col-0 | 12.3% | 50.8% | 87.8× | 215 | 7.03 | 123.52 |
| Cvi-0 | 12.1% | 48.5% | 91.9× | 163 | 5.61 | 122.35 | ||
| H. sapiens | 72.3× | NA12891 | 0.9% | 43.2% | 31.3× | 7,252 | 1.18 | 2,743.25 |
| NA12892 | 1.0% | 44.2% | 31.9× | 7,388 | 1.17 | 2,749.17 | ||
| B. taurus | 135× | Angus | 2.9% | 49.3% | 66.6× | 1,747 | 26.65 | 2,573.81 |
| Brahman | 4.8% | 49.6% | 67.0× | 1,585 | 23.26 | 2,678.77 |
Total cov: total F1 sequencing coverage relative to the haploid genome size. Haplotype: F1 parental haplotype. Haplotype k-mers: fraction of diploid genome k-mers specific to this haplotype (16-mers for A. thaliana and 21-mers for H. sapiens and B. taurus). Assigned bases: percent of all sequencing read bases assigned to this haplotype. Coverage: depth of coverage for all reads assigned to this haplotype. Haplotig NG50: half of the haplotype is contained in haplotigs of this size or larger (based on haploid genome size estimate for each species).
Read classification accuracy depends not only on the zygosity of the offspring, but also on sequencing read length and error rate (Figure 2). Due to the high error rates in current long-read technologies, the k-mer size is also important. It must be long enough to be unique in the genome but short enough that it will not be corrupted by sequencing errors (e.g. k≈21 for a 3 Gbp genome). Given current long-read sequencing characteristics (read N50 >15 kb and read accuracy >85%), it is possible to bin and assemble nearly all of a human genome. A small fraction of reads will remain unclassified, but in the three datasets analyzed here, these reads were typically short and derived from either homozygous alleles or identically heterozygous alleles (i.e. both parents share the same heterozygous genotype). The former reads, being homozygous, can be co-assembled with both haplotype bins, while the latter are a limitation of trios and would require additional linkage information to be assigned correctly. However, current read lengths typically exceed the size of such alleles and unclassifiable reads are rare in practice.
Figure 2. Effect of data characteristics on trio binning.
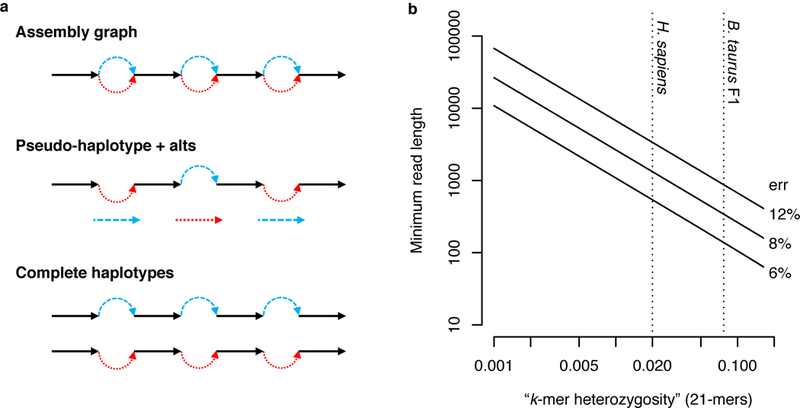
a) Diploid assembly representations shown with homozygous alleles in black and heterozygous alleles (called “bubbles”) colored by haplotype. Graphical representations typically collapse homozygous alleles into a single sequence. A pseudo-haplotype is a path through the diploid graph that separates heterozygous alleles but does not preserve phase between loci. Complete haplotypes represent all alleles and preserve phase across the entire genome. Ability to assign sequencing reads to a haplotype depends on the zygosity of the genome, the sequencing read length, and the sequencing error rate. b) Log-log plot of minimum required read length (y-axis) such that there is a 99% probability of observing at least one haplotype-specific 21-mer per read (negative binomial distribution, Methods), dependent on the sequencing error rate (labels) and fraction of haplotype-specific 21-mers in the genome (x-axis). Dotted vertical lines mark the fraction of heterozygous 21-mers for H. sapiens and the B. taurus F1 cross.
Validation on an Arabidopsis cross
The published description of FALCON-Unzip provided a valuable dataset for benchmarking diploid assembly algorithms 20. The authors crossed two well-characterized strains of Arabidopsis thaliana, Col-0 and Cvi-0, and generated both long-read PacBio and short-read Illumina sequencing reads for the F1 hybrid. Because the parental strains are both highly inbred, recombination is inconsequential and the F1 haplotypes are expected to match the parental genomes, providing a truth set for validation. No short-read data was available for the parental lines, so we inferred haplotype-specific k-mers directly from the assemblies. The heterozygosity was estimated to be 1.36% 30, or one variant every 73 bases, representing a best-case scenario for diploid assembly.
TrioCanu successfully classified the A. thaliana F1 reads by haplotype, resulting in unimodal k-mer distributions for the read bins, and an assembly that fully resolved both parental haplotypes (Figure 3, Supplementary Note and Supplementary Fig. 1). In contrast, rather than reporting complete haplotypes, FALCON-Unzip produces pseudo-haplotypes (“primary contigs”) along with a set of alternate alleles (“associated haplotigs”) that represent haplotype variants. Thus, homozygous alleles are only represented once and there can be considerable haplotype switching within the pseudo-haplotypes. To compare these results to the TrioCanu haplotigs, the FALCON-Unzip primary contigs and associated haplotigs were aligned to the parental genomes to infer the correct mapping between assembly and haplotype.
Figure 3. Read and assembly k-mer statistics for an Arabidopsis thaliana F1 hybrid.
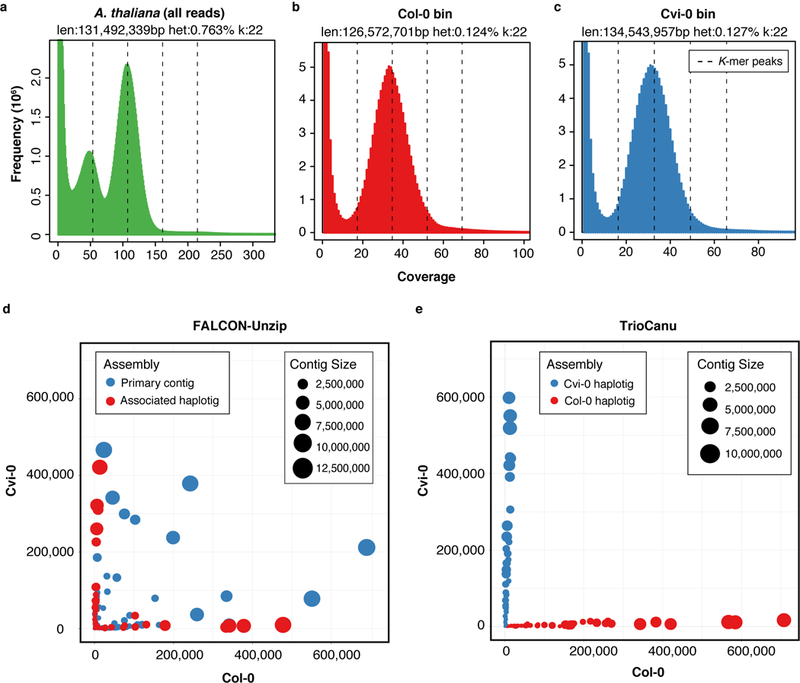
a) GenomeScope 30 k-mer count distributions for the F1 PacBio data corrected by Canu, and partitioned by haplotype and corrected by TrioCanu for the b) Col-0 and c) Cvi-0 haplotypes. GenomeScope reports an estimated genome size and SNP heterozygosity based on a model fit to the histogram. The dashed lines show k-mer peaks identified by GenomeScope, from left to right they are the 1-copy (heterozygous), 2-copy (homozygous), 3-copy, and 4-copy (repeats). The k-mer distribution for all reads shows two clear peaks, characteristic of a diploid read set. In comparison, the binned PacBio data shows a normal k-mer count distribution, characteristic of a haploid read set. d) Counts of Col-0 (x-axis) and Cvi-0 (y-axis) haplotype-specific k-mers in FALCON-Unzip and e) TrioCanu contigs (colored circles). FALCON-Unzip primary contigs switch between haplotypes, resulting in a mix of k-mers from both parents, whereas the FALCON-Unzip associated haplotigs are smaller but preserve local phase information. In comparison, TrioCanu haplotigs contain sequence from only a single haplotype and are automatically sorted into two complete haplotypes.
The TrioCanu haplotypes covered 99.50% and 99.00% of the Col-0 and Cvi-0 parental genomes, respectively, and the alignment identity for both was 99.97%. This exceeds the FALCON-Unzip result of 98.47% and 98.53% coverage and 99.94% and 99.92% identity. The NG50 size of the TrioCanu F1 haplotigs was 7.0 Mbp and 5.6 Mbp for the Col-0 and Cvi-0 haplotypes, respectively, compared to 7.4 Mbp and 6.1 Mbp for the inbred parental genomes (NG50 such that 50% of the haploid genome is contained in haplotigs of this size or greater). The NG50 for each haplotype of the diploid TrioCanu assembly was far better than a Canu assembly of the combined haplotypes (0.6 Mbp) and of comparable quality to the haploid parental genome assemblies (Supplementary Table 2). In comparison, the NG50 of the FALCON-Unzip pseudo-haplotype was slightly longer (8.0 Mbp) but contained substantial haplotype switching (Figure 3d). Compared to either TrioCanu haplotype, the FALCON-Unzip pseudo-haplotype also contained fourfold more duplicated BUSCO genes 31 and the associated haplotigs were missing fourfold more BUSCO genes (Supplementary Table 2), suggesting that some alternate alleles were incorrectly included in the FALCON-Unzip primary contig set.
While convenient for some applications, pseudo-haplotypes must be split prior to reconstructing complete haplotypes with the assistance of additional linkage information. This is a difficult and error-prone process that will propagate any errors in the pseudo-haplotype (e.g. collapsed heterozygosity or erroneous duplications). In contrast, TrioCanu haplotigs are inherently partitioned by haplotype and, if desired, can be recombined into a pseudo-haplotype after assembly for greater contiguity. This was simulated by aligning the assemblies of both F1 haplotypes to the A. thaliana Col-0 reference and merging alternative haplotigs found to overlap on the reference by >10 kbp, which effectively defines alignment paths through the diploid genome graph. Since this process is alignment based, the corresponding pseudo-haplotype NGA50 metric is useful for measuring both contiguity and structural accuracy of the assembly (NGA50 such that 50% of the reference genome is covered by continuous graph alignments of this size or greater 32). By this measure, the TrioCanu assembly scored better than FALCON-Unzip (NGA50 7.1 Mbp vs. 6.7 Mbp), illustrating that the larger FALCON-Unzip contigs are often due to the fact that it reports a pseudo-haplotype rather than completely resolved haplotypes (Supplementary Table 3).
Accurate structural variant detection from complete haplotypes simply requires a whole-genome alignment of the two. To demonstrate this approach and the accuracy of the TrioCanu haplotypes, we used Nucmer 33 and Assemblytics 34 to identify a total of 4,828 structural variants (SVs) between the Col-0 reference genome (TAIR10 35) and a de novo assembly of the Cvi-0 genome 20. Assemblytics classifies SVs in the range ≥50 bp and <10 kbp. These SVs were then compared to those identified by aligning the TrioCanu and FALCON-Unzip F1 assemblies against the Col-0 reference genome. Positive predictive value (PPV) and sensitivity for the TrioCanu Cvi-0 haplotype was 99.1% and 99.2%, respectively, compared to 96.99% and 98.81% for the FALCON-Unzip assembly (primary contigs plus associated haplotigs). However, the FALCON-Unzip PPV is artificially high in this case because variants are only being discovered on one haplotype (i.e. the other haplotype matches the reference and induces no variants). As expected, the TrioCanu F1 Col-0 haplotype showed good agreement with the Col-0 reference genome, differing on average by less than 2 variants per 10,000 bases and 108 SVs, which could represent errors in the assembly, errors in the reference, or true intra-strain variation.
A personal, diploid human genome
We next evaluated trio binning on a human trio of European descent (father: NA12891, mother: NA12892, and daughter: NA12878 25), and compared against Supernova 10x Genomics (linked reads) 21 and FALCON-Unzip (PacBio) assemblies of NA12878. Due to historical population bottlenecks, human genomes typically have a heterozygosity of ~0.1%, which was confirmed for NA12878 via k-mer analysis (Supplementary Fig. 2). This presents a challenge for haplotype recovery, because heterozygous variants are sparse (1 per 1,000 bases on average) and long-range linking information is required to preserve phase. A trio-based approach overcomes this problem because variants can be associated with the parent from which they were inherited, preserving phase across the entire genome.
A TrioCanu assembly of NA12878 from 72× PacBio coverage produced a haplotig NG50 of 1.2 Mbp and separate 2.7 Gbp assemblies for each parental haplotype (Table 1 and Supplementary Note). By comparison, the Supernova assembly from 41× linked-read coverage had a smaller contig NG50 of 103 kbp and phase block NG50 of 4.2 Mbp. The FALCON-Unzip pseudo-haplotype had a larger contig NG50 of 8.7 Mbp but a shorter phase block NG50 of 0.4 Mbp. TrioCanu and FALCON-Unzip pseudo-haplotype NGA50 sizes were 3.0 Mbp and 4.2 Mbp, respectively (Supplementary Table 3). Because TrioCanu generates complete haplotypes, the entire genome is in phase and all haplotigs are assigned to the parent from which they were inherited (Supplementary Fig. 3). For example, the TrioCanu paternal haplotype correctly assembled a known CYP2C19 substitution 27. Comparing the two TrioCanu NA12878 haplotypes using Assemblytics yielded 6,674 structural variants affecting 3.4 Mbp of the genome, including 12 inversions with an average size of 19 kbp. The alignment included 2.67 million single-nucleotide substitutions, matching the expected heterozygosity. Insertions and deletions (indels) between the haplotypes were well balanced, with an enrichment for 300 bp and 6 kbp events, corresponding to human Alu and LINE elements, respectively (Figure 4a).
Figure 4. Haplotype variation in a diploid human genome.
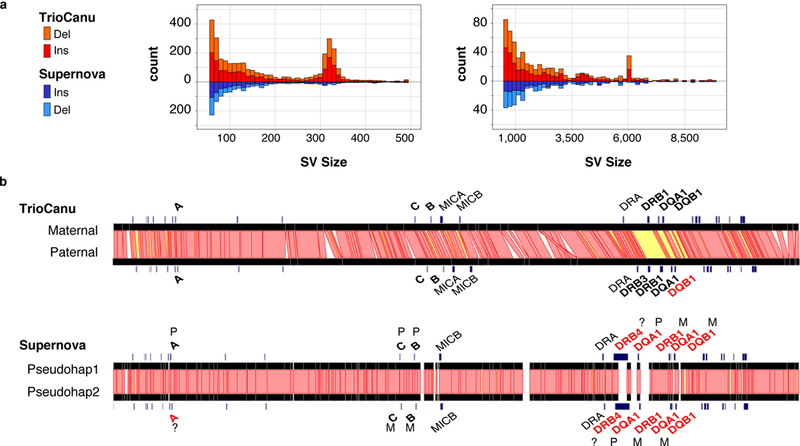
a) Counts of structural variants between NA12878 haplotypes across the entire genome as reported by Assemblytics 34. Canu haplotypes (top, red) showed a balance of insertions and deletions, with peaks at ~300 bp and ~6 kbp corresponding to human Alu and LINE elements, respectively. In comparison, the Supernova pseudo-haplotypes (bottom, blue) were missing these larger structural variants. b) Ribbon visualization 56 of MHC haplotypes for human reference sample NA12878 as assembled by TrioCanu from PacBio data (top) and Supernova from 10X Genomics data (bottom). Red bands indicate >95% identity between haplotypes; yellow bands <95% identity; and unaligned in white (gaps and indels). Genes are annotated in black if matching the known truth without error. TrioCanu captured more haplotype variation than Supernova, especially in the highly variable MHC class II region, which contains a long stretch of high sequence divergence (yellow). In addition to phasing the entire region, TrioCanu perfectly reconstructed all typed MHC genes on both haplotypes, with the exception of the paternal DQB1, which contained a single base indel (Supplementary Table 4). Supernova produced an overly homozygous reconstruction that incorrectly assembled a majority of genes and introduced false gene duplications (Supplementary Table 5). FALCON-Unzip correctly reconstructed the MHC genes but with a higher edit distance than TrioCanu (Supplementary Table 6). Canu (without binning) correctly reconstructed the more heterozygous class II genes but collapsed the class I genes (Supplementary Table 7).
To measure accuracy, we compared individual SNPs extracted from the TrioCanu and Supernova assemblies against a gold standard variant call set for NA12878 27. Considering only genomic positions covered in both assemblies, sensitivity of TrioCanu was 91.2% versus 90.9% for Supernova and the PPV was 90.2% versus 93.4%. FALCON-Unzip had lower sensitivity (73.13%) and PPV (70.26%) due to incomplete phasing (e.g. the associated haplotigs summed to only 65% of the primary assembly length). The slightly lower TrioCanu PPV versus Supernova is likely due to residual consensus errors in the PacBio assembly. A k-mer analysis also showed that the TrioCanu assembly is missing some homozygous alleles due to assembly gaps and/or sequencing errors (Supplementary Fig. 4). A higher coverage of long reads, so that each haplotype approaches 50× coverage, could be expected to reduce both consensus errors and missing alleles. Despite the 10x Genomics assembly having longer input fragments than PacBio (mean 51 kbp vs. 12 kbp), the NG50 perfect phase block for Supernova was 4.3 Mbp versus 5.6 Mbp for TrioCanu. The few TrioCanu phase errors originate from regions where both parents have identical heterozygous genotypes, which cannot be resolved by the trio method alone without longer read lengths or additional linkage information (Supplementary Note).
The TrioCanu assembly was more structurally accurate than the Supernova assembly. In particular, Supernova missed many larger variants, and assembled fewer Alu and LINE indels relative to TrioCanu (Figure 4a). To better understand the structural accuracy of these assemblies, we examined the Major Histocompatibility Complex (MHC), which is a highly repetitive and heterozygous region of the genome that presents a serious challenge for de novo assembly. This region contains the human leukocyte antigen (HLA) genes, which have been well characterized for NA12878 36 and serve as a quality check. Supernova did not accurately assemble either MHC haplotype, failed to capture an HLA-DRB3 gene insertion in the paternal haplotype, and incorrectly reported the majority of the MHC class II region as homozygous (Figure 4b). By comparison, TrioCanu correctly assembled both MHC haplotypes, as demonstrated by perfect HLA typing results and only a single base error in the typing genes. FALCON-Unzip also correctly assembled both MHC haplotypes, but with an additional three errors in the typing genes (Supplementary Tables 4–7 and Supplementary Note).
Reference assembly of two cattle breeds using an F1 hybrid
Using trio binning, we sought to generate high-quality, breed-specific reference genomes for Angus and Brahman cattle (examples of the Bos taurus taurus and Bos taurus indicus subspecies, respectively). We collected ~60× Illumina coverage each for an Angus bull and a Brahman cow, and 134× PacBio coverage in reads ≥1 kbp for their male F1 offspring. Heterozygosity of the F1 was estimated to be 0.9% (Supplementary Fig. 5). 98.9% of all F1 bases were assigned to a parental haplotype. Unassigned reads were short and not enriched for any particular region of the genome. A separate assembly of these unassigned reads resulted in no contigs over a few thousand base pairs, suggesting that all regions of the genome were successfully partitioned by haplotype.
TrioCanu successfully resolved both F1 haplotypes with a haplotig NG50 exceeding 20 Mbp for each (NG50: Angus 26.6 Mbp, Brahman 23.3 Mbp) (Supplementary Note). This far surpasses previous B. taurus taurus 37 and B. taurus indicus 38 reference genomes, both of which have contig NG50s <100 kbp. The TrioCanu haplotype assemblies were also more contiguous than a Canu assembly of the unbinned data due to heterozygous branching in the assembly graph (NG50: 15.6 Mbp). A FALCON-Unzip assembly of the combined data achieved an impressive NG50 of 31.4 Mbp for its pseudo-haplotype, but with substantial switch error (Supplementary Fig. 6) and a pseudo-haplotype NGA50 similar to TrioCanu (4.19 Mbp vs. 4.20 Mbp, Supplementary Table 3). Further analysis of k-mer distributions in the FALCON-Unzip assembly show less complete haplotype separation, with more homozygous k-mers (expected to be 2-copies) occurring either as 1-copy (over-collapsed) or >2-copy (over-split) (Figure 5ab, Supplementary Fig. 7). For example, FALCON-Unzip over-collapsed roughly twice as many k-mers as TrioCanu (Supplementary Table 8). Each TrioCanu haplotype was polished using only the haplotype-assigned PacBio reads, and the quality of the final assembly was estimated to be QV47 (accuracy 99.998%, Supplementary Note), supporting our contention that higher coverage can overcome the limitations of PPV and missing homozygous alleles observed for the lower-coverage human sample. In addition, polishing only the Brahman haplotype using reads from both haplotypes increased the total number of errors more than twofold, despite the increased coverage, due to artifacts introduced by the Angus haplotype. This highlights the advantage of binning reads by haplotype for accurate consensus generation (Supplementary Note).
Figure 5. Diploid assembly of a Bos taurus F1 hybrid.
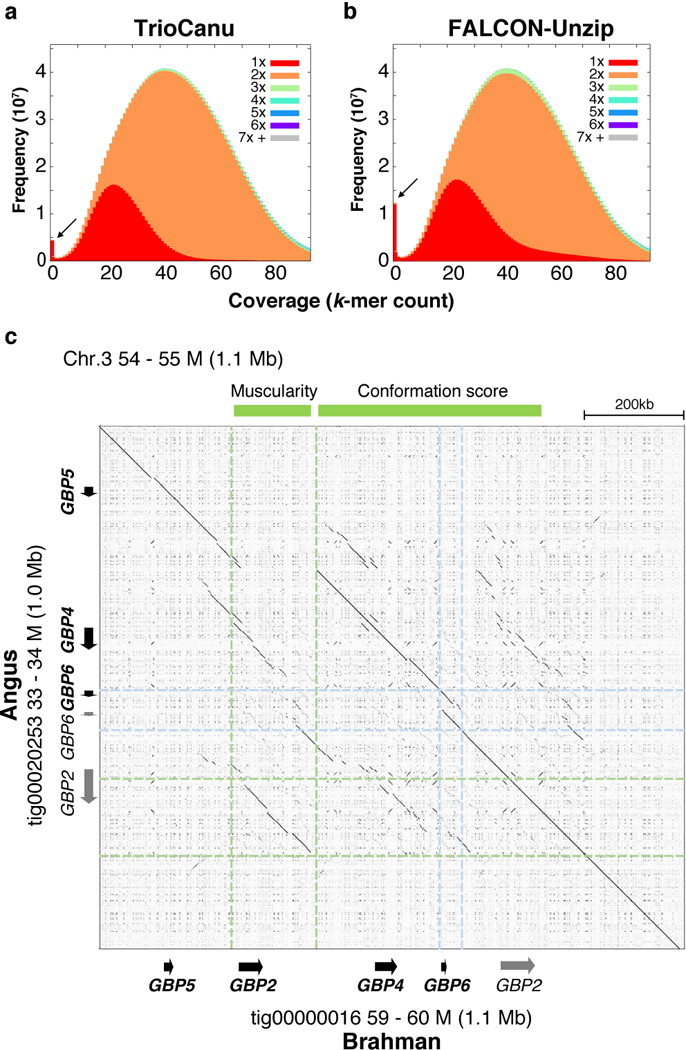
Stacked k-mer histograms from KAT 57 comparing a) TrioCanu and b) FALCON-Unzip k-mer counts to an independent Illumina dataset of the same individual. The x-axis bins are k-mer coverage in the Illumina dataset, and the y-axis is the frequency of those k-mers in the Illumina set colored by copy number in the assembly. The FALCON-Unzip distribution has more k-mers that do not appear in the Illumina data (arrows), a longer tail of 1-copy k-mers (red, collapsed haplotype), and slightly more 3-copy k-mers (green, duplicated haplotype). c) Alignment dotplot of the TrioCanu Angus and Brahman haplotypes in a highly heterozygous region containing multiple guanylate binding protein (GBP) genes. Relative to Brahman, the Angus haplotype is missing a ~140 kbp region containing GBP2, previously reported to be associated with muscularity (light green). The Angus haplotype also has a duplicated GBP6-like sequence (light blue) in a region associated with conformation score (genes marked in grey are highly divergent from known transcripts). The FALCON-Unzip assembly confirms the TrioCanu structure but is split into five primary contigs and four associated haplotigs of mixed origin (Supplementary Fig. 9).
The Angus and Brahman haplotypes aligned to one another with 99.35% identity and contained 25,245 haplotype-specific structural variants and 124 inversion breakpoints. Common SV sizes corresponded to known retrotransposon families in the Bos taurus lineage, including the three most common elements: tRNA-Core-RTE (214 bp), RTE-BovB (1,650 bp), and L1 (5,981 bp) (Supplementary Fig. 8). One of the most heterozygous regions between the two haplotypes contained notable copy number variations (CNVs) of GBP2~GBP6 (Figure 5c). Notably, the Angus haplotype has a large (~140 kb) deletion containing GBP2, while Brahman includes a complete version of GBP2 transcript variant X8. In addition, GBP6 is partially duplicated only on the Angus haplotype. The FALCON-Unzip assembly is structurally consistent with TrioCanu but breaks this region into 9 haplotype-mixed contigs (5 primary, 4 associated) rather than 2 complete haplotypes (Supplementary Fig. 9). These regions overlap with a previously reported association of quantitative traits for muscularity and visual conformation scores 39, suggesting our breed-specific haplotypes will be important for understanding growth traits in cattle.
BUSCO 31 reported 92.6% and 93.4% complete universal single-copy orthologs and a low rate of duplication (1.0% and 1.1%) for the TrioCanu Angus and Brahman haplotigs, respectively, which is consistent with 93.7% completeness and 1.3% duplication for the current B. taurus taurus Hereford UMD3.1.1 reference 37. As with the other genomes, FALCON-Unzip shows more duplicated and fewer complete BUSCO genes (Supplementary Table 2). To further measure accuracy of the assemblies, we aligned the probe sequence for 735,636 autosomal markers from Illumina’s BovineHD BeadChip to both haplotypes. Only 333 marker loci did not align to either of the TrioCanu haplotypes, and 2,726 and 3,718 were absent from Angus and Brahman, respectively. The 333 marker loci missing from both haplotypes also had low evidence in the parental Illumina data, suggesting that their absence is real in the parental genotypes and not due to incomplete assembly (Supplementary Fig. 10). The majority of marker sequences missing in one haplotype were also depleted in the corresponding parent’s short read data, but not the other parent, indicating these are haplotype-specific loci correctly phased by the assembly (Supplementary Figs. 11 and 12). Switch error between the haplotypes was roughly estimated at 0.68% using independent Hi-C data from the F1 (Supplementary Note and Supplementary Table 9).
The Angus and Brahman haplotype assemblies cover 94.2% and 96.2% of the UMD3.1.1 reference genome, respectively, with the Brahman dam haplotype containing the X chromosome and mitochondrial genome (Supplementary Figs. 13 and 14). Surprisingly, we identified 3,178 inversions shared by both haplotypes with respect to the reference (mean 9,447 bp, median 4,385 bp, Supplementary Table 10). Most of these inversions (94.5%) corresponded with reference scaffold gaps, and the inverted sequences were fully contained within TrioCanu haplotigs. To validate the TrioCanu reconstruction, we used NGM-LR and Sniffles 40 to identify structural variants in the combined F1 PacBio read set versus the UMD3.1.1 reference assembly and both TrioCanu haplotypes. This identified 3,354 inversions in the UMD3.1.1 assembly, versus just 11 and 20 the Angus and Brahman haplotigs, respectively. Thus, it appears the current cattle reference genome contains systematic inversion errors within its scaffolds. Sniffles also identified over 4-fold more SVs in the UMD3.1.1 assembly versus our haplotigs and 1.8-fold more deletions than insertions (Supplementary Table 10), which was also evident from the Assemblytics output (Supplementary Figs. 15 and 16), suggesting artefactual duplications in the UMD3.1.1 assembly. Comparison against a new long-read reference sequence ARS-UCDv1.0.11 (B. Rosen, personal communication) of the same Hereford animal used for UMD3.1.1, showed no apparent indel bias and returned 3-fold and 2-fold fewer variants versus the Angus and Brahman assemblies, respectively, further supporting error in the UMD3.1.1 assembly (Supplementary Figs. 17 and 18). A comparison between our Angus and Brahman haplotypes mirrored a comparison between ARS-UCDv1.0.11 and the Brahman haplotype. In contrast, the existing short-read B. taurus indicus genome contains few variants over 500 bp (Supplementary Fig. 19), and likely inherits assembly errors from UMD3.1.1 due to use of a reference-guided assembly approach 38 (Supplementary Fig. 20).
Discussion
Here we have demonstrated that trio binning facilitates complete haplotype assembly for heterozygous diploid genomes, including human. This strategy has several advantages over traditional approaches. First, trio binning recovers the true haplotypes of a viable organism. Both haplotypes of our diploid cattle assembly achieved >20 Mbp NG50 haplotig sizes, matching the best contiguities previously reported for homozygous human cell lines sequenced to similar PacBio coverage (e.g. CHM1 and CHM13 9). Trio binning is also applicable to organisms that have long generation times or are otherwise recalcitrant to inbreeding. Second, by isolating haplotype variation prior to assembly, the resulting assembly graphs are simplified. As a result, haplotype-specific assemblies can exceed the continuity of merged diploid assemblies. After assembly, the resulting haplotypes can be recombined to form a diploid genome graph or contiguous pseudo-haplotype. Third, our approach is able to accurately reconstruct structurally heterozygous alleles that can be important factors in adaptation and immunity (e.g. MHC genes) and have previously been linked to quantitative traits (e.g. GBP genes). We have shown that such sequences are often mis-assembled by alternative approaches, and the accurate representation of haplotypes is essential for studies of intraspecific variation, chromosome evolution, and allele-specific expression.
We evaluated trio binning on a variety of long-read PacBio coverages, ranging from 70× to 180×. Linked-read assemblies typically require 50× Illumina coverage but do not accurately assemble complex structural variants. Standard PacBio assemblies typically require 60× coverage but are less accurate for identifying small variants, and so are typically combined with Illumina data to maximize base accuracy. In contrast, trio binning accurately identifies both SNPs and SVs. We currently recommend a minimum of 40× PacBio coverage per haplotype to achieve accurate consensus sequences, plus an additional 30× Illumina coverage per parent to identify haplotype-specific k-mers. This parental Illumina data can also be used to verify and possibly polish the final assembly. For highly repetitive or less heterozygous genomes, additional long-read coverage may be required to maximize contiguity. Trio binning is compatible with any long-read sequencing technology, such as Oxford Nanopore 41, and the resulting assemblies will mirror the error characteristics of the chosen platform.
Because trio binning outputs two sets of haplotype-specific reads, it is compatible with any long-read assembler 20, 42, 43 and repeat separation technique 44 for assembling the individual haplotypes. Unlike graph-based assembly representations, which require a specialized bioinformatics toolchain, linear haplotypes can be easily analyzed with existing methods. For example, the partitioned read sets can be reused for haplotype-specific gap filling 45 and consensus polishing 46, 47, and we have shown that polishing with haplotype-specific reads achieves a more accurate consensus sequence. Given sufficient haplotype divergence and read lengths, nearly all reads are assigned to the correct haplotype. However, for genomes with lower heterozygosity, long homozygous alleles may receive lower coverage and quality due to a lack of assigned reads. In these cases, homozygous reads can be assigned to both haplotypes to boost coverage at the risk of masking some true variants. Additional processing after assembly could correct for this, for example, by mapping the parental short read data to identify missed variants and correct switch error. Alternatively, the accuracy of long-read binning could be improved by more sophisticated classification (e.g. using spaced k-mers 48) or the integration of additional data types (e.g. Hi-C). The latter option may allow partial haplotype binning without the use of a trio.
Long-read trio binning, as described here, is the first method able to assemble complete haplotypes from a heterozygous genome and has immediate applications to reference genome construction as well as human and agricultural genomics. New reference genomes will benefit from the improved assembly accuracy and continuity of this approach. For agricultural genomics, trio binning can be used to study breed diversity and has the advantage of producing two reference-quality haplotypes from a single individual. Our assembly of an outbred F1 resulting from a cross between Angus and Brahman cattle produced two breed-specific haplotypes that improve upon and correct the current best reference genomes for both subspecies. These haplotype-specific reference sequences provide an important resource for understanding genetic variation in cattle. The more general idea of haplotype binning should also work well for polyploid plant genomes (e.g. bread wheat) by utilizing species markers (rather than parental markers) to pre-partition reads by haplotype. For human genomics, our approach is a viable method for reconstructing complete, personalized haplotypes, and could be used to generate a more complete database of human haplotype variation.
Online Methods
Haplotype k-mer identification
TrioCanu automates k-mer counting, thresholding, and set operations to identify haplotype-specific k-mers. All k-mers are counted using Meryl, a sort-based k-mer counter used within Canu that allows linear-time k-mer set operations. First, a k-mer frequency distribution is obtained by counting k-mers in the parental genomes. This distribution is examined to eliminate k-mers likely to be erroneous (low copy) or from genomic repeats (high copy), leaving only k-mers from unique homozygous or heterozygous genome sequences 49. For k-mer coverage x and frequency y, the optimal low coverage threshold is determined by finding the first critical point and its corresponding coverage x0 and frequency y0. The same frequency cutoff y0 is used to determine the high coverage threshold x1. A k-mer set D for each haplotype is drawn from all haplotype k-mers. For two parental haplotypes i and j, haplotype-specific k-mer sets are then constructed as and . Classification k-mers with coverage are selected from and from .
Smaller k-mers are more likely to avoid sequencing error in the reads, so it is preferable to choose a small value for k. However, k must be large enough to minimize random k-mer collisions in the genome. For example, the total space of 16-mers is only 416 or 4.29 billion, close to the total number of k-mers in a 3 Gbp mammalian genome, increasing the chance that some k-mer may occur multiple times simply by chance (and not homology). Given a genome size G and tolerable collision rate p, an appropriate k can be computed as 50. According to this formula, we used k=16 for A. thaliana and k=21 for H. sapiens and B. taurus.
A. thaliana, H. sapiens, and B. taurus were assembled prior to TrioCanu automation, and haplotype-specific k-mer thresholds were identified manually as described in Supplementary Note. For A. thaliana, which was lacking Illumina data for the parents, k-mers were collected from assemblies of the parents, excluding repetitive k-mers occurring more than 10 times. For H. sapiens and B. taurus, haplotype-specific k-mers were collected from unassembled, short read sequencing of the parents. Low and high k-mer coverage thresholds were chosen manually as x0=30 and x1=160 for H. sapiens and x0=11 and x1=100 for B. taurus. Retrospective application of the automated thresholding method selected similar thresholds of {[25,143], [27,147]} and {[10,57], [10,67]} for H. sapiens and B. taurus, respectively.
Haplotype binning
Haplotype binning is a general strategy for partitioning a read set into haplotype groups prior to assembly. The number of haplotypes is not necessarily limited to two. Given N haplotypes, the goal is to identify haplotype-specific k-mers that are exclusive to one haplotype. Given a database of haplotype-specific k-mers, the number of specific k-mers from each haplotype is counted in each read. It is expected that k-mers in a single read will be from the same haplotype, but due to sequencing errors it is possible to observe spurious k-mers from a different haplotype. Therefore, the observed haplotype-specific k-mer counts are normalized by the database size to control for the different k-mer set sizes of the parents. Reads are then assigned to the haplotype with the most matching haplotype-specific k-mers. In the event of a tie or too few haplotype-specific k-mers, the read is marked as ambiguous. Finally, the N read bins are passed to Canu for assembly, with the option to include the ambiguous reads in all bins.
Whether a read can be correctly classified is a function of the k-mer heterozygosity h, read length l, read error rate e, and k-mer size k. For simplicity of modeling, errors and haplotype differences are assumed to be random point mutations, and heterozygosity h is defined as the fraction of genomic k-mers that are haplotype specific. It is assumed that k is large enough to avoid chance collisions. A read of length l contains k-mers. The probability of a single k-mer surviving uncorrupted is , and the expected number of uncorrupted k-mers in a read is . The expected number of haplotype-specific k-mers in a read is , and the number of surviving haplotype specific k-mers in a read is . Thus, for a typical long sequence read with e=0.12 and l=15,000, and k-mer heterozygosity h=0.001, the expected number of surviving haplotype-specific 16-mers is 2 and 21-mers is 1. Increasing divergence to h=0.01 increases the expected number of 16-mers to 19 and 21-mers to 10.
Validation
Classification accuracy was evaluated using a truth set of A. thaliana parental reads. The simple majority-wins classification heuristic showed a good sensitivity/specificity trade off, exceeding 80% true positive rate (TPR) with <20% false positive rate (FPR) (Supplementary Fig. 21). We further simulated increased heterozygosity within each parent to measure the effect on k-mer classification. Read classification is more difficult with increasing heterozygosity in the parents, and performance dropped to 74% TPR with <28% FPR when parental heterozygosity was increased to 2% (Supplementary Fig. 21). False positives include homozygous reads which do not affect the resulting assembly, and a small fraction of mis-classified heterozygous reads. These will be outvoted by the majority of correctly classified reads when building the haplotype consensus. If high specificity is required, the classifier can be tuned to require more than a simple majority of haplotype-specific k-mers.
Assembly alignments were performed with MUMmer 3.23 33 with the commands
nucmer -maxmatch -l 100 -c 500 ref.fa asm.fa dnadiff -d out.delta
GRCh38 51 excluding ALT loci was used for H. sapiens. TAIR10 was used for A. thaliana. GCF_000003055.6 with chromosome Y from NC_016145.1 was used for B. taurus and AGFL00000000.1 for B. indicus. A genome size of 119,667,750 was used for A. thaliana (TAIR 10 length), 3,098,794,149 for H. sapiens (GRCh38 primary assembly excluding alternates), and 2,713,423,491 for B. taurus (the UMD 3.1.1 reference plus the Y chromosome).
NGA50 statistics for individual assemblies were computed using MUMmer’s dnadiff tool. One-to-one alignment intervals for the contigs versus the reference (1coords output) were filtered to only include those intervals >10 kbp and ≥97% identity. To ignore small structural variants versus the reference, same-strand alignments within 2000 bp of each other were merged. For TrioCanu and FALCON-Unzip assemblies, this process was repeated for the combined assemblies (all haplotigs from both haplotypes) to compute a pseudo-haplotype NGA50. In this case, same-strand alignments between alterative haplotigs that overlapped by more than 10 kbp on the reference were merged to represent a path through the diploid genome graph.
Parent-specific k-mers were used to estimate switch error within assembly contigs. MHC typing was run as previously described 41 with the truth set from Dilthey et al. 52. B. taurus markers used in BovineHD BeadChip (Illumina Inc., San Diego, CA) were used to identify missing regions in the assemblies as well as haplotype-specific sequences. Illumina data was used to estimate QV by mapping with BWA-MEM 53 and identifying variants with Freebayes 54. Repeats in the Bos taurus genome were downloaded from the UCSC genome browser 55 (Supplementary Note).
Sample preparation and sequencing of the cattle trio
The animals used were part of the Davies Epigenetics and Genetics Resource at the University of Adelaide, Australia, and were established and sampled using procedures approved by the animal ethics committee of the University. A two-year-old cow of the Brahman breed (subspecies Bos taurus indicus) was bred by artificial insemination using semen from a five-year-old bull of the Angus breed (Bos taurus taurus). The Brahman female had been previously typed for mitochondrial DNA haplotype to verify the maternal lineage as indicus-specific. At day 153 post-insemination, the animal was sacrificed and the fetus removed for dissection. The fetal lung was removed immediately into liquid nitrogen, and DNA was extracted using a salting out procedure. Briefly, approximately 100 mg of tissue was ground under liquid nitrogen to a powder and transferred to a tube containing 2.26 mL of nuclei lysis mixture (2 mL buffer NFB composed of 10 mM Tris-HCL pH 8.0, 0.4 M NaCl, 2 mM EDTA, plus 0.2 mL 10% SDS, plus 0.06 mL 10 mg/mL RNase A). Tissue and solution were mixed by inversion for 2 minutes, then set to shake slowly at 37°C 1 hour. Protein digestion was performed by adding 0.025 mL Proteinase K (20 mg/mL) and returning to the shaker overnight (approximately 16 hours). Protein was removed by addition of 1.25 mL of saturated NaCl, followed by vigorous hand shaking for 15 seconds and centrifugation 2250 x g, 20 minutes, 4 °C. The clarified supernatant was transferred to a tube containing 8 mL of cold 100% ethanol, and DNA was precipitated by gentle rocking of the solution. The DNA was transferred using a glass rod and washed twice in tubes containing 5 mL of 70% ethanol. The pellet was then transferred to a 1.5 mL tube and air dried for 10 minutes at room temperature. DNA was removed from the glass rod by dissolving in 0.25 mL of solution containing 10 mM TrisHCl pH 8.0 and 0.1 mM EDTA overnight at 4 °C. Parental DNA samples were extracted using standard phenol-chloroform based procedures.
Sequence libraries for the parents and the fetus were prepared with TruSeq PCR-free preparation kits as directed by the manufacturer (Illumina, San Diego, CA). The three libraries were sequenced in separate runs, with no other libraries present in the flow cell, on a NextSeq500 instrument using 2×150 paired end reads with High Output Kit v2 chemistry. The libraries employed unique indexes and, despite being in separate runs, only reads with appropriate indexes for the library were used for analysis to prevent any cross-contamination between the sire, dam, or fetal library data.
Libraries for SMRT sequencing were constructed as recommended by the manufacturer (Procedure P/N 100–286-000–07, Pacific Biosciences, Menlo Park, CA), using a 15 kb cutoff for size selection on the BluePippin instrument (Sage Science, Beverly, MA). A total of 12 library preparations were used, nine of which were sequenced using P6/C4 chemistry on an RSII instrument (Pacific Biosciences, Menlo Park, CA) which generated approximately 152 Gb of sequence, and the other three libraries were sequenced on a Sequel instrument which generated another 205 Gb.
Supplementary Material
Acknowledgements
We thank William Thompson, Kristen Kuhn, Kelsey McClure, and Robert Lee for technical assistance, and Tina Graves-Lindsay and Washington University in St. Louis for public release of the PacBio NA12878 data. SK, AR, BPW, and AMP were supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health. SH and JLW are funded from the JS Davies bequest to the University of Adelaide. TPLS was supported by USDA-ARS Project 3040–31000-100–00D. DMB was supported by USDA-ARS Project 5090–31000-026–00-D. This research was also supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI17C2098). This work utilized the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov). Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement.
Footnotes
Competing financial interests
SBK is a current employee of Pacific Biosciences. All other authors declare no competing interests.
Data availability
Sequencing data for the cattle trio is available under NCBI BioProject PRJNA432857. All other sequencing data was obtained from public sources. Data accessions, software versions, and commands used to produce the described results are provided in Supplementary Note. Assembly files, accession numbers, and other miscellaneous information can be found at https://gembox.cbcb.umd.edu/triobinning/index.html.
Code availability
Prototype code used to build k-mer sets, subtract parental k-mers, and classify reads is available at https://github.com/skoren/triobinningScripts and as Supplementary Code. TrioCanu is implemented as a module of Canu v1.7 and freely available at https://github.com/marbl/canu
References
- 1.Phillippy AM New advances in sequence assembly. Genome Res 27, xi–xiii (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koren S et al. Reducing assembly complexity of microbial genomes with single-molecule sequencing. Genome Biol 14, R101 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Korlach J et al. De novo PacBio long-read and phased avian genome assemblies correct and add to reference genes generated with intermediate and short reads. Gigascience 6, 1–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Myers EW et al. A whole-genome assembly of Drosophila. Science 287, 2196–2204 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Mouse Genome Sequencing Consortium et al. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002). [DOI] [PubMed] [Google Scholar]
- 6.International Human Genome Sequencing Consortium et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Cao H et al. De novo assembly of a haplotype-resolved human genome. Nat Biotechnol 33, 617–622 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Steinberg KM et al. Single haplotype assembly of the human genome from a hydatidiform mole. Genome research 24, 2066–2076 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider VA et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome research 27 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaisson MJ et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517, 608–611 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Batzoglou S et al. ARACHNE: a whole-genome shotgun assembler. Genome Res 12, 177–189 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vinson JP et al. Assembly of polymorphic genomes: algorithms and application to Ciona savignyi. Genome Res 15, 1127–1135 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy S et al. The diploid genome sequence of an individual human. PLoS Biol 5, e254 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patterson M et al. WhatsHap: Weighted Haplotype Assembly for Future-Generation Sequencing Reads. J Comput Biol 22, 498–509 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Edge P, Bafna V & Bansal V HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res 27, 801–812 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larkin DM et al. Whole-genome resequencing of two elite sires for the detection of haplotypes under selection in dairy cattle. Proc Natl Acad Sci U S A 109, 7693–7698 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang H, Chen X & Wong WH Completely phased genome sequencing through chromosome sorting. Proc Natl Acad Sci U S A 108, 12–17 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falconer E & Lansdorp PM Strand-seq: a unifying tool for studies of chromosome segregation. Semin Cell Dev Biol 24, 643–652 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Selvaraj S, J, R.D., Bansal, V. & Ren, B. Whole-genome haplotype reconstruction using proximity-ligation and shotgun sequencing. Nat Biotechnol 31, 1111–1118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chin CS et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods 13, 1050–1054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weisenfeld NI, Kumar V, Shah P, Church DM & Jaffe DB Direct determination of diploid genome sequences. Genome Res 27, 757–767 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seo JS et al. De novo assembly and phasing of a Korean human genome. Nature 538, 243–247 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Porubsky D et al. Dense and accurate whole-chromosome haplotyping of individual genomes. Nat Commun 8, 1293 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matthews BJ et al. Improved Aedes aegypti mosquito reference genome assembly enables biological discovery and vector control. bioRxiv (2017). [Google Scholar]
- 25.International HapMap Consortium. The International HapMap Project. Nature 426, 789–796 (2003). [DOI] [PubMed] [Google Scholar]
- 26.The 1000 Genomes Project Consortium et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eberle MA et al. A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res 27, 157–164 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malinsky M, Simpson JT & Durbin R trio-sga: facilitating de novo assembly of highly heterozygous genomes with parent-child trios. bioRxiv (2016). [Google Scholar]
- 29.Koren S et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vurture GW et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waterhouse RM et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salzberg SL et al. GAGE: A critical evaluation of genome assemblies and assembly algorithms. Genome research 22, 557–567 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurtz S et al. Versatile and open software for comparing large genomes. Genome Biol 5, R12–R12 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nattestad M & Schatz MC Assemblytics: a web analytics tool for the detection of variants from an assembly. Bioinformatics 32, 3021–3023 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamesch P et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res 40, D1202–1210 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dilthey AT et al. High-Accuracy HLA Type Inference from Whole-Genome Sequencing Data Using Population Reference Graphs. PLoS Comput Biol 12, e1005151 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zimin AV et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol 10, R42 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Canavez FC et al. Genome sequence and assembly of Bos indicus. The Journal of heredity 103, 342–348 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Zhou Y et al. Genome-wide CNV analysis reveals variants associated with growth traits in Bos indicus. BMC Genomics 17, 419 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sedlazeck FJ et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 15, 461–468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jain M et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kolmogorov M, Yuan J, Lin Y & Pevzner P Assembly of Long Error-Prone Reads Using Repeat Graphs. bioRxiv, 247148 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Chaisson MJ, Mukherjee S, Kannan S & Eichler EE in International Conference on Research in Computational Molecular Biology 117–133 (Springer, 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.English AC et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS One 7, e47768 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chin CS et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10, 563–569 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Loman NJ, Quick J & Simpson JT A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods 12, 733–735 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Ma B, Tromp J & Li M PatternHunter: faster and more sensitive homology search. Bioinformatics 18, 440–445 (2002). [DOI] [PubMed] [Google Scholar]
References for Online Methods
- 49.Kajitani R et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res 24, 1384–1395 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fofanov Y et al. How independent are the appearances of n-mers in different genomes? Bioinformatics 20, 2421–2428 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Schneider VA et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 27, 849–864 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dilthey A, Cox C, Iqbal Z, Nelson MR & McVean G Improved genome inference in the MHC using a population reference graph. Nat Genet 47, 682–688 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.399 7 (2013). [Google Scholar]
- 54.Garrison E & Marth G Haplotype-based variant detection from short-read sequencing. arXiv preprint arXiv:1207.3907 (2012). [Google Scholar]
- 55.Casper J et al. The UCSC Genome Browser database: 2018 update. Nucleic Acids Res 46, D762–D769 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nattestad M, Chin C-S & Schatz MC Ribbon: Visualizing complex genome alignments and structural variation. bioRxiv (2016). [Google Scholar]
- 57.Mapleson D, Garcia Accinelli G, Kettleborough G, Wright J & Clavijo BJ KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 33, 574–576 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.